

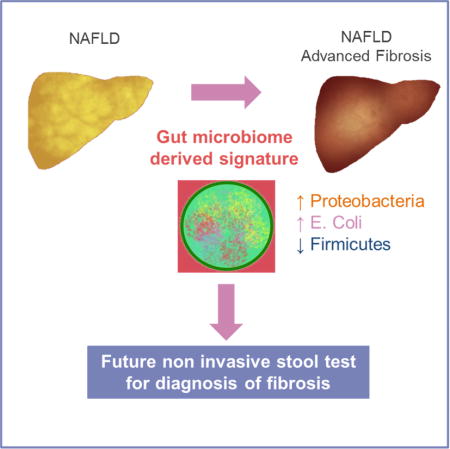
SUMMARY
The presence of advanced fibrosis in nonalcoholic fatty liver disease (NAFLD) is the most important predictor of liver mortality. There are limited data on the diagnostic accuracy of gut microbiota derived signature for predicting the presence of advanced fibrosis. In this prospective study, we characterized the gut microbiome compositions using whole-genome shotgun sequencing of DNA extracted from stool samples. This study included 86 uniquely well-characterized patients with biopsy-proven NAFLD, 72 of which had mild/moderate (stage 0–2 fibrosis) NAFLD, and 14 had advanced fibrosis (stage 3 or 4 fibrosis). We identified a set of forty features (p-value <0.006), which included 37 bacterial species that were used to construct a Random Forest classifier model to distinguish mild/moderate NAFLD from advanced fibrosis. The model had a robust diagnostic accuracy (AUC 0.936) for detecting advanced fibrosis. This study provides preliminary evidence for a novel fecal-microbiome derived metagenomic signature to detect advanced fibrosis in NAFLD.
Keywords: NASH, cirrhosis, biomarker, microbiome, non-invasive, fatty liver, hepatic steatosis, liver disease, hepatitis
Graphical abstract
INTRODUCTION
Nonalcoholic fatty liver disease (NAFLD) is the most common cause of chronic liver disease in the United States, affecting approximately 80–100 million Americans. (Loomba and Sanyal, 2013; Rinella, 2015; Vernon et al., 2011) NAFLD is broadly sub-divided into two phenotypes: nonalcoholic fatty liver (NAFL), which is considered the non-progressive subtype, and nonalcoholic steatohepatitis (NASH), which is considered the progressive subtype that can lead to cirrhosis, hepatocellular carcinoma and liver-related death. (Adams et al., 2005; Bhala et al., 2011; Matteoni et al., 1999; Singh et al., 2015; Wong et al., 2010) The presence of advanced fibrosis has consistently been identified as the most important predictor for liver-related events and complications in NAFLD, and therefore represents the most clinically meaningful determinant of long-term outcomes. (Angulo et al., 2015; Dulai et al., 2017; Ekstedt et al., 2015; Younossi et al., 2015) Early identification of the presence of advanced fibrosis using non-invasive modalities is a major unmet need in the field. (Decaris et al., 2016; Dulai et al., 2016)
There has been an increased interest in understanding the role of the microbiome in metabolic disorders, with studies trying to elucidate the functional significance of stool microbiome in the progression of liver disease in NAFLD and other chronic liver diseases. (Anand et al., 2016; Gill et al., 2006; Human Microbiome Jumpstart Reference Strains et al., 2010; Qin et al., 2014; Zhu et al., 2013) Specifically, a dysbiotic microbiome is often observed among obese individuals, and is considered to be one of the major risk factors for NAFLD. (Turnbaugh et al., 2009) Both obesity and NAFLD are associated with a higher proportion of Gram-negative bacterial species in the gut microbiome. (Zhu et al., 2013) Microbial populations of NASH patients have been suggested to have a higher ability to produce alcohol,(Qin et al., 2014) and NASH has been associated with disrupted bile acid profiles in serum and feces, a finding which is thought to be due to reduced bacterial diversity and loss of gut microbiota members that are responsible for the generation of secondary bile acids. (Kakiyama et al., 2013) In addition, some members of the gut microbiota can convert choline to trimethylamine, which can induce liver injury leading to steatohepatitis. (Chen et al., 2016) Therefore, changes in gut microbiome have been linked to NAFLD, and NASH. (Betrapally et al., 2016; Henao-Mejia et al., 2012)
Qin and colleagues (Qin et al., 2014) recently reported on a Chinese cirrhosis cohort and observed that a specific gut microbiome signature is present in individuals with cirrhosis. However, this study included diverse etiologies of cirrhosis (alcoholic liver disease, hepatitis B and hepatitis C), and did not provide gut microbiome signatures that are specific to NAFLD, and NAFLD related cirrhosis. It is likely that the gut microbiome signatures of advanced fibrosis in patients with NAFLD who are residing in the United States would be very different than gut microbiome signature of patients with cirrhosis predominantly due to hepatitis B who are residing in China.
Given the importance of advanced fibrosis in NAFLD, and the association between specific microbial populations and NASH, a strong rationale exists for the development of a panel of gut-microbiome derived biomarkers that can be used to predict the presence of advanced fibrosis in NAFLD. Therefore, we studied the stool microbiome and serum metabolome of a well-characterized, prospective cohort of patients with biopsy-proven NAFLD. Our aim was to develop a panel of gut-microbiome derived biomarkers for the non-invasive diagnosis of advanced fibrosis in NAFLD.
RESULTS
Baseline characteristics of the study cohort
This prospective study included 86 patients (female 56%) with biopsy-proven NAFLD, 72 patients had stage 0–2 fibrosis and were classified as mild/moderate NAFLD (Group G1), and 14 patients had stage 3–4 fibrosis and were classified as advanced NAFLD (Group G2). Table 1 provides a detailed demographic, clinical, biochemical and metabolic profile of the entire cohort classified by the advanced fibrosis status. Patients with advanced fibrosis were more likely to be older, Hispanic, diabetic, and had higher ALT, higher AST, lower platelet count, and a higher HbA1c than those without advanced fibrosis. In addition, although the two groups had similar BMI, patients with advanced fibrosis had higher waist circumference. Table 2 provides detailed histologic differences in the study cohort classified by the advanced fibrosis status. Patients with advanced fibrosis were more likely to have more severe lobular and portal inflammation and ballooning than those without advanced fibrosis.
Table 1.
Baseline Characteristics of patients with biopsy-proven NAFLD
| Characteristics | All patients N=86 | Stage 0–2 NAFLD N=72 | Stage 3–4 Advanced Fibrosis N=14 | p-value (Student’s t-test) |
|---|---|---|---|---|
| Demographics | ||||
| Age (mean ± SD) | 48 ± 1.4 | 49.3 ± 12.6 | 63.4 ± 3 | 1.5e-12 |
| Male n (%) | 38 (44.2%) | 36 (50 %) | 2 (14.3%) | 0.030 |
| White n (%) | 40 (46.5%) | 33 (40.2%) | 7 (50%) | 1.000 |
| Hispanic n (%) | 29 (33.7%) | 23 (31.9%) | 6 (42.9%) | 0.630 |
| Clinical | ||||
| Type 2 diabetes n (%) | 20 (23.3%) | 14 (19.4%) | 6 (42.9%) | 0.126 |
| Anthropometric (mean ± SD) | ||||
| Body mass index (kg/m2) | 31.2 ± 5.5 | 31.0 ± 5.4 | 32.2 ± 6.0 | 0.503 |
| Waist circumference (cm) | 102.4 ± 16.3 | 101.5 ± 19.2 | 107.1 ± 17.3 | 0.823 |
| Hepatology panel (mean ± SD) | ||||
| AST (U/L) | 41.0 ± 30.0 | 35 ± 24.5 | 72 ± 36.8 | 0.002 |
| ALT (U/L) | 57.0 ± 55.2 | 53.8 ± 54.3 | 73.8 ± 55.2 | 0.253 |
| AST/ALT | 0.72 | 0.65 | 0.98 | |
| Bilirubin, direct (mg/dL) | 0.16 ± 0.12 | 0.13 ± 0.06 | 0.29 ± 0.23 | 0.033 |
| Hematology and other laboratory studies (mean ±SD) | ||||
| White blood cells (1000/mm3) | 6.3 ± 1.7 | 6.3 ± 1.6 | 6.2 ± 2.2 | 0.843 |
| Platelet count (1000/mm3) | 250.5 ± 79.6 | 254.5 ± 64.3 | 230.2 ± 135.2 | 0.521 |
| Total cholesterol (mg/dL) | 190.5 ± 42.3 | 193.9 ± 42.2 | 173.0 ± 39.6 | 0.089 |
| HDL cholesterol (mg/dL) | 48.9 ± 16.0 | 48.9 ± 15.9 | 48.6 ± 17.1 | 0.942 |
| LDL cholesterol (mg/dL) | 112.4 ± 36.2 | 116 ± 34.7 | 94.9 ± 39.8 | 0.178 |
| Triglycerides (mg/dL) | 159.9 ± 95.8 | 160.6 ± 98.3 | 156.6 ± 84.1 | 0.565 |
| HbA1c (%) | 6.2 ± 0.9 | 6.0 ± 0.9 | 6.7 ± 0.8 | 0.016 |
| Fasting serum insulin (lU/mL) | 28.1 ± 26.1 | 25.1 ± 22 | 43.9 ± 39.1 | 0.130 |
| Ferritin (ng/mL)* | 199.8 ± 180.2 | 210 ± 189.4 | 132 ± 73.2 | 0.032 |
Abbreviations: SD: standard deviation, AST: aspartate aminotransferase, ALT: alanine aminotransferase, HDL: high density lipoprotein, LDL: low density lipoprotein.
ferritin data were missing in 14 patients including 6 with advanced fibrosis.
Table 2.
Histological Features of patients with NAFLD by fibrosis status
| Histological Feature* | Definition | Score/Code | Stage 0–2 Healthy, Moderate Fibrosis N=72 | Stage 3–4 Advanced Fibrosis N=14 | p-value (χ2) | Stage 0–2 Fibrosis N=XX |
|---|---|---|---|---|---|---|
| Steatosis: | 2.6e-14 | |||||
| Grade | Low- to medium-power evaluation of parenchymal involvement by steatosis | |||||
| <5% | 0 | 4 (5.6%) | 1 (7.1%) | |||
| 5%–33% | 1 | 25 (34.7%) | 9 (64.3%) | |||
| >33%–66% | 2 | 29 (40.3%) | 1 (7.1%) | |||
| >66% | 3 | 13 (18.1%) | 2 (14.3%) | |||
| Inflammation: | ||||||
| Lobular inflammation | Overall assessment of all inflammatory foci (no. foci per 200X field) | 2.6e-14 | ||||
| No foci | 0 | 4 (5.6%) | 0 (0%) | |||
| <2 foci | 1 | 39 (54.2%) | 2 (14.3%) | |||
| 2–4 foci | 2 | 26 (36.1%) | 9 (64.3%) | |||
| >4 foci | 3 | 2 (2.8%) | 0 (0%) | |||
| Portal Inflammation | Assessed from low magnification | 1.9e-11 | ||||
| None | 0 | 15 (20.8%) | 2 (14.3%) | |||
| Mild | 1 | 42 (58.3%) | 3 (21.4%) | |||
| Greater than mild | 2 | 3 (4.2%) | 4 (28.6%) | |||
| Liver cell injury: | 4.2e-06 | |||||
| Ballooning‡ | None | 0 | 26 (36.1%) | 1 (7.1%) | ||
| Few balloon cells | 1 | 32 (44.4%) | 4 (28.6%) | |||
| Many cells/prominent ballooning | 2 | 8 (11.1%) | 7 (50%) |
Determination of histological features from centrally reviewed biopsy using the NASH Clinical Research Network Scoring System (Kleiner et al, Hep 2005)
Ballooning classification: few indicates rare but definite ballooned hepatocytes, as well as cases that are diagnostically borderline
The “None to rare” category is meant to alleviate the need for time-consuming searches for rare examples or deliberation over diagnostically borderline changes. If the feature is identified after a reasonable search, it should be coded as “many.”
Differences in the taxonomic composition of stool derived metagenomes between mild/moderate NAFLD versus advanced fibrosis
Gut microbiome compositions of the patients were determined using whole-genome shotgun sequencing of DNA extracted from their stool samples. The 86 stool samples yielded an average of 6.58 × 109 bases per sample (after trimming low-quality bases and removing human sequences). At the phylum level, the gut microbiomes in both groups were dominated by members of Firmicutes and Bacteroidetes, followed by Proteobacteria and Actinobacteria in much lower abundances (Table 3). Furthermore, both Firmicutes and Proteobacteria were differentially abundant across the two groups (p-value < 0.05), with Firmicutes being higher in mild/moderate NAFLD (G1) while Proteobacteria was higher in advanced fibrosis (G2). At the species level, Eubacterium rectale (2.5% median relative abundance) and Bacteroides vulgatus (1.7%) were the most abundant organisms in mild/moderate NAFLD (G1) while B. vulgatus (2.2%) and Escherichia coli (1%) were the most abundant in advanced fibrosis (G2). Ruminococcus obeum CAG: 39, R. obeum, and E. rectale were significantly lower in advanced fibrosis than mild/moderate NAFLD.
Table 3. Taxonomic Composition.
Median and standard deviations of relative abundances of top 4 phyla found in all 86 samples and representative species from the first 3 phyla.
| Phylum | G1 Median (SD) | G2 Median (SD) | p-value |
|---|---|---|---|
|
|
|||
| Firmicutes | 58.81% (20.8) | 42.61% (23.9) | 0.01520 |
| Proteobacteria | 1.85% (15.3) | 4.54% (22.9) | 0.04004 |
| Bacteroidetes | 23.62% (18.1) | 28.46% (27.4) | 0.57840 |
| Actinobacteria | 2.67% (4.1) | 2.02% (7.4) | 0.78340 |
| Species | |||
| Ruminococcus obeum CAG:39 | 0.06% (0.54) | 0.01% (0.02) | 0.00005* |
| Ruminococcus obeum | 0.29% (0.90) | 0.11% (0.15) | 0.00009* |
| Eubacterium rectale | 2.56% (5.66) | 0.12% (1.35) | 0.00009* |
| Faecalibacterium prausnitzii | 1.63% (4.07) | 0.34% (3.07) | 0.01961 |
| Escherichia coli | 0.29% (15.8) | 0.99% (25.3) | 0.44330 |
| Bacteroides vulgatus | 1.76% (4.56) | 2.19% (7.04) | 0.85610 |
Significant p-value after multiple test correction
Diagnostic accuracy of the metagenomics derived gut microbiome model for the detection of advanced fibrosis
The RF model selected 37 species together with Shannon diversity, Age, and BMI as the most important features. Age was observed to be the top predictor in nearly all of the RFs in the training phase. The forty selected features were determined from the feature elimination step and the best performing model was selected as the final model. This model had a robust and statistically significant diagnostic accuracy of AUC 0.936 (Figures 1 and 2).
Figure 1.
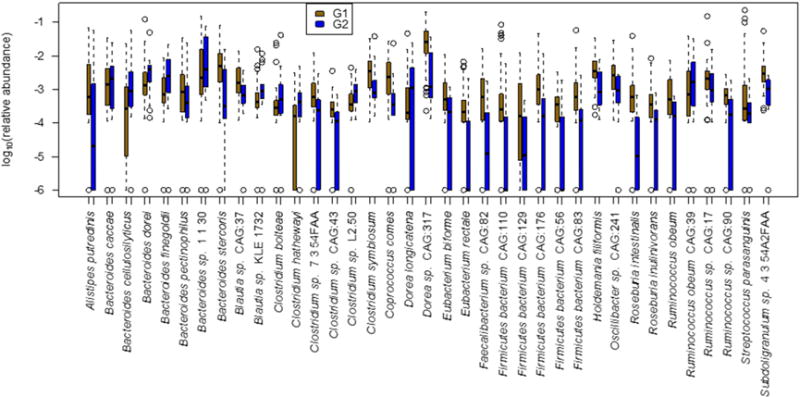
Boxplots of the relative abundances of 37 species selected for the random forest model used to distinguish samples in the mild/moderate group (G1) from those in the advanced fibrosis group (G2). Sample diversity and patient age and BMI were also selected as important features by the random forest model (boxplots not shown).
Figure 2.
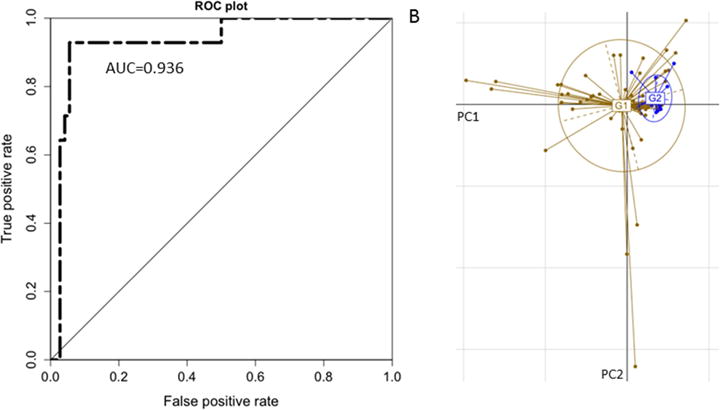
Performance of the Random Forest model and Ordination of the NAFLD samples. (A) Receiver Operating Characteristic (ROC) curve of the final Random Forest model constructed using the relative abundances of the 37 selected species together with Shannon diversity, age, and BMI, which were also selected as important features by the training process. (B) Principal Component Analysis ordination of the NAFLD samples. Samples from the mild/moderate group (G1) are in brown while samples from the advanced fibrosis group (G2) are in blue. The samples were plotted using the first two principal components PC1 and PC2. These components, respectively, account for 34.3% and 19.6% of the total variance.
From the 37 species selected by the optimized model, eight species were more than two-fold more abundant in advanced fibrosis (G2) compared to mild/moderate NAFLD (G1), while 22 species were more than two-fold abundant in mild/moderate NAFLD (G1) compared to advanced fibrosis (G2) (Table 4). The orthogonal machine learning method resulted in a model whose final feature set had a high degree of concordance with the features in the RF based model and with a similarly high AUC (Figure S1).
Table 4. Important species selected by Random Forest.
Species list, mean decrease in Gini index and log ratios of median species abundances in G2 and G1. The log ratio was not determined (ND) for a few species due to zero median values in G2.
| Species | MeanDecreaseGini | log2 (G2/G1) |
|---|---|---|
| Dorea sp. CAG:317 | 0.06 | 2.50 |
| Bacteroides cellulosilyticus | 0.11 | 1.86 |
| Bacteroides finegoldii | 0.31 | 1.77 |
| Bacteroides dorei | 0.18 | 1.59 |
| Streptococcus parasanguinis | 0.14 | 1.49 |
| Clostridium symbiosum | 0.15 | 1.35 |
| Clostridium sp. 7_3_54FAA | 0.16 | 1.34 |
| Clostridium bolteae | 0.36 | 1.03 |
| Clostridium hathewayi | 0.14 | 0.88 |
| Bacteroides stercoris | 0.12 | 0.87 |
| Bacteroides caccae | 0.10 | 0.68 |
| Eubacterium biforme | 0.06 | −0.50 |
| Subdoligranulum sp. 4_3_54A2FAA | 0.05 | −1.00 |
| Bacteroides sp. 1_1_30 | 0.09 | −1.05 |
| Faecalibacterium sp. CAG:82 | 0.10 | −1.16 |
| Clostridium sp. L2–50 | 0.07 | −1.16 |
| Blautia sp. KLE 1732 | 0.12 | −1.22 |
| Clostridium sp. CAG:43 | 0.14 | −1.38 |
| Firmicutes bacterium CAG:56 | 0.14 | −1.39 |
| Ruminococcus sp. CAG:17 | 0.15 | −1.46 |
| Ruminococcus obeum | 0.56 | −1.47 |
| Alistipes putredinis | 0.09 | −1.48 |
| Roseburia inulinivorans | 0.22 | −1.53 |
| Ruminococcus sp. CAG:90 | 0.10 | −1.64 |
| Bacteroides pectinophilus | 0.35 | −1.89 |
| Roseburia intestinalis | 0.19 | −2.05 |
| Coprococcus comes | 0.18 | −2.10 |
| Oscillibacter sp. CAG:241 | 0.36 | −2.26 |
| Firmicutes bacterium CAG:83 | 0.27 | −2.69 |
| Dorea longicatena | 0.24 | −2.77 |
| Firmicutes bacterium CAG:129 | 0.25 | −3.00 |
| Ruminococcus obeum CAG:39 | 2.37 | −3.53 |
| Blautia sp. CAG:37 | 0.11 | −3.82 |
| Eubacterium rectale | 0.68 | −4.40 |
| Firmicutes bacterium CAG:176 | 0.05 | ND |
| Firmicutes bacterium CAG:110 | 0.13 | ND |
| Holdemania filiformis | 0.21 | ND |
Microbial metabolism and function
The comparison between metabolites detected and metabolites predicted from the microbial pathways reconstructed from stool metagenome data yielded 89 metabolites (Table S1, Figure S2) and included several known to be produced by both host and microbes. A differential analysis identified 11 metabolites whose abundances (peak intensities) are significantly different between mild/moderate NAFLD (G1) and advanced fibrosis (G2) (Wilcoxon rank sum corrected for FDR and α = 0.05). (Figure 3) In this set, two metabolites (associated with nucleoside metabolism) were enriched in mild/moderate NAFLD (G1), while nine metabolites (associated with amino acids and carbon metabolism) were enriched in advanced fibrosis (G2). Though its differential abundance was not statistically significant (Table S1), the metabolite with the highest fold increase in advanced fibrosis (G2) was 3-phenylpropanoate, a metabolite produced by anaerobic bacteria. (Moss et al., 1970; Wikoff et al., 2009)
Figure 3.
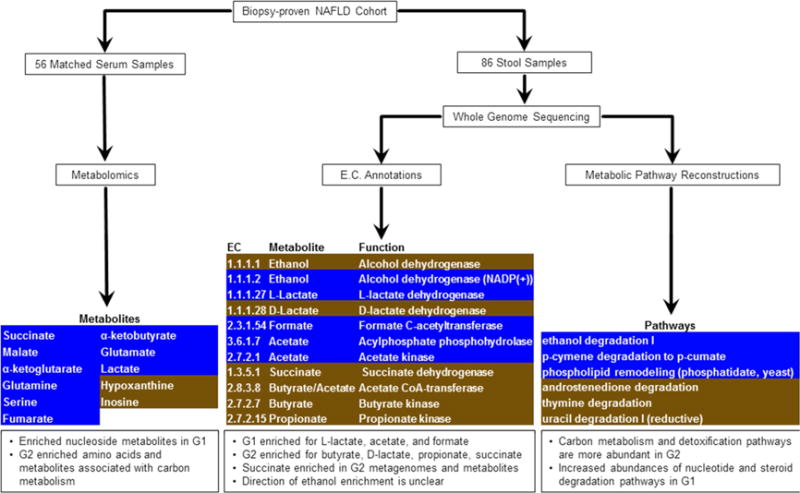
Overview of metabolomic and metagenomic analyses of Biopsy-proven NAFLD patients. Serum and stool samples from a cohort of 86 patients were analyzed for their metabolic and functional content. [Left] The metabolic profiles of 56 serum samples detected several differentially abundant metabolites, after multiple test correction. These are highlighted, with G1 enriched in brown and G2 enriched in blue. [Center] ORF sequences identified from whole genome sequencing of 86 stool samples were used to compute relative abundances of enzymes involved in SCFA production. Several enzymes were enriched in either G1 (brown) or G2 (blue), though they were not statistically significant after multiple test correction. [Right] Metabolic pathways were reconstructed from whole genome sequencing of 86 stool samples. Pathway abundance was calculated by summing the abundances of species in which the pathway was reconstructed. Several pathways were enriched in G1 (brown) or G2 (blue), though these were not statistically significant after multiple test correction.
We did not identify any pathways, protein families, or enzymes whose differential abundances across mild/moderate NAFLD (G1) and advanced fibrosis (G2) were statistically significant (after multiple test correction). However, an examination of pathway abundances showed that advanced fibrosis (G2) had an increased abundance of pathways associated with carbon metabolism and detoxification, while mild/moderate NAFLD (G1) had an increased abundance of pathways associated with nucleotide and steroid degradation (Figure 3, Tables S2–S3). An evaluation of the protein families and enzymes associated with Short-Chain Fatty Acid (SCFA) production suggested that mild/moderate NAFLD (G1) had higher abundances of enzymes associated with lactate, acetate, and formate, while advanced fibrosis (G2) had higher abundances of enzymes for butyrate, D-lactate, propionate, and succinate (Figure 3, Tables S2–S3). The trend for the abundances of ethanol metabolism enzymes in G1 or G2 was not as clear, with enzyme EC 1.1.1.1 (Alcohol dehydrogenase) increased in G2, while enzyme EC 1.1.1.2 (Alcohol dehydrogenase NADP(+)) was increased in G1.
Validation of model and microbial signature
The resulting AUC of the models made by the trained RF on data from the 16 healthy older twin individuals was 0.81. Furthermore, the presence of a strong microbial signature was found by two orthogonal methods. We built a new RF model for Cohort B (NASH cirrhosis/advanced fibrosis [n=16] and control [33] samples) who all were 60 years or older. The patient data and species abundances were used to train this model in the same manner described and had an AUC of 0.88 after a feature elimination step (9 features selected, p-value < 0.0001). From the nine microbial species selected by this model (Table S4), seven overlap with the 37 species selected by our original model and this overlap was statistically significant (p-value < 0.0008). A similar microbial signature was further validated by applying SVM to the original dataset, and looking for the overlap of selected features. The trained SVM selected 18 species as the most important predictors (Table S5) and 12 of those species overlapped with the species found as features in the original RF model (p-value < 0.00006).
Sensitivity analyses
We conducted sensitivity analyses after adjustment for diabetes and the results remained unchanged. We also assessed HbA1c as a continuous trait and the models remained robust and consistent. We also assessed whether presence of metformin use had any significant effect on the model as recent studies have suggested that it may modify the gut microbiome (Forslund et al., 2015; Mardinoglu et al., 2016). The results remained consistent even after adjustment for metformin use with adjusted AUROC 0.94.
DISCUSSION
Utilizing this well-characterized cohort, we describe the diagnostic test accuracy of a panel of gut microbiome-derived biomarkers for the detection of advanced fibrosis in NAFLD. The novelty of this study is as follows: approaches to non-invasive detection of advanced fibrosis are major unmet need and here we present proof of concept data to support the development of stool based tests to screen for advanced fibrosis or cirrhosis in future. In this study, we found that the gut microbiomes in NAFLD is dominated by members of Firmicutes and Bacteroidetes, followed by Proteobacteria and Actinobacteria in much lower abundances (Table 3). However, as the disease progresses from mild/moderate NAFLD to advanced fibrosis, the Proteobacteria phylum has a statistically significant increase in abundance while the Firmicutes phylum decrease.
At the species level, E. rectale (2.5% median relative abundance) and B. vulgatus (1.7%) were the most abundant organisms in mild/moderate NAFLD while B. vulgatus (2.2%) and E. coli (1%) were the most abundant in advanced fibrosis. None of the patients with advanced fibrosis had ascites or any evidence of hepatic decompensation but still had higher E. coli abundance (though the abundance increase was not statistically significant). This increased abundance of E. coli in advanced fibrosis has potential clinical implications. To our astonishment, these data suggest that E. coli dominance occurs much earlier in the stage of fibrosis progression and supports the hypothesis that dysbiosis may precede development of portal hypertension. Although this provides evidence of temporal association between E. coli and portal hypertension it does not imply causality.
We observed a decrease of Gram-positive Firmicutes and an increase of Gram-negative Proteobacteria (including E.coli) in patients with advanced NASH fibrosis. This suggests that the microbiota shifts toward more Gram-negative microbes. Fecal transplantation of Gram-negative bacteria including Proteobacteria resulted in a significant increase cholestatic liver fibrosis when compared with mice transplanted with Gram-positive bacteria. (De Minicis et al., 2014) In addition, several preclinical studies mechanistically showed that LPS, a cell wall component of Gram-negative bacteria, causes progression of liver fibrosis. (Affo et al., 2014; Bai et al., 2016; Liu et al., 2016; Seki et al., 2007) Lelouvier and colleagues have also recently reported that Proteobacteria are increased in morbidly obese individuals undergoing bariatric surgery who have presence of fibrosis(Lelouvier et al., 2016). Thus, dysbiosis with predominant Gram-negative bacteria might contribute to liver fibrosis.
Our study builds on previous seminal studies conducted by other independent groups. Using quantitative genomics (similar to this study), Qin and colleagues characterized the gut microbiome of 98 Chinese patients with various etiologies of cirrhosis (predominantly viral hepatitis and alcoholic cirrhosis) of the liver that included both patients with compensated and decompensated cirrhosis, and compared it with 83 healthy controls. (Qin et al., 2014) They demonstrated that oral bacteria take over the gut microbiome in patients with cirrhosis with and without portal hypertension. Bajaj and colleagues have characterized the association of gut microbiome with hepatic decompensation in patients with diverse etiologies of liver disease. (Bajaj et al., 2014) Using 16S rRNA gene sequencing, Boursier and colleagues characterized the association between severity of NAFLD and gut dysbiosis in patients with biopsy-proven NAFLD. (Boursier et al., 2016) They imputed metagenomic functions from the 16S sequence data. The novelty of our study is that we utilized quantitative metagenomic sequencing in well-characterized patients with biopsy-proven NAFLD to examine the diagnostic test accuracy of microbiome-derived signature for the presence of advanced fibrosis, and then we also assessed serum metabolites and integrated the stool microbiome data with the serum metabolomics. We aimed to specifically address the role of gut dysbiosis in the progression of liver disease to advanced fibrosis and cirrhosis before the onset of clinically significant portal hypertension.
Qin and colleagues studied metagenomics profiling in Chinese patients with cirrhosis due to diverse etiologies but mainly included patients with viral hepatitis and alcoholic liver disease with a small subset of patients who had NAFLD cirrhosis. (Qin et al., 2014) Liver histology data and detailed liver disease characterization were not available in that study, and no advanced MR imaging assessment or serum metabolomics was performed. Bajaj and colleagues conducted studies in a diverse group of patients with cirrhosis with or without hepatic decompensation and assessed association with 16S profiling. (Bajaj et al., 2014) Boursier and colleagues conducted a study using 16S RNA sequencing platform in patients with biopsy-proven NAFLD and imputed signature of the 16S derived profiling and assessed differences in NAFLD patients with either NASH and stage 2 or higher fibrosis versus those with stage 0–1 fibrosis. (Boursier et al., 2016) These seminal studies have provided novel insights into the understanding of microbiome in liver disease. Our study fills the gap in knowledge by providing a metagenomics-based profiling in extremely well-characterized patients with biopsy-proven NAFLD who were further phenotyped using advanced MR imaging to examine the diagnostic accuracy of a panel of stool microbiome derived biomarker panel to detect advanced fibrosis. We then integrated the stool microbiome with metabolomics conducted in serum of these patients. This is the first study in NAFLD to integrate stool metagenomic profiling with serum metabolomics in this population. Therefore, albeit the results are preliminary, the study is novel in it’s design and innovative in characterization of NAFLD.
We acknowledge the following strengths and limitations of our study. Limitations of the study include single center study with expertise in clinical investigation of NAFLD that may potentially limit generalizability of the findings, relatively small cohort size for a clinical study, and hurdles in development of an easy to use diagnostic test using these methods to diagnose advanced fibrosis. It is possible that there are additional microbial species that could be used as indicators of disease status that were missed by virtue of the chosen cohort or the depth of sequencing performed in this study, and finally, the association does not suggest causality. We acknowledge that this study may be underpowered despite the fact that till date it is the largest metagenomics stool microbiome profiling in patients with biopsy-proven NAFLD. Therefore, further multicenter studies including larger number of patients with biopsy-proven NAFLD are needed to validate these findings.
Strengths of the study included prospective study design, detailed phenotyping of the biopsy-proven NAFLD cohort as well as the age-balanced subset assessment, utilization of metagenomics sequencing rather than 16S rRNA gene sequencing (which is known to have major limitations in terms of data interpretation) with serum metabolomics, and assessment of accuracy using AUROC. Here, we describe an investigation of stool metagenomes from a well phenotyped NAFLD cohort and identify 37 microbial species that are differentially present in the different stages of the disease.
It is also plausible that some of the initial findings were reflecting differences in age, and may not be specific to fibrosis stage. This study only provides preliminary evidence of an association between microbiome and advanced fibrosis in NAFLD. These data do not suggest causality. Further studies are needed to assess the how and if these microbial species play a role in gut permeability, perturbing liver inflammation, and/or cross-talk with serum metabolites to induce liver injury to affect disease progression in NAFLD.
Implications for future research and clinical practice
The results suggest that microbial biomarkers can be used to diagnose metabolic and fibrotic diseases and present an adjunct tool to current invasive approaches to determine stage of liver disease. We believe that our study sets the stage to explore the potential role of a stool-based test to detect advanced fibrosis in the future. The metagenomics signature may also be used in conjunction with other noninvasive serum/plasma or imaging based tests to detect fibrosis, advanced fibrosis and cirrhosis. It is plausible that further studies on the bacterial dysbiosis may lead to new approaches to inhibit bacterial derived pathways that in turn lead to disease progression in NAFLD. Further studies are needed to validate the clinical utility of the proposed microbiome-derived signature to detect advanced fibrosis as well as candidacy for anti-fibrotic treatment trials in NAFLD.
STAR METHODS
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Rohit Loomba (roloomba@ucsd.edu).
Experimental Model and Subject Details
Human subject
UCSD NAFLD Cohort: Training set
86 patients with biopsy-proven NAFLD were included. The baseline characteristics are detailed in Table 1. Based on histology assessment, NAFLD patients were classified into two groups: Group 1 (G1) (n=72) – mild/moderate NAFLD patients with stage 0–2 fibrosis, and Group 2 (G2) (n=14) – advanced fibrosis NAFLD patients with stage 3–4 fibrosis. Serum samples from 56 individuals (50 from Group 1 and 6 from Group 2) were used to generate metabolite profiles.
Normal Cohort Derived from Older Twins: age validation
16 healthy adult controls derived from the twin study who are 60 years or older.
Cohort B
Cohort B was created to address the observed skew in age for patients with advanced fibrosis, of patients that are all 60 years or older from multiple cohorts. The 49 patients in Cohort B consist of 17 G1 and 14 G2 patients from the UCSD NAFLD Cohort, 16 healthy patients from the normal cohort derived from older twins (single twin from each pair), and two biopsy-proven cirrhotic patients from a familial cirrhosis study. The numbers of healthy and NAFLD cirrhosis patients were 33 and 16, respectively.
Sample size and subject allocation to experimental groups
This is a pilot proof of concept study including 86 patients with biopsy-proven NAFLD (72 in mild/moderate group and 14 in advanced fibrosis group. We were able to detect clinically meaningful differences between the sub-populations. The validation cohort (B) including 49 subjects had mild/moderate NAFLD and 14 with advanced fibrosis. Furthermore, 33 patients with cirrhosis/advanced fibrosis and 16 normal controls were included.
Patient consent
All patients provided a written informed consent and the study protocol was approved by the UCSD Institutional Review Board (approval number: UCSD IRB111298).
Inclusion criteria for UCSD NAFLD Cohort
Participants were included in the study in they met the following criteria: 1. 18 years or older, 2. Fat accumulation in the liver (steatosis) involving at least 5% of hepatocytes on routine stains, 3. No evidence of other acute or chronic liver disease, 4. Absence of regular or excessive use of alcohol. Regular or excessive alcohol is defined as an average alcohol intake of more than 14 drinks of alcohol/week in men or more than 7 drinks of alcohol/week in women.
Exclusion criteria for UCSD NAFLD Cohort
Participants were excluded from the study if they met any of the following criteria 1. Clinical or histological evidence of alcoholic liver disease, 2. Total parenteral nutrition for more than 1 month within a 6 month period before baseline liver biopsy, 3. Short bowel syndrome, 4. History of gastric or jejunoileal bypass preceding the diagnosis of NAFLD. Bariatric surgery performed following enrollment is not exclusionary. Liver biopsies obtained during bariatric surgery cannot be used for enrollment because of the associated surgical or anesthetic acute changes and the weight loss efforts that precede bariatric surgery, 5. History of biliopancreatic diversion, 6. Evidence of advanced liver disease defined as a Child-Pugh-Turcotte score equal to or greater than 10, 7. Evidence of chronic hepatitis B as marked by the presence of HBsAg in serum (participants with isolated antibody to hepatitis B core antigen, anti-HBc total, arenot excluded), 8. Evidence of chronic hepatitis C as marked by the presence of anti-HCV or HCV RNA in serum, 9. Low alpha-1-antitrypsin level and ZZ phenotype, 9. Wilson’s disease, 10. Known glycogen storage disease, 11 Known dysbetalipoproteinemia or known phenotypic hemochromatosis (HII greater than 1.9 or removal of more than 4 g of iron by phlebotomy) or prominent bile duct injury (florid duct lesions or periductal sclerosis) or bile duct paucity or chronic cholestasis or vascular lesions (vasculitis, cardiac sclerosis, acute or chronic Budd-Chiari, hepatoportal sclerosis, peliosis) or Iron overload greater than 3+, 12. Zones of confluent necrosis, infarction, massive or sub-massive, pan-acinar necrosis, 13. Multiple epithelioid granulomas, 14. Congenital hepatic fibrosis, 15. Polycystic liver disease, other metabolic or congenital liver disease, 16. Evidence of systemic infectious disease, 17. Known HIV positive, 18. Disseminated or advanced malignancy, 19. Concomitant severe underlying systemic illness that in the opinion of the investigator would interfere with completion of follow-up.
Inclusion criteria for Normal Cohort Derived from Older Twins
Participants were included in the study: (1) if they were twins at least 18 years old who provided written informed consent. The zygosity of the majority of twin pairs as monozygotic (MZ) or dizygotic (DZ) had been previously confirmed via genetic testing before the participants enrolled in the study. (2) Aged 60 years or older. (3) MRI-PDFF estimated liver fat content of less than 5% (therefore, documenting absence of NAFLD). (4) MRE estimated liver stiffness of less than 3 Kpa (therefore, documenting absence of any fibrosis).
Exclusion criteria for Normal Cohort Derived from Older Twins
Participants were excluded from the study if they met any of the following criteria: (1) significant alcohol intake (>10 grams/day in females or >20 grams/day in males) for at least three consecutive months over the previous 12 months, or if the quantify of alcohol consumption could not be reliably ascertained; (2) clinical or biochemical evidence of liver diseases other than NAFLD, including hepatitis B, hepatitis C, alpha-1 antitrypsin deficiency, hemochromatosis, Wilson’s disease, autoimmune hepatitis, polycystic liver diseases, cholestatic liver diseases, and vascular liver diseases; (3) chronic illnesses associated with hepatic steatosis, including human immunodeficiency virus infection, type I diabetes mellitus, celiac disease, cystic fibrosis, lipodystrophy, dysbetalipoproteinemia, and glycogen storage diseases; (4) use of drugs known to cause hepatic steatosis, including amiodarone, glucocorticoids, methotrexate, L-asparaginase, and valproic acid for at least three out of the previous six months; (5) history of bariatric surgery, including roux-en-Y gastric bypass and gastroplasty; (6) presence of systemic infectious illnesses; (7) females who were pregnant or nursing at the time of the study; (8) contraindications to MRI, including metal implants, claustrophobia, and body circumference greater than that of the imaging chamber; (9) any other condition(s) which, based on the principal investigator’s opinion, may significantly affect the participant’s compliance, competence, or ability to complete the study.
Method Details
Study design and recruitment
This is a prospective cohort study of consecutive biopsy-proven NAFLD patients who were participating in a biobanking initiative at the University of California at San Diego NAFLD Research Center between January 2012 to December 2013. Patients in this biobank had a confirmed diagnosis of NAFLD based on clinical, Magnetic Resonance, and histologic assessments. Patients underwent routine research visits at which time a detailed history, physical exam, assessment of alcohol use, fasting laboratory assessment, advanced Magnetic Resonance examination, and liver biopsy were performed, as per standard of care. At the time of each research visit, patients provided stool and fasting serum samples. These were collected and immediately stored in a −80*C freezer.
Liver histology
Liver histology assessment was done using the NASH CRN Histologic Scoring System by an experienced blinded GI pathologist. All biopsies were assessed for the following three parameters: Steatosis was graded 0–3, lobular inflammation was graded 0–3, ballooning was graded 0–2. Presence of NASH was defined as a pattern that was consistent with steatohepatitis including presence of steatosis, lobular inflammation and ballooning with or without peri-sinusoidal fibrosis. Fibrosis stage was classified into five staged from 0–4. Advanced fibrosis was defined as stage 3 (bridging fibrosis) or stage 4 (cirrhosis).
DNA extraction
A 3-mL volume of lysis buffer (20 mM Tris-HCl pH 8.0, 2 mM Sodium EDTA 1.2% Triton X-100) was added to 0.5 grams of stool sample, and the sample vortexed until homogenized. A 1.2 mL volume of homogenized sample and 15 μL of Proteinase K (Sigma Aldrich, PN. P2308) enzyme was aliquoted to a 1.5 mL tube with garnet beads (Mo Bio PN. 12830-50-BT). Bead tubes were then incubated at 65°C for 10 minutes and then 95°C for 15 minutes. Tubes were then placed in a Vortex Genie 2 to perform bead beating for 15 minutes and the sample subsequently spun in an Eppendorf Centrifuge 5424. 800 μL of supernatant was then transferred to a deep well block and DNA extracted and purified using a Chemagic MSM I (Perkin Elmer) following the manufacturer’s protocol. Zymo Onestep Inhibitor Removal kit was then performed following manufacturer’s instructions (Zymo Research PN. D6035). DNA samples were then quantified using Quant-iT on an Eppendorf AF2200 plate reader.
Primary outcome
Primary outcome measure was to examine the diagnostic accuracy of a gut-microbiota derived metagenomic signature for the presence of advanced fibrosis in NAFLD.
The rationale for advanced fibrosis as the primary outcome is that advanced fibrosis is associated with significantly increased risk of all-cause as well as liver-related mortality, hepatocellular carcinoma and need for liver transplantation. (Angulo et al., 2015; Ekstedt et al., 2015; Younossi et al., 2015)
Library Preparation and Sequencing
Nextera XT libraries were prepared manually following the manufacturer’s protocol (Illumina, PN. 15031942). Briefly, samples were normalized to 0.2 ng/μl DNA material per library using a Quant-iT picogreen assay system (Life Technologies, PN. Q33120) on an AF2200 plate reader (Eppendorf), then fragmented and tagged via tagmentation. Amplification was performed by Veriti 96 well PCR (Applied Biosystems) followed by AMPure XP bead cleanup (Beckman Coulter, PN. A63880). Fragment size for all libraries were measured using a Labchip GX Touch Hi Sens. Sequencing was performed on an Illumina HiSeq 2500 using SBS kit V4 chemistry.
Metagenome data annotation
Microbiome sequence data were processed as previously described. (Jones et al., 2015) The annotation pipeline generated relative genome abundance estimates of the constituent microbes in the samples and relative abundances of protein families (COGs, Pfams, TIGRFAMs, and ECs). As part of the annotation process, data from each metagenomic sample was also assembled to generate contigs. Contigs were assigned taxonomy and organized into species bins. The annotation information was then used to carry out metabolic reconstructions of the assembled species using Pathway Tools. (Karp et al., 2002) ORFs were generated from assembled contigs and unassembled singleton reads using MetaGene (Noguchi et al., 2006). The relative abundance of a protein family is sum of ORF abundances. The relative abundance of a pathway is defined to be the sum of relative abundances of all species where that pathway was reconstructed.
Metabolite Profiles
Metabolites were identified using Metabolon’s mass spectrometry based metabolic profiling of serum samples (Guo et al., 2015). Serum samples from 56 individuals (50 from Group 1 and 6 from Group 2) were used to generate metabolite profiles.
Quantification and Statistical Analysis
Development of a model utilizing stool derived metagenome profiles to predict advanced fibrosis
To build a model capable of distinguishing samples belonging to mild/moderate NAFLD from those of advanced fibrosis, we developed a custom machine learning process that employed Random Forest (RF) analysis (Breiman, 2001; Liaw, 2002). The set of input features for model building consisted of metagenome features and patient metadata features. Features from metagenome data consisted of the number (richness) and relative abundances of 152 constituent species, and microbiome diversity (Shannon diversity). The patient metadata consisted of age, gender, race, and BMI. The first step in building an RF model consisted of training 300 RFs and then selecting the top features from the top-performing model. A feature elimination step was then done to optimize the performance of subsequent RF models. The statistical significance of the final set of selected features was assessed by Monte Carlo simulation using 10,000 models that were each trained on 40 randomly selected features and comparing their predictive value on the dataset.
Analysis of microbial function using metagenome and metabolome data
Next, we explored the plausible function of the metagenome derived gut microbiota profile of advanced fibrosis in NAFLD. Metagenome data were used to assess the functional and metabolic potential of the microbial communities associated with the two groups, via a quantification of the relative abundances of protein families and enzymes in the samples and the relative abundances of the pathways reconstructed from species bins generated from assembled data. These data were integrated with serum metabolite data to evaluate microbial metabolism. Metabolites detected in serum samples include those that are endogenous or of microbial origin. (Guo et al., 2015) To further evaluate those metabolites that may be of microbial origin, the full set of metabolites detected from the 56 serum samples were intersected with the set of metabolites predicted from the microbial pathways reconstructed from the stool metagenome data.
Model Validation
We used an additional source of data to validate the performance of the metagenome derived model to differentiate mild/moderate NAFLD from advanced fibrosis. Within our original data set, age was determined to be a possible confounder masking the microbial signature of advanced fibrosis. In order to show that the metagenome-derived model was not biased by age, we applied the RF model to a previously published and well-phenotyped twin cohort dataset. (Loomba et al., 2015) A priori, we selected a single twin (as twins are known to have a significantly shared microbiome) from a pair of twins who were 60 years of age or older and healthy based upon a normal liver fat content without hepatic steatosis as determined by MRI PDFF <5% (no NAFLD) and absence of fibrosis as determined by an MRE < 3 Kpa (no fibrosis) (N=16, Table S1)(Cui et al., 2016; Loomba et al., 2015; Zarrinpar et al., 2016).
Microbial Signature Validation
In order to further validate the existence of a signature to distinguish between mild/moderate NAFLD (G1) and advanced fibrosis (G2), we used an orthogonal machine learning method based on Support Vector Machine to build a classifier from the same input feature set.
Random Forest Analysis
The Random Forest algorithm was used for two purposes: 1) to model microbial signatures of liver fibrosis; and 2) to select important species that may contribute most to the progression of liver fibrosis. Species relative abundances and patient data, also referred to as features, were analyzed using the Random Forest package in R (Breiman, 2001; Liaw, 2002). A forest is trained by supervised learning in which each tree in the forest finds an ideal split for a set of randomly chosen features such that the predicted outcome of each sample is the same as the expected outcome. The data partition found by every tree in a forest is used to vote on a predicted overall outcome of each sample. The voting strategy of Random Forest is documented in the literature to avoid the over fitting of data due to the random sampling of features by each tree. Using every tree to vote on an outcome prevents any single tree that may have memorized the data from having a dominant prediction. For our study, outcomes are disease or no disease. AUC or Area Under the Receiver-Operator Curve measured the accuracy of trained forests. AUC is a widely used estimator of true positive and false positive prediction rates. Variable or species importance lists from those forests with the highest AUCs were selected for further analysis.
Training Data
Our dataset consisted of sample diversity, sample richness, and the relative genome abundances of species detected in 86 stool samples collected from patients in a Registry Cohort. Age, Gender, Race, and BMI of each patient were also included in the training set. For this study, individuals were categorized into two groups based on the severity of fibrosis. The first group (Group 1) consisting of individuals with mild/moderate fibrosis (Stage 0–2) and the second group (Group 2) consisting of individuals with advanced fibrosis (Stage 3–4). Most patients (72) were in Group 1 and 14 patients were part of Group 2. To reduce the level of noise that may be present in the relative abundance data, abundances that were less than 10−4 were set to zero and a species had to be present in more than 70% of the patient stool samples to be considered as an input feature.
To reduce the effect that correlated data may have on training we further filtered the species abundance data by hierarchical clustering. We used the cor function in R to calculate the Spearman correlation coefficients from species abundance data. The correlation matrix was converted to a dissimilarity matrix before using the hclust function for a complete linkage clustering of the dissimilarity matrix. The cor and hclust functions are part of the R STATS package. The resulting tree from the clustering was cut at a height of 0.1 and the species that was the closest to all other species within a cluster was chosen as a representative species from that cluster. When this procedure was applied to the initial set of 152 species, it resulted in 136 representative species, which were subsequently used for the training phase. (See Table S6 for a list of the species clusters generated by our procedure.)
We developed a series of steps to train a Random Forest with the best overall accuracy of classification, which we report as AUC. We trained 300 forests, containing 1001 trees each, with the relative genome abundances of species that passed abundance and prevalence filtering as previously described. In addition, the Shannon Diversity Index and richness of each sample, and the age, BMI, gender, and race of each patient were also included in the training set. Due to the small number of patients in Group 2 in comparison to Group 1, training was done with stratified sampling in which features from an equal number of samples from each group were randomly sampled and used to train each tree. A trained forest produces a variable importance list based on mean decrease in Gini index. For our dataset the variable importance list is a list of species, sample indices, or patient measurements that contributed most to the correct classification or the correct group assignment of every sample. The species importance list from the forest with the highest AUC is selected for Iterative Feature Elimination, which is described next.
Iterative Feature Elimination (IFE) and Forest/Feature Selection
Features (species, sample indices, and patient data) from the feature importance list described in the previous section were iteratively eliminated to find a set of features that trains a forest with the highest overall accuracy of sample classification. The feature importance list was ordered from highest to lowest Mean Decrease in Gini index and the least important species was removed. A random forest was trained with the remaining features in the feature importance list and an AUC is calculated. Removing least important features, training a forest with the remaining features, and calculating an AUC was continued until all of the features from the importance list were removed. The features used to train a forest with the highest AUC were used as the final feature importance list. In the case where there are two or more forests with the highest AUC, the forest with the largest number of features was chosen. The species that trained the forest with the highest AUC after the feature elimination step are reported in the final model.
Statistical Significance of Species Selection
To determine the significance of the final species importance list, we used a Monte-Carlo simulation approach in which we created a null distribution of AUCs from forests trained on randomly chosen features. The number of randomly chosen features is the same number of features found by the Iterative Feature Elimination step as described in the previous section. AUCs are calculated for 10,000 forests trained on randomly selected features and is used to form a null distribution from which to compare against the significance of the top features selected by iterative feature elimination (IFE features). A p-value associated with the IFE features is the fraction of times that the AUC of forests trained on randomly selected sets of features were higher than the AUC of the forest trained by the IFE features.
Linear Support Vector Machine
Linear support vector machine (linear SVM) is used for two procedures: (1) feature selection, i.e. selection of important patient data and microbial species, and (2) classifier training with selected features. Feature selection is done with L1 norm regularization and classifier training is done with L2 norm regularization. Dataset used for linear SVM is the same as for Random Forest classification. Group 1 with mild/moderate fibrosis is assigned with class label “−1” and Group 2 with advanced fibrosis is assigned with class label “1”. Feature set consists of patient data, including sex, age, BMI, race (White, Asian, Hispanic) and referred to as metadata, and microbial species present in more than 70% of the 86 samples in the registry cohort. Linear SVM module sklearn.svm.LinearSVC from Python is applied and a grid search for penalty parameter C in range 2−5 to 25 is performed to pick the best estimator parameters. Stratified 2-fold cross-validation is used to configure training and testing datasets. ROC-AUC is used as the scoring method to evaluate accuracy of the classifier on testing dataset.
Feature Selection with L1 Norm
Linear SVM with L1 norm penalty is used for feature selection on feature set containing numeric metadata (age, BMI), binary metadata (female, Hispanic, Asian, White), and log-transformed relative abundances of 152 microbial species. 24 features are selected with non-zero coefficients under L1 regularization, including 4 metadata (age, female, Asian, Hispanic) and 20 microbial species. These selected features are used as new feature set for the next step training of linear SVM classifier.
Significance of SVM Selected Feature Set
To determine the significance of the set of selected features, a null distribution of ROC-AUC scores is created in the following procedure: (1) randomly choose 20 microbial species from 152 species list, (2) combine 4 metadata and 20 random microbial species as a new feature set, (3) train linear SVM with L2 norm using the new feature set, (4) calculate AUCs using stratified 2-fold cross-validation, (5) repeat random species selection 10,000 times to form the null distribution. P-value is obtained by comparing AUC of the selected feature set to the null distribution (Figure S1).
Concordance of RF and SVM models on the biopsy proven NAFLD cohort
The trained SVM selected 18 species as the most important predictors (Table S1) and 12 of those species overlapped with the species selected by the Random Forest method.
Statistical test for difference in relative abundance
Wilcoxon Rank Sum test was used to assess differential abundance. Multiple test correction was used when appropriate and tests were controlled for false discovery rate at significance level of 0.05.
Age-Balanced Dataset
We observed that all patients in the advanced stages (stages 3 and 4) of fibrosis from the biopsy-proven registry cohort (present cohort, 86 patients), were 60 years of age or older. The skew in age was not as extreme for patients in Group 1 such that a wider range of ages was observed for patients with either Stage 0, 1, or 2. To address the observed skew in age for patients with advanced fibrosis, we created a second cohort, referred to as Cohort B, of patients that are all 60 years or older from multiple cohorts. The 49 patients in Cohort B consist of 17 G1 and 14 G2 patients from the present cohort, 16 healthy patients from a cohort of twins (single twin from each pair), and two biopsy-proven cirrhotic patients from a familial cirrhosis study. The numbers of healthy and NAFLD cirrhosis patients are 33 and 16, respectively, Table S4).
Data and Software Availability
Data resources
The metagenomic sequence data were deposited at NCBI under Bioproject accession PRJNA373901, available from https://www.ncbi.nlm.nih.gov/bioproject/373901.
Supplementary Material
Table S1. List of 89 metabolites detected in serum samples and associated with pathways reconstructed in species bins, Related to Figure 3.
Acknowledgments
The authors are grateful to Alan Hofmann, MD for his insightful comments and suggestions.
Funding Support: The study was conducted at the Clinical and Translational Research Institute, University of California at San Diego. RL is supported in part by the American Gastroenterological Association (AGA) Foundation – Sucampo – ASP Designated Research Award in Geriatric Gastroenterology and by a T. Franklin Williams Scholarship Award. PSD is supported in part by the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) training grant 5T32DK007202. Funding provided by: Atlantic Philanthropies, Inc, the John A. Hartford Foundation, OM, the Association of Specialty Professors, and the American Gastroenterological Association and grant K23-DK090303, and R01-DK106419. The microbiome sequencing was performed and funded by Human Longevity, Inc. Research reported in this publication was supported by the National Institute of Environmental Health Sciences of the National Institutes of Health under Award Number P42ES010337. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Role of funding agencies: Funding agencies did not have any role in the design and conduct of the study, collection, management, analysis or interpretation of the data; preparation, review, or approval of the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interests: The authors report no conflict of interests.
AUTHOR CONTRIBUTIONS
RL and KEN study design. RL patient recruitment. MBJ and WB sample processing. VS, WL, TL, NK, SKH, MBJ, SY, KEN data analysis. All co-authors manuscript generation.
The top 11 metabolites (blue) in the list have significantly different abundances across groups G1 and G2, after correcting for multiple testing,
References
- Adams LA, Lymp JF, St Sauver J, Sanderson SO, Lindor KD, Feldstein A, Angulo P. The natural history of nonalcoholic fatty liver disease: a population-based cohort study. Gastroenterology. 2005;129:113–121. doi: 10.1053/j.gastro.2005.04.014. [DOI] [PubMed] [Google Scholar]
- Affo S, Morales-Ibanez O, Rodrigo-Torres D, Altamirano J, Blaya D, Dapito DH, Millan C, Coll M, Caviglia JM, Arroyo V, et al. CCL20 mediates lipopolysaccharide induced liver injury and is a potential driver of inflammation and fibrosis in alcoholic hepatitis. Gut. 2014;63:1782–1792. doi: 10.1136/gutjnl-2013-306098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anand G, Zarrinpar A, Loomba R. Targeting Dysbiosis for the Treatment of Liver Disease. Semin Liver Dis. 2016;36:37–47. doi: 10.1055/s-0035-1571276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angulo P, Kleiner DE, Dam-Larsen S, Adams LA, Bjornsson ES, Charatcharoenwitthaya P, Mills PR, Keach JC, Lafferty HD, Stahler A, et al. Liver Fibrosis, but No Other Histologic Features, Is Associated With Long-term Outcomes of Patients With Nonalcoholic Fatty Liver Disease. Gastroenterology. 2015;149:389–397 e310. doi: 10.1053/j.gastro.2015.04.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai L, Kong M, Zheng Q, Zhang X, Liu X, Zu K, Chen Y, Zheng S, Li J, Ren F, et al. Inhibition of the translocation and extracellular release of high-mobility group box 1 alleviates liver damage in fibrotic mice in response to D-galactosamine/lipopolysaccharide challenge. Mol Med Rep. 2016;13:3835–3841. doi: 10.3892/mmr.2016.5003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bajaj JS, Heuman DM, Hylemon PB, Sanyal AJ, White MB, Monteith P, Noble NA, Unser AB, Daita K, Fisher AR, et al. Altered profile of human gut microbiome is associated with cirrhosis and its complications. J Hepatol. 2014;60:940–947. doi: 10.1016/j.jhep.2013.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betrapally NS, Gillevet PM, Bajaj JS. Changes in the Intestinal Microbiome and Alcoholic and Nonalcoholic Liver Diseases: Causes or Effects? Gastroenterology. 2016;150:1745–1755 e1743. doi: 10.1053/j.gastro.2016.02.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhala N, Angulo P, van der Poorten D, Lee E, Hui JM, Saracco G, Adams LA, Charatcharoenwitthaya P, Topping JH, Bugianesi E, et al. The natural history of nonalcoholic fatty liver disease with advanced fibrosis or cirrhosis: an international collaborative study. Hepatology. 2011;54:1208–1216. doi: 10.1002/hep.24491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boursier J, Mueller O, Barret M, Machado M, Fizanne L, Araujo-Perez F, Guy CD, Seed PC, Rawls JF, David LA, et al. The severity of nonalcoholic fatty liver disease is associated with gut dysbiosis and shift in the metabolic function of the gut microbiota. Hepatology. 2016;63:764–775. doi: 10.1002/hep.28356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breiman L. Random forests. Mach Learn. 2001;45:5–32. [Google Scholar]
- Chen YM, Liu Y, Zhou RF, Chen XL, Wang C, Tan XY, Wang LJ, Zheng RD, Zhang HW, Ling WH, et al. Associations of gut-flora-dependent metabolite trimethylamine-N-oxide, betaine and choline with nonalcoholic fatty liver disease in adults. Sci Rep. 2016;6:19076. doi: 10.1038/srep19076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui J, Chen CH, Lo MT, Schork N, Bettencourt R, Gonzalez MP, Bhatt A, Hooker J, Shaffer K, Nelson KE, et al. Shared genetic effects between hepatic steatosis and fibrosis: A prospective twin study. Hepatology. 2016;64:1547–1558. doi: 10.1002/hep.28674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Minicis S, Rychlicki C, Agostinelli L, Saccomanno S, Candelaresi C, Trozzi L, Mingarelli E, Facinelli B, Magi G, Palmieri C, et al. Dysbiosis contributes to fibrogenesis in the course of chronic liver injury in mice. Hepatology. 2014;59:1738–1749. doi: 10.1002/hep.26695. [DOI] [PubMed] [Google Scholar]
- Decaris ML, Li KW, Emson CL, Gatmaitan M, Liu S, Wang Y, Nyangau E, Colangelo M, Angel TE, Beysen C, et al. Identifying nonalcoholic fatty liver disease patients with active fibrosis by measuring extracellular matrix remodeling rates in tissue and blood. Hepatology. 2016 doi: 10.1002/hep.28860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dulai PS, Singh S, Patel J, Soni M, Prokop LJ, Younossi Z, Sebastiani G, Ekstedt M, Hagstrom H, Nasr P, et al. Increased risk of mortality by fibrosis stage in non-alcoholic fatty liver disease: Systematic Review and Meta-analysis. Hepatology. 2017 doi: 10.1002/hep.29085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dulai PS, Sirlin CB, R SL. MRI and MRE for non-invasive quantitative assessment of hepatic steatosis and fibrosis in NAFLD and NASH: Clinical trials to clinical practice. J Hepatol. 2016 doi: 10.1016/j.jhep.2016.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekstedt M, Hagstrom H, Nasr P, Fredrikson M, Stal P, Kechagias S, Hultcrantz R. Fibrosis stage is the strongest predictor for disease-specific mortality in NAFLD after up to 33 years of follow-up. Hepatology. 2015;61:1547–1554. doi: 10.1002/hep.27368. [DOI] [PubMed] [Google Scholar]
- Forslund K, Hildebrand F, Nielsen T, Falony G, Le Chatelier E, Sunagawa S, Prifti E, Vieira-Silva S, Gudmundsdottir V, Krogh Pedersen H, et al. Disentangling type 2 diabetes and metformin treatment signatures in the human gut microbiota. Nature. 2015;528:262–266. doi: 10.1038/nature15766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill SR, Pop M, Deboy RT, Eckburg PB, Turnbaugh PJ, Samuel BS, Gordon JI, Relman DA, Fraser-Liggett CM, Nelson KE. Metagenomic analysis of the human distal gut microbiome. Science. 2006;312:1355–1359. doi: 10.1126/science.1124234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo L, Milburn MV, Ryals JA, Lonergan SC, Mitchell MW, Wulff JE, Alexander DC, Evans AM, Bridgewater B, Miller L, et al. Plasma metabolomic profiles enhance precision medicine for volunteers of normal health. Proc Natl Acad Sci U S A. 2015;112:E4901–4910. doi: 10.1073/pnas.1508425112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henao-Mejia J, Elinav E, Jin C, Hao L, Mehal WZ, Strowig T, Thaiss CA, Kau AL, Eisenbarth SC, Jurczak MJ, et al. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature. 2012;482:179–185. doi: 10.1038/nature10809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Human Microbiome Jumpstart Reference Strains, C. Nelson KE, Weinstock GM, Highlander SK, Worley KC, Creasy HH, Wortman JR, Rusch DB, Mitreva M, Sodergren E, et al. A catalog of reference genomes from the human microbiome. Science. 2010;328:994–999. doi: 10.1126/science.1183605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones MB, Highlander SK, Anderson EL, Li W, Dayrit M, Klitgord N, Fabani MM, Seguritan V, Green J, Pride DT, et al. Library preparation methodology can influence genomic and functional predictions in human microbiome research. Proc Natl Acad Sci U S A. 2015;112:14024–14029. doi: 10.1073/pnas.1519288112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakiyama G, Pandak WM, Gillevet PM, Hylemon PB, Heuman DM, Daita K, Takei H, Muto A, Nittono H, Ridlon JM, et al. Modulation of the fecal bile acid profile by gut microbiota in cirrhosis. J Hepatol. 2013;58:949–955. doi: 10.1016/j.jhep.2013.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karp PD, Paley S, Romero P. The Pathway Tools software. Bioinformatics. 2002;18(Suppl 1):S225–232. doi: 10.1093/bioinformatics/18.suppl_1.s225. [DOI] [PubMed] [Google Scholar]
- Lelouvier B, Servant F, Paisse S, Brunet AC, Benyahya S, Serino M, Valle C, Ortiz MR, Puig J, Courtney M, et al. Changes in blood microbiota profiles associated with liver fibrosis in obese patients: A pilot analysis. Hepatology. 2016;64:2015–2027. doi: 10.1002/hep.28829. [DOI] [PubMed] [Google Scholar]
- Liaw A. Classification and Regression by randomForest. R news. 2002;2:18–22. [Google Scholar]
- Liu H, Pathak P, Boehme S, Chiang JY. Cholesterol 7alpha-hydroxylase protects the liver from inflammation and fibrosis by maintaining cholesterol homeostasis. J Lipid Res. 2016;57:1831–1844. doi: 10.1194/jlr.M069807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loomba R, Sanyal AJ. The global NAFLD epidemic. Nat Rev Gastroenterol Hepatol. 2013;10:686–690. doi: 10.1038/nrgastro.2013.171. [DOI] [PubMed] [Google Scholar]
- Loomba R, Schork N, Chen CH, Bettencourt R, Bhatt A, Ang B, Nguyen P, Hernandez C, Richards L, Salotti J, et al. Heritability of Hepatic Fibrosis and Steatosis Based on a Prospective Twin Study. Gastroenterology. 2015;149:1784–1793. doi: 10.1053/j.gastro.2015.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mardinoglu A, Boren J, Smith U. Confounding Effects of Metformin on the Human Gut Microbiome in Type 2 Diabetes. Cell Metab. 2016;23:10–12. doi: 10.1016/j.cmet.2015.12.012. [DOI] [PubMed] [Google Scholar]
- Matteoni CA, Younossi ZM, Gramlich T, Boparai N, Liu YC, McCullough AJ. Nonalcoholic fatty liver disease: a spectrum of clinical and pathological severity. Gastroenterology. 1999;116:1413–1419. doi: 10.1016/s0016-5085(99)70506-8. [DOI] [PubMed] [Google Scholar]
- Moss CW, Lambert MA, Goldsmith DJ. Production of hydrocinnamic acid by clostridia. Appl Microbiol. 1970;19:375–378. doi: 10.1128/am.19.2.375-378.1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noguchi H, Park J, Takagi T. MetaGene: prokaryotic gene finding from environmental genome shotgun sequences. Nucleic Acids Res. 2006;34:5623–5630. doi: 10.1093/nar/gkl723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin N, Yang F, Li A, Prifti E, Chen Y, Shao L, Guo J, Le Chatelier E, Yao J, Wu L, et al. Alterations of the human gut microbiome in liver cirrhosis. Nature. 2014;513:59–64. doi: 10.1038/nature13568. [DOI] [PubMed] [Google Scholar]
- Rinella ME. Nonalcoholic fatty liver disease: a systematic review. JAMA. 2015;313:2263–2273. doi: 10.1001/jama.2015.5370. [DOI] [PubMed] [Google Scholar]
- Seki E, De Minicis S, Osterreicher CH, Kluwe J, Osawa Y, Brenner DA, Schwabe RF. TLR4 enhances TGF-beta signaling and hepatic fibrosis. Nat Med. 2007;13:1324–1332. doi: 10.1038/nm1663. [DOI] [PubMed] [Google Scholar]
- Singh S, Allen AM, Wang Z, Prokop LJ, Murad MH, Loomba R. Fibrosis progression in nonalcoholic fatty liver vs nonalcoholic steatohepatitis: a systematic review and meta-analysis of paired-biopsy studies. Clin Gastroenterol Hepatol. 2015;13:643–654 e641. 649. doi: 10.1016/j.cgh.2014.04.014. quiz e639–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnbaugh PJ, Hamady M, Yatsunenko T, Cantarel BL, Duncan A, Ley RE, Sogin ML, Jones WJ, Roe BA, Affourtit JP, et al. A core gut microbiome in obese and lean twins. Nature. 2009;457:480–484. doi: 10.1038/nature07540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vernon G, Baranova A, Younossi ZM. Systematic review: the epidemiology and natural history of non-alcoholic fatty liver disease and non-alcoholic steatohepatitis in adults. Aliment Pharmacol Ther. 2011;34:274–285. doi: 10.1111/j.1365-2036.2011.04724.x. [DOI] [PubMed] [Google Scholar]
- Wikoff WR, Anfora AT, Liu J, Schultz PG, Lesley SA, Peters EC, Siuzdak G. Metabolomics analysis reveals large effects of gut microflora on mammalian blood metabolites. Proc Natl Acad Sci U S A. 2009;106:3698–3703. doi: 10.1073/pnas.0812874106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong VW, Wong GL, Choi PC, Chan AW, Li MK, Chan HY, Chim AM, Yu J, Sung JJ, Chan HL. Disease progression of non-alcoholic fatty liver disease: a prospective study with paired liver biopsies at 3 years. Gut. 2010;59:969–974. doi: 10.1136/gut.2009.205088. [DOI] [PubMed] [Google Scholar]
- Younossi ZM, Koenig AB, Abdelatif D, Fazel Y, Henry L, Wymer M. Global Epidemiology of Non-Alcoholic Fatty Liver Disease-Meta-Analytic Assessment of Prevalence, Incidence and Outcomes. Hepatology. 2015 doi: 10.1002/hep.28431. [DOI] [PubMed] [Google Scholar]
- Zarrinpar A, Gupta S, Maurya MR, Subramaniam S, Loomba R. Serum microRNAs explain discordance of non-alcoholic fatty liver disease in monozygotic and dizygotic twins: a prospective study. Gut. 2016;65:1546–1554. doi: 10.1136/gutjnl-2015-309456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu L, Baker SS, Gill C, Liu W, Alkhouri R, Baker RD, Gill SR. Characterization of gut microbiomes in nonalcoholic steatohepatitis (NASH) patients: a connection between endogenous alcohol and NASH. Hepatology. 2013;57:601–609. doi: 10.1002/hep.26093. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. List of 89 metabolites detected in serum samples and associated with pathways reconstructed in species bins, Related to Figure 3.