

Abstract
Previous studies have suggested that macrophage-mediated chronic inflammation is involved in the development of rotator cuff muscle atrophy and degeneration following massive tendon tears. Increased RhoA signaling has been reported in chronic muscle degeneration, such as muscular dystrophy. However, the role of RhoA signaling in macrophage infiltration and rotator muscle degeneration remains unknown. Using a previously established rat model of massive rotator cuff tears, we found RhoA signaling is upregulated in rotator cuff muscle following a massive tendon-nerve injury. This increase in RhoA expression is greatly potentiated by the administration of a potent RhoA activator, lysophosphatidic acid (LPA), and is accompanied by increased TNFα and TGF-β1 expression in rotator cuff muscle. Boosting RhoA signaling with LPA significantly worsened rotator cuff muscle atrophy, fibrosis, and fatty infiltration, accompanied with massive monocytic infiltration of rotator cuff muscles. Co-staining of RhoA and the tissue macrophage marker CD68 showed that CD68+ tissue macrophages are the dominant cell source of increased RhoA signaling in rotator cuff muscles after tendon tears. Taken together, our findings suggest that LPA-mediated RhoA signaling in injured muscle worsens the outcomes of atrophy, fibrosis, and fatty infiltration by increasing macrophage infiltraion in rotator cuff muscle. Clinically, inhibiting RhoA signaling may represent a future direction for developing new treatments to improve muscle quality following massive rotator cuff tears.
Keywords: rotator cuff tear, muscle, LPA, macrophage, atrophy, fatty infiltration
Rotator cuff (RC) tears represent a growing disease burden for our aging population, with a full-thickness tear prevalence of 22% in patients 65 and older.1 While small tears respond well to surgical repair, large tears often have poor outcomes marked by failed repair, persistent pain, and retear. RC muscle quality is an important predictor of RC repair outcomes, with atrophy and fatty infiltration identified as independent predictors of poor outcomes.2 Currently, muscle degeneration after massive RC tears is thought to be largely irreversible.
Chronic inflammation has been identified as an important factor responsible for muscle atrophy and degeneration in muscular dystrophy,3 chronic obstructive pulmonary disease (COPD),4 cachexia, and sarcopenia.5 Recently, Gumucio et al. showed increased inflammatory cytokines are accompanied by accretion of macrophages in areas of fat accumulation in RC muscles in a rat RC tear model.6 These data suggest that macrophage-mediated chronic inflammation is also involved in the development of RC muscle atrophy and degeneration.
Lysophosphatidic acid (LPA) is a small biologically active phospholipid derivative, which binds to its G-protein-coupled receptors, LPAR1, 2, and 3, and leads to increased cell division, migration, and invasion.7–9 Through these effects on cells, LPA plays diverse roles in the normal development of bone10 as well as the nervous system.11 A previous study demonstrated that LPA is a major serum noncytokine survival factor for macrophages.12 Endogenous LPA has also been implicated in the development of pulmonary13 and renal fibrosis.14 A recent study showed that LPA-induced renal fibrosis was mediated through LPA2 receptors and Gαq proteins, resulting in transactivation of latent TGF-β in a Rho/Rho-kinase (ROCK)-dependent manner,15 suggesting a potential role for both TGF-β and RhoA signaling in the downstream effects of LPA.
RhoA is a member of the small GTPase-Rho family, which also includes Rho, Rac and Cdc42.16 LPA is an important activator of RhoA17 through Akt/IκBα signaling.18 The RhoA signaling pathway is regulated via G protein-coupled receptors and is activated by the interaction between the receptors and ECM and/or soluble factors.16,19 RhoA activates RhoA kinase (ROCK) to increase turnover of the actin cytoskeleton and has been shown to contribute to monocyte transendothelial migration into tissue.20 Increased RhoA signaling has been reported in chronic muscle degeneration, such as muscular dystrophy.21 However, the role of RhoA signaling in RC muscle atrophy and degeneration remains unknown.
The goal of this study was to investigate LPA-induced RhoA signaling in RC muscle degeneration in a rat model. We hypothesized that persistent upregulation of RhoA signaling will lead to a chronic inflammatory state with enhanced macrophage infiltration, leading to increased muscle atrophy, fibrosis, and fatty infiltration.
MATERIALS AND METHODS
Animal Surgery
Sixteen adult female Sprague–Dawley rats underwent complete supraspinatus and infraspinatus tendon transection (TT) with a 5 mm ipsilateral suprascapular nerve resection (DN), as previously described.22,23 Sham surgery was performed on the contralateral side of each animal. Our previous power analyses determined that N=4 animals/group is sufficient to detect a significant change in muscle weight and protein expression (α=0.05, β 0.2).22,24 Thus, we employed four animals per group per time point in this study. All procedures were approved by the San Francisco Veterans Affairs Health Care System (SFVAHCS) Institutional Animal Care and Use Committee (IACUC).
LPA Treatment
Lyosophosphatidic acid was dissolved in 0.1% BSA in PBS and delivered to the animals at a dose of 40 μg/kg via intraperitoneal injection. Injections with dissolved LPA or vehicle (0.1% BSA in PBS) were conducted for 5 out of every 7 days per week in a volume of 0.6 ml for either 2 or 6 weeks. The first dose was given immediately after surgery.
Muscle Harvest
Animals were sacrificed 2 or 6 weeks after surgery. Supraspinatus and infraspinatus muscles were harvested. Supraspinatus muscles were weighed and flash frozen in liquid nitrogen-cooled isopentane for histological processing, as previously described.24 Infraspinatus muscles were homogenized in Trizol reagent for RNA extraction.
Oil Red-O and Trichrome Staining
Sections were obtained from the muscle belly, away from the musculotendinous junction, at −20°C with a thickness of 10 μm. Total extracellular matrix collagen was assessed using Masson’s trichrome staining (Fisher Scientific). Fatty infiltration was assessed with oil red-O staining. The amount of collagen or fat per slide was quantified with Adobe Photoshop as previously described.25,26 Fibrosis and fat indices were calculated using the equation: Index (%)=(100* (Total area of collagen or fat)/(Total area of the section)). Four serial sections from the muscle belly from each of four animals were used to calculate the indices above.
α-Naphthyl Acetate Esterase and Naphthol AS-D Chloroacetate Esterase Staining
A double esterase staining kit (Sigma–Aldrich, St. Louis, MO) for naphthol AS-D chloroacetate esterase and α-napthyl acetate esterase was used to identify granulocyte-and monocyte-derived cells, respectively, according to manufacturer instructions. Granulocytes were identified as red in color, while cells of the monocytic lineage stained black. Esterase positive cells were counted for four whole muscle sections from each animal (N=4/time point/group). Granulocyte-and monocyte-derived cell density was calculated as number of cells/mm2 muscle section.
Immunohistofluorescence
CD68 and RhoA co-staining was performed on tissue sections to determine RhoA activity in macrophages. Sections were incubated overnight in rabbit anti-rat RhoA antibody (1:100 dilution, Sigma–Aldrich) with mouse anti-rat CD68 (1:50 dilution, Thermo Scientific, Middletown, VA) and then incubated with goat anti-rabbit Alexa Fluor® 488 and donkey anti-mouse Alexa Fluor® 568-conjugated secondary antibodies at a dilution of 1:5,000 and mounted with VectaShield® Antifade Mounting Medium with DAPI. Slides were visualized with Zeiss Axiovert 200 M fluorescent microscope (Zeiss Inc, Germany).
Real-Time Reverse Transcript Polymerase Chain Reaction (RT-PCR)
Total RNA was isolated using Trizol reagent (Invitrogen, Burlington, ONT, Canada) according to manufacturer instructions. cDNA was synthesized using Transcriptor First Strand cDNA Synthesis Kit (Roche Applied Bioscience, Indianapolis, IN). Real-time qRT-PCR was performed to quantify the expression of RhoA, MIF, GM-CSF, IL-1a, IL-6, IL-10, TNFα, and TGF-β1 in muscle samples using a SYBR Green I Master kit (Roche Applied Bioscience) with the primers found in Table 1. Gene expression was normalized to the housekeeping gene, RPS26. Fold change in mRNA expression was calculated by using ΔΔCT as described previously.25,26
Table 1.
Sequences of Primers Used for Real-time qRT-PCR
| Primer | Forward (5′ → 3′) | Reverse (5′ → 3′) |
|---|---|---|
| RhoA | GTAGAGTTGGCTTTATGGGACAC | TGFAGTCCATTTTTCTGGGATG |
| TGF-β1 | GGAGCCACTGCCCATCGTCTACTAC | GGAGCGCACGATCATGTTGGAC |
| IL-1a | AAGACAAGCCTGTGTTGCTGAAGG | TCCCAGAAGAAAATGAGGTCGGTC |
| IL-6 | TCCTACCCCAACTTCCAATGCTC | TTGGATGGTCTTGGTCCTTAGCC |
| IL-10 | CAATAACTGCACCCACTTCC | ATTCTTCACCTGCTCCACTGC |
| TNFα | AAATGGGCTCCCTCTCATCAGTTC | TCTGCTTGGTGGTTTGCTACGAC |
| GMCSF | TAAATGACATGCGTGCTCTGG | ATGAAATCCTCAAAGGTGGTG |
| MIF | CCATGCCTATGTTCATCGTG | GAACAGCGGTGCAGGTAAGTG |
| RPS26 | AAGGAGAAACAACGGTCGTG | GCAGGTCTGAATCGTGGTG |
Statistical Analysis
Student’s two-tailed t-test was used for all statistical analysis between LPA and vehicle treatment groups. Significance was defined as p<0.05. Data are presented as the mean±standard error.
RESULTS
RhoA Is Elevated in Injured RC Muscle
At 2 weeks after injury in vehicle-treated rats, we observed significantly increased RhoA staining in injured muscle, compared to minimal staining in sham-side muscle (Fig. 1). Expression of RhoA was determined to be upregulated fourfold compared to sham at 2 weeks (p<0.05) and 1.5-fold at 6 weeks after injury (n.s.) (Fig. 2).
Figure 1.
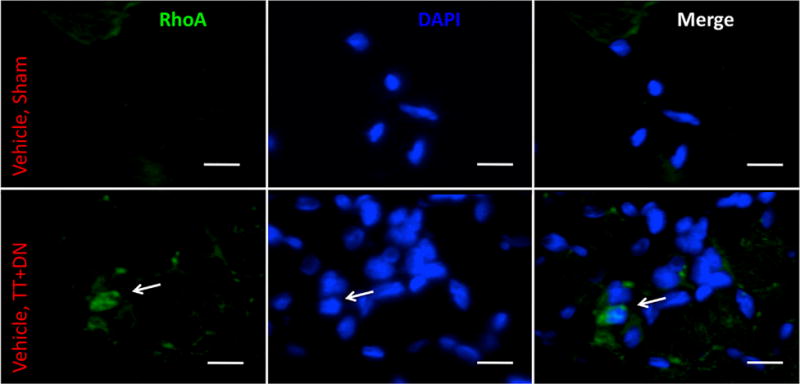
Representative immunohistofluorescence demonstrating increase in total RhoA expression in injured muscle (bottom row) compared to uninjured muscle (top row). White arrow indicates RhoA-positive cell. Scale bar=10 μm.
Figure 2.
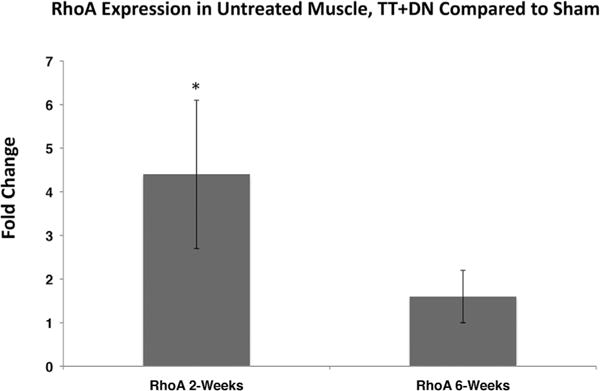
Real-time qRT-PCR demonstrating increase in total RhoA expression at 2 and 6 weeks after injury and initiation of treatment, TT+DN ΔCT values normalized to mean sham ΔCT values to calculate ΔΔCT. Student’s two-tailed t-test was performed between ΔCT values of injured and sham muscle, *p<0.05.
LPA Increases RhoA and Chronic Inflammatory Marker mRNA Expression in Injured Muscle
We found that at both 2 and 6 weeks after injury, total RhoA mRNA levels were significantly elevated in the injured muscle of mice treated with LPA compared to vehicle (Fig. 3). No change in RhoA expression between uninjured sides was observed at either time point.
Figure 3.
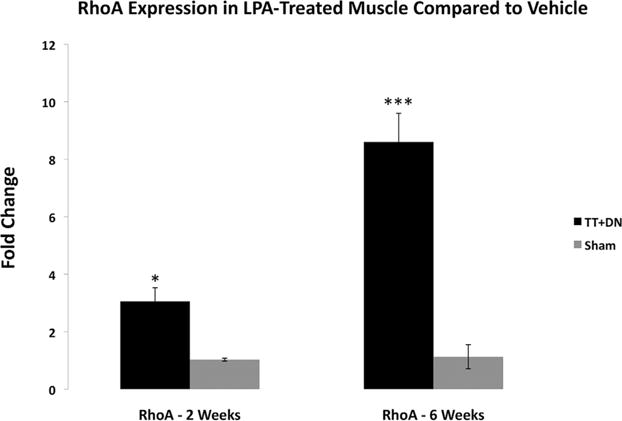
Real-time qRT-PCR comparing the effects of LPA vs. vehicle on total RhoA expression at 2 and 6 weeks after injury and initiation of treatment, LPA-treatment ΔCT values normalized to those for the vehicle group for both TT+DN and sham muscle to calculate ΔΔCT. Student’s two-tailed t-test was performed between ΔCT values of LPA treatment and vehicle groups for both injured and sham muscle, *p<0.05, ***p<0.001.
We next assessed a panel of acute and chronic inflammatory markers at 2 and 6 weeks and found that TNFα was expressed at higher levels in injured muscle of LPA-treated rats at 6 weeks along with a near-significant increase in TGF-β1 expression (p=0.06) (Fig. 4). Other inflammatory cytokines, including IL-1a, IL-6, and IL-10 did not show significant differences between LPA and vehicle groups at either time point. The markers of M1 macrophage polarization, MIF, and GM-CSF, likewise did not show significant differences in expression at either time point. We did not observe any differences in the expression of inflammatory markers in the uninjured muscle.
Figure 4.
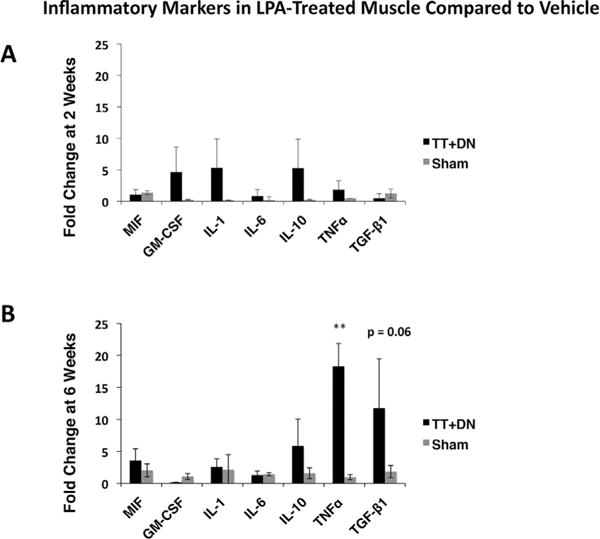
Real-time qRT-PCR comparing the effects of LPA versus vehicle on inflammatory markers at 2 and 6 weeks after injury and initiation of treatment, LPA-treatment ΔCT values normalized to those for the vehicle group for both TT DN and sham muscle to calculate ΔΔCT. Student’s two-tailed t-test was performed between ΔCT values of LPA treatment and vehicle groups for both injured and sham muscle, *p<0.01.
LPA Increases Inflammatory Cells Within RC Muscles
In order to assess the general inflammatory milieu in RC muscles, we performed α-naphtyl acetate esterase and naphthol AS-D-chloroacetate esterase double staining for monocytic and granulocytic cells. At 2 weeks, we observed a significant spike in the number of tissue macrophages present in injured muscle of rats treated with LPA compared to those that received vehicle (Fig. 5A and C). In LPA-treated, injured muscle, the number of α-naphthyl acetate esterase positive cells outnumbered those staining positive for naphthol AS-D chloroacetate esterase, contrary to what was observed in the injured muscle of rats treated with vehicle. There was no significant difference in the number of naphthol AS-D chloroacetate esterase-positive granulocytes present at 2 weeks in injured muscle between LPA and vehicle groups. At 6 weeks, the number of inflammatory cells present of both the granulocytic and monocytic lineages was increased in the injured muscle of rats that received LPA compared to vehicle, though the numbers of all inflammatory cell types had decreased significantly compared to 2 weeks (Fig. 5B and D).
Figure 5.
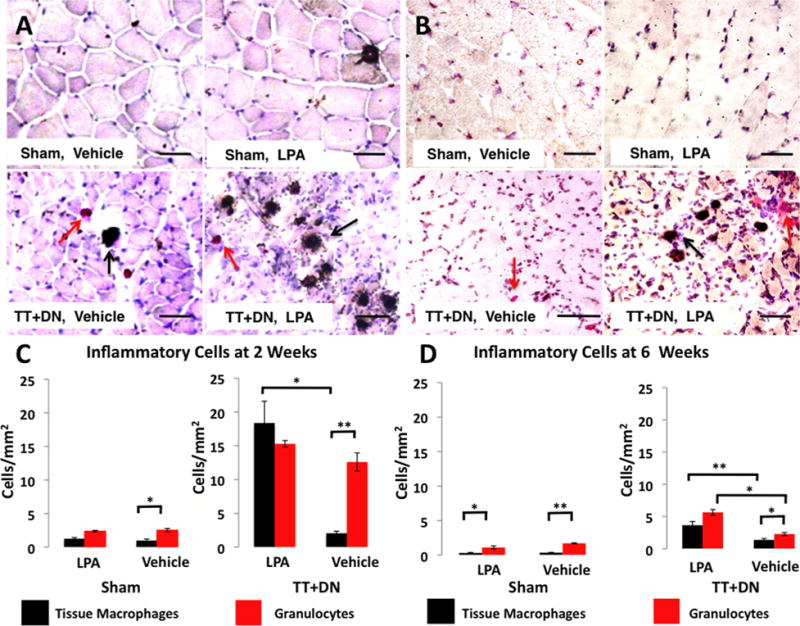
Representative double-esterase staining at 2 weeks (A and C) and 6 weeks (B and D) after injury. Black arrows indicate α-naphthyl acetate esterase positive, monocyte-derived cells; red arrows indicate naphthol ASD-chloroacetate esterase positive, granulocyte-derived cells. Scale bar=50 μm. Cell density shown in C–D, *p<0.05, *p<0.01.
RhoA Expression Co-Localizes With CD68+ Macrophages in Injured Muscle
Given the high number of monocyte-derived inflammatory cells present in injured RC muscles, we performed immunohistofluorescence to assess the expression patterns of CD68, a macrophage marker, as well as RhoA. We found that at 2 weeks after injury, CD68+ cells strongly co-expressed RhoA at much higher levels than background muscle tissue in both vehicle and LPA-treated tissue (Fig. 6). We noted minimal CD68 or RhoA staining in the uninjured muscle of either group.
Figure 6.
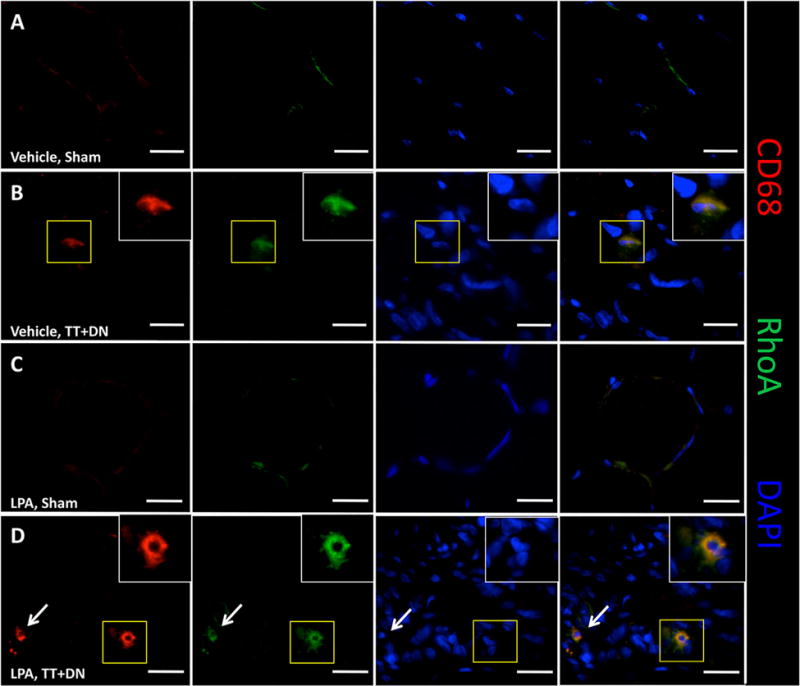
Representative immunohistofluorescence showing co-expression of CD68 and RhoA in tissue macrophages in rotator cuff muscles. Area within yellow box is enlarged in white box. Scale bar=20μm. White arrow (bottom row) indicates an additional CD68+/RhoA+ cell.
LPA Treatment Worsens Muscle Atrophy, Fibrosis, and Fatty Infiltration After Injury
We assessed the gross morphological characteristics of muscles at 6 weeks after injury, at which time point we have previously observed the development of significant muscle atrophy and fibrosis, and early fatty infiltration. At 6 weeks after TT+DN injury, rats treated with LPA demonstrated significantly lower supraspinatus muscle weights on the injury side than those that received vehicle (Fig. 7). This trend was also noted on the sham-side muscle, though was not found to be statistically significant (p=0.08).
Figure 7.
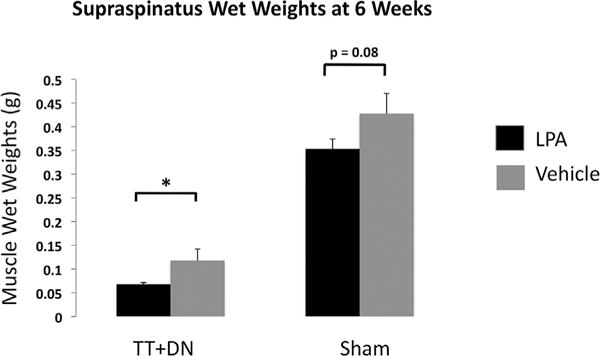
LPA treatment significantly reduced muscle wet weights (g) at 6 weeks after TT+DN injury, but not after sham surgery.
We next performed Masson’s trichrome staining to determine the fibrosis index of injured muscle, and found that LPA significantly worsens fibrosis of injured muscle with an index of 31.3±5.5% compared to 18.8±3.9% in rats that received vehicle (Fig. 8). Oil red-O staining revealed significantly increased fatty infiltration in the injured muscle of rats that received LPA, with a fat index of 6.3±0.7% compared to 2.6±0.7% in rats that received vehicle (Fig. 9). We observed minimal collagen deposition and no fat in the sham-side muscle of both groups.
Figure 8.
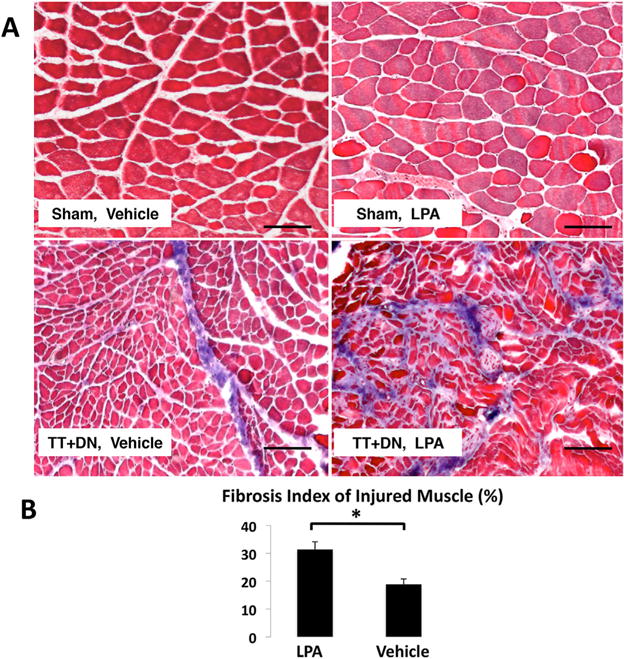
(A) Representative Masson’s trichrome stain at 6 weeks after injury (Scale bar=100 μm.). (A) Quantitative analysis of fibrosis index showed LPA treatment significantly increased fibrosis of rotator cuff muscles after TT DN injury (*p<0.05).
Figure 9.
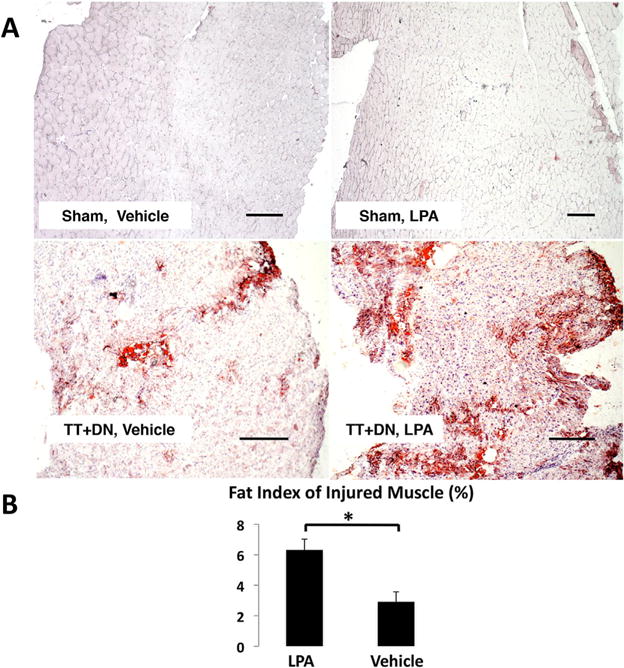
(A) Representative oil red-O stain at 6 weeks after injury (Scale bar=500 μm). (B) Quantitative analysis of fatty infiltration index showed LPA treatment significantly increased fatty infiltration of rotator cuff muscles after TT DN injury (*p<0.05).
DISCUSSION
In this study, we have demonstrated that administration of LPA led to increased RhoA signaling and macrophage infiltration in RC muscles after massive RC tears in our rat model. We further showed that LPA administration led to accelerated RC muscle atrophy, fibrosis, and fatty infiltration. This process was associated with an increase in RhoA expression that co-localized with CD68+ macrophages in injured tissue, suggesting that increased RhoA expression may play a key role in inflammatory macrophage infiltration that occurs after a massive RC tear.
Previous studies of the RhoA pathway in injured muscle have suggested that RhoA inhibition with small molecule inhibitors such as Y-27632 leads to improved histologic outcomes compared to vehicle.20 Our current study contributes further to the literature by demonstrating that LPA-induced inflammation and increased RhoA expression worsen muscle atrophy, fibrosis, and fatty infiltration following massive RC tears in rats.
Although RhoA is significantly increased with LPA in RC muscles after injury, we did not observe a strong difference in expression of inflammatory markers between treatment and vehicle groups, including MIF and GM-CSF, markers of M1 macrophage polarization27 at an early stage (2 weeks) after tendon tears. However, at a later stage (by week 6) after tendon injury, we observed an increase in the expression of the inflammation marker, TNFα. TNFα has been proposed as an intermediate signal in the activation of RhoA by LPA,28 and has been shown to upregulate RhoA expression via NF-κB signaling in smooth muscle.29 Additionally, TNFα is elevated in human RC muscle samples of patients with RC tendinopathy and inhibition of TNFα in rats has been shown to improve tendon-to-bone healing following RC tears in rats.30 That LPA further upregulates TNFα signaling in injured rat muscle compared to vehicle suggests that it accentuates a chronic inflammatory environment analogous to that seen with human RC tears. The near-significant increase in TGF-β1 expression seen with LPA treatment may be related to the increase in monocyte-derived inflammatory cells present in injured muscle. Macrophages are thought to be a key source of TGF-β1 in chronically injured muscle,31 and TGF-β1 signaling has been linked to increased fibrosis within the RC of rats and increased fibrofatty infiltration in mice.23,32 Inhibition of TGF-β1 signaling in mice has likewise shown a reduction in RC muscle atrophy and fibrofatty infiltration by decreasing the number of fibro/adipogenic progenitor cells present in injured muscle.32 Interestingly, a recent study showed that LPA-induced renal fibrosis was mediated through LPA2 receptors and Gαq proteins via transactivation of TGF-β in a Rho/Rho-kinase (ROCK)-dependent manner, suggesting another possible connection between LPA signaling and a pathway previously linked to muscle degeneration.15 Taken together, this suggests that LPA-induced TGF-β expression may be one mechanism by which we observed increases in fibrosis and fatty infiltration compared to vehicle in this study.
We next sought to characterize the pattern of inflammatory cells present in injured RC muscle at early (2 weeks) and later (6 week) stages after RC injury. We were surprised to find a significant increase in the number of monocyte-derived, α-naphthyl acetate esterase-positive cells at 2 weeks after injury in rats treated with LPA compared to those seen in rats that received vehicle. This difference in inflammatory cell number in injured muscle between LPA and vehicle groups persisted at 6 weeks, although inflammatory cells were globally decreased at this later time point compared to 2 weeks. Having observed a striking increase in α-naphthyl acetate esterase-positive cells at 2 weeks in the LPA treatment group compared to vehicle, we next characterized the expression pattern of CD68, a surface marker of tissue macrophages, in the context of the elevated RhoA expression seen with LPA treatment. We found that CD68+ cells in injured muscle strongly co-expressed RhoA in nearly all cases observed. This pattern was not observed in uninjured muscle, in which there was little expression of CD68 or RhoA. A previous study noted co-localization of CD68+ microglia and RhoA in the setting of multiple sclerosis,33 but this is the first study, to our knowledge, to describe this finding in the setting of a tendon-nerve injury in muscle. Worthylake et al. have shown that inhibiting RhoA within monocytes inhibits their ability to undergo transendothelial migration,34 preventing them from maturing into tissue macrophages. We postulate that activated monocytes and macrophages express RhoA in response to an injury stimulus, and that LPA, by increasing activation and expression levels of RhoA, potentiates the migration and polarization of macrophages in response to injury, leading to a greater number of tissue macrophages in injured muscle. In chronic muscle injury, this likely leads to degenerative changes including fibrosis and fatty infiltration and thus is a potential future pharmacologic target to improve muscle function after RC repair.
This study has certain inherent limitations. First, our sample size is relatively small, though sufficient to detect statistically significant differences between groups in this study. Second, only two time points were adopted in this study. Inflammation is a highly dynamic process that cannot be fully analyzed from two time points alone, and future studies would benefit from the addition of time points earlier than 2 weeks as well as time points later than 6 weeks. In spite of this, we are still able to draw strong conclusions about the impact of LPA and chronic inflammation on injured muscle from the time points observed in this study. Additionally, we did not perform inhibition of RhoA signaling in this study, though this may be the focus of future experiments. Differentiating the fraction of GTP-bound, active RhoA from total RhoA levels was also beyond the scope of this study, but is of interest for future experiments. Finally, we did not quantify the number of CD68+ positive cells that co-expressed RhoA on histology, though co-expression was observed for nearly all CD68+ cells in both the LPA and vehicle groups.
We have observed a marked connection between muscle inflammation, RhoA+ macrophage infiltration, and worsened muscle atrophy, fibrosis, and fatty infiltration in a pre-clinical model of massive RC tear. Our findings suggest that LPA-mediated inflammation worsens RC muscle degeneration in rats, and thus further characterizing the role of RhoA and other mediators of inflammation following RC injury is of great clinical and therapeutic importance.
Acknowledgments
This material is based upon work supported by the U.S. Department of Veterans Affairs, Veterans Health Administration, Office of Research and Development, Biomedical Laboratory Research, and Development Merit Review Grant (1I01BX002680), NIH/NIAMS (RO3 AR060871-02) and the Orthopaedic Research and Education Foundation (OREF) Career Development grant.
Grant sponsor: U.S. Department of Veterans Affairs; Grant sponsor: Veterans Health Administration; Grant sponsor: Office of Research and Development; Grant sponsor: Biomedical Laboratory Research and Development Merit Review; Grant number: 1I01BX002680; Grant sponsor: NIH/NIAMS; Grant number: RO3 AR060871-02; Grant sponsor: Orthopaedic Research and Education Foundation (OREF) Career Development; Grant sponsor: Orthopaedic Research and Education Foundation.
Footnotes
AUTHORS’ CONTRIBUTIONS
Designed Experiments: MD, BF, HK, XL; Performed Experiments: MD, LL, XL; Funded Research: BF, HK, XL; Wrote Manuscript: MD, LL, BF, XL; Read and Approved Manuscript: MD, LL, BF, HK, XL.
References
- 1.Fehringer EV, Sun J, VanOeveren LS, et al. Full-thickness rotator cuff tear prevalence and correlation with function and co-morbidities in patients sixty-five years and older. J Shoulder Elbow Surg. 2008;17:881–885. doi: 10.1016/j.jse.2008.05.039. [DOI] [PubMed] [Google Scholar]
- 2.Gladstone JN, Bishop JY, Lo IK, et al. Fatty infiltration and atrophy of the rotator cuff do not improve after rotator cuff repair and correlate with poor functional outcome. Am J Sports Med. 2007;35:719–728. doi: 10.1177/0363546506297539. [DOI] [PubMed] [Google Scholar]
- 3.De Paepe B, De Bleecker JL. Cytokines and chemokines as regulators of skeletal muscle inflammation: presenting the case of Duchenne muscular dystrophy. Mediators Inflamm. 2013;2013:540370. doi: 10.1155/2013/540370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Londhe P, Guttridge DC. Inflammation induced loss of skeletal muscle. Bone. 2015;80:131–142. doi: 10.1016/j.bone.2015.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Argilés JM, Busquets S, Stemmler B, et al. Cachexia and sarcopenia: mechanisms and potential targets for intervention. Curr Opin Pharmacol. 2015;22:100–106. doi: 10.1016/j.coph.2015.04.003. [DOI] [PubMed] [Google Scholar]
- 6.Gumucio JP, Korn MA, Saripalli AL, et al. Agingassociated exacerbation in fatty degeneration and infiltration after rotator cuff tear. J Shoulder Elbow Surg. 2014;23:99–108. doi: 10.1016/j.jse.2013.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Moolenaar WH, van Meeteren LA, Giepmans BN. The ins and outs of lysophosphatidic acid signaling. Bioessays. 2004;26:870–881. doi: 10.1002/bies.20081. [DOI] [PubMed] [Google Scholar]
- 8.Bian D, Mahanivong C, Yu J, et al. The G12/13-RhoA signaling pathway contributes to efficient lysophosphatidic acid-stimulated cell migration. Oncogene. 2006;25:2234–2244. doi: 10.1038/sj.onc.1209261. [DOI] [PubMed] [Google Scholar]
- 9.Hwang YS, Hodge JC, Sivapurapu N, et al. Lysophosphatidic acid stimulates PC-3 prostate cancer cell matrigel invasion through activation of RhoA and NF-κB activity. Mol Carcinog. 2006;45:518–529. doi: 10.1002/mc.20183. [DOI] [PubMed] [Google Scholar]
- 10.Blackburn J, Mansell JP. The emerging role of lysophosphatidic acid (LPA) in skeletal biology. Bone. 2012;50:756–762. doi: 10.1016/j.bone.2011.12.002. [DOI] [PubMed] [Google Scholar]
- 11.Fotopoulou S, Oikonomou N, Grigorieva E, et al. ATX expression and LPA signalling are vital for the development of the nervous system. Dev Biol. 2010;339:451–464. doi: 10.1016/j.ydbio.2010.01.007. [DOI] [PubMed] [Google Scholar]
- 12.Koh JS, Lieberthal W, Heydrick S, et al. Lysophosphatidic acid is a major serum noncytokine survival factor for murine macrophages which acts via the phosphatidylinositol 3-kinase signaling pathway. J Clin Invest. 1998;102:716. doi: 10.1172/JCI1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tager AM, LaCamera P, Shea BS, et al. The lysophosphatidic acid receptor LPA1 links pulmonary fibrosis to lung injury by mediating fibroblast recruitment and vascular leak. Nat Med. 2008;14:45–54. doi: 10.1038/nm1685. [DOI] [PubMed] [Google Scholar]
- 14.Pradére JP, Gonzalez J, Klein J, et al. Lysophosphatidic acid and renal fibrosis. Biochim Biophys Acta (BBA) 2008;1781:582–587. doi: 10.1016/j.bbalip.2008.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Geng H, Lan R, Singha PK, et al. Lysophosphatidic acid increases proximal tubule cell secretion of profibrotic cytokines PDGF-B and CTGF through LPA2-and Gαq-mediated Rho and αvβ6 integrin-dependent activation of TGF-β. Am J Pathol. 2012;181:1236–1249. doi: 10.1016/j.ajpath.2012.06.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nobes CD, Hall A. Rho, rac, and cdc42 GTPases regulate the assembly of multimolecular focal complexes associated with actin stress fibers, lamellipodia, and filopodia. Cell. 1995;81:53–62. doi: 10.1016/0092-8674(95)90370-4. [DOI] [PubMed] [Google Scholar]
- 17.Xiang SY, Dusaban SS, Brown JH. Lysophospholipid receptor activation of RhoA and lipid signaling pathways. Biochim Biophys Acta. 2013;1831:213–222. doi: 10.1016/j.bbalip.2012.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hwang YS, Lee J, Zhang X, et al. Lysophosphatidic acid activates the RhoA and NF-kappaB through Akt/IkappaBalpha signaling and promotes prostate cancer invasion and progression by enhancing functional invadopodia formation. Tumour Biol. 2016;37:6775–6785. doi: 10.1007/s13277-015-4549-x. [DOI] [PubMed] [Google Scholar]
- 19.Lutz S, Shankaranarayanan A, Coco C, et al. Structure of Gαq-p63RhoGEF-RhoA complex reveals a pathway for the activation of RhoA by GPCRs. Science. 2007;318:1923–1927. doi: 10.1126/science.1147554. [DOI] [PubMed] [Google Scholar]
- 20.Mu X, Usas A, Tang Y, et al. RhoA mediates defective stem cell function and heterotopic ossification in dystrophic muscle of mice. FASEB J. 2013;27:3619–3631. doi: 10.1096/fj.13-233460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Honing H, van den Berg TK, van der Pol SM, et al. RhoA activation promotes transendothelial migration of monocytes via ROCK. J Leukoc Biol. 2004;75:523–528. doi: 10.1189/jlb.0203054. [DOI] [PubMed] [Google Scholar]
- 22.Liu X, Manzano G, Kim HT, et al. A rat model of massive rotator cuff tears. J Orthop Res. 2011;29:588–595. doi: 10.1002/jor.21266. [DOI] [PubMed] [Google Scholar]
- 23.Liu X, Joshi SK, Ravishankar B, et al. Upregulation of transforming growth factor-β signaling in a rat model of rotator cuff tears. J Shoulder Elbow Surg. 2014;23:1709–1716. doi: 10.1016/j.jse.2014.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Joshi SK, Liu X, Samagh SP, et al. MTOR regulates fatty infiltration through SREBP-1 and PPARγ after a combined massive rotator cuff tear and suprascapular nerve injury in rats. J Orthop Res. 2013;31:724–730. doi: 10.1002/jor.22254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Davies MR, Ravishankar B, Laron D, et al. Rat rotator cuff muscle responds differently from hindlimb muscle to a combined tendon-nerve injury. J Orthop Res. 2015;33:1046–1053. doi: 10.1002/jor.22864. [DOI] [PubMed] [Google Scholar]
- 26.Dahab GM, Kheriza MM, El-Beltagi HM, et al. Digital quantification of fibrosis in liver biopsy sections: description of a new method by Photoshop software. J Gastroenterol Hepatol. 2004;19:78–85. doi: 10.1111/j.1440-1746.2004.03183.x. [DOI] [PubMed] [Google Scholar]
- 27.Wang N, Liang H, Zen K. Molecular mechanisms that influence the macrophage m1–m2 polarization balance. M1/M2 Macrophages: the Arginine Fork in the Road to Health and Disease. 2015:230. doi: 10.3389/fimmu.2014.00614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wojciak-Stothard B, Ridley AJ. Rho GTPases and the regulation of endothelial permeability. Vascul Pharmacol. 2002;39:187–199. doi: 10.1016/s1537-1891(03)00008-9. [DOI] [PubMed] [Google Scholar]
- 29.Goto K, Chiba Y, Sakai H, et al. Tumor necrosis factor-. ALPHA. (TNF-. ALPHA.) induces upregulation of RhoA via NF-. KAPPA. B activation in cultured human bronchial smooth muscle cells. J Pharmacol Sci. 2009;110:437–444. doi: 10.1254/jphs.09081fp. [DOI] [PubMed] [Google Scholar]
- 30.Gulotta LV, Kovacevic D, Cordasco FA, et al. TNF-alpha blockade improves early tendon-to-Bone healing in a rat rotator cuff repair model (SS-10) Arthroscopy. 2009;25:e5–e6. doi: 10.1016/j.arthro.2011.03.076. [DOI] [PubMed] [Google Scholar]
- 31.Lemos DR, Babaeijandaghi F, Low M, et al. Nilotinib reduces muscle fibrosis in chronic muscle injury by promoting TNF-mediated apoptosis of fibro/adipogenic progenitors. Nat Med. 2015;21:786–794. doi: 10.1038/nm.3869. [DOI] [PubMed] [Google Scholar]
- 32.Davies MR, Liu X, Lee L, et al. TGF-β small molecule inhibitor SB431542 reduces rotator cuff muscle fibrosis and fatty infiltration by promoting Fibro/Adipogenic progenitor apoptosis. PLoS ONE. 2016;11:e0155486. doi: 10.1371/journal.pone.0155486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang Z, Schittenhelm J, Meyermann R, et al. Lesional accumulation of RhoA+cells in brains of experimental autoimmune encephalomyelitis and multiple sclerosis. Neuropathol Appl Neurobiol. 2008;34:231–240. doi: 10.1111/j.1365-2990.2007.00892.x. [DOI] [PubMed] [Google Scholar]
- 34.Worthylake RA, Lemoine S, Watson JM, et al. RhoA is required for monocyte tail retraction during transendothelial migration. J Cell Biol. 2001;154:147–160. doi: 10.1083/jcb.200103048. [DOI] [PMC free article] [PubMed] [Google Scholar]