

Abstract
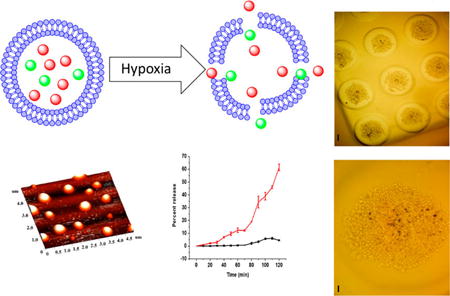
Solid tumors are often poorly irrigated due to structurally compromised microcirculation. Uncontrolled multiplication of cancer cells, insufficient blood flow, and the lack of enough oxygen and nutrients lead to the development of hypoxic regions in the tumor tissues. As the partial pressure of oxygen drops below the necessary level (10 psi), the cancer cells modulate their genetic makeup to survive. Hypoxia triggers tumor progression by enhancing angiogenesis, cancer stem cell production, remodeling of the extracellular matrix, and epigenetic changes in the cancer cells. However, the hypoxic regions are usually located deep in the tumors and are usually inaccessible to the intravenously injected drug carrier or the drug. Considering the designs of the reported nanoparticles, it is likely that the drug is delivered to the peripheral tumor tissues, close to the blood vessels. In this study, we prepared lipid nanoparticles (LNs) comprising the synthesized hypoxia-responsive lipid and a peptide–lipid conjugate. We observed that the resultant LNs penetrated to the hypoxic regions of the tumors. Under low oxygen partial pressure, the hypoxia-responsive lipid undergoes reduction, destabilizing the lipid membrane, and releasing encapsulated drugs from the nanoparticles. We demonstrated the results employing spheroidal cultures of the pancreatic cancer cells BxPC-3. We observed that the peptide-decorated, drug encapsulated LNs reduced the viability of pancreatic cancer cells of the spheroids to 35% under hypoxic conditions.
INTRODUCTION
Stimuli-responsive nanoparticles show tremendous potential to deliver anticancer chemotherapeutics to the targeted tumor site with enhanced efficacy and reduced side effects.1 Liposomes are the most popular lipid-based bilayer vesicles approved by the U.S. Food and Drug Administration (FDA) for the treatment of various cancers.2 Compared to the conventional chemotherapy, the anticancer drug encapsulated, polyethylene glycol-coated (PEGylated) liposomes show reduced side effects.3 However, the slow release of the encapsulated drugs from the clinically approved liposomal formulations have stimulated intense research on tumor microenvironment responsive, PEGylated drug carriers.
After intravenous administration, the PEGylated nanoparticles accumulate in the tumor tissues due to the leaky vasculature and poor lymphatic drainage (the enhanced permeation and retention or EPR effect).4 The abnormal tumor microenvironment (such as reduced pH, elevated enzyme and reducing agent levels, high cell surface receptor density) acts as a trigger for stimuli-responsive nanoparticles to release the encapsulated drugs.5 In-vivo studies demonstrate better control over tumor growth by the drug-encapsulated, stimuli-responsive nanoparticles compared to the conventional treatment.6 Considering the designs of the reported nanoparticles, it is likely that the drug is delivered to the peripheral tumor tissues, close to the blood vessels.
The solid tumors are often poorly irrigated due to the structurally compromised microcirculation.7 Uncontrolled multiplication of cancer cells, insufficient blood flow, and the lack of enough oxygen and nutrients lead to the development of hypoxic regions in the tumor tissues. As the partial pressure of oxygen drops below the necessary level (10 psi), the cancer cells modulate their genetic makeup to survive.8 Hypoxia triggers tumor progression by enhancing angiogenesis, cancer stem cell production, remodeling of the extracellular matrix, and epigenetic changes in the cancer cells.9 However, the hypoxic regions are usually located deep in the tumors and are usually inaccessible to the intravenously injected drug carrier or the drug.10 The cyclic iRGD peptide is reported to interact with the overexpressed integrin and neuropilin receptors11—increasing the penetration of nanoparticles in tumor tissues.12
In this study, we prepared LNs comprising a synthesized hypoxia-responsive, PEGylated lipid and an iRGD peptide conjugated lipid. We hypothesized that the resultant LNs would penetrate to the hypoxic regions of the tumors. Under low oxygen partial pressure, the hypoxia responsive lipid will undergo reduction, destabilize the lipid membrane, and release the encapsulated drugs from the LNs. We validated our hypothesis employing spheroidal cultures of the pancreatic cancer cells BxPC-3. The BxPC-3 cells form coherent spheroids with hypoxic cores—making them ideal for in vitro studies.13 We observed that the iRGD peptide-decorated, drug encapsulated LNs reduced the viability of pancreatic cancer cells of the spheroids to 35% under hypoxic conditions. To the best of our knowledge, this is the first report of the hypoxia-triggered release of liposomal anticancer drugs.
RESULTS AND DISCUSSION
Hypoxia alters the tumor microenvironment through the overexpressed HIF-1α and contributes to the overall cancer progression and metastasis.14,15 The azobenzene group is reduced in the presence of elevated levels of reducing enzymes in the hypoxic tumor microenvironment (Figure 1).16 We synthesized a hypoxia-sensitive lipid by conjugating POPE to polyethylene glycol (PEG1900) through the azobenzene linker (Scheme 1). We incubated a solution of the purified lipid under simulated hypoxic conditions (100 µM NADPH, rat liver microsomes, and nitrogen gas bubbling) for 2 h. The rat liver microsomes contain cytochrome P450 enzymes.17 Reduction of azobenzene takes place under hypoxic conditions in the presence of reducing enzymes and a hydrogen donor source. Hence, for in vitro studies we have used rat liver microsomes as a source of reducing enzymes and NADPH as a proton donor for the reduction of azobenzene. We observed that the fluorescence emission intensity from the azobenzene group was substantially reduced after the hypoxic treatment. We did not observe any reduction in the emission intensity of the lipid under normoxic conditions (100 µM NADPH, rat liver microsomes, and nitrogen gas bubbling).
Figure 1.
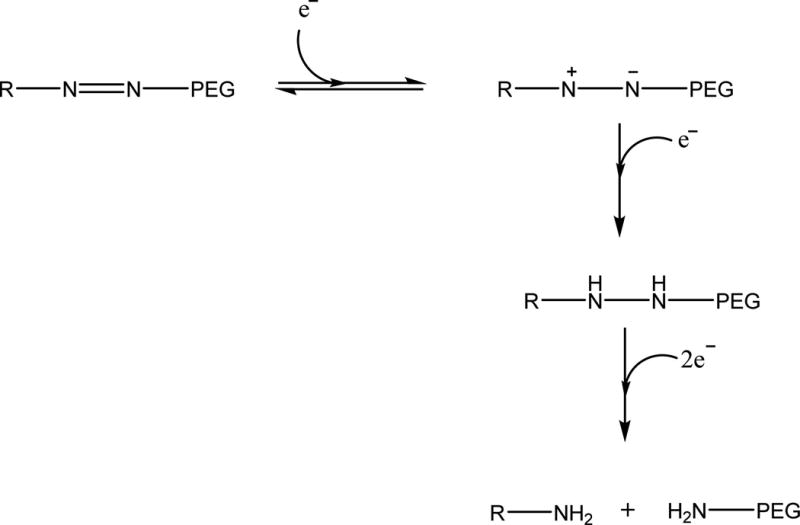
Mechanism of reduction of azobenzene under hypoxic reducing environment, where R represents POPE.16
Scheme 1. Synthesis of the Hypoxia Responsive POPE-Azobenzene-PEG1900 Lipida.
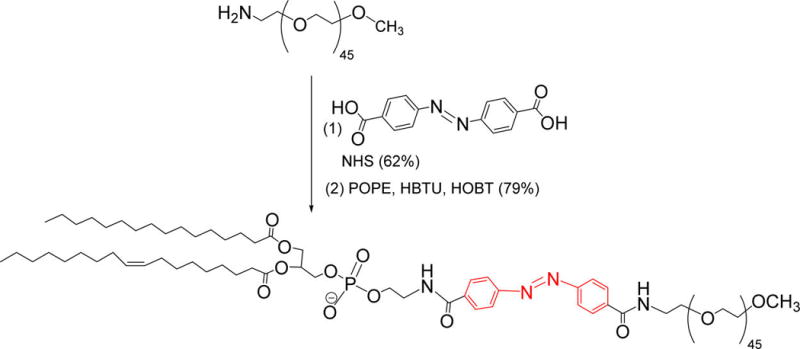
aThe hypoxia-responsive linker is indicated in red.
Tumor tissues develop hypoxia in the regions with inadequate blood flow due to the irregularly formed vasculature.15 These areas are usually less accessible to passively targeted drug carriers. The cyclic iRGD peptide has been previously used to enhance accumulation of the drugs or nanoparticles in the tumor tissues.12,18 The iRGD peptide functionalized nanoparticles preferentially accumulate and penetrate deep into the tumor tissue.19
We synthesized the hexynoic acid conjugated iRGD peptide by employing a microwave assisted, solid-phase peptide synthesizer. The alkyne of hexynoic acid allowed conjugation of the synthesized peptide to the surface of the LNs by “Click” chemistry. To confirm the cyclization of the peptide, we recorded the CD spectrum in the absence and presence of 10 mM glutathione. Thiol-sulfhydryl exchange and the reduction of the disulfide bond in the peptide resulted in a shift of the negative peak in the CD spectra from 199 to 195 nm (Supporting Information, Figure S2). The peptide was then conjugated to the saturated lipid DSPE-PEG-N3 by using the copper catalyzed [2 + 3]-cycloaddition reaction (Scheme 2). The product was confirmed by CD spectroscopy (Supporting Information, Figure S3). This lipid was then incorporated into the LNs to enhance the tissue penetration depth of the vesicles.
Scheme 2. Synthesis of iRGD Peptide Conjugated DSPE Lipida.
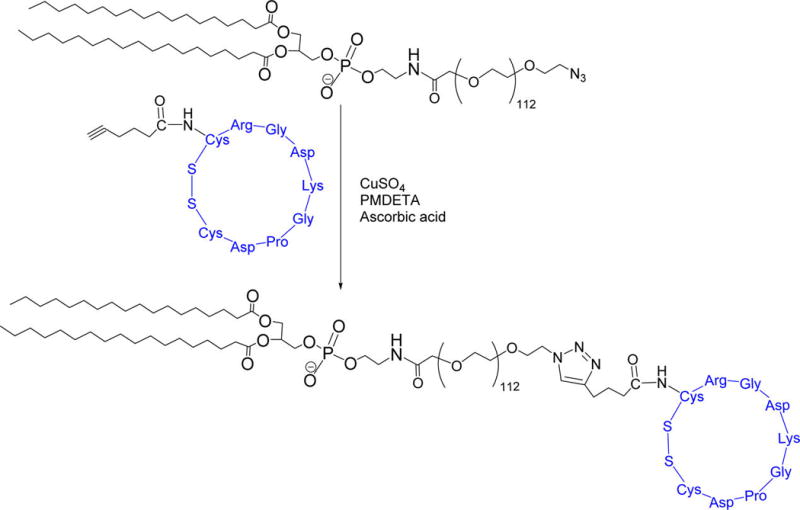
aThe iRGD peptide is shown in blue.
The stability of the nanoparticles in the absence of a stimulus is critical for drug delivery applications. We have previously observed that liposomes prepared from a proper combination of unsaturated and saturated lipids are structurally stable in the circulation, although the lipids phase-separate in the bilayer. However, in the presence of a stimulus, the vesicles become unstable and rapidly release the contents.6 Based on the precedent, we prepared the LNs incorporating DSPC, DSPE-PEG-iRGD, and the hypoxia-sensitive lipid containing the unsaturated POPE group. We optimized the relative ratio of these lipids such that the LNs are stable in normoxic circulatory conditions but disintegrate in the hypoxic environment to release the encapsulated contents. We encapsulated the self-quenching dye carboxyfluorescein in the LNs and monitored its release employing fluorescence spectroscopy (excitation: 485 nm, emission: 515 nm). The optimum stability and release characteristics were observed with the lipid composition DSPC: POPE-azobenzene-PEG1900: DSPE-PEG-iRGD in the molar percentage of 40:50:10. Under normoxic conditions, the release from these LNs was observed to be less than 5% in 2 h (Figure 2, black squares). However, under hypoxic conditions, the LNs released 66% of the encapsulated dye within 2 h (Figure 2, red triangles). As another control, we prepared carboxyfluorescein-encapsulated liposomes from the DSPC lipid and subjected them to the hypoxic conditions. We observed less than 20% of contents release in 2 h (Figure S4, Supporting Information). We anticipate that the reduction of the azobenzene linker will separate the PEG and the POPE fragments of the hypoxia-sensitive lipid (Scheme 1). The resultant shift in the hydrophilic–hydrophobic balance of the lipids will likely destabilize the nanoparticles, leading to the release of the encapsulated contents.6
Figure 2.
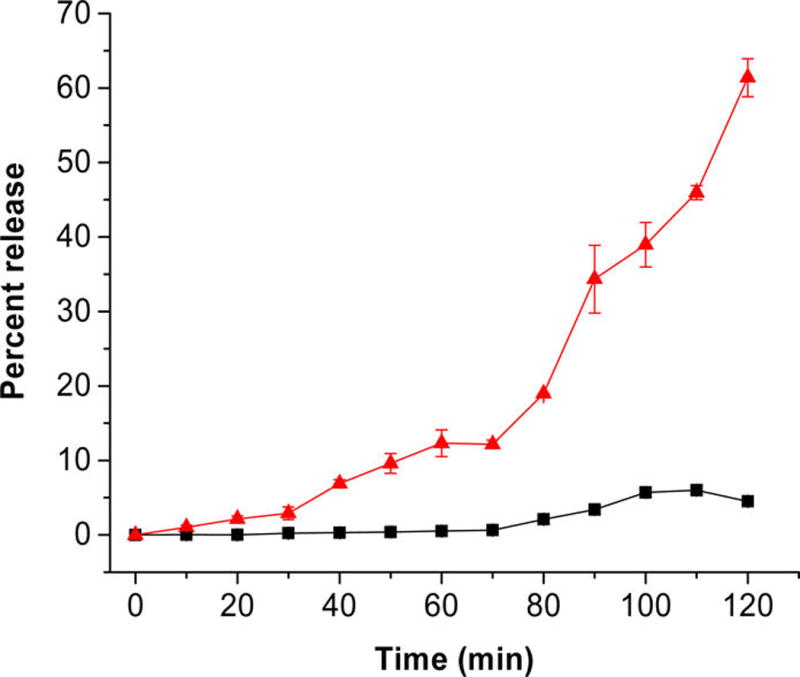
Dye release profiles from the LNs in hypoxic (red triangles) and normoxic (black squares) environment. The lines connecting the observed data points are shown (n = 3).
The LNs were less than 250 nm in size with a polydispersity index (PDI) of less than 0.3. A typical size distribution pattern, as measured by dynamic light scattering, is shown in Figure 3. Table 1 shows the size analysis of the LNs as determined by dynamic light scattering.
Figure 3.
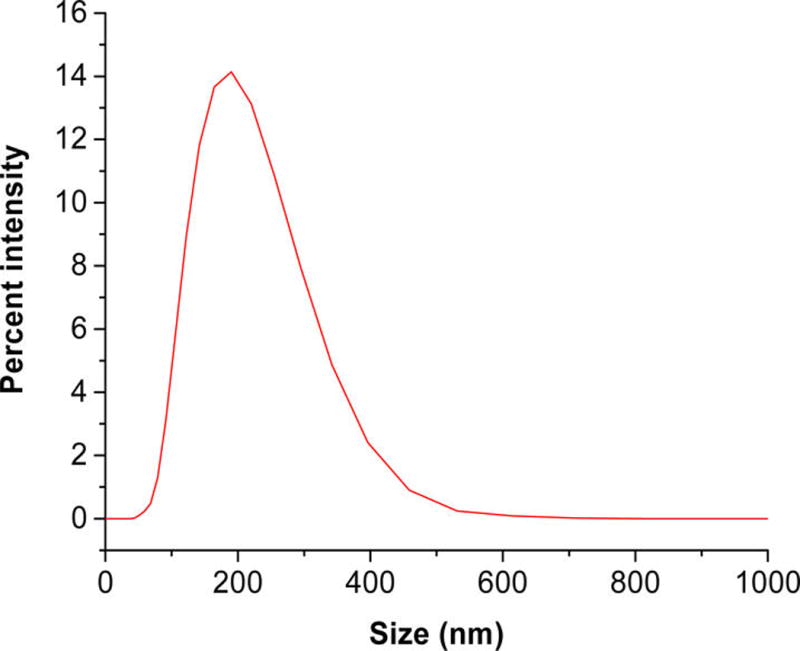
Size distribution of LNs as determined by the dynamic light scattering. The mean diameter was (180 ± 3) nm with a polydispersity index of 0.23 ± 0.01.
Table 1.
Size Analysis by Dynamic Light Scattering for the Test and the Control LNs
| lipid composition (molar percentage) | encapsulated content | size (nm) ± std dev | PDI ± std dev | |
|---|---|---|---|---|
| DSPC | DSPC (100) | gemcitabine | 178 ± 3.6 | 0.15 ± 0.02 |
| Test LNs | POPE-azobenzene-PEG (50): | gemcitabine | 180 ± 3.4 | 0.23 ± 0.01 |
| DSPC (39): | ||||
| DSPE-PEG-iRGD (10): | ||||
| Lissamine rhodamine (1) | ||||
| LNs without iRGD | POPE-AZB-PEG (50): | gemcitabine | 121 ± 1.5 | 0.12 ± 0.01 |
| DSPC (49): | ||||
| Lissamine rhodamine (1) | ||||
| Test LNs | POPE-AZB-PEG (50): | carboxyfluorescein | 235 ± 2.3 | 0.20 ± 0.04 |
| DSPC (40): | ||||
| DSPE-PEG-iRGD (10): |
We imaged the LNs by atomic force microscopy (AFM) to determine any structural changes in the hypoxic environment. In this endeavor, we prepared the LNs in 25 mM HEPES buffer (pH 7.4) and added NADPH and rat liver microsomes. The control samples were exposed to normal atmospheric oxygen levels while nitrogen gas was bubbled through the test reaction mixture for 2 h. We observed that the LNs under normoxic conditions retained the spherical structures (Figure 4, Panel A). However, hypoxic conditions distorted the shapes of the vesicles (Figure 4, Panel B), indicating structural changes in the lipid bilayer. We conducted the AFM studies by placing the LNs on a mica surface, drying, and subsequent imaging. We note that some inadvertent structural perturbations may be introduced due to the sample preparation procedure. However, multiple reports indicate that liposomes and other lipid nanoparticles retain their structure after drying on a mica surface.20,21
Figure 4.
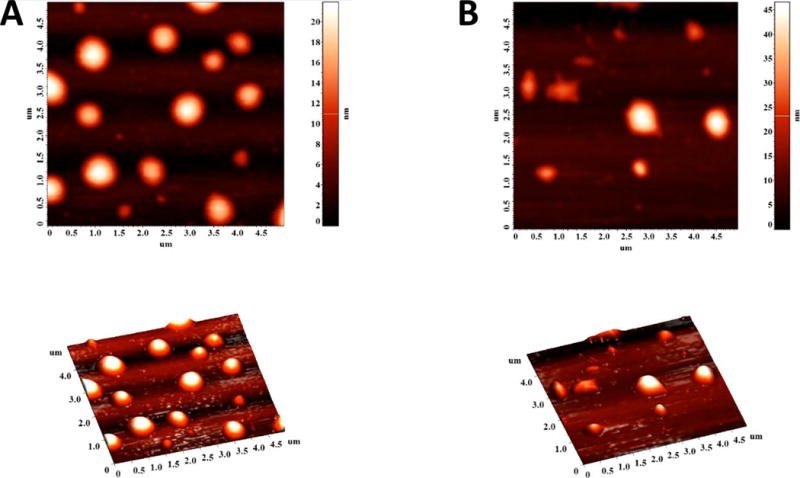
AFM images of LNs under normal oxygen levels (A) and after 2 h of hypoxia treatment in the presence of NADPH and rat liver microsomes (B).
After confirming the hypoxia responsive characteristics of the LNs, we encapsulated the anticancer drug gemcitabine by the active loading method (as a model hydrophilic drug).6 Gemcitabine is the first line of treatment for pancreatic cancer. Since gemcitabine in a solution is colorless, we incorporated 1% of the fluorescent lipid 1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine-N-(lissamine rhodamine B sulfonyl) (lissamine rhodamine lipid) in the LNs (molar ratio of DSPC:POPE-azobenzene-PEG1900:DSPE-PEG-iRGD:lissamine rhodamine lipid 39:50:10:1) for visualization. To encapsulate gemcitabine, a pH gradient was developed across the lipid bilayer with inside pH of 4 and outside pH of 7.4. After the active loading, we used gel filtration to remove any unencapsulated drug. The entrapment efficiency for these LNs was found to be about 40% with a drug loading capacity of 8% (by absorption spectroscopy).
The effectiveness of the drug encapsulated LNs was tested using monolayer and three-dimensional spheroid cultures of the pancreatic cancer cells BxPC-3. We observed that the viability of cells in monolayer cultures decreased in the presence of LNs encapsulating gemcitabine under both normoxic and hypoxic conditions. However, the response of three-dimensional tumor tissues cannot be predicted from monolayer cells cultures.22 Hence, we cultured the BxPC-3 cells as spheroids in agarose scaffolds (Figure 5).
Figure 5.
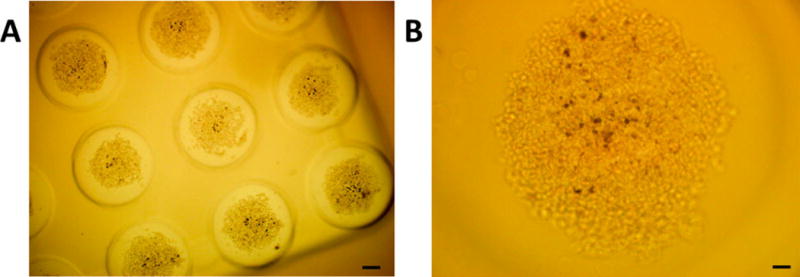
Optical microscopic images of the cultured spheroids of BxPC-3 cells in agarose molds at (A) 4× magnification (scale bar: 200 µm), and (B) 10× magnification (scale bar: 50 µm).
We divided the BxPC-3 cell spheroids into two groups and cultured them in normoxic and hypoxic (1% oxygen) conditions for 72 h. Both groups received treatments with gemcitabine encapsulating iRGD functionalized hypoxia-sensitive LNs, gemcitabine encapsulating LNs without the iRGD peptide on the surface, free gemcitabine, and gemcitabine encapsulating LNs devoid of hypoxia responsive lipid (prepared with DSPC). We measured the cell viability by the Alamar Blue assay.6 To make a valid comparison between the groups, we ensured the gemcitabine concentration was 20 µM for all the treatments. Results indicated that the cell viability in monolayer cultures (Figure 6A) was unaffected after treatment with drug encapsulated LNs under normoxic and hypoxic conditions. However, we observed significant decrease in cell viability for the cells treated with iRGD conjugated LNs under hypoxia compared to the normoxic conditions (p ≤ 0.05). Cell viability in spheroids treated with DSPC LNs, free gemcitabine, and LNs devoid of iRGD peptide showed a decrease in cell viability under normoxic as well as hypoxic conditions (Figure 6B). The reduction in cell viability under normoxic spheroids may be due to reduction of azobenzene and release of gemcitabine in hypoxic core of the BxPC-3 cell spheroids. The treatment with iRGD conjugated, hypoxia-responsive LNs decreased the cell viability in hypoxic spheroidal culture to 25% (Figure 6B) showing a significant decrease in cell viability compared to the spheroidal cultures treated under normoxic conditions. We speculate that the enhanced cytotoxicity is due to the penetration ability of iRGD conjugated LNs. As the LNs penetrate deep inside the core of the spheroids, they are likely to encounter a hypoxic microenvironment and release the encapsulated drugs. The reduced effectiveness of the other liposomal formulations and the free gemcitabine is probably due to less penetration in the spheroids.
Figure 6.
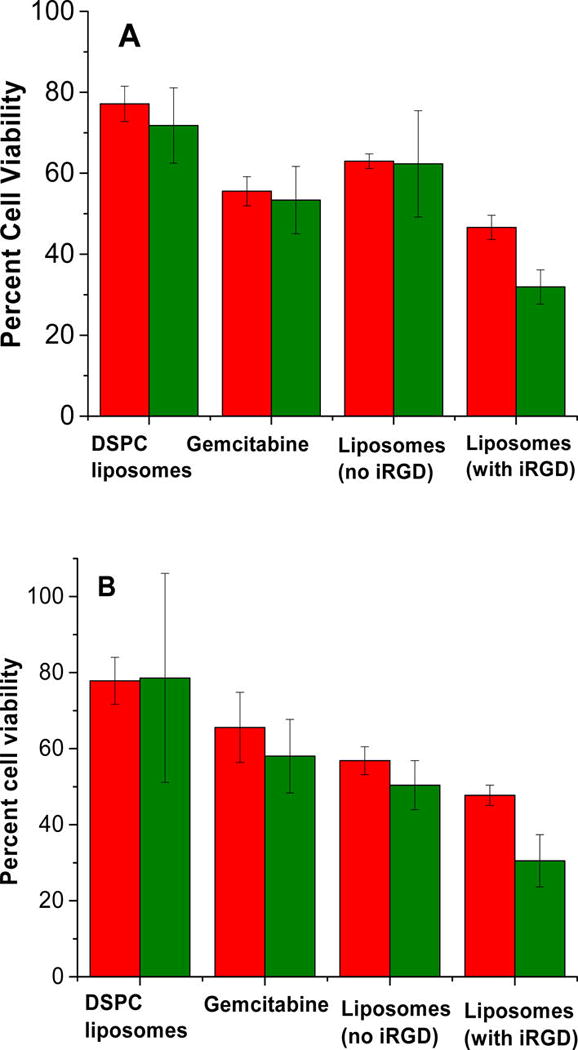
Viability of BxPC-3 cells cultured as monolayers (A) and three-dimensional spheroids (B) after treatment with DSPC LNs encapsulating gemcitabine (20 µM), free gemcitabine (20 µM), gemcitabine encapsulated hypoxia-responsive LNs devoid of surface iRGD peptide, and the hypoxia-sensitive LNs with the iRGD under normoxic (red bars) and hypoxic conditions (green bars).
The cell viability studies in the spheroidal cultures indicated that the hypoxia-responsive LNs were able to release the encapsulated drug in the hypoxic BxPC-3 cells. To observe the penetration depth facilitated by iRGD peptide on the surface, we cultured the BxPC-3 cells in a paper stack as a tumor tissue23 using our 3D printed apparatus (Materials and Methods). We delivered 20 µL of carboxyfluorescein encapsulated, hypoxia-sensitive LNs through the hollow tube of the paper stack insert, and incubated for 2 h under normoxic and hypoxic conditions. Subsequently, the apparatus was disassembled, the filter papers were separated, and imaged under a fluorescence microscope. We observed very low fluorescence under normoxic conditions after 2 h, even from the top filter paper (Figure 7A, Normoxic Panel). However, under hypoxic conditions, considerably higher fluorescence intensity was observed (Figure 7B, Hypoxic Panel) from the top filter paper, indicating the release of the LN encapsulated carboxyfluorescein dye. We also noted that the LNs conjugated to the iRGD peptide penetrated deeper into the cultured tumor tissues. LNs devoid of the iRGD peptide were unable to reach the eighth layer of the stack under hypoxic conditions (Figure 7A, Hypoxic Panel). However, LNs functionalized with the iRGD peptide penetrated deeper into the stack, reaching the 12th layer of the culture from the top (approximately 1.8 mm in depth, Figure 7B, Hypoxic Panel).
Figure 7.
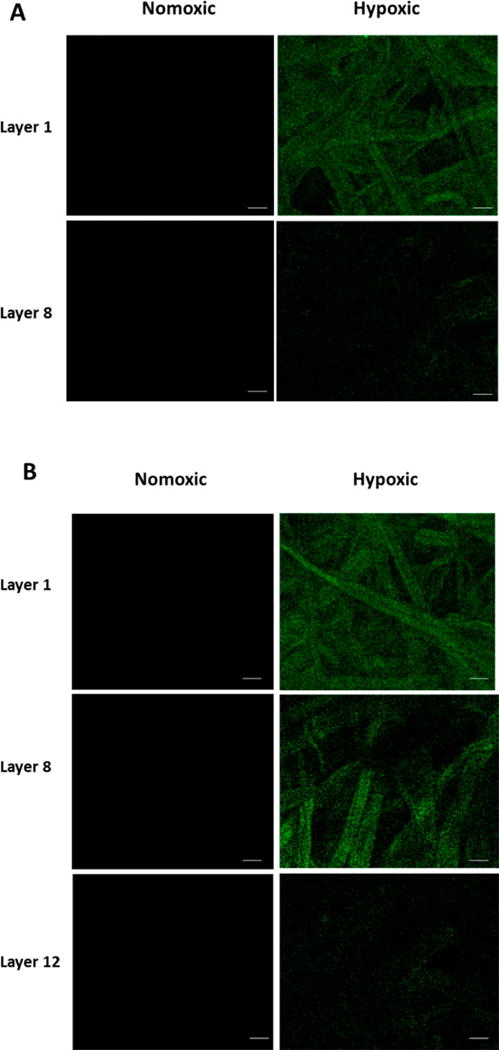
Fluorescence microscopic images of layers of cells indicating carboxyfluorescein release from the LNs under normoxic and hypoxic conditions without (A) and with (B) the iRGD peptide functionalization (scale bar: 100 µm).
In conclusion, we have successfully synthesized a hypoxia-sensitive lipid and prepared iRGD peptide functionalized, hypoxia-responsive LNs encapsulating the anticancer drug gemcitabine in the aqueous core. The LNs released 65% of the encapsulated contents under hypoxic conditions in 2 h. The pancreatic cancer cells BxPC-3 treated with the LNs showed decreased cell viability compared to the free drug and DSPC LNs. The iRGD peptide on the surface allowed the LNs to penetrate deeper and deliver the anticancer drug to the hypoxic cores, resulting in enhanced cytotoxicity for the cultured pancreatic cancer cell spheroids.
MATERIALS AND METHODS
Materials
The amino acids and the resins for peptide synthesis were purchased from Peptides International. The lipids, 1,2-distearoyl-sn-glycero-3-phosphocholine (DSPC), 1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine-N-lissamine rhodamine B sulfonyl ammonium salt (rhodamine lipid), and palmitoyl oleoylphosphatidylethanolamine (POPE) were purchased from Avanti Polar Lipids. The 1,2-distearoyl-sn-glycero-3-phosphoethanolamine (DSPE)-PEG-N3 (molecular weight 5000) was purchased from Nanocs. The pancreatic cancer cell line BxPC-3 was purchased from ATCC. The cell culture media, fetal bovine serum, and antibiotics were purchased from Lonza. All other chemicals used were laboratory grade.
Synthesis and Characterization of Hypoxia Responsive Lipid PEG–Azobenzene–POPE
Following a reported procedure,24 the amine terminated mPEG2000 was conjugated with the azobenzene-4,4′ dicarboxylic acid. NMR spectrum confirmed formation of the product.24 1H NMR (400 MHz, chloroform-d) δ ppm 0.00–0.01 (m), 1.08–1.36 (m), 3.37–3.38 (m), 3.53–3.63 (m), 3.77–3.91 (m), 5.25–5.38 (m), 7.27 (s) 7.93–8.07 (m). The resultant polymer attached diphenyl azacarboxylate (100 mg, 0.046 mmol) was reacted with POPE (35 mg, 0.048 mmol) employing HBTU (19 mg, 0.048 mmol), HOBt (7 mg, 0.050 mmol), and diisopropylethyl amine (18 mg, 0.138 mmol) in DMF (20 mL) under inert conditions overnight. After removal of the solvent under reduced pressure, the semisolid was dissolved in dichloromethane, washed with 10% citric acid solution, and 0.5 N NaOH solution. The clear organic phase was dried over sodium sulfate and evaporated under reduced pressure. The resulting semisolid was subjected to chromatographic purification (Rf = 0.7 in 10% methanol in dichloromethane) affording 105 mg (79%) of the final product. 1H NMR (400 MHz, chloroform-d) δ ppm 0.80–0.92 (m), 1.16–1.40 (m), 1.52–1.64 (m), 1.97–2.22 (m), 2.24–2.32 (m), 2.91–3.08 (m), 3.35–3.50 (m), 3.54–3.75 (m), 3.90–4.07 (m), 4.09–4.22 (m), 5.19–5.38 (m), 7.27 (s), 7.91–8.03 (m). 13C NMR (100 MHz, chloroform-d) δ ppm 14.12, 22.69, 27.22, 29.32, 29.53, 29.71, 45.60, 59.04, 70.57, 71.95, 76.71, 77.03, 77.35, 119.07, 128.51, 133.30, 153.08, 167.2, 173.79.
Synthesis and Characterization of the iRGD Peptide
The peptide was synthesized using a microwave-assisted, solid-phase peptide synthesizer (Liberty Blue with Discover microwave unit, CEM Corporation) employing the CLEAR Amide resin as the solid support (0.2 g, 0.1 mmol/g). The sequence hexynoic acid-Cys(Acm)-Arg(Pbf)-Gly-Asp(OBut)-Lys(Boc)-Gly-Pro-Asp(OBut)-Cys(Acm)-OH was synthesized without the final deprotection. To cyclize the product, thallium trifluoroacetate (55 mg, 0.1 mmol) in DMF (5 mL) was stirred with the resin conjugated protected peptide for 3 h. The resin was washed with DMF (3×) and dichloromethane (3×). Subsequently, the peptide was cleaved from the resin with trifluoroacetic acid (19 mL), and distilled water (0.5 mL) for 2 h. The resin was filtered through a Whatman filter paper. To the filtrate, 15 mL cold diethyl ether was added, and the obtained precipitate was dried in a vacuum desiccator. The obtained product was characterized by MALDI-TOF mass spectrometry (observed mass: 1042.36, expected mass: 1042.43, Supporting Information, Figure S1) and circular dichroism spectroscopy. CD spectra were recorded using 1 mg/mL solution of the iRGD peptide in phosphate buffer (4 mM, pH = 7.5, Supporting Information, Figure S2).
Synthesis of iRGD Peptide–Lipid Conjugate
The lipid DSPE-PEG-N3 was reacted with the hexynoic acid conjugated iRGD peptide using the “Click” reaction. To conjugate the alkyne of the peptide to the azide group of the lipid, the compounds were reacted in 1:2 molar ratio. The hexynoic acid conjugated peptide (40 mg) and DSPE-PEG-N3 (50 mg) were dissolved in 3 mL water. The copper complex was prepared by mixing copper(II) sulfate with N,N,N′,N′,N″-pentamethyl diethylenetriamine (PMDETA) for 2 h at room temperature. The ascorbic acid solution was prepared in water. The copper complex (53 mM CuSO4 solution in water and 2 mmol PMDETA stirred for 2 h) and sodium ascorbate (1.4 µmol) were added to the reaction mixture and stirred for 24 h at room temperature. The sample was then transferred to a dialysis cassette with a molecular weight cutoff of 3000. The reaction mixture was dialyzed against water for 72 h to remove the catalyst and unreacted iRGD peptide. The product was then analyzed by CD spectroscopy (Supporting Information, Figure S3).
Preparation of LNs for Release Studies
LNs were prepared by the thin film hydration method. DSPC, hypoxia-sensitive POPE-azobenzene-PEG, and iRGD conjugated DSPE lipid were dissolved in chloroform in a round-bottom flask (molar ratio 50:40:10 respectively). Chloroform was then evaporated using a rotary evaporator to form a thin film at the bottom of the flask. The thin film was dried overnight in a vacuum desiccator. The dry film was hydrated with a carboxyfluorescein (100 mM) solution (prepared in 25 mM HEPES buffer, pH 7.4) for 1 h at 60 °C. The multilamellar LNs formed were then subjected to ultrasonication for 1 h at level 9 on a bath sonicator (Aquasonic, model 250D). The formed LNs were then extruded 11 times through 0.8 µm Whatman filter paper using an extruder (Avanti Polar Lipids). To reduce the size, the LNs were again extruded 11 times through 0.2 µm Whatman filter. Excess of carboxyfluorescein (unencapsulated) was removed by a Sephadex G100 size exclusion column. The eluted LNs were used for release studies.
Size Analysis
The hydrodynamic diameter of the LNs was measured by dynamic light scattering, using a Malvern Zetasizer. All measurements were performed employing 1 mL sample with 6 runs and 6 repeats.
Release Studies
LNs (20 µL of 1 mg/mL total lipid concentration) encapsulating carboxyfluorescein were taken in 25 mM HEPES buffer of pH 7.5 (180 µL). Rat liver microsomes were isolated and (10 µL of 790 µg/mL) were added to this solution along with NADPH (100 µM) as a cofactor.25 The LNs were subjected to normoxic and hypoxic conditions to measure the release of the encapsulated dye. Normoxic conditions were maintained by bubbling air through the buffer containing the LNs. Hypoxic conditions were created by bubbling nitrogen through the reaction mixture. Emission intensity of the mixture was measured at 515 nm as a function of time (excitation: 485 nm).
Atomic Force Microscopic Imaging
An atomic force microscope (AFM) was used to image the LNs on a mica surface. The AFM measurements were carried out in noncontact mode at a scanning rate of 0.7 Hz and a resonance frequency of 145 kHz using an NT-MDT NTEGRA (NT-MDT America, Tempe, AZ). The cantilever was made of silicon nitride and was 100 µm long. The scanning areas were 5 × 5 or 20 × 20 µm2 at the resolution of 512 or 1024 points per line, respectively. The LN solution was dropped on top of a freshly cleaved mica surface and kept for 10 min at room temperature. The remaining solution was rinsed with water and dried extensively with an air blow gun. The mica substrate was glued on top of a sapphire substrate (sample holder) using Scotch double-sided tape, and cleaved with Scotch tape to obtain a debris-free and flat surface. The images were flattened by a first-order line correction and a first-order plane subtraction to compensate for the sample tilt.
Preparation of Gemcitabine Encapsulated LNs
Gemcitabine was encapsulated in LNs by the active loading method.6 The lipid DSPC, hypoxia-sensitive POPE-azobenzene-PEG, iRGD conjugated DSPE, and POPE-lissamine rhodamine were dissolved in chloroform in a round-bottom flask (molar ratio of 49:40:10:1 respectively). The lipid thin film was prepared as described before. The thin film was hydrated with 25 mM citrate buffer (pH 4) for 1 h at 60 °C. Subsequently, the formed LNs were sonicated in a bath sonicator (Aquasonic 250D, Power level 9) for 1 h. The LNs were then extruded through a 0.2 µm Whatman Nucleopore membranes for 11 times and were passed through Sephadex G100 column to build the pH gradient across the membrane. Gemcitabine was encapsulated in these LNs by incubating the drug (lipid:drug ratio 9:1) with the LNs for 3 h at 60 °C. The absorbance spectrum of the LNs was recorded. From a standard curve, the amount of gemcitabine was determined. Percent entrapment and encapsulation efficiency for gemcitabine were calculated using the following formula:
Cellular Studies
The cellular studies were carried out with the pancreatic cancer cell line BxPC-3 in RPMI media (supplemented with 10% FBS and 1% antibiotics). The cells were grown as a monolayer and as three-dimensional spheroids. Spheroids grown on 96 well agarose microwell plate were used for cell viability assay. To study the penetration ability of the LNs, BxPC-3 cells were grown on stacks of wet strengthened Whatman filter paper (number 114).
Cell Viability Studies in Monolayer Cultures
In a 96 well clear bottom plate, the BxPC-3 cells (2000 per well) were seeded. The cells were allowed to attach to the surface and grow for 24 h at 37 °C in an incubator supplemented with 5% CO2. The cells received treatment with hypoxia responsive, iRGD peptide-decorated LNs encapsulating gemcitabine (20 µM) for 3 days. LNs devoid of hypoxia-sensitive lipid and without iRGD conjugated lipid were used as controls. Gemcitabine (20 µM) in aqueous solution was used as a positive control. After 3 days of treatment, the conditioned media was removed from the plate (to discard the fluorescent LNs) and was replaced with 200 µL of fresh medium supplemented with 20 µL Alamar Blue (Invitrogen). The plates were incubated for 2 h, and fluorescence was measured (excitation wavelength 560 nm, emission wavelength 590 nm).
Cell Viability Study in Spheroid Cultures
The BxPC-3 cell spheroids were prepared by seeding the 2 × 106 cells in each of the 96 well agarose scaffolds. To make the scaffold, an aqueous agarose solution (2% w/v) was sterilized in an autoclave for 45 min. The mold was purchased from Microtissues, and the scaffolds were prepared using manufacturer’s protocol. The cells were seeded on 36 scaffolds and were incubated for 7 days. Media on the scaffolds was changed every 2 days. The scaffolds were then divided equally into two groups (18 scaffolds per group). One group of scaffold was allowed to grow under normoxic conditions, and another group was incubated in a hypoxic chamber maintained at an oxygen level of 1% (hypoxic conditions) for 24 h. After establishing normoxic and hypoxic cultures, the scaffolds were treated with gemcitabine, gemcitabine encapsulated hypoxia-sensitive LNs, gemcitabine encapsulated iRGD functionalized hypoxia responsive LNs, and gemcitabine encapsulating LNs devoid of iRGD and stimuli responsive lipids. Treatment with buffer and buffer encapsulating LNs was used as a control. The scaffolds were divided equally into each of these treatment groups (3 scaffolds in each treatment group). The cell spheroids were treated under normoxic and hypoxic conditions for 72 h. After the treatment, the cells in each spheroid scaffold were dissociated by using a Tryple solution (1 mL for each scaffold) for 10 min. The scaffolds were then washed with 3 mL of cell culture medium to harvest the cells. The harvested cells from each scaffold were then dissociated and plated directly in 6 wells of 96 well plate. This step converted the three-dimensional cell culture to a monolayer culture. It was crucial to keep the dilution of the cells exactly same for each treatment group to keep the relative ratios of cell viability in each treatment group. The cell viability was then measured by the Alamar Blue assay.
Cellular Uptake in Spheroid Cultures
To facilitate the hypoxic microenvironment of tumors, we cultured the BxPC-3 cells as layers. To grow the layered cells, a stack of wet strengthened Whatman filter paper (No. 114) was used.23 This setup allowed us to image the depth of penetration of the iRGD peptide-decorated LNs. We cut these papers as 1-in.-diameter circles using a commercially available hole punch. The cut circles were then wrapped in aluminum foil and were autoclaved for 45 min. We designed a culture apparatus (using the CAD software Creo Parametric) to hold the paper stack in place. The assembly was then 3D printed using poly(lactic acid) as the “ink”. Our apparatus had four components—base, paper stack insert, paper press insert attached to a media transfer tube, and a cover (Figure 8).
Figure 8.
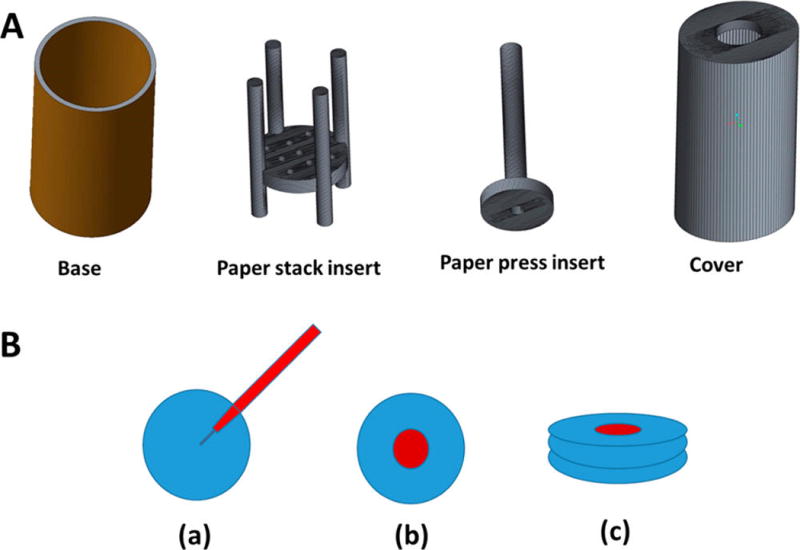
(A) 3D printed parts for the cell culture apparatus to hold the stacks of Whatman filter paper. (B) (a) Sterilized filter papers were inoculated with BxPC-3 cells embedded in agarose and sodium alginate, (b) stacked with other filter papers, and (c) incubated together for layered cell culture. The paper stack inert holds the cells inoculated filter papers. The paper insert press allows holding the paper stack in place. The hollow tube inside the paper press insert allows addition of culture media to the growing cells without disturbing the cell layers. These inserts were contained in a chamber with the help of base and cover.
The trypsinized BxPC-3 cells were centrifuged, and 100 000 cells were suspended in 500 µL culture medium. A solution of agarose and sodium alginate (1:2) was autoclaved, cooled to 40 °C, and the cells suspension was added. After mixing, 20 µL of this suspension was applied at the center of each paper circle. The cell-seeded papers were stacked together on the 3D printed paper stack insert with the paper press insert on the top. The assembly was inserted in the base, and the cover was placed. Two apparatus were then incubated under normoxic conditions, and two were incubated in 1% oxygen for 3 days. The cell stacks in normoxic and hypoxic incubators received treatments with 20 µL of carboxyfluorescein encapsulated LNs (delivered through the hollow tube of the paper press insert) with and without the iRGD on the surface. The cell stacks were treated for 2 h in respective incubation conditions (normoxic/hypoxic), the paper racks were removed, and were submerged in HBSS solution to wash excess of carboxyfluorescein. After washing the stacks 3 times, they were again immersed in cell culture medium before imaging. To image the cells with a fluorescence microscope, the papers from the stack were peeled one at a time and fluorescence was measured in each paper layer. The fluorescence intensity and the depth of penetration of polymersomes were measured using an Olympus laser scanning confocal microscope. The images were analyzed with the ImageJ software.
Acknowledgments
This research was supported by NSF grant DMR 1306154, and NIH grant 1 R01GM 114080 to S.M. P.K. was supported by a Doctoral Dissertation Award (IIA-1355466) from the North Dakota EPSCoR (National Science Foundation). Y.C. acknowledges support from the NDSU startup funds, the NSF EPSCoR New Faculty Award, and the ND NASA EPSCoR RID Grant.
Footnotes
ASSOCIATED CONTENT
Supporting Information
- MALDI mass spectrum of hexynoic acid conjugated iRGD, CD spectra of hexynoic acid conjugated iRGD peptide, CD spectrum of iRGD functionalized DSPE lipid, release profile of the control DSPC liposomes (PDF)
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
References
- 1.Ganta S, Devalapally H, Shahiwala A, Amiji M. A review of stimuli-responsive nanocarriers for drug and gene delivery. J. Controlled Release. 2008;126:187–204. doi: 10.1016/j.jconrel.2007.12.017. [DOI] [PubMed] [Google Scholar]
- 2.Sharma A, Sharma US. Liposomes in drug delivery: progress and limitations. Int. J. Pharm. 1997;154:123–140. [Google Scholar]
- 3.O’brien M, Wigler N, Inbar M, Rosso R, Grischke E, Santoro A, Catane R, Kieback D, Tomczak P, Ackland S. Reduced cardiotoxicity and comparable efficacy in a phase III trial of pegylated liposomal doxorubicin HCl (CAELYX/Doxil®) versus conventional doxorubicin for first-line treatment of metastatic breast cancer. Ann. Oncol. 2004;15:440–449. doi: 10.1093/annonc/mdh097. [DOI] [PubMed] [Google Scholar]
- 4.Torchilin V. Tumor delivery of macromolecular drugs based on the EPR effect. Adv. Drug Delivery Rev. 2011;63:131–135. doi: 10.1016/j.addr.2010.03.011. [DOI] [PubMed] [Google Scholar]
- 5.Danhier F, Feron O, Préat V. To exploit the tumor microenvironment: passive and active tumor targeting of nanocarriers for anti-cancer drug delivery. J. Controlled Release. 2010;148:135–146. doi: 10.1016/j.jconrel.2010.08.027. [DOI] [PubMed] [Google Scholar]
- 6.Kulkarni PS, Haldar MK, Nahire RR, Katti P, Ambre AH, Muhonen WW, Shabb JB, Padi SK, Singh RK, Borowicz PP, et al. MMP-9 Responsive PEG cleavable nanovesicles for efficient delivery of chemotherapeutics to pancreatic cancer. Mol. Pharmaceutics. 2014;11:2390–2399. doi: 10.1021/mp500108p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Höckel M, Vaupel P. Tumor hypoxia: definitions and current clinical, biologic, and molecular aspects. J. Natl. Cancer Inst. 2001;93:266–276. doi: 10.1093/jnci/93.4.266. [DOI] [PubMed] [Google Scholar]
- 8.Vaupel P, Kallinowski F, Okunieff P. Blood flow, oxygen and nutrient supply, and metabolic microenvironment of human tumors: a review. Cancer Res. 1989;49:6449–6465. [PubMed] [Google Scholar]
- 9.Höckel M, Schlenger K, Aral B, Mitze M, Schäffer U, Vaupel P. Association between tumor hypoxia and malignant progression in advanced cancer of the uterine cervix. Cancer Res. 1996;56:4509–4515. [PubMed] [Google Scholar]
- 10.Brown JM, Giaccia AJ. The unique physiology of solid tumors: opportunities (and problems) for cancer therapy. Cancer Res. 1998;58:1408–1416. [PubMed] [Google Scholar]
- 11.Kadonosono T, Yamano A, Goto T, Tsubaki T, Niibori M, Kuchimaru T, Kizaka-Kondoh S. Cell penetrating peptides improve tumor delivery of cargos through neuropilin-1-dependent extravasation. J. Controlled Release. 2015;201:14–21. doi: 10.1016/j.jconrel.2015.01.011. [DOI] [PubMed] [Google Scholar]
- 12.Sugahara KN, Teesalu T, Karmali PP, Kotamraju VR, Agemy L, Girard OM, Hanahan D, Mattrey RF, Ruoslahti E. Tissue-penetrating delivery of compounds and nanoparticles into tumors. Cancer Cell. 2009;16:510–520. doi: 10.1016/j.ccr.2009.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Longati P, Jia X, Eimer J, Wagman A, Witt M-R, Rehnmark S, Verbeke C, Toftgård R, Löhr M, Heuchel RL. 3D pancreatic carcinoma spheroids induce a matrix-rich, chemoresistant phenotype offering a better model for drug testing. BMC Cancer. 2013;13:1. doi: 10.1186/1471-2407-13-95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brown JM, Wilson WR. Exploiting tumour hypoxia in cancer treatment. Nat. Rev. Cancer. 2004;4:437–447. doi: 10.1038/nrc1367. [DOI] [PubMed] [Google Scholar]
- 15.Vaupel P, Harrison L. Tumor hypoxia: causative factors, compensatory mechanisms, and cellular response. Oncologist. 2004;9:4–9. doi: 10.1634/theoncologist.9-90005-4. [DOI] [PubMed] [Google Scholar]
- 16.Kiyose K, Hanaoka K, Oushiki D, Nakamura T, Kajimura M, Suematsu M, Nishimatsu H, Yamane T, Terai T, Hirata Y. Hypoxia-sensitive fluorescent probes for in vivo real-time fluorescence imaging of acute ischemia. J. Am. Chem. Soc. 2010;132:15846–15848. doi: 10.1021/ja105937q. [DOI] [PubMed] [Google Scholar]
- 17.Guengerich FP, Martin MV. Purification of cytochrome P-450, NADPH-cytochrome P-450 reductase, and epoxide hydratase from a single preparation of rat liver microsomes. Arch. Biochem. Biophys. 1980;205:365–379. doi: 10.1016/0003-9861(80)90119-8. [DOI] [PubMed] [Google Scholar]
- 18.Ruoslahti E. Peptides as targeting elements and tissue penetration devices for nanoparticles. Adv. Mater. 2012;24:3747–3756. doi: 10.1002/adma.201200454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Teesalu T, Sugahara KN, Ruoslahti E. Tumor-penetrating peptides. Front. Oncol. 2013;3:b3. doi: 10.3389/fonc.2013.00216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Egawa H, Furusawa K. Liposome adhesion on mica surface studied by atomic force microscopy. Langmuir. 1999;15:1660–1666. [Google Scholar]
- 21.Teschke O, De Souza E. Liposome structure imaging by atomic force microscopy: verification of improved liposome stability during adsorption of multiple aggregated vesicles. Langmuir. 2002;18:6513–6520. [Google Scholar]
- 22.Lee J, Lilly GD, Doty RC, Podsiadlo P, Kotov NA. In vitro toxicity testing of nanoparticles in 3D cell culture. Small. 2009;5:1213–1221. doi: 10.1002/smll.200801788. [DOI] [PubMed] [Google Scholar]
- 23.Derda R, Laromaine A, Mammoto A, Tang SK, Mammoto T, Ingber DE, Whitesides GM. Paper-supported 3D cell culture for tissue-based bioassays. Proc. Natl. Acad. Sci. U. S. A. 2009;106:18457–18462. doi: 10.1073/pnas.0910666106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Perche F, Biswas S, Wang T, Zhu L, Torchilin V. Hypoxia-Targeted siRNA Delivery. Angew. Chem. Int. Ed. 2014;53:3362–3366. doi: 10.1002/anie.201308368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kamath S, Kummerow F, Narayan KA. A simple procedure for the isolation of rat liver microsomes. FEBS Lett. 1971;17:90–92. doi: 10.1016/0014-5793(71)80571-9. [DOI] [PubMed] [Google Scholar]