

Abstract
Ipomoeassin F is a plant-derived macrocyclic glycolipid with single-digit nanomolar IC50 values against cancer cell growth. In previous structure–activity relationship studies, we have demonstrated that certain modifications around the fucoside moiety did not cause significant cytotoxicity loss. To further elucidate the effect of the fucoside moiety on the biological activity, we describe here the design and synthesis of several fucose-truncated monosaccharide analogues of ipomoeassin F. Subsequent biological evaluation strongly suggests that the 6-membered ring of the fucoside moiety is essential to the overall conformation of the molecule, thereby influencing bioactivity.
Keywords: Resin glycosides, Ipomoeassin F, Monosaccharide analogues, Macrolide, Cytotoxicity
Graphical abstract
Resin glycosides are amphipathic secondary metabolites unique to the morning glory family, Convolvulaceae. Their broad bioactivities, such as anticancer, antibacterial, biosurfactant, ionophoretic, purgative and plant growth controlling activities, have attracted more and more attention from phytochemists and pharmaceutical chemists. The common macrolide structure of resin glycosides contains a hydrophobic C-l 1 hydroxylated fatty acid aglycone and a hydrophilic oligosaccharide. The latter usually comprises two to six sugar units, part or entire portion of which forms the ring structure with the fatty acid.1 Although monosaccharide cyclic glycolipids have been isolated from other families of plants, no resin glycosides containing a single carbohydrate moiety have been reported to date.
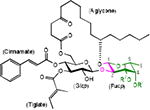
Ipomoeassins are a family of resin glycosides containing an embedded disaccharide. They were discovered by Kingston's group in 2005 and 2007.2 Whereas most resin glycosides showed micromolar cytotoxicity, several members of the ipomoeassin family exhibited low to single-digit nanomolar IC50 values against several cancer cell lines. Moreover, the naturally most abundant member of the family, ipomoeassin A, was screened against the NCI-60 tumor cell lines and its cytotoxicity profile is well distinguished from those of other anticancer agents in the database.3 Therefore, the ipomoeassins quickly inspired synthetic chemists to tackle their total syntheses. In particular, ipomoeassin F (Table 1) has been an attractive synthetic target due to its highest potency.4,5
Table 1. Structures and IC50 Values of Ipomoeassin F and Its Analogues.
|
Compounds | structure | IC50(nM) | |||||
|
|
|
|||||||
| R1 | R2 | MDA-MB-231 | MCF7 | HeLa | U937 | Jurkat | ||
|
| ||||||||
| ipomoeassin F | Ac | H | 6.5 | 43.7 | 16.4 | 5.4 | 6.1 | |
| 1 | H | H | 131 | 86.7 | 133 | 72.5 | 139 | |
| 2 | Ac | Ac | 16.5 | 216 | 138 | 79.8 | 82.6 | |
Recently, we developed new efficient gram-scale syntheses of ipomoeassin F and conducted its most systematic structure-activity relationship (SAR) studies to date.6–8 During the studies, we found that two α,β-unsaturated esters in the (3-D-glucoside moiety, that is, cinnamate and tiglate (Table 1), are the most critical to the cytotoxicity of ipomoeassin F. On the other hand, modifications of the (3-D-fucoside moiety,7 i.e. removal of the acetyl group from 4-OH-Fucp (analogue 1, Table 1) or introduction of an acetyl group to 3-OH-Fucp (analogue 2, Table 1), did not cause a dramatic cytotoxicity loss for the five tested cancer cell lines (2-23 fold loss for 1 and 2-14 fold loss for 2, respectively). The IC50 values of both 1 and 2 are largely still below 150 nM. Therefore, the contribution of the whole fucoside moiety to the cytotoxicity is of great interest. In addition, given the high price of D-fucose ($427/5g, Sigma-Aldrich), relatively potent monosaccharide analogues (IC50 < 0.5 μM) of ipomoeassin F without the fucoside moiety would significantly decrease the production cost and also shorten the synthetic route, thereby benefiting future ipomoeassin research in drug discovery and chemical biology.
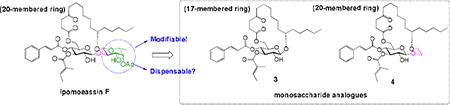
In our earlier studies, we proved that some peripheral modifications of the fucoside moiety could be well withstood (analogues 1 and 2, Table 1). To further elucidate function of the fucoside moiety, analogue 3 (Figure 1) was first designed, in which the entire fucoside moiety is removed. This analogue also changes the ring size from 20-membered ring in ipomoeassin F to 17-membered ring. Because ring size may have a great impact on biological activity, we also designed analogue 4 (Figure 1) in which the fucoside moiety is partially truncated; therefore, 4 maintains the same ring size as ipomoeassin F. Using both analogues 3 and 4, we hope to address the question of whether the fucoside ring is dispensable or not.
Figure 1.
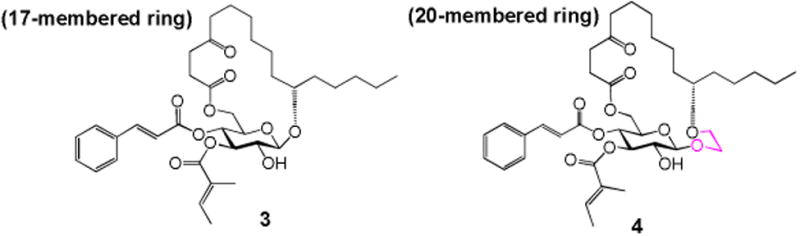
Design of the monosaccharide analogues 3 and 4.
The two monosaccharide analogues 3 and 4 can be straightforwardly synthesized by using the same strategy we developed for the total synthesis of ipomoeassin F (Scheme 1).6 From the diene intermediate 6a/b, ring-closing metathesis (RCM) and hydrogenation were still adopted to construct the saturated ring structure in 5a/b, to which cinnamate could be introduced, followed by removal of the TBS group, to give the desired monosaccharide analogues 3 and 4. To control the β-linkage in 6a/b, the glucosyl donor 76 with Alloc as the neighboring participation group was chosen to couple with the alcohol acceptor 8a/b. After that, replacement of the Alloc group with TBS, followed by removal of isopropylidene and then chemoselective esterification with 4-oxo-8-nonenoic acid 9,9 would lead to the key diene intermediate 6a/b as the RCM precursor. Synthesis of the acceptor 8b could be achieved by alkylation of alcohol 8a4 with bromoacetic acid, followed by the esterification and reduction procedure.
Scheme 1.
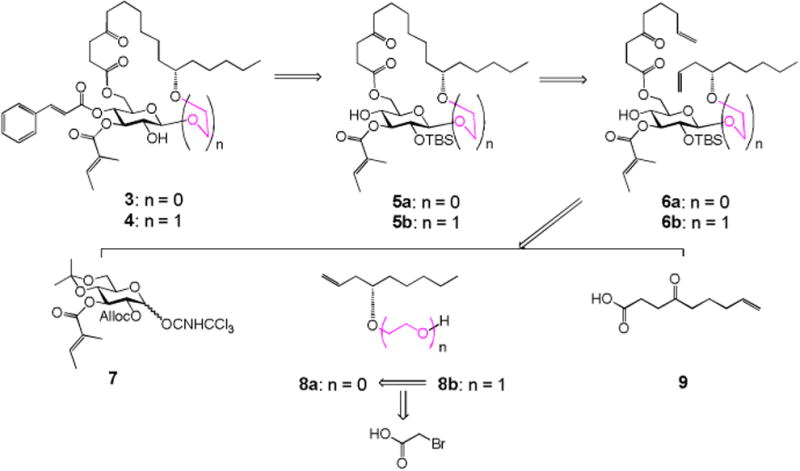
Retrosynthesis of the monosaccharide analogues 3 and 4.
The synthesis of 8b is outlined in Scheme 2. Alkylation of the other acceptor 8a4 with bromoacetic acid in the presence of NaH afforded acid 10 in moderate yield. Direct reduction of acid 10 using LiAlH4 was not successful. Then we converted acid 10 to ethyl ester 11 first, followed by reduction with LiAlH4 to give 8b in 62% overall yield over two steps.
Scheme 2.
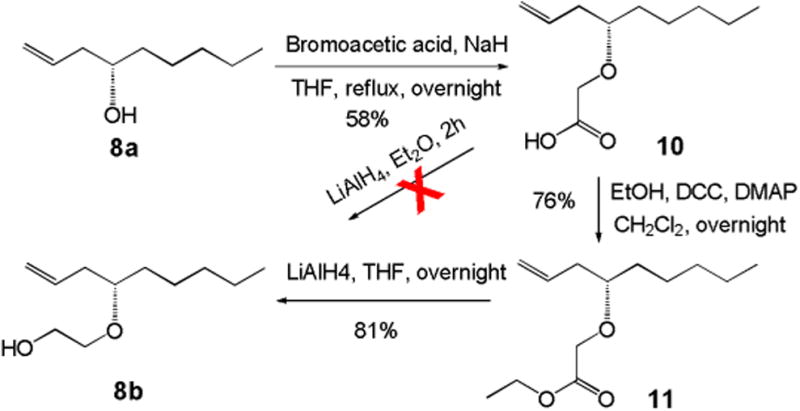
Synthesis of the acceptor 8b.
The glucosyl donor 7 was synthesized by following the route we developed previously. With both glucosyl donor and two acceptors in hand, the RCM precursor 6a/b was synthesized as shown in Scheme 3. Schmidt glycosylation of 7 with acceptor 8a/b promoted by TMSOTf in cold CH2Cl2 gave β-linked glycoside 12a in 62% yield or 12b in 46% yield. Alloc group was then selectively removed in the presence of CH3COONH4, Pd[P(C6H5)3]4, and NaBH4 in excellent yield to give alcohol 13a/b within 4 min.10 Alcohol 13a/b was then treated with tert-butyldimethylsilyl triflate (TBSOTf) in the presence of 2,6-lutidine to give silyl ether 14a in 84% yield or 14b in 97% yield. Removal of isopropylidene using camphorsulfonic acid (CSA) in MeOH afforded diol 15a/b without any problem. The subsequent chemoselective Steglich esterification of diol 15a/b with 4-oxo-8-nonenoic acid 99 to give diene 6a/b as the key RCM precursor.
Scheme 3.
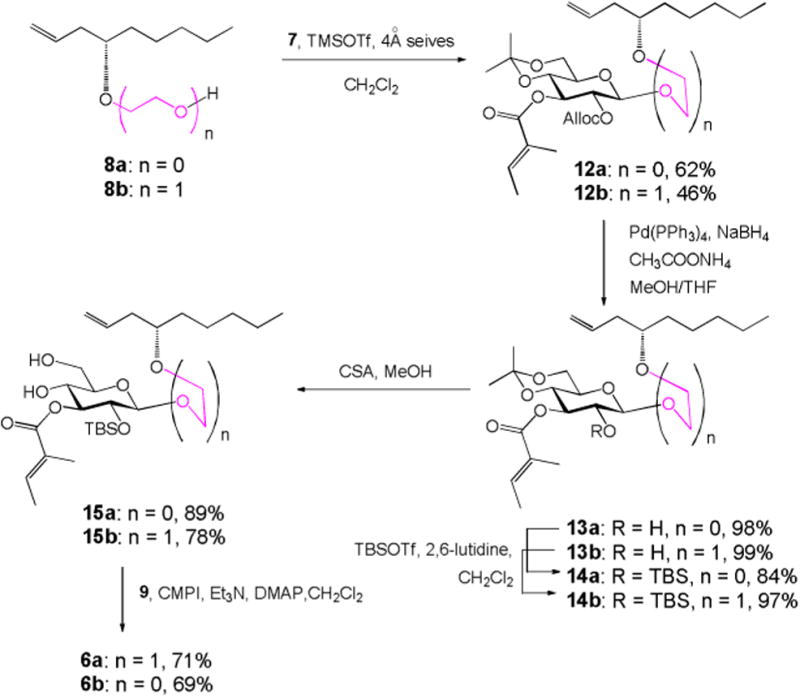
Synthesis of diene 6a/b.
RCM of 6a/b using Hoveyda-Grubbs catalyst (II)11 (10 mol%) under refluxing in CH2Cl2, followed by hydrogenation of the resulting alkene isomers over Wilkinson's catalyst, constructed the desired ring structure (76% for 5a and 62% for 5b) over two steps (Scheme 4). Cinnamate ester was then introduced to 5a/b through Mukaiyama esterification to afford the fully protected compound 16a/b in high yield. In the last step, removal of TBS using TBAF in cold THF/MeOH was first tried on substrate 16a. Unfortunately, instead of forming the desired monosaccharide analogue 3, the acyl-migrated compound 17 was obtained as the major product (42% yield) along with 18 as the minor product (29% yield). The structures of compounds 17 and 18 were supported by 1H, 13C, COSY and HMBC NMRs (see the Supporting Information). Treatment of 16a/b with TBAF and acetic acid in THF/MeOH finally delivered the desired monosaccharide analogues in good yield (79% for 3 and 77% for 4). The structures of compounds 3 and 4 were confirmed by 1H, 13C, COSY and HMBC NMRs. Key correlations in COSY and HMBC are illustrated in Figure 2.
Scheme 4.
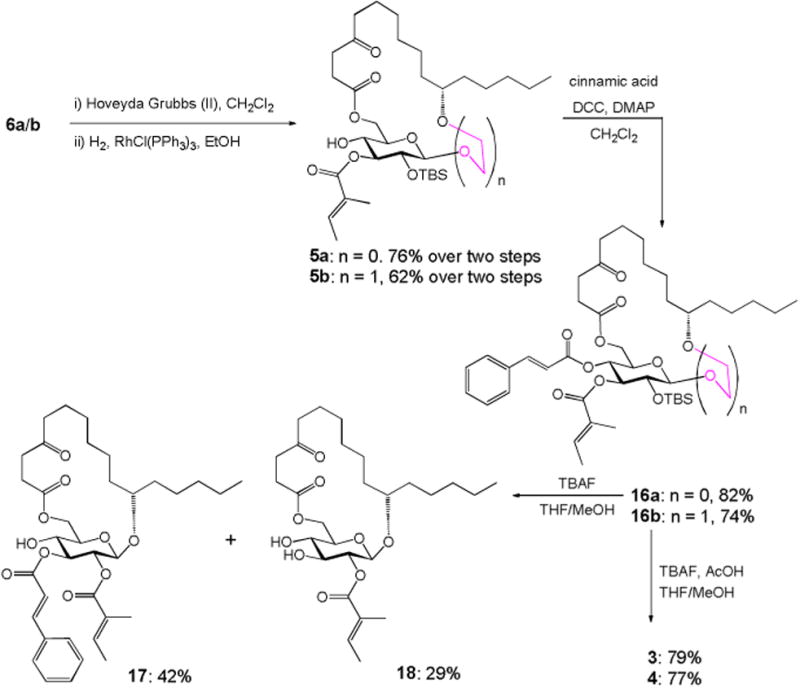
Synthesis of monosaccharide analogues 3 and 4.
Figure 2.
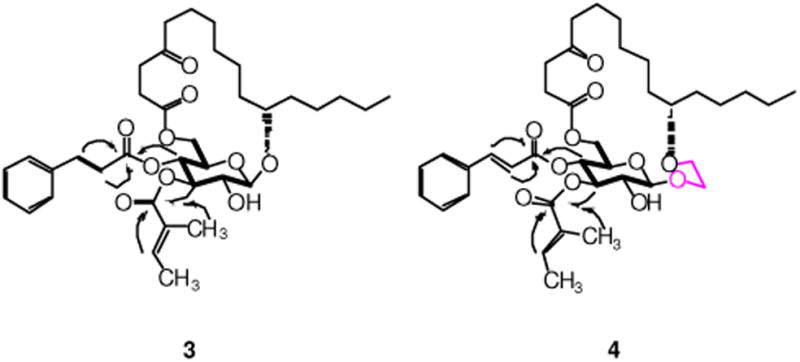
Key COSY (bold) and HMBC (arrows) correlations in 3 and 4.
Using the fluorescent alarmarBlue or colorimetric MTT assay, the cytotoxicity of monosaccharide analogues 3 and 4, together with the unexpected analogue 17, was evaluated against two breast cancer cell lines (MDA-MB-231 and MCF7) using ipomoeassin F as the positive control and vehicle-treated cells as the negative control. Compared to ipomoeassin F, all the three analogues showed no appreciable or very weak inhibition of cell growth at 10 μM (Figure 3), demonstrating that the fucoside moiety is indispensable for the cytotoxicity of the ipomoeassins.
Figure 3.
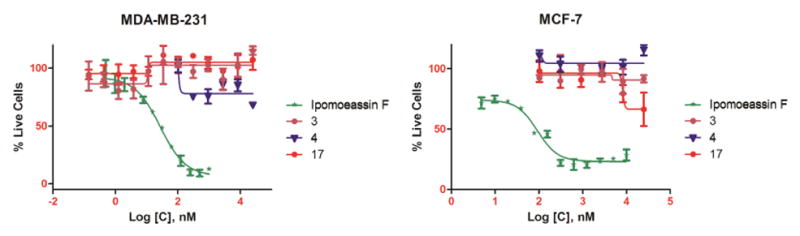
IC50 curves of ipomoeassin F and analogues 3, 4 and 17 against the MDA-MB-231 and MCF-7 cell line.
In summary, we report here the synthesis and biological evaluation of three monosaccharide analogues of ipomoeassin F, 3, 4, and 17, consisting of a truncated carbohydrate core. The fucoside moiety was completely or partially removed from the disaccharide portion of the original natural product. Evaluation of these compounds against two breast cancer cell lines unambiguously revealed the vital role of the pyranose ring of the fucoside moiety in the biological activity of ipomoeassin F, very likely through conformational control. Further studies of using other economical carbohydrate building blocks to replace expensive D-fucose are planned to consolidate and enrich the SAR data while improving the production efficiency. In this regard, D-galactose is the most attractive to us (Figure 4). The small 6-OH-Galp may provide an anchor point for introduction of new functional moieties to better biological activities and/or facilitate the discovery of molecular targets.
Figure 4.
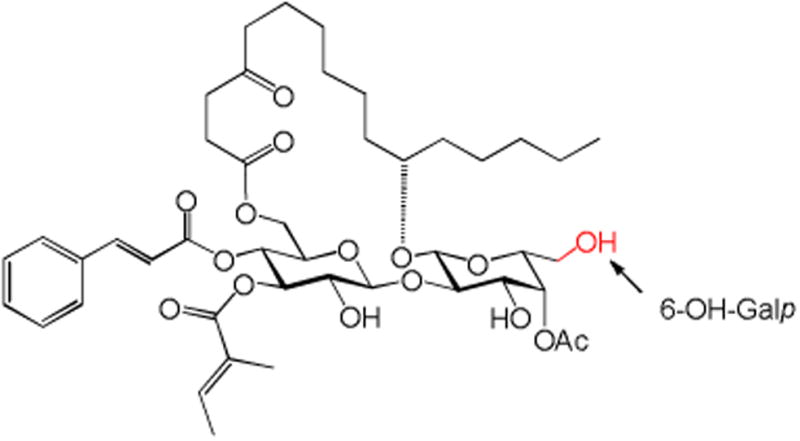
D-galactose-containing analogue of ipomoeassin F.
Supplementary Material
Acknowledgments
This work was primarily supported by the startup funds from the University of Arkansas and also in part by Grant Number P30GM103450 and R15GM116032 from the National Institute of General Medical Sciences of the National Institutes of Health (NIH) and by the seed money from the Arkansas Biosciences Institute (ABI). MH is currently the recipient of a Distinguished Doctoral Fellowship from the University of Arkansas. CM was the recipient of the Student Undergraduate Research Fellowship (SURF) from the Arkansas Department of Higher Education.
Footnotes
Notes: The authors report no conflicts of interest.
Supplementary Data: Supplementary data associated with this article can be found, in the online version, at link.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Pereda-Miranda R, Rosas-Ramirez D, Castaneda-Gomez J. Prog Chem Org Nat Prod. 2010;92:77–153. doi: 10.1007/978-3-211-99661-4_2. [DOI] [PubMed] [Google Scholar]
- 2.Cao S, Norris A, Wisse Jan H, Miller James S, Evans R, Kingston David GI. Nat Prod Res. 2007;21:872–876. doi: 10.1080/14786410600929576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kingston DGI. J Org Chem. 2008;73:3975–3984. doi: 10.1021/jo800239a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nagano T, Pospisil J, Chollet G, et al. Chem - Eur J. 2009;15:9697–9706. doi: 10.1002/chem.200901449. [DOI] [PubMed] [Google Scholar]
- 5.Postema MHD, TenDyke K, Cutter J, Kuznetsov G, Xu Q. Org Lett. 2009;11:1417–1420. doi: 10.1021/ol900086b. [DOI] [PubMed] [Google Scholar]
- 6.Zong G, Barber E, Aljewari H, et al. J Org Chem. 2015;80:9279–9291. doi: 10.1021/acs.joc.5b01765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zong G, Aljewari H, Hu Z, Shi WQ. Org Lett. 2016;18:1674–1677. doi: 10.1021/acs.orglett.6b00555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zong G, Whisenhunt L, Hu Z, Shi WQ. J Org Chem. 2017 doi: 10.1021/acs.joc.7b00409. Ahead of Print. http://pubs.acs.org/doi/abs/10.1021/acs.joc.7b00409. [DOI] [PMC free article] [PubMed]
- 9.Lhommet G, Freville S, Thuy V, Petit H, Celerier JP. Synth Commun. 1996;26:3897–3901. [Google Scholar]
- 10.Zong GH, Yan SQ, Liang XM, Zhang JJ, Wang DQ, Kong FZ. Chin Chem Lett. 2009;20:127–130. [Google Scholar]
- 11.Garber SB, Kingsbury JS, Gray BL, Hoveyda AH. J Am Chem Soc. 2000;122:8168–8179. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.