

Significance
Germline mutations of BRCA2 associate with tissue-specific cancers, such as familial pancreatic cancer. However, it is unclear why BRCA2 mutation causes tissue-specific cancer. Here, we show that pancreas-specific reactive nitrogen species (RNS) causes oxidative damage and DNA double-strand breaks. Lacking BRCA2, cells could not repair DNA lesions, which led to genomic instability and pancreatic tumorigenesis. On the basis of the mechanistic analysis, we treated Brca2-deficient mice with antioxidant. Interestingly, the antioxidant treatment suppressed RNS-induced lesions and delayed the onset of pancreatic tumorigenesis. Thus, our study reveals a potential approach for the eradication of BRCA2 mutation-associated pancreatic cancer.
Keywords: DNA damage response, DNA damage repair, BRCA2, RNS, pancreatic cancer
Abstract
Germline mutation of BRCA2 induces hereditary pancreatic cancer. However, how BRCA2 mutation specifically induces pancreatic tumorigenesis remains elusive. Here, we have examined a mouse model of Brca2-deficiency–induced pancreatic tumors and found that excessive reactive nitrogen species (RNS), such as nitrite, are generated in precancerous pancreases, which induce massive DNA damage, including DNA double-strand breaks. RNS-induced DNA lesions cause genomic instability in the absence of Brca2. Moreover, with the treatment of antioxidant tempol to suppress RNS, not only are DNA lesions significantly reduced, but also the onset of pancreatic cancer is delayed. Thus, this study demonstrates that excess RNS are a nongenetic driving force for Brca2-deficiency–induced pancreatic tumors. Suppression of RNS could be an important strategy for pancreatic cancer prevention.
Pancreatic cancer is one of the deadliest human cancers (1, 2). The median survival of patients with pancreatic cancer is merely 6 mo (3, 4). Thus, it is crucial to understand the molecular mechanism of pancreatic cancer, which will facilitate cancer prevention, diagnosis, and therapy.
Accumulated evidence suggests that genetic alterations on activating oncogenes such as KRas, as well as inactivating tumor suppressor genes such as p53 and BRCA2, induce pancreatic tumorigenesis (1, 5, 6). Most of these genetic alterations are somatic mutations during tumorigenesis. However, BRCA2 is often mutated in germ cells (1, 7). BRCA2 mutation carriers are predisposed to familial pancreatic cancer (8). Moreover, PALB2, a functional partner of BRCA2 during homologous recombination (HR), is involved in familial pancreatic cancer suppression (9, 10). It suggests BRCA2 and its dependent pathway play key roles to suppress hereditary pancreatic cancer.
The BRCA2 gene product is a nuclear polypeptide with 3,418 residues and is involved in DNA damage repair (11–13). After DNA double-strand breaks (DSBs), BRCA2 is recruited to DNA lesions via PALB2 and facilitates the loading of RAD51, the key recombinase for HR (14–18). Loss of BRCA2 abolishes DSB repair, and thus induces genetic instability (19, 20). In addition to pancreatic cancer, mutations of BRCA2 are also associated with several other types of cancer, such as breast cancer, ovarian cancer, prostate cancer, and lymphoma (21–25). However, compared with the mutation of KRas or p53-induced tumor spectrum, the BRCA2 mutation-induced tumor spectrum is much narrower (26, 27). Because the major known function of BRCA2 is to repair DNA lesions, we hypothesize that a type of carcinogen is specifically accumulated in pancreas and induces massive DNA damage. Without BRCA2, DNA lesions are not repaired, which causes genomic instability and pancreatic tumorigenesis.
The specific carcinogen inducing pancreatic cancer has not been revealed yet. Interestingly, it has been shown that pancreatitis, including both acute and chronic pancreatitis, is associated with pancreatic cancer, especially hereditary pancreatic cancer (28–30). Pancreatitis is an inflammation of the pancreas, which draws phagocytic macrophages and neutrophils to secrete a large amount of nitric oxide (NO) and superoxide (O2˙) for the eradication of microbial pathogens and processing dead cells (31, 32). NO and O2˙ are unstable and react with each other to generate peroxynitrite (ONOO−) at the site of inflammation (33, 34). ONOO− itself is not a free radical. However, pancreatic acinar cells secrete high concentrations (up to 140 mM in pancreatic duct) of bicarbonate (HCO3−) for neutralizing acidic chyme from stomach (35, 36). ONOO− can react with HCO3− from pancreatic acinar cells to generate nitrosoperoxycarbonate (ONOOCO2−), a major reactive nitrogen species (RNS) in vivo that homolyzes to form carbonate radical (CO3˙ −) and nitrite (NO2˙) (34, 37).
Both CO3˙ − and NO2˙ are free radicals and extremely dangerous for the integrity of genomic DNA (37, 38). Among four different DNA bases, deoxyguanine (dG) has the least reduction potential (37, 39), which makes it the most easily oxidized base by CO3˙ − and NO2˙. Although most DNA oxidation damage is quickly repaired by the base excision repair (BER) pathway, a small amount of lesions may escape from surveillance and induce DNA single-strand breaks or DSBs (40, 41). In the absence of DSB repair, such as BRCA2-dependent homologous recombination, these lesions will ultimately lead to genomic instability in pancreas and then induce pancreatic tumorigenesis. Here, we have tested our hypothesis in vivo and found that excessive RNS induces massive DNA damage in the absence of BRCA2, which may lead to genomic instability and tumorigenesis. Suppression of RNS by antioxidant reduces DNA lesions and delays the onset of pancreatic tumorigenesis. Thus, this study reveals a molecular mechanism of pancreatic tumorigenesis and provides a potential approach to suppressing tumor development.
Results
Development of Pancreatic Ductal Adenocarcinoma in the KPB Mouse Model.
To examine the molecular mechanism of pancreatic cancer, we set up a pancreatic ductal adenocarcinoma (PDAC) mouse model, as PDAC is the most common type of pancreatic cancer. On the basis of previous animal studies (27, 42), we generated the Ptf1a-Cre; KrasG12D; p53flox/+; Brca2flox/flox mice, in which the Ptf1a-Cre allele drives Cre recombinase expression to conditionally activate endogenous oncogenic KrasG12D (K) and knockout one p53 allele (P) and the exon 11 of both Brca2 alleles (B) in pancreas (Fig. S1A), also named the KPB mice. The exon 11 of Brca2 encodes the Rad51-binding region, and loss of this region abolishes the function of Brca2 in DNA damage repair (43, 44). During the generation of the KPB mice, we also maintained the littermates Ptf1a-Cre; KrasG12D; p53flox/+; Brca2+/+ mice (KP mice) and mice without oncogenic alterations (WT mice). The median life span of the KPB mice was 62 d, whereas the average KP mice survived 157 d (Fig. 1A). All the KPB mice developed large, solid pancreatic tumors (Fig. 1B), frequently accompanied by gallbladder distension, bowel obstruction, cachexia, and jaundice (Fig. S1 B and C), and 44.4% of the KPB mice suffered abdominal distension, randomly with the accumulation of hemorrhagic ascites (Fig. S1D). Similar tumor phenotype with delayed onset was observed in the KP mice. To validate pancreatic tumorigenesis in the KPB mice, we examined the survival KPB mice at the age of 4–5 wk and found a full spectrum of pancreatic intraepithelial neoplasia (PanINs). By the age of 7–8 wk, cancer invasion and adenocarcinoma formation were readily observed in the pancreases of the KPB mice (Fig. 1C). Thus, in agreement with previous study, loss of Brca2 remarkably promotes pancreatic tumorigenesis in vivo (27).
Fig. S1.

The KPB mice develop PDAC. (A) Generation of Ptf1a-Cre; KrasG12D; p53flox/+; Brca2flox/flox mice (the KPB mice) and Ptf1a-Cre; KrasG12D; p53flox/+; Brca2+/+mice (the KP mice) by cross-mating the Ptf1a-Cre, LSL-KrasG12D, p53flox/+, and Brca2flox/+ mice. Ptf1a-Cre shows a single band in agarose gel. Mutant KrasG12D and wild-type Kras were separated by electrophoresis. Floxed alleles of Brca2 and p53 and their wild-type alleles were separated in agarose gel as well. (B) Anatomical KPB mouse with gallbladder distension (Upper) and bowel obstruction (Lower). (C) Anatomical KPB mouse with jaundice. (D) Percentage of abdominal distension in the KP and KPB mice (Left); anatomical photograph of hemorrhagic ascites of the KPB mice (Right).
Fig. 1.

Development of PDAC in the KPB mice. (A) Kaplan-Meier analysis for comparing overall survival of the WT, KP, and KPB mice. The median survival of the KP and KPB mice is 157 and 62 d, respectively (P < 0.001, log-rank test, for each pairwise combination). (B) Anatomic photograph of KPB mouse with PDAC. Outline denotes the tumor. Note that the distensible gallbladder (marked by *) is a common symptom accompanied with PDAC. Gb, gallbladder; L, liver; Sp: spleen; T, tumor. (C) Histomorphology of normal pancreas in the WT mice and different stages of PanINs and invasive PDAC in the KPB mice. Arrowheads indicate the representative morphology in different stages of PanINs. (Scale bars, 50 µm.) (D) Immunostaining of γH2AX and 53BP1 in pancreatic tissues of the WT mice, and precancerous or cancerous pancreases of the KP and KPB mice. (Scale bars, 50 µm.) (E) Representative mitotic spreads of the WT and KPB pancreatic cell. Chromosomes were stained with Giemsa. Chromosome breaks from pancreatic tumors of the KPB mice are shown. Sixteen mitotic spreads in each sample were examined. Percentage of chromosome breaks in each sample is summarized. Means and SDs were plotted. ***P < 0.001.
Because BRCA2 plays a key role in HR repair for DSBs, we next examine whether DSBs are elevated in pancreases in the absence of Brca2. We collected both the precancerous pancreatic tissues and pancreatic cancer tissues from the KPB and KP mice, and the normal pancreases from WT mice, followed by the immunofluorescent examination of γH2AX, a surrogate maker of DSBs. In the pancreatic tissues from the KPB mice including both precancerous pancreas and pancreatic tumor, γH2AX was positively stained, suggesting DSBs occur in the pancreases of the KPB mice (Fig. 1D). However, in the presence of Brca2, we did not observe obvious DSBs in the pancreatic tissues at precancerous or tumor stages from the KP mice, or in the normal pancreases from the WT mice. Moreover, we also found that the expression of 53BP1, another important DSB repair machinery, was increased only in the pancreatic tissues from the KPB mice, further indicating that DSBs exist in these mice. In addition, we examined the mitotic spreads of tumor samples from the KPB mice and found DNA breaks did exist in these pancreatic tumors (Fig. 1E). As a control, we also examined Ptf1a-Cre; Brca2flox/flox mice. However, the mice did not develop pancreatic tumors, which is consistent with previous studies (27, 45). Meanwhile, we did not observe obvious DSBs in the pancreatic tissues of these mice (Fig. S2). The results indicate that loss of BRCA2 alone is insufficient for driving tumorigenesis.
Fig. S2.

DNA DSBs from pancreatic cells of the Ptf1a-Cre; Brca2flox/flox mice are not detectable. (A) The pancreatic tissues from the Ptf1a-Cre; Brca2flox/flox mice (Brca2 mice) were immunostained with anti-γH2AX and anti-53BP1 antibodies. The pancreatic tissues from the wild-type (WT) and the PDAC from the KPB mice were used as negative and positive controls. (B) Mitotic breaks were examined in the cells from pancreatic tissues of the Ptf1a-Cre; Brca2flox/flox mice.
RNS and RNS-Induced DNA Adducts in the Pancreases of the KPB Mice.
Next, we examine the cause of the elevated DSBs in the KPB mice. As mentioned earlier, we hypothesize that massive RNS are generated in the pancreases of the KPB mice. These RNS attack dG and cause DNA lesions, which consequently leads to DSBs and genomic instability (Fig. 2A). To examine the RNS in vivo, we examined the level of endogenous nitrite based on an established protocol (46). Precancerous pancreases from KPB and KP mice and normal pancreas from WT mouse were harvested, followed by the detection of endogenous nitrite. Compared with that in pancreases of the WT mice, the level of nitrite was around sixfold and 10-fold higher in pancreases of the KP and KPB mice, respectively (Fig. 2B). Moreover, the RNS react with dG in vivo and form two major DNA adducts: 8-oxo-guanine (8-oxo-G) and 8-nitro-guanine (8-nitro-G) (33, 47). Because 8-oxo-G can also be generated by other free radicals such as reactive oxidative species, we only focused on 8-nitro-G. The relative level of 8-nitro-G increased 22.4-fold and 29-fold in the pancreases of the KP and KPB mice compared with that in the pancreases of WT mice (Fig. 2C). 8-nitro-G is not stable and is quickly depurinated from the genomic DNA, during which abasic sites, aka AP (apurinic/apyrimidinic) sites, are generated (38). Thus, we examined AP sites by fluorescence staining and found that AP sites remarkably increased in both the precancerous and tumor tissues of pancreases from the KP and KPB mice (Fig. 2D). We also examined AP endonuclease 1 (APE1), the major enzyme to repair AP sites, by immunostaining. The expression level of APE1 is elevated in pancreatic tissues from the KP and KBP mice (Fig. 2E). Collectively, these results suggest that a high level of RNS causes extensive oxidative damage in the pancreases of the KP and KPB mice.
Fig. 2.

RNS and RNS-induced DNA adduct in the KPB pancreases. (A) Schematic diagram of RNS and RNS-induced DNA adducts. (B) The level of nitrite in the pancreases of the WT, KP, and KPB mice was measured. (C) The relative level of 8-nitro-G in pancreases of the WT and KPB mice was examined by mass spectrometry. The molecular weight of the reference of 8-nitro-G and 8-nitro-G isolated from pancreases of the WT and KPB mice was determined by Q-TOF mass spectrometry (Left). Peak value of 8-nitro-G from different samples in the mass spectrometry is shown (Right). (D and E) AP sites (D) and APE1 (E) in the pancreatic tissues of the WT, KP, and KPB mice were immunostained. (Scale bars, 50 µm.) Means and SDs were plotted. ***P < 0.001.
RNS Induce DSBs.
Next, we examined whether massive oxidative lesions can be converted to DSBs. Primary pancreatic acinar cells were isolated from the KP and KPB mice and treated with ONOOCO2−. As expected, we found that the level of 8-nitro-G was significantly increased in the cells after the treatment with ONOOCO2− (Fig. 3A). Moreover, the AP sites were induced in both the KP and KPB cells (Fig. 3B). Most of the oxidative lesions were quickly repaired, as we could not detect the obvious AP sites after 24 h recovery from the treatment with ONOOCO2−. However, if many oxidative lesions occur simultaneously, some lesions may not be timely repaired, which can be converted into DSBs. Thus, we examined DSBs by immunostaining γH2AX. Immediately after the treatment with ONOOCO2−, we found that DSBs occurred in both the KP and KPB cells. After 24 h recovery from the RNS insults, most DSBs in the KP cells were repaired, as we could only find sporadic and weak γH2AX foci. However, in the KPB cells, strong and big γH2AX foci were observed under the same assay condition, suggesting DSBs still exist in the KPB cells (Fig. 3C). Moreover, we performed the neutral comet assay to examine DSB repair after the treatment with ONOOCO2−. With the increased dose of ONOOCO2−, DNA breaks were clearly detected in the KPB cells (Fig. 3D). Taken together, these results show that RNS treatment could induce DSBs in pancreatic cells. In the absence of Brca2, the cells lose the last protection system to correct DSBs, which may induce genomic instability and tumorigenesis.
Fig. 3.

RNS induce DNA lesions in vitro. (A) ONOOCO2− treatment induces 8-nitro-G. Primary pancreatic acinar cells from the KP and KPB mice were treated with or without ONOOCO2−. Mass spectrometry analysis shows the molecular weight of 8-nitro-G isolated from the KPB cells (Left). Peak value of 8-nitro-G in mass spectrometry is shown. ND, nondetectable (Right). (B) ONOOCO2− treatment induces AP sites in pancreatic cells. Primary pancreatic acinar cells from the KP or KPB mice were treated with ONOOCO2−, followed by AP site staining. Alternatively, cells were recovered from ONOOCO2− treatment for 24 h before AP site staining. (C) Immunostaining of γH2AX in pancreatic cells. Primary pancreatic acinar cells were treated with ONOOCO2− followed by γH2AX staining. Alternatively, cells were recovered for 24 h followed by γH2AX staining. (Scale bars, 10 µm.) (D) RNS-induced DSBs is examined by neutral comet assay. Primary pancreatic acinar cells from the KP and KPB mice were treated with mock or ONOOCO2− (10, 20, and 50 µM) followed by 24-h recovery and neutral comet assay. Means and SDs were plotted. **P < 0.01; ***P < 0.001.
Antioxidant Treatment Suppresses RNS-Induced DNA Damage and Tumorigenesis.
If RNS induce DNA damage and genomic instability in pancreatic cells, treatment with antioxidants should be able to suppress DNA lesions and tumorigenesis. To test our hypothesis, we treated the primary pancreatic acinar cells from the KPB mice with tempol (4-hydroxy-2,2,6,6-tetramethylpiperidine-N-oxyl), a membrane-permeable free radical scavenger. To confirm the antioxidative efficiency of tempol, we first examined the nitrite level of the KPB cells in the presence of ONOOCO2−. As expected, tempol decreased the cellular level of nitrite significantly, and this effect was shown in a dose-dependent manner (Fig. 4A). Pretreatment of tempol remarkably reduced RNS-induced 8-nitro-G and the AP sites in these cells (Fig. 4 B and C). Moreover, ONOOCO2−-induced foci of γH2AX and 53BP1 were repressed in the KPB cells in the presence of tempol (Fig. 4D). Consistently, with the increased level of tempol, DSBs were also suppressed (Fig. 4E).
Fig. 4.

Tempol treatment suppresses RNS-induced DNA lesions in vitro. (A and B) Tempol treatment suppresses cellular nitrite (A) and 8-nitro-G (B) induced by ONOOCO2−. Primary pancreatic acinar cells from the KPB mice were preincubated with mock or tempol (20, 50, and 100 µM) followed by the treatment of ONOOCO2−. The level of 8-nitro-G was examined by mass spectrometry. (C) AP sites are remarkably reduced in the presence of tempol. Primary pancreatic acinar cells from KPB mice were preincubated with or without tempol followed by the treatment of ONOOCO2−. (D and E) Tempol treatment suppresses RNS-induced DSBs. (D) Foci of γH2AX and 53BP1 in the primary pancreatic acinar cells from the KPB mice were examined. (Scale bars, 10 µm.) Foci numbers of γH2AX and 53BP1 in each cell are shown in the graph. (E) Primary pancreatic acinar cells from the KPB mice were preincubated with or without tempol (20, 50, and 100 µM). Neutral comet assays were performed to examine ONOOCO2−-induced DSBs. Tail moments of the cells treated with different doses of tempol are shown in the histogram. Means and SDs were plotted. *P < 0.1; **P < 0.01; ***P < 0.001.
Because tempol is a powerful antioxidant in vitro, we next examined whether tempol could suppress oxidative damage and pancreatic tumorigenesis in vivo. The pancreatic cancer developed very early in the KPB mice and induced the lethality as early as 5 wk. Tempol treatment after weaning only slightly suppressed the tumor onset of the KPB mice (Fig. 5A). Thus, we treated pregnant female mice with tempol and kept administrating tempol to the newborn mice after weaning. Interestingly, tempol treatment significantly delayed onset of pancreatic tumor and increased the life span of the KPB mice (Fig. S3 and Fig. 5A). The median survival of the KPB mice was increased from 62 to 114 d by tempol treatment. Moreover, the level of nitrite and 8-nitro-G were significantly reduced in vivo (Fig. 5 B and C). Along with the reduction of RNS, both AP sites and DSBs were dramatically decreased in the precancerous pancreases or pancreatic tumors after tempol treatment (Fig. 5 D and E). In addition, the tempol treatment for pregnant female mice and the newborn mice showed a trend toward increased survival of the KP mice, but there is no statistical significance (Fig. 5F). Thus, tempol can be used as a potent suppressor of RNS-induced DNA damage and tumorigenesis in pancreas.
Fig. 5.

Tempol treatment suppresses RNS-induced DNA damage in vivo and extends the life span of the KPB mice. (A) Kaplan-Meier analysis comparing overall survivals of the KPB mice treated with or without tempol. Median survivals of the groups of the nontreated (Mock), tempol treatment after weaning, and tempol treatment before birth are 62, 73, and 114 d, respectively. Log-rank test was performed to examine the efficacy of tempol treatment: nontreated vs. tempol treatment after weaning (P > 0.1); nontreated vs. tempol treatment before birth (P < 0.001). (B) The level of nitrite in precancerous pancreases of the KPB mice was measured. Mice were treated with or without tempol. (C) Peak values of 8-nitro-G in mass spectrometry are shown. (D and E) Tempol treatment suppresses AP sites (D) and DSBs (E) in the precancerous and cancerous pancreases of the KPB mice. Immunostaining of γH2AX and 53BP1 indicate DSBs in the pancreatic issues. (Scale bar, 50 µm.) Means and SDs were plotted. ***P < 0.001. (F) Kaplan-Meier analysis comparing overall survivals of the KP mice treated with or without tempol, displaying the median survivals of 157 and 184 d, respectively (P > 0.1, log-rank test).
Fig. S3.
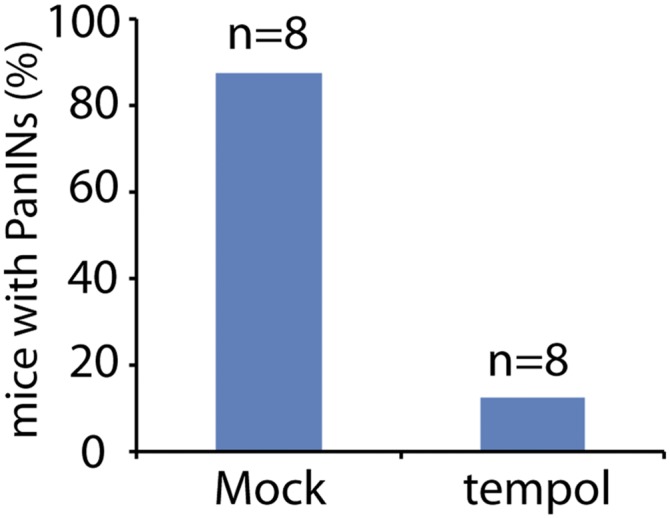
Tempol treatment delays the onset of PanINs in the KPB mice. The onsets of PanINs were examined by the H&E staining of pancreases from the KPB mice at the age of 4 wk with or without the treatment of tempol.
Discussion
In this study, we demonstrate that RNS is a critical nongenetic factor for BRCA2 deficiency-induced pancreatic tumorigenesis. The RNS is likely to be generated by pancreatic acinar cells, macrophages, and neutrophils. Acinar cells uniquely secrete a large amount of HCO3− into pancreatic ducts, which protects the surrounding cells from acid chyme (35, 36). However, this physiological process is a double-edged sword, as HCO3− can react with ONOO− from macrophages and neutrophils and generate ONOOCO2−, a dangerous RNS that releases free radicals to damage genomic DNA (34, 37). Both macrophages and neutrophils are recruited by inflammation (31, 32). It has been shown that oncogenic Kras regulates and facilitates inflammation in pancreas (48–50). Consistently, we found that in the Ptf1a-Cre; KrasG12D mice, RNS have been largely generated (Fig. S4). Accumulated evidence has shown that RNS causes DNA lesions such as base damage, which potentially induces genomic instability.
Fig. S4.
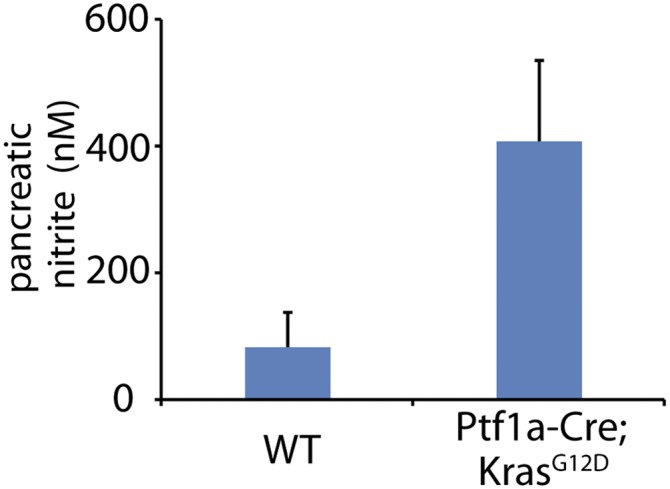
The level of nitrite in the pancreases of the Ptf1a-Cre; KrasG12D mice. The level of nitrite in pancreases of the WT and Ptf1a-Cre; KrasG12D mice at the age of 10 wk.
Under physiologically relevant conditions, a low level of oxidative damage should be easily repaired by BER in most tissues (41, 51). However, as the pancreas is the largest organ that secretes HCO3− (35, 36), more RNS are likely to be generated once inflammation occurs. If DNA lesions are too many to be timely repaired by BER, these lesions will be duplicated during DNA replication, and are converted into DSBs. Alternatively, if two single-strand breaks during BER in the complement strands locate close to each other, DSBs may also occur naturally (52). Thus, BRCA2-dependent DSB repair is the last protection for cells to maintain genomic stability in pancreas. Hence, loss of BRCA2 induces the accumulation of DSBs and accelerates genomic instability. In agreement with this mechanism, we and others observed that BRCA2 deficiency significantly shortens tumor latency (27).
BRCA2 is an important tumor suppressor expressed in most types of cells and tissues. However, mutations of BRCA2 only cause tissue-specific tumors, indicating that tissue-specific carcinogens may exist to induce DNA lesions. Without BRCA2, the lesions will be accumulated, and eventually induce genomic stability and tumorigenesis. Thus, this proposed mechanism may explain the tissue-specific tumors induced by BRCA2 mutations. Unlike other oncogenic mutations, it is rare to find the somatic mutations of BRCA2 in spontaneous tumors. Most BRCA2 mutations are germline mutations that mainly induce familial breast, ovarian, and pancreatic cancers (21, 22, 53). A high level of HCO3−-induced RNS in pancreas could be the unique factor for BRCA2 mutation carriers to predispose to tumorigenesis in pancreas. With similar mechanisms, other tissue-specific carcinogens such as oxidized estrogen could induce DSBs that lead to breast and ovarian tumorigenesis in BRCA2 mutation carriers.
Our study also provides a feasible chemoprevention approach for familial pancreatic cancer. The antioxidant treatment significantly reduces the RNS, DNA oxidative adducts, and DNA lesions. More important, the antioxidant treatment significantly delays tumor onset and nearly doubles the life span of the KPB mice. To our knowledge, this chemoprevention strategy is one of the most effective approaches to extend life span in a mouse pancreatic cancer model. Moreover, for BRCA2 mutation or other familial pancreatic tumor suppressor gene (such as PALB2) mutation carriers, our study has provided the molecular mechanism for a possible chemoprevention trial. In addition, although there is no statistical significance, the antioxidant treatment also showed a trend to extend the life span of the KP mice. However, because of the limited animal strains examined in this study, it is unclear whether the Kras and p53 mutations induce oxidative damage in other tissues. Additional studies on other tumor animal models will reveal whether the antioxidant treatment could be an effective chemoprevention approach for other Kras and p53 mutation-induced tumors.
Methods
Chemicals and Antibodies.
All chemicals were purchased from Sigma except those specifically mentioned. Anti-γH2AX antibody was purchased from Cell Signaling Technology; Anti-53BP1 and APE1 antibodies were purchased from Novus Biologicals.
Animal Strains and Maintenance.
The Ptf1a-Cre, LSL-KrasG12D mice were kindly provided by Dr. Magliano at the University of Michigan Medical School, Ann Arbor, MI. The p53flox/+ mice were provided by Dr. Yuan at the University of Michigan Medical School, Ann Arbor, MI. The Brca2flox/+ mice were obtained from NCI Mouse Repository. The p53flox/+ mice were intercrossed with Brca2flox/+ mice to create the p53flox/+; Brca2flox/+ mice, which were further crossed with the Ptf1a-Cre, LSL-KrasG12D mice to generate Ptf1a-Cre; KrasG12D; p53flox/+; Brca2flox/flox mice (KPB mice) and Ptf1a-Cre; KrasG12D; p53flox/+; Brca2+/+ mice (KP mice). All experiments were performed using the littermates from a mixed but uniform genetic background. The animals were maintained in a specific pathogen-free environment under a 12-h light/dark cycle. All experiments were performed in accordance with national and University of Michigan institutional guidelines. The study was approved by the ethical review committee of the University of Michigan.
Genotyping.
Genomic DNA was isolated from mouse tails using the DNeasy Blood and Tissue Kit (Qiagen) and genotyped by PCR. Reaction conditions for Cre, Kras, p53, and Brca2 were 40 cycles of 94 °C for 30 s, 60 °C for 30 s, and 72 °C for 1 min. PCR products were then run in the 1.5% agarose gel.
Histological Staining and Immunofluorescence Staining.
Precancerous pancreatic tissues and pancreatic cancer tissues were harvested from the KP or KPB mice at the age of 50 d. Normal pancreatic tissues were harvest from the WT mice at the age of 50 d as well. Explanted tissues were fixed in 10% neutral-buffered formalin solution for at least 16 h and gradually transferred to 70% ethanol. Then the tissues were embedded in paraffin at University of Michigan Microscopy & Image Analysis Core, cut in 5-µm sections on poly-lysine-coated slides, and deparaffinized, rehydrated, and stained with H&E. Images were taken by an Olympus IX 71 microscope with the CellSens software. For tissue immunofluorescence, after deparaffinization and rehydration, sections were unmasked in 10 mM citric acid (pH 6.0) in a microwave for 20 min. Then the samples were subjected to standard immunofluorescence staining. Briefly, tissues were incubated in primary antibody overnight at 4 °C. After being washed in PBS three times, the samples were incubated in FITC-conjugated or Rho-conjugated IgG for 1 h at room temperature. After washing and staining with DAPI, tissues were mounted in antifade and observed under the Olympus IX 71 microscope with the CellSens software. For immunofluorescence staining of cells, cells were fixed in 3% paraformaldehyde for 25 min and permeabilized in 0.5% Triton X-100 for 5 min at room temperature. Samples were blocked with 5% goat serum and then incubated in primary antibody for 1 h. Samples were then washed with PBS three times and incubated with fluorescence-conjugated secondary antibody for 40 min. After washing with PBS, nuclei were stained with DAPI. The samples were visualized by the fluorescence microscope and analyzed by CellSens software.
AP Site Assay.
After the standard deparaffinization, rehydration, and unmasking described earlier, sections were stained for AP sites, using DNA damage-AP site assay kit (abcam, ab133076) according to the standard protocol for fluorescence microscopy. AP sites in the tissues were labeled with Avidin-FITC and detected by the Olympus IX 71 microscope.
Nitrite Measurement.
Nitrite was analyzed with the OxiSelect Nitrite Assay Kit (Cell Biolabs, STA-802), and 200 mg frozen tissues were homogenized in cellular lysis buffer for 20 min, using a Cornless Polytron homogenizer, followed by centrifugation at 14,000 × g for 15 min at 4 °C. The supernatant was filtered through a 10-kDa MWCO ultrafilter to reduce protein interference and turbidity. Nitrite was examined according to the provided protocol.
Genomic DNA Isolation from Tissues.
Genomic DNA from mouse pancreas was isolated according the previous study, with minor modification (54). Briefly, 200 mg frozen tissue were homogenized in 5 mL cellular lysis buffer containing an antioxidant (0.1 mM desferrioxamine) for 20 min, using a Cornless Polytron homogenizer. The homogenates were digested with proteinase K (17 μL, 20 mg/mL) at 65 °C for 1 h, followed by the addition of DNase-free RNase A (200 μL, 25 mg/mL) for an additional 30 min incubation at 37 °C. Proteins were removed by adding 2.1 mL protein precipitation solution followed by 10 min incubation on ice and centrifugation at 14,000 × g for 15 min at 4 °C. The supernatants were carefully transferred to a fresh tube, and DNA was recovered by precipitation in 200 mM NaCl and 2.5 volumes 100% ethanol. The floating DNA filament was recovered with a micropipette tip, washed twice with 70% cold ethanol, air-dried at room temperature, and resuspended in water. DNA concentration was determined by UV spectroscopy, and samples were stored at −80 °C until used.
Isolation and Analysis of 8-Nitro-G.
Twenty micrograms genomic DNA isolated from pancreas was digested with DNA Degradase Plus at 37 °C for 4 h. The enzyme was then removed by passing the solution through a 10-kDa MWCO ultrafilter, and the filtrates were separated by HPLCy. Filtrates were injected directly onto a preparative reverse-phase Nucleoside C18 column. 8-nitro-G was eluted according with the commercial 8-nitro-G (Santa Cruz, sc-202439). The purified 8-nitro-G was then subjected to a Q-TOF mass spectrometry with the following parameters: auto sampler temperature, 4 °C; injection volume, 4 µL; ESI mode and voltage, 4,000 V (+) ion mode; pump solvent, 50% H2O and 50% acetonitrile; flow rate, 0.5 mL/min. Data were acquired at a rate of 2.5 spectra/s with a stored mass range of m/z 50–1,500. Data were collected and analyzed using Agilent MassHunter Workstation Data software. The peak values of 8-nitro-G showed the relative abundance of 8-nitro-G derived from different samples. Unmodified guanine was used as the loading control.
Isolation and Culture of Mouse Primary Pancreatic Acinar Cells.
Isolation and culture of mouse primary pancreatic acinar cells were performed according to the previous report (55). Briefly, mouse pancreas was mechanically cut into pieces and digested in 1× HBSS containing 10 mM Hepes, 200 U/mL collagenase IA, and 0.25 mg/mL trypsin inhibitor for around 30 min at 37 °C. The enzymatic reaction was stopped by adding 10 mL cold washing solution (HBSS 1× containing 5% FBS and 10 mM Hepes). After washing and suspension, cell mixture was filtered by a 100-µm filter to retain the nondigested fragments. The isolated acini were seeded in a 6-well plate and cultured at 37 °C with 5% (vol/vol) CO2. Twenty-four hours after seeding, acini (in suspension) were transferred to the type I collagen-coated 6-well dish to eliminate the contaminants, and were cultured in the same condition as described earlier. The cells adhered to the type I collagen substrate after 2 d culture. Culture medium was changed every 3 d.
Cell Treatment.
For the ONOOCO2− treatment, 50 µM ONOO− was mixed with 1 mM sodium bicarbonate in cell culture medium according to the previous studies (56, 57). ONOOCO2−-treated cells were then subjected for other assays. For the recovery from ONOOCO2− treatment, cells were washed with fresh culture medium and cultured for 24 h. For the tempol treatment, primary pancreatic acinar cells were incubated in the presence of 100 µM tempol or tempol with the indicated concentrations during the culture.
Comet Assays.
Single-cell gel electrophoretic comet assay was performed under neutral condition to detect DSBs. Cells were collected and rinsed twice with ice-cold PBS; 2 × 104/mL cells were combined with 1% LMAgarose at 40 °C at a ratio of 1:3 (vol/vol) and immediately pipetted onto slides. For cellular lysis, the slides were immersed in the neutral lysis solution (2% sarkosyl, 0.5M Na2EDTA, 0.5 mg/mL proteinase K at pH 8.0) overnight at 37 °C in the dark, followed by washing in the rinse buffer (90 mM Tris buffer, 90 mM boric acid, 2mM Na2EDTA at pH 8.5) for 30 min with two repeats. The slides were then subjected to electrophoresis at 20 V (0.6 V/cm) for 25 min and stained with 2.5 µg/mL propidium iodide for 20 min. All images were taken with a fluorescence microscope and analyzed by Comet Assay IV software.
In Vivo Tempol Treatment.
Tempol treatment after weaning: tempol was added in the drinking water [0.05% (wt/vol)] for the newborn mice after weaning. Tempol treatment before birth: tempol was added in the drinking water [0.05% (wt/vol)] for the pregnant female mice until the pups were weaning. After weaning, tempol was directly administered to the pups through drinking water.
Statistical Analyses.
All experiments were performed in triplicate unless indicated otherwise. Means and SDs were plotted. Student’s t test was used for statistical analyses. The log-rank test was performed on the Kaplan-Meier survival curves.
Acknowledgments
We thank Dr. Marina Magliano for providing reagents and Dr. Muzaffer Kassab for proofreading the manuscript. This work was supported by grants from the National Institutes of Health (CA132755, CA130899, and CA187209, to X.Y.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1702156114/-/DCSupplemental.
References
- 1.Hezel AF, Kimmelman AC, Stanger BZ, Bardeesy N, Depinho RA. Genetics and biology of pancreatic ductal adenocarcinoma. Genes Dev. 2006;20:1218–1249. doi: 10.1101/gad.1415606. [DOI] [PubMed] [Google Scholar]
- 2.Jemal A, Siegel R, Xu J, Ward E. Cancer statistics, 2010. CA Cancer J Clin. 2010;60:277–300. doi: 10.3322/caac.20073. [DOI] [PubMed] [Google Scholar]
- 3.Warshaw AL, Fernández-del Castillo C. Pancreatic carcinoma. N Engl J Med. 1992;326:455–465. doi: 10.1056/NEJM199202133260706. [DOI] [PubMed] [Google Scholar]
- 4.Hidalgo M. Pancreatic cancer. N Engl J Med. 2010;362:1605–1617. doi: 10.1056/NEJMra0901557. [DOI] [PubMed] [Google Scholar]
- 5.Cowgill SM, Muscarella P. The genetics of pancreatic cancer. Am J Surg. 2003;186:279–286. doi: 10.1016/s0002-9610(03)00226-5. [DOI] [PubMed] [Google Scholar]
- 6.Caldas C, Kern SE. K-ras mutation and pancreatic adenocarcinoma. Int J Pancreatol. 1995;18:1–6. doi: 10.1007/BF02825415. [DOI] [PubMed] [Google Scholar]
- 7.Rustgi AK. Familial pancreatic cancer: Genetic advances. Genes Dev. 2014;28:1–7. doi: 10.1101/gad.228452.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Murphy KM, et al. Evaluation of candidate genes MAP2K4, MADH4, ACVR1B, and BRCA2 in familial pancreatic cancer: Deleterious BRCA2 mutations in 17% Cancer Res. 2002;62:3789–3793. [PubMed] [Google Scholar]
- 9.Jones S, et al. Exomic sequencing identifies PALB2 as a pancreatic cancer susceptibility gene. Science. 2009;324:217. doi: 10.1126/science.1171202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tischkowitz MD, et al. Analysis of the gene coding for the BRCA2-interacting protein PALB2 in familial and sporadic pancreatic cancer. Gastroenterology. 2009;137:1183–1186. doi: 10.1053/j.gastro.2009.06.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Venkitaraman AR. Cancer susceptibility and the functions of BRCA1 and BRCA2. Cell. 2002;108:171–182. doi: 10.1016/s0092-8674(02)00615-3. [DOI] [PubMed] [Google Scholar]
- 12.Scully R, Livingston DM. In search of the tumour-suppressor functions of BRCA1 and BRCA2. Nature. 2000;408:429–432. doi: 10.1038/35044000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wooster R, et al. Identification of the breast cancer susceptibility gene BRCA2. Nature. 1995;378:789–792. doi: 10.1038/378789a0. [DOI] [PubMed] [Google Scholar]
- 14.Zhang F, et al. PALB2 links BRCA1 and BRCA2 in the DNA-damage response. Curr Biol. 2009;19:524–529. doi: 10.1016/j.cub.2009.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xia B, et al. Control of BRCA2 cellular and clinical functions by a nuclear partner, PALB2. Mol Cell. 2006;22:719–729. doi: 10.1016/j.molcel.2006.05.022. [DOI] [PubMed] [Google Scholar]
- 16.Pellegrini L, et al. Insights into DNA recombination from the structure of a RAD51-BRCA2 complex. Nature. 2002;420:287–293. doi: 10.1038/nature01230. [DOI] [PubMed] [Google Scholar]
- 17.Sy SM, Huen MS, Chen J. PALB2 is an integral component of the BRCA complex required for homologous recombination repair. Proc Natl Acad Sci USA. 2009;106:7155–7160. doi: 10.1073/pnas.0811159106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xia B, et al. Fanconi anemia is associated with a defect in the BRCA2 partner PALB2. Nat Genet. 2007;39:159–161. doi: 10.1038/ng1942. [DOI] [PubMed] [Google Scholar]
- 19.Roy R, Chun J, Powell SN. BRCA1 and BRCA2: Different roles in a common pathway of genome protection. Nat Rev Cancer. 2011;12:68–78. doi: 10.1038/nrc3181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Moynahan ME, Pierce AJ, Jasin M. BRCA2 is required for homology-directed repair of chromosomal breaks. Mol Cell. 2001;7:263–272. doi: 10.1016/s1097-2765(01)00174-5. [DOI] [PubMed] [Google Scholar]
- 21.King MC, Marks JH, Mandell JB. New York Breast Cancer Study Group Breast and ovarian cancer risks due to inherited mutations in BRCA1 and BRCA2. Science. 2003;302:643–646. doi: 10.1126/science.1088759. [DOI] [PubMed] [Google Scholar]
- 22.Gayther SA, et al. Variation of risks of breast and ovarian cancer associated with different germline mutations of the BRCA2 gene. Nat Genet. 1997;15:103–105. doi: 10.1038/ng0197-103. [DOI] [PubMed] [Google Scholar]
- 23.Tryggvadóttir L, et al. Prostate cancer progression and survival in BRCA2 mutation carriers. J Natl Cancer Inst. 2007;99:929–935. doi: 10.1093/jnci/djm005. [DOI] [PubMed] [Google Scholar]
- 24.Howlett NG, et al. Biallelic inactivation of BRCA2 in Fanconi anemia. Science. 2002;297:606–609. doi: 10.1126/science.1073834. [DOI] [PubMed] [Google Scholar]
- 25.Couch FJ, et al. BRCA2 germline mutations in male breast cancer cases and breast cancer families. Nat Genet. 1996;13:123–125. doi: 10.1038/ng0596-123. [DOI] [PubMed] [Google Scholar]
- 26.Goggins M, Hruban RH, Kern SE. BRCA2 is inactivated late in the development of pancreatic intraepithelial neoplasia: Evidence and implications. Am J Pathol. 2000;156:1767–1771. doi: 10.1016/S0002-9440(10)65047-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Skoulidis F, et al. Germline Brca2 heterozygosity promotes Kras(G12D) -driven carcinogenesis in a murine model of familial pancreatic cancer. Cancer Cell. 2010;18:499–509. doi: 10.1016/j.ccr.2010.10.015. [DOI] [PubMed] [Google Scholar]
- 28.Guerra C, et al. Chronic pancreatitis is essential for induction of pancreatic ductal adenocarcinoma by K-Ras oncogenes in adult mice. Cancer Cell. 2007;11:291–302. doi: 10.1016/j.ccr.2007.01.012. [DOI] [PubMed] [Google Scholar]
- 29.Mujica VR, Barkin JS, Go VL. Study Group Participants Acute pancreatitis secondary to pancreatic carcinoma. Pancreas. 2000;21:329–332. doi: 10.1097/00006676-200011000-00001. [DOI] [PubMed] [Google Scholar]
- 30.Lowenfels AB, et al. International Pancreatitis Study Group Pancreatitis and the risk of pancreatic cancer. N Engl J Med. 1993;328:1433–1437. doi: 10.1056/NEJM199305203282001. [DOI] [PubMed] [Google Scholar]
- 31.Taylor EL, Megson IL, Haslett C, Rossi AG. Nitric oxide: A key regulator of myeloid inflammatory cell apoptosis. Cell Death Differ. 2003;10:418–430. doi: 10.1038/sj.cdd.4401152. [DOI] [PubMed] [Google Scholar]
- 32.Bogdan C. Nitric oxide and the immune response. Nat Immunol. 2001;2:907–916. doi: 10.1038/ni1001-907. [DOI] [PubMed] [Google Scholar]
- 33.Sawa T, Ohshima H. Nitrative DNA damage in inflammation and its possible role in carcinogenesis. Nitric Oxide. 2006;14:91–100. doi: 10.1016/j.niox.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 34.Lim CH, Dedon PC, Deen WM. Kinetic analysis of intracellular concentrations of reactive nitrogen species. Chem Res Toxicol. 2008;21:2134–2147. doi: 10.1021/tx800213b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Steward MC, Ishiguro H, Case RM. Mechanisms of bicarbonate secretion in the pancreatic duct. Annu Rev Physiol. 2005;67:377–409. doi: 10.1146/annurev.physiol.67.031103.153247. [DOI] [PubMed] [Google Scholar]
- 36.Lee MG, Ohana E, Park HW, Yang D, Muallem S. Molecular mechanism of pancreatic and salivary gland fluid and HCO3 secretion. Physiol Rev. 2012;92:39–74. doi: 10.1152/physrev.00011.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Geacintov NE, Broyde S, editors. The Chemical Biology of DNA Damage. Wiley-VCH; Weinheim, Germany: 2010. pp. 7–12. [Google Scholar]
- 38.Wiseman H, Halliwell B. Damage to DNA by reactive oxygen and nitrogen species: Role in inflammatory disease and progression to cancer. Biochem J. 1996;313:17–29. doi: 10.1042/bj3130017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jortner J, Bixon M, Langenbacher T, Michel-Beyerle ME. Charge transfer and transport in DNA. Proc Natl Acad Sci USA. 1998;95:12759–12765. doi: 10.1073/pnas.95.22.12759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Caldecott KW. Single-strand break repair and genetic disease. Nat Rev Genet. 2008;9:619–631. doi: 10.1038/nrg2380. [DOI] [PubMed] [Google Scholar]
- 41.Maynard S, Schurman SH, Harboe C, de Souza-Pinto NC, Bohr VA. Base excision repair of oxidative DNA damage and association with cancer and aging. Carcinogenesis. 2009;30:2–10. doi: 10.1093/carcin/bgn250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hingorani SR, et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell. 2003;4:437–450. doi: 10.1016/s1535-6108(03)00309-x. [DOI] [PubMed] [Google Scholar]
- 43.Davies AA, et al. Role of BRCA2 in control of the RAD51 recombination and DNA repair protein. Mol Cell. 2001;7:273–282. doi: 10.1016/s1097-2765(01)00175-7. [DOI] [PubMed] [Google Scholar]
- 44.Wong AK, Pero R, Ormonde PA, Tavtigian SV, Bartel PL. RAD51 interacts with the evolutionarily conserved BRC motifs in the human breast cancer susceptibility gene brca2. J Biol Chem. 1997;272:31941–31944. doi: 10.1074/jbc.272.51.31941. [DOI] [PubMed] [Google Scholar]
- 45.Rowley M, et al. Inactivation of Brca2 promotes Trp53-associated but inhibits KrasG12D-dependent pancreatic cancer development in mice. Gastroenterology. 2011;140:1303–1313. doi: 10.1053/j.gastro.2010.12.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Marzinzig M, et al. Improved methods to measure end products of nitric oxide in biological fluids: Nitrite, nitrate, and S-nitrosothiols. Nitric Oxide. 1997;1:177–189. doi: 10.1006/niox.1997.0116. [DOI] [PubMed] [Google Scholar]
- 47.Ohshima H, Sawa T, Akaike T. 8-nitroguanine, a product of nitrative DNA damage caused by reactive nitrogen species: Formation, occurrence, and implications in inflammation and carcinogenesis. Antioxid Redox Signal. 2006;8:1033–1045. doi: 10.1089/ars.2006.8.1033. [DOI] [PubMed] [Google Scholar]
- 48.Collins MA, et al. Oncogenic Kras is required for both the initiation and maintenance of pancreatic cancer in mice. J Clin Invest. 2012;122:639–653. doi: 10.1172/JCI59227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Daniluk J, et al. An NF-κB pathway-mediated positive feedback loop amplifies Ras activity to pathological levels in mice. J Clin Invest. 2012;122:1519–1528. doi: 10.1172/JCI59743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pinho AV, Chantrill L, Rooman I. Chronic pancreatitis: A path to pancreatic cancer. Cancer Lett. 2014;345:203–209. doi: 10.1016/j.canlet.2013.08.015. [DOI] [PubMed] [Google Scholar]
- 51.David SS, O’Shea VL, Kundu S. Base-excision repair of oxidative DNA damage. Nature. 2007;447:941–950. doi: 10.1038/nature05978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature. 2009;461:1071–1078. doi: 10.1038/nature08467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hahn SA, et al. BRCA2 germline mutations in familial pancreatic carcinoma. J Natl Cancer Inst. 2003;95:214–221. doi: 10.1093/jnci/95.3.214. [DOI] [PubMed] [Google Scholar]
- 54.Pang B, et al. Lipid peroxidation dominates the chemistry of DNA adduct formation in a mouse model of inflammation. Carcinogenesis. 2007;28:1807–1813. doi: 10.1093/carcin/bgm037. [DOI] [PubMed] [Google Scholar]
- 55.Gout J, et al. Isolation and culture of mouse primary pancreatic acinar cells. J Vis Exp. 2013;78:50514. doi: 10.3791/50514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yermilov V, Yoshie Y, Rubio J, Ohshima H. Effects of carbon dioxide/bicarbonate on induction of DNA single-strand breaks and formation of 8-nitroguanine, 8-oxoguanine and base-propenal mediated by peroxynitrite. FEBS Lett. 1996;399:67–70. doi: 10.1016/s0014-5793(96)01288-4. [DOI] [PubMed] [Google Scholar]
- 57.Yun BH, Geacintov NE, Shafirovich V. Generation of guanine-thymidine cross-links in DNA by peroxynitrite/carbon dioxide. Chem Res Toxicol. 2011;24:1144–1152. doi: 10.1021/tx200139c. [DOI] [PMC free article] [PubMed] [Google Scholar]