

Abstract
Privileged structures inspire compound library design in medicinal chemistry. We performed a comprehensive analysis of 1.4 million bioactive compounds, with the aim of assessing the prevalence of certain molecular frameworks. We used the Shannon entropy formalism to quantify the promiscuity of the most frequently observed atom scaffolds across the annotated target families. This analysis revealed an apparent inverse relationship between hydrogen‐bond‐acceptor count of a scaffold and its potential promiscuity. The results further suggest that chemically easily accessible scaffolds can serve as templates for the generation of bespoke compound libraries with differing degrees of multiple target engagement, and heterocyclic, sp3‐rich frameworks are particularly suited for target‐focused library design. The outcome of our study enables us to place some of the many narratives surrounding the concept of privileged structures into a critical context.
Keywords: cheminformatics, combinatorial chemistry, medicinal chemistry, polypharmacology, Shannon entropy
In 1988, Evans et al. observed that “[…]certain ‘privileged structures’ are capable of providing useful ligands for more than one receptor and that judicious modification of such structures could be a viable alternative in the search for new receptor agonists and antagonists.”1 Generally speaking, a privileged structure may be considered to possess geometries suitable for decoration with side chains, such that the resulting products bind to different target proteins. Herein, we refer to such molecular frameworks as privileged scaffolds, to avoid confusion with other terminologies. In 2002, we introduced the related concept of frequent hitters2 for compounds that generate readouts in multiple activity assays.3, 4, 5 Some of these compounds are undesired false positives.6 Others, however, are truly promiscuous ligands that potently, specifically, reversibly, but not selectively, bind to members of different macromolecular target families (Figure 1).
Figure 1.

The concepts of frequent hitters and privileged structures.
Privileged scaffolds may be considered desirable for the bespoke design of compound screening libraries.7 It is important to realize that screening hits from a privileged‐scaffold library do not necessarily have to be promiscuous with regard to their target; they can, in fact, represent useful starting points for hit‐to‐lead expansion, with a well‐defined mode of action and selective target engagement. In 2010, Welsch et al. published a broad list of privileged scaffolds compiled from medicinal chemistry literature.8 Popular examples of molecular frameworks considered promiscuous are indoles, quinolines, coumarines, isoxazolidines, benzimidazoles, thiazolopyrimidines, and arylaminopyrazole.9 Evidently, the scaffold can contribute to bioactivity directly, via shape or binding interactions, or indirectly, via functionalization potential.10 Often, the designated privileged scaffolds are inspired by, or derived from, natural products.11
In this present study, we have computationally analyzed the scaffold promiscuity of the ChEMBL2212 compound database. The hypothetical idealized privileged structure, according to Evans’ definition, would be found in a ligand that potently interacts with one (selective binder) or many target receptors (promiscuous binder), and with a collection of such compounds containing the privileged structure being able to address all target families. We quantified the ability of a scaffold to interact with members from different target families in terms of its Shannon entropy Η [Eq. (1)].13a This value corresponds to the divergence between the distribution of the reported activities from the idealized equal distribution across all target families. The Shannon entropy was computed based on the set of compounds containing a certain atom scaffold. It may be considered a set property of the common scaffold.
| (1) |
where p i is the observed fraction of actives for target family i. Information I may be defined as the difference between the maximal and the actual entropy [Eq. (2)].13b Accordingly, high values of I designate target‐selective scaffolds.
| (2) |
We additionally computed the relative entropy to account for the unequal target activity distribution in ChEMBL22 [Kullback–Leibler divergence, KLD; Eq. (3)].13c
| (3) |
where q corresponds to the background (prior) distribution of actives for the different target families considered.
In this present study, ten target families according to the IUPHAR definition14 with disparate, non‐overlapping targets were considered (GPCR, enzyme, kinase, proteinase, nuclear receptor, catalytic receptor, ion channel, transporter, protein, unidentified). The Shannon entropy value for a scaffold is maximal for the equal distribution of activity annotations across the ten target families (Η max=log2(10)=3.32 bit). Accordingly, the perfectly target‐promiscuous compound library would assume this value, while the ideal target‐family‐selective library would be designated by Η=0 bit and I=3.32 bit, respectively.
Database analysis was performed using KNIME15 workflows with RDkit16 functions. ChEMBL22 contains 1 397 535 compounds with standardized activity annotations (K d/i/b and EC/IC50 values), and a total of 181 888 scaffolds (atom frameworks, “Murcko” scaffolds17). By definition, such a scaffold represents the union of all rings and their connecting atoms in a given molecular graph. These atom frameworks do not include side chains. To reduce the risk of artefacts, we exclusively focused on the most potent compounds (pActivity≥6, 677 044 compounds) and the most abundant scaffolds with at least 100 compound samples each (585 scaffolds).
The most frequent scaffolds overall were a single phenyl ring (1.6 % of all potently active compounds) and acyclic frameworks (0.4 %). False‐positive spotting according to Rishton3 and Hann et al.4 showed most warnings for acyclic scaffolds (up to 37 % substructure warnings with n≥2 flags) and compounds containing a single phenyl ring scaffold (17 %). Otherwise, we observed low false‐positive potential for these scaffold‐focused ligand sets according to the substructure list of Hann et al., with generally more warnings based on the Rishton list (see the Supporting Information).
We subsequently analyzed the activity annotations for all scaffold sets with regard to target‐family bias. The quinoline compound set 1 a turned out to possess the most balanced activity spectrum (Η=2.75 bit, I=0.57 bit, KLD=0.37 bit), followed by diphenylmethanes 2 a (Η=2.54 bit, I=0.78 bit, KLD=0.47 bit) and phenylether 3 a derivatives (Η=2.46 bit, I=0.86 bit, KLD=0.87 bit; Table 1). Four of the ten most promiscuous scaffolds (bisphenylether, benzyloxybenzene, phenylbenzamide, benzylindole) are also part of Welsch's list of traditional privileged scaffolds. Overall, among the set of high‐entropy scaffolds, we observed a prevalence of small scaffolds containing two arene rings connected by a short linker (e.g., methyl, ether, amine, amide). Whilst this finding might appear somewhat trivial at first glance, it adds a novel perspective on the concept of privileged structures, coming, as it does, from an entirely target‐driven vantage point. Quinn and co‐workers recently reported a similar observation for natural products.18 Furthermore, 12 % of the 1 822 molecular structures with a molecular weight less than or equal to 2000 g mol−1, which were approved by the U.S. Food and Drug Administration between 1939 and 2016, contain this substructure motif (data retrieved from the e‐Drug3D database19). Accordingly, it seems prudent to compile general screening compound decks based on scaffolds with low structural intricacy, to increase hit rates and facilitate swift hit‐to‐lead expansion.20
Table 1.
Top‐ranking scaffolds according to their promiscuity expressed as Shannon entropy Η (1 a–5 a), and according to maximal information content I (1 b–5 b). N=number of potent (pActivity≥6) compounds containing only the respective atom scaffold and no other ring system.

|
In stark contrast to the architecture of these promiscuous scaffolds, the most information‐rich scaffolds retrieved from ChEMBL22 were sp3‐rich heterocyclic molecular frameworks (1 b–5 b, Table 1). In fact, several of them are annotated as target‐family selective (Η=0 bit, I=3.32 bit, KLD=1.44–5.14 bit; Table 1, see the Supporting Information). The presence of several of these scaffolds on the Welsh list reveals a dual perception of privileged structures among medicinal chemists. However, according to Evans’ original definition of the term,1 target‐selective scaffolds are not “privileged”. For some of these examples, the ligand–protein complexes have been solved by X‐ray crystallography and exposed directed interactions of the scaffolds with the respective binding pocket, e.g., with kinase hinge residues in the case of scaffold 3 b (PDB ID: 4N7021). Hypothesizing different degrees of target promiscuity, it will be worthwhile to comprehensively analyze ligand–protein interaction patterns with regard to the scaffold promiscuity measures introduced in this present study.22
A second computational analysis focused on the predicted average promiscuity score of the scaffold‐centered ligand sets. In contrast to the scaffold Shannon entropy, this index is computed for each compound individually. We employed a neural network model for this purpose, which we had previously trained to distinguish between undesired (potential false positives) and target‐promiscuous frequent hitters.23 We found a lack of correlation (Pearson r=0.05, 0.03) between the two entropy measures (Η, KLD) and the average neural network score. This result shows that high Shannon entropy (low relative entropy) of the scaffold does not necessarily reflect the binding promiscuity of the individual ligands.
Importantly, correlation analysis suggested an inverse relationship between scaffold target promiscuity (Η) and both the sp3 atom count (r=−0.24) and the number of hydrogen‐bond acceptors (r=−0.28). Accordingly, scaffolds with few hydrogen‐bond acceptors have a greater promiscuity potential than scaffolds with many such interaction points. This hypothesis was strengthened when focusing on scaffolds with at least 300 active compound samples each (r sp3=−0.38; r H‐bond acceptors=−0.53; Figure 2). This finding is in line with former studies of the influence of scaffold structure and complexity on hit‐to‐drug progression.19, 24 Notably, we did not observe a meaningful correlation between Η or KLD and the logP of the compounds (r=0.07).
Figure 2.
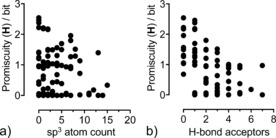
Relationship between the target promiscuity of scaffolds (expressed as Shannon entropy Η, based on ChEMBL22 activity annotations) and the number of sp3‐hybridised centers (a) and number of hydrogen‐bond acceptors (b). Only scaffolds with more than 300 potent ligands were considered (87 scaffolds).
This present study showcases a straightforward information‐theoretical approach for the quantification of scaffold promiscuity, which could assist medicinal chemists in scaffold prioritization and screening library design. However, there are several caveats to keep in mind when interpreting the results presented here. Evidently, the information presented merely reflects the current status of the ChEMBL activity annotations, and there may be many more hitherto‐unknown ligand activities.25 In addition, ChEMBL may not reflect the true diversity of pharmacologically relevant compound structures, since the majority of the proprietary hits and lead compounds from industry are not contained in the ChEMBL database. Although we considered sp3 hybridization, our analysis does not explicitly account for the three‐dimensionality of scaffolds and compounds. It will now be worthwhile to systematically apply the quantitative Shannon entropy concept to analyze the relationship between the shape and flexibility of a scaffold and its promiscuity.26
The Shannon concept has been used before to assess chemical scaffold diversity.27 Our present analysis complements these chemical‐diversity analyses by contributing a quantitative target‐oriented vantage point. Promiscuity seems to be a property of the full compound, and for most of the published examples (ChEMBL) cannot be attributed to the scaffold alone. More specifically, the results of this present study do not corroborate the existence of an apparent generalizable relationship between the size of a molecular scaffold and the ability of a compound to bind to members from different target families. However, the observed inverse correlation between the number of sp3‐hybridised centers and hydrogen‐bond acceptors present in a scaffold and the promiscuity of the respective scaffold‐based compound libraries qualifies the use of certain scaffolds and fragments for target‐focused hit discovery. Our study further revealed that chemically easily accessible scaffolds can serve as templates for the generation of compounds that could bind to almost all target families (i.e., scaffolds with high Shannon entropy). Together with the neural network score, this information could be utilized to generate custom‐made combinatorial screening decks, depending on the intended target(s) or disease.
Conflict of interest
P. S. and G. S. are the founders of inSili.com LLC, Zurich.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This research was financially supported by the OPO‐Foundation Zurich.
P. Schneider, G. Schneider, Angew. Chem. Int. Ed. 2017, 56, 7971.
References
- 1. Evans B. E., Rittle K. E., Bock M. G., DiPardo R. M., Freidinger R. M., Whitter W. L., Lundell G. F., Veber D. F., Anderson P. S., Chang R. S. L., Lotti V. J., Cerino D. J., Chen T. B., Kling P. J., Kunkel K. A., Springer J. P., Hirshfieldt J., J. Med. Chem. 1988, 31, 2235–2246. [DOI] [PubMed] [Google Scholar]
- 2.
- 2a. Roche O., Schneider P., Zuegge J., Guba W., Kansy M., Alanine A., Bleicher K., Danel F., Gutknecht E. M., Rogers-Evans M., Neidhart W., Stalder H., Dillon M., Sjögren E., Fotouhi N., Gillespie P., Goodnow R., Harris W., Jones P., Taniguchi M., Tsujii S., von der Saal W., Zimmermann G., Schneider G., J. Med. Chem. 2002, 45, 137–142; [DOI] [PubMed] [Google Scholar]
- 2b. Schneider G., Schneider P., in: Chemogenomics in Drug Discovery (Eds.: H. Kubinyi, G. Müller), Wiley-VCH, Weinheim, 2004, pp. 341–376. [Google Scholar]
- 3. Rishton G. M., Drug Discovery Today 1997, 2, 382–384. [Google Scholar]
- 4. Hann M., Hudson B., Lewell X., Lifely R., Miller L., Ramsden N., J. Chem. Inf. Comput. Sci. 1999, 39, 897–902. [DOI] [PubMed] [Google Scholar]
- 5. Walters W. P., Namchuk M., Nat. Rev. Drug Discovery 2003, 2, 259–266. [DOI] [PubMed] [Google Scholar]
- 6.
- 6a. Seidler J., McGovern S. L., Doman T. N., Shoichet B. K., J. Med. Chem. 2003, 46, 4477–4486; [DOI] [PubMed] [Google Scholar]
- 6b. Baell J., Walters M. A., Nature 2014, 513, 481–483; [DOI] [PubMed] [Google Scholar]
- 6c. Nissink J. W., Blackburn S., Future Med. Chem. 2014, 6, 1113–1126; [DOI] [PubMed] [Google Scholar]
- 6d. Dahlin J. L., Walters M. A., Future Med. Chem. 2014, 6, 1265–1290; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6e. Irwin J. J., Duan D., Torosyan H., Doak A. K., Ziebart K. T., Sterling T., Tumanian G., Shoichet B. K., J. Med. Chem. 2015, 58, 7076–7087; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6f. Papadatos G., Gaulton A., Hersey A., Overington J. P., J. Comput. Aided Mol. Des. 2015, 29, 885–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.
- 7a. Zhao H., Dietrich J., Expert Opin. Drug Discovery 2015, 10, 781–790; [DOI] [PubMed] [Google Scholar]
- 7b. Song Y., Chen W., Kang D., Zhang Q., Zhan P., Liu X., Comb. Chem. High Throughput Screening 2014, 17, 536–553; [DOI] [PubMed] [Google Scholar]
- 7c. DeSimone R. W., Currie K. S., Mitchell S. A., Darrow J. W., Pippin D. A., Comb. Chem. High Throughput Screening 2004, 7, 473–494. [DOI] [PubMed] [Google Scholar]
- 8. Welsch M. E., Snyder S. A., Stockwell B. R., Curr. Opin. Chem. Biol. 2010, 14, 347–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.
- 9a. Sravanthi T. V., Manju S. L., Eur. J. Pharm. Sci. 2016, 91, 1–10; [DOI] [PubMed] [Google Scholar]
- 9b. Berthet M., Cheviet T., Dujardin G., Parrot I., Martinez J., Chem. Rev. 2016, 116, 15235–15283; [DOI] [PubMed] [Google Scholar]
- 9c. Marinozzi M., Marcelli G., Carotti A., Mini-Rev. Med. Chem. 2015, 15, 272–299; [DOI] [PubMed] [Google Scholar]
- 9d. Kaur G., Kaur M., Silakari O., Mini-Rev. Med. Chem. 2014, 14, 747–767; [DOI] [PubMed] [Google Scholar]
- 9e. Lal S., Snape T. J., Curr. Med. Chem. 2012, 19, 4828–4837. [DOI] [PubMed] [Google Scholar]
- 10. Yang J. J., Ursu O., Lipinski C. A., Sklar L. A., Oprea T. I., Bologa C. G., J. Cheminf. 2016, 8, 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.
- 11a. Rodrigues T., Reker D., Schneider P., Schneider G., Nat. Chem. 2016, 8, 531–541; [DOI] [PubMed] [Google Scholar]
- 11b. de Sá Alves F. R., Barreiro E. J., Fraga C. A., Mini-Rev. Med. Chem. 2009, 9, 782–793; [DOI] [PubMed] [Google Scholar]
- 11c. Polanski J., Kurczyk A., Bak A., Musiol R., Curr. Med. Chem. 2012, 19, 1921–1945. [DOI] [PubMed] [Google Scholar]
- 12. Bento A. P., Gaulton A., Hersey A., Bellis L. J., Chambers J., Davies M., Kruger F. A., Light Y., Mak L., McGlinchey S., Nowotka M., Papadatos G., Santos R., Overington J. P., Nucleic Acids Res. 2014, 42, D1083–D1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.
- 13a. Shannon C. E., Bell Syst. Tech. J. 1948, 27, 379–423; [Google Scholar]
- 13b. Wiener N., Cybernetics or Control and Communication in the Animal and the Machine, 2 nd ed, MIT Press, Cambridge, MA, 1948, p. 18; [Google Scholar]
- 13c. Kullback S., Leibler R. A., Ann. Math. Stat. 1951, 22, 79–86. [Google Scholar]
- 14. Southan C., Sharman J. L., Benson H. E., Faccenda E., Pawson A. J., Alexander S. P. H., Buneman O. P., Davenport A. P., McGrath J. C., Peters J. A., Spedding M., Catterall W. A., Fabbro D., Davies J. A., Nucleic Acids Res. 2016, 44, D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Berthold M. R., Cebron N., Dill F., Kötter T. R., Meinl T., Ohl P., Sieb C., Thiel K., Wiswedel B., in: Studies in Classification, Data Analysis, and Knowledge Organization, Springer, Heidelberg, 2007, pp. 319–326. [Google Scholar]
- 16.RDKit: Cheminformatics and Machine Learning Software, 2013, http://www.rdkit.org.
- 17. Bemis G. W., Murcko M. A., J. Med. Chem. 1996, 39, 2887–2893. [DOI] [PubMed] [Google Scholar]
- 18. Pascolutti M., Campitelli M., Nguyen B., Pham N., Gorse A. D., Quinn R. J., PLoS One 2015, 10, e0120942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Pihan E., Colliandre L., Guichou J. F., Douguet D., Bioinformatics 2012, 28, 1540–1541. [DOI] [PubMed] [Google Scholar]
- 20. Hann M. M., Leach A. R., Harper G., J. Chem. Inf. Comput. Sci. 2001, 41, 856–864. [DOI] [PubMed] [Google Scholar]
- 21. Burger M. T., Han W., Lan J., Nishiguchi G., Bellamacina C., Lindval M., Atallah G., Ding Y., Mathur M., McBride C., Beans E. L., Muller K., Tamez V., Zhang Y., Huh K., Feucht P., Zavorotinskaya T., Dai Y., Holash J., Castillo J., Langowski J., Wang Y., Chen M. Y., Garcia P. D., ACS Med. Chem. Lett. 2013, 4, 1193–1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.
- 22a. Bajorath J., Mol. Inf. 2016, 35, 583–587; [DOI] [PubMed] [Google Scholar]
- 22b. Glinca S., Klebe G., J. Chem. Inf. Model. 2013, 53, 2082–2092; [DOI] [PubMed] [Google Scholar]
- 22c. Weisel M., Kriegl J. M., Schneider G., ChemBioChem 2010, 11, 556–563; [DOI] [PubMed] [Google Scholar]
- 22d. Weisel M., Proschak E., Kriegl J. M., Schneider G., Proteomics 2009, 9, 451–459; [DOI] [PubMed] [Google Scholar]
- 22e. Abagyan R., Kufareva I., Methods Mol. Biol. 2009, 575, 249–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Schneider P., Röthlisberger M., Reker D., Schneider G., Chem. Commun. 2016, 52, 1135–1138. [DOI] [PubMed] [Google Scholar]
- 24.
- 24a. Lovering F., Bikker J., Humblet C., J. Med. Chem. 2009, 52, 6752–6756; [DOI] [PubMed] [Google Scholar]
- 24b. Ortholand J. Y., Ganessan A., Curr. Opin. Chem. Biol. 2004, 8, 271–280. [DOI] [PubMed] [Google Scholar]
- 25.
- 25a. Hu Y., Bajorath J., PLoS One 2015, 10, e0126838; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25b. Hu Y., Bajorath J., Drug Discovery Today 2013, 18, 644–650. [DOI] [PubMed] [Google Scholar]
- 26.
- 26a. Meyers J., Carter M., Mok N. Y., Brown N., Future Med. Chem. 2016, 8, 1753–1767; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26b. Müller G., Berkenbosch T., Benningshof J. C., Stumpfe D., Bajorath J., Chemistry 2017, 23, 703–710; [DOI] [PubMed] [Google Scholar]
- 26c. Morley A. D., Pugliese A., Birchall K., Bower J., Brennan P., Brown N., Chapman T., Drysdale M., Gilbert I. H., Hoelder S., Jordan A., Ley S. V., Merritt A., Miller D., Swarbrick M. E., Wyatt P. G., Drug Discovery Today 2013, 18, 1221–1227; [DOI] [PubMed] [Google Scholar]
- 26d. Wirth M., Sauer W. H., Mol. Inf. 2011, 30, 677–688. [DOI] [PubMed] [Google Scholar]
- 27.
- 27a. Yan B. B., Xue M. Z., Xiong B., Liu K., Hu D. Y., Shen J. K., Acta Pharmacol. Sin. 2009, 30, 251–258; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27b. Medina-Franco J. L., Martínez-Mayorga K., Bender A., Scior T., QSAR Comb. Sci. 2009, 28, 1551–1560; [Google Scholar]
- 27c. Langdon S. R., Brown N., Blagg J., J. Chem. Inf. Model. 2011, 51, 2174–2185; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27d. Hu Y., Wassermann A. M., Loukine E., Bajorath J., J. Med. Chem. 2010, 53, 752–758; [DOI] [PubMed] [Google Scholar]
- 27e. Erlanson D. A., Fesik S. W., Hubbard R. E., Jahnke W., Jhoti H., Nat. Rev. Drug Discovery 2016, 15, 605–619. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary