

Abstract
Resistance of Neisseria gonorrhoeae to expanded-spectrum cephalosporins such as ceftriaxone and cefixime has increased markedly in the past decade. The primary cephalosporin-resistance determinant is a mutated penA gene, which encodes the essential peptidoglycan transpeptidase, penicillin-binding protein 2 (PBP2). Decreased susceptibility and resistance can be conferred by mosaic penA alleles containing upwards of 60 amino acid changes relative to wild-type PBP2, or by non-mosaic alleles with relatively few mutations, the most important of which occurs at Ala501 located near the active site of PBP2. Recently, fully cefixime- and ceftriaxone-resistant clinical isolates were identified that harbored a mosaic penA allele with an A501P mutation. To examine the potential of mutations at Ala501 to increase resistance to expanded-spectrum cephalosporins, we randomized codon-501 in a mosaic penA allele and transformed N. gonorrhoeae to increased cefixime resistance. Interestingly, only five substitutions of Ala501 (A501V, A501T, A501P, A501R, A501S) were isolated that increased resistance and preserved essential transpeptidase function. To understand their structural implications, these mutations were introduced into the non-mosaic PBP2-6140CT, which contains four C-terminal mutations present in PBP2 from the penicillin-resistant strain FA6140. The crystal structure of PBP2-6140CT-A501T was solved and revealed ordering of a loop near the active site and a new hydrogen bond involving Thr501 that connects the loop and the SxxK conserved active-site motif. The structure suggests that increased rigidity in the active site region is a mechanism for cephalosporin resistance mediated by Ala501 mutations in PBP2.
Graphical abstract
Neisseria gonorrhoeae is the causative agent of the sexually transmitted infection gonorrhea. Gonorrhea is the second most common bacterial sexually transmitted infection worldwide, with an estimated 78 million cases per year in 2012(1). Gonorrhea is mainly an infection of the lower urogenital tract, although it can also colonize the mucous membranes of the eyes, rectum and pharynx. If left untreated, infections in females can ascend to the upper genital tract and cause pelvic inflammatory disease and the associated increased risks of ectopic pregnancies and infertility. The efficacy of ceftriaxone, hitherto the most effective antibiotic against gonorrhea, is now being threatened by a large increase in infections over the past decade that are caused by cephalosporin intermediate-resistant strains (CephI), and most recently, by a small number of strains exhibiting high-level cephalosporin resistance (CephR). This has resulted in verified treatment failures with cefixime in many countries worldwide and sporadic treatment failures with ceftriaxone in Japan, Australia, Sweden, and Slovenia(2–4). The inexorable rise in ceftriaxone resistance portends a future with no effective antibiotics against gonococcal infections.
N. gonorrhoeae undergoes natural transformation by importing DNA from the extracellular milieu and inserting it into its genome by homologous recombination. The extracellular DNA can come from other Neisserial species (including other strains of N. gonorrhoeae) that share the same host niche, and is the predominant means of acquiring resistance to antibiotics. The major determinant mediating resistance to cephalosporins is a mutated penA gene encoding penicillin-binding protein 2 (PBP2)(5). PBP2 is an essential peptidoglycan transpeptidase (TPase) involved in cell division, and the lethal target of β-lactam antibiotics used to treat gonorrhea. β-lactam antibiotics bind to PBPs and form a stable acyl-enzyme complex that inactivates the enzyme and prevents peptidoglycan cross-linking(6). The role of PBP2 in resistance was first noted in penicillin-resistant N. gonorrhoeae strains, which harbored a penA allele with both an Asp345a insertion and 4–8 mutations located within the carboxyl terminal domain(7). In the early 2000s, strains emerged in Japan and the Western Pacific Rim with much higher minimum inhibitory concentrations (MICs) of the expanded-spectrum cephalosporins, ceftriaxone and cefixime, as well as of other oral cephalosporins that were in common use in Japan at that time(2, 5, 8). These strains contained so-called mosaic penA alleles with up to 60 amino acid mutations in the PBP2 coding sequence compared to a wild-type allele(5, 9). Strains harboring mosaic penA alleles often contain the mtrR and penB resistance determinants, which increase expression of the MtrCDE efflux pump and decrease diffusion of antibiotics across the outer membrane, respectively(10). The combination of these three determinants markedly increases resistance to ceftriaxone and cefixime(11, 12).
Mutations that confer decreased susceptibility to expanded-spectrum cephalosporins were also identified in non-mosaic mutant alleles of penA(13, 14). These mutations were mostly confined to a single amino acid, Ala501, which is located on the β3-β4 loop of PBP2. In particular, mutations of Ala501 to Val and Thr were associated with increased MICs of expanded-spectrum cephalosporins in non-mosaic alleles, but at the time had not been observed in mosaic penA alleles(13–15). In anticipation of Ala501 mutations appearing in mosaic penA alleles, we showed previously that introduction of an A501V mutation into the penA35 allele of the CephI strain 35/02 increased the MICs of ceftriaxone and cefixime by ~3-fold. When introduced into FA6140, a penicillin-resistant but cephalosporin-susceptible strain that carries mtrR and penB mutations, the MIC increased to levels above the resistance breakpoint for the two antibiotics (MIC=0.25 μg ml−1 for both antibiotics)(16). Subsequently, we reported on a clinical isolate from France with a mosaic penA allele harboring an A501P mutation that conferred high-level resistance to cefixime and ceftriaxone. This same penA allele was reported in two isolates a year later in Spain(17, 18).
To understand the molecular mechanism of how mutations of Ala501 increase resistance, here we identified a set of six mutations that, when incorporated into mosaic and non-mosaic penA alleles, increased resistance to cephalosporins while retaining essential transpeptidase activity of PBP2. Structural analysis was initiated with the Ala501 mutants of the non-mosaic PBP2 variant, PBP2-6140CT, and of these, PBP2-6140CT-A501T crystallized and was solved at 2.4 Å. The primary difference of this protein compared to PBP2-6140CT is that the mutation causes the β3-β4 loop to become ordered, whereas the same loop is disordered in both wild-type PBP2 and PBP2-6140CT. These data suggest that increased rigidity of the β3-β4 loop is the primary mechanism by which these mutations confer resistance to expanded-spectrum cephalosporins.
Experimental Procedures
Strains and Plasmids
N. gonorrhoeae strain FA19, a penicillin- and cephalosporin-susceptible strain, was used for all transformations of mutant penA alleles. Gonococci were grown on GC Medium Base (GCB) agar plates containing Supplements I and II(19) at 37°C and 4% CO2 or in liquid GCB with Supplements I and II, 10 mM MgCl2, and 10 mM sodium bicarbonate in a shaking incubator. E. coli strain MC1061 was used for routine cloning experiments and was grown in Lysogeny Broth (LB) or on LB plates. Purified PCR amplification products for all penA mutants containing the 10 bp gonococcal uptake sequence(20) on one end of the fragment were used directly in transformation experiments.
Mutagenesis and Transformation
A library of penA35 mutants in which the codon for Ala501 was randomized was constructed by overlap-extension PCR(21). Briefly, the first fragment was amplified with a 5′-primer complementary to codons 42–48 (and with the gonococcal uptake sequence GCCGTCTGAA at its 5′end) and a 3′-primer complementary to codons 495 to 500, three random bases, and codons 502–508. The second fragment was amplified with a 5′-primer comprising codons 502–508 and a 3′-primer that hybridizes ~200 bp downstream of the penA stop codon. A plasmid with penA35 cloned into pUC18us (pUC18 containing the gonococcal uptake sequence) was used as template. The resulting fragments were isolated, combined, and a second amplification was carried out with the 5′-primer of the first reaction and the 3′-primer from the second reaction. The resulting PCR product was sequenced to confirm randomization of codon 501, and then used to transform FA19 directly. Overlap-extension mutagenesis with primers encoding A501G, A501K, A501L, and A501I mutations was used in later experiments to insert specific mutations of Ala501 into the penA35 allele.
Transformations were carried out exactly as previously described(16). Pilot experiments revealed that transformants of FA19 with penA35 could be selected at concentrations of cefixime between 0.02 and 0.08 μg ml−1, but decreased to nearly zero transformants at higher concentrations. Thus, FA19 bacteria transformed with the randomized PCR product were plated on GCB agar containing 0.1 μg ml−1 cefixime in the first set of experiments, and 0.2 μg ml−1 cefixime in later experiments to identify alleles conferring the highest levels of resistance. Transformants with increased resistance were isolated, and bacteria were boiled in water to provide template for PCR reactions using the two outside primers from above. DNA fragments were purified with a PCR purification kit (Bio Basic, Amherst, NY) and sequenced (Genewiz, South Plainfield, NJ).
For the crystallization experiments described below, we introduced the five Ala501 mutations identified in the randomized screen into PBP2-6140CT, which is a construct of PBP2 containing four mutations (relative to wild-type) in the C-terminal region that decrease the acylation rate of the protein with penicillin G by 5-fold(22). The same PCR protocol was used as described above, except that primers with the specific codons were used in the PCR reactions.
MIC determinations, growth curves, and competitive co-culture
FA19 transformants harboring penA alleles with mutations at Ala501 were grown overnight on GCB plates, resuspended in GCB broth with supplements I and II, 10 mM MgCl2, and 20 mM sodium bicarbonate at a final OD600 of 0.018 (~1 × 107 colony forming units (CFU) ml−1). Aliquots (5 μl) of each strain were spotted onto GCB plates containing antibiotics at <2-fold dilutions. The MIC was defined as the lowest concentration of antibiotic on which fewer than 5 colonies appeared after 24 hrs of incubation.
For assessment of growth kinetics, bacterial colonies with a non-piliated morphology were harvested from GCB agar plates after 18–20 hours incubation and inoculated into 35 ml of supplemented GC broth with 5 mM sodium bicarbonate at an initial optical density at 600 nm (OD600) of 0.08. Bacteria were grown in untreated T75 tissue culture flasks for 8 hours shaking at 220 rpm and 37°C. The OD600 values were recorded every hour, and every two hours aliquots were removed for determination of viable bacteria (CFU ml−1). To avoid potential problems with clumping and to obtain more consistent CFU counts, bacteria were diluted in GCB + 0.05% saponin prior to plating.
For competitive co-culture experiments, similar numbers of FA19 bacteria and FA19 bacteria harboring a penA35-A501 mutant allele were inoculated into GC broth as above and aliquots quantitatively cultured every two hours on non-selective GC agar (total number of CFU ml−1) and GC agar with either 0.016 μg ml−1 or 0.031 μg ml−1 of ceftriaxone for selection (number of mutant CFU ml−1). The higher ceftriaxone concentration was used to select for FA19 bacteria having an A501P or A501R mutation. The competitive index was determined at each time point, which is the ratio of mutant to wild-type bacteria divided by the ratio of mutant to wild-type bacteria in the initial inoculation. All growth experiments were repeated at least twice and gave similar results.
Acylation Rate Constants
The second order rate constants (k2/KS) of acylation with [14C]penicillin G (Moravek, Brea, CA) for PBP-35/02 with and without four Ala501 mutations (the A501S mutant was not assessed) were determined using purified protein, as described previously(22–24). The k2/KS values for ceftriaxone and cefixime were determined indirectly by determining the concentrations of the cephalosporins that inhibited the binding of [14C]penicillin G by 50% and using the following equation(24):
Protein purification and crystallization
The constructs encoding PBP2-6140CT with A501 mutations comprise residues 44–581 of PBP2 fused in-frame to the C-terminal end of His6-tagged maltose-binding protein (MBP) with an intervening tobacco etch virus (TEV) protease site. The protein was expressed at 18°C, the cells lysed, and the fusion protein was purified from the soluble fraction on a Ni2+-NTA column (GE Healthcare, Piscataway, NJ). The peak fractions were pooled and dialyzed at 4 °C at a 10:1 molar ratio of MBP-PBP2 to His6-TEV, which resulted in nearly complete digestion of the fusion protein. The mutant PBP2 variants were then rerun over a Ni2+-NTA column to remove His6-MBP, uncleaved fusion protein, and His6-TEV. The purified proteins were concentrated to ~20 mg ml−1 in 20 mM Tris•HCl, pH 8.0, 500 mM NaCl, 10% glycerol and stored at −80°C until ready for use.
Crystals of PBP2-6140CT-A501T were grown by vapor diffusion at 24°C in hanging drops, where the reservoir contained 2.2 M ammonium sulfate buffered with 100 mM Tris pH 8.25. Crystals were cryo-protected by passage through mother liquor to which glycerol had been added to a final concentration of 30% (v/v), followed by flash freezing in liquid nitrogen.
Data collection and refinement
Data were collected on the SER-CAT beam line ID22 in 1° oscillations, with a crystal-to-plate distance of 200 mm and an exposure time of 3 sec at 40% transmission. Data were collected for 180° and processed using HKL2000(25). The space group and unit dimensions are the same as for wild-type PBP2(22) and there are two molecules in the asymmetric unit. The beginning model for refinement was the crystal structure of PBP2-6140CT (PDB code 3EQV). The model was improved by iterative rounds of manual building using the program O(26) and REFMAC refinement(27). |Fo|-|Fc| difference maps were used to build the β3-β4 loops that are ordered in this structure. The final model was refined to 2.4 Å and contains 39 water molecules, 3 glycerols and 12 sulfates (Table 1). In both molecules of the asymmetric unit, positive electron density next to the side chain hydroxyl indicates that Thr465 is phosphorylated. This was also observed in both wild-type and 6140CT structures of PBP2 and is presumed to be an artifact resulting from protein expression in the cytoplasm of E. coli(22).
Table 1. X-ray diffraction data and refinement statistics.
Rmerge=Σ|Ii − Im |/ΣIi, where Ii is the intensity of the measured reflection, and Im is the mean intensity for all observations of that reflection. Numbers within parentheses are for the outer resolution shell of data.
| Data collection: | |
|---|---|
| Cell dimensions: a, b, c (Å) | 57.6, 137.2, 229.8 |
| Resolution range (Å) | 39.0 – 2.40 (2.44–2.40) |
| Rmerge* (%) | 10.7 (99.8) |
| Completeness (%) | 98.9 (97.6) |
| Redundancy | 5.9 (5.5) |
| <I>/<σI> | 26.0 (2.5) |
| No. of unique reflections | 71,874 (3,435) |
| Refinement: | |
| Resolution (Å) | 39.0 – 2.4 |
| No. of sulfates | 12 |
| No. of glycerols | 3 |
| No. of water molecules | 39 |
| Rcryst/Rfree (%) | 0.218/0.244 |
| RMS deviations from ideal stereochemistry:- | |
| bond lengths (Å) | 0.013 |
| bond angles (°) | 1.72 |
| Ramachandran plot: | |
| Residues in most favored region (%) | 92.2 |
| Residues in disallowed region (%) | 7.8 |
| Residues in generously allowed regions | 0.0 |
| Residues in disallowed regions | 0.0 |
| PDB code | 5KSH |
Circular dichroism
PBP2, PBP2-6140CT and four PBP2-6140CT mutants (A501R, A501P, A501V, A501T) were each diluted to a concentration of 0.5–1 μM in a buffer containing 10 mM Tris-HCl, pH 8.0, 250 mM NaCl and 10% glycerol. Melting curves for each protein were measured in an AVIV Biomedical, Inc. (Lakewood, NJ) Model 400 circular dichroism (CD) spectrophotometer at 206 nm, which corresponds to the minimum in a 260 to 195 nm wavelength scan. Data were recorded every degree across the temperature range of 25 to 75°C with 30 seconds of temperature equilibration time at each step. For each protein, at least 3 experiments were performed using a new sample each time and then averaged. All spectra were corrected against a control spectrum collected from buffer only. The molar ellipticity (ME) was calculated using AVIV software. The melting temperature (Tm) for each construct was calculated as the midpoint of the slope between the ME values where the protein was folded and unfolded.
Results
Random selection of Ala501 mutants
To investigate the mechanism(s) by which mutations at Ala501 of PBP2 confer resistance to expanded-spectrum cephalosporins and to define the types of amino acids that confer increased resistance, we randomized codon-501 in the mosaic penA35 allele, transformed the library of randomized PCR products into the wild-type strain FA19, and selected for transformants with increased resistance to cefixime. We initially used 0.1 μg ml−1 cefixime for selection, and obtained hundreds of transformants with increased resistance; of these, we sequenced the penA genes from 65 strains (Table 2). Three mutations (A501R, A501V, and A501T) comprised 90% of the sequenced clones and were obtained with similar frequency, whereas two other mutations, A501S and A501P, were isolated at much lower frequencies. The identification of three of these mutations (A501V, A501T, and A501P) was expected, as these mutations have been observed in either non-mosaic penA alleles from strains with decreased susceptibility to cephalosporins (A501V and A501T)(13, 28–31), or in a mosaic penA allele from ceftriaxone-resistant strains (A501P)(17, 18). The isolation of just one A501P mutation in our screen was surprising given its prior emergence in the penA alleles of the ceftriaxone-resistant F89(17) and the Spanish gonococcal isolates(18). In contrast, the appearance of both A501R and A501S was novel, as these mutations have not yet been reported in a penA gene from any gonococcal clinical isolate.
Table 2. Random mutagenesis of Ala501 in PBP2 revealed 5 amino acids that increase resistance to expanded-spectrum cephalosporins.
Transformants were selected at the indicated concentrations of cefixime, and the penA genes were amplified and sequenced. Data are from three independent transformations.
| Cefixime concentration used for selection | Ala501 mutations | Number of isolates |
|---|---|---|
|
| ||
| 0.1 μg ml−1 | Arg | 21 |
| Ser | 5 | |
| Thr | 20 | |
| Val | 18 | |
| Pro | 1 | |
|
| ||
| 0.2 μg ml−1 | Arg | 24 |
To increase the probability of isolating transformants with Ala501 mutations that confer higher levels of resistance, we increased the stringency of selection by plating transformants at 0.2 μg ml−1 cefixime, which preliminary MIC data suggested would eliminate A501S, A501T, and A501V mutations, but select for A501R and A501P and any other mutations conferring higher levels of resistance. Under these selection conditions, only A501R mutations were obtained in 24 isolates sequenced (Table 2).
MICs of strains harboring penA35-A501 mutant alleles
The MICs of cefixime, ceftriaxone and penicillin G for FA19 strains containing the penA35 allele with each of the five Ala501 mutations were determined (Fig. 1). For the cephalosporin MICs, these strains fell into two groups. The fold-increases in the MICs for FA19 penA35-A501V, -A501T, and -A501S relative to that of FA19 penA35 were very similar, with increases of 2- to 3-fold for cefixime and ~2-fold for ceftriaxone. In contrast, the MICs for FA19 penA35-A501R and -A501P were higher, with increases of ~5-fold for both antibiotics compared to that of FA19 penA35. These data are consistent with the increased resistance of strains such as F89 that harbor a mosaic penA allele with an A501P mutation(17), but also suggest that A501R mutations would be just as effective at increasing resistance if they appear in clinical isolates in the future. In contrast to the MICs of the extended-spectrum cephalosporins, the Ala501 mutations decreased the MIC of penicillin compared to the parental penA35 allele. Four of the mutants (A501S, A501T, A501V and A501R) conferred a 2-fold decrease in the MIC, while the A501P mutant conferred a 10-fold decrease in the MIC.
Figure 1. MICs of cefixime, ceftriaxone, and penicillin G for Neisseria gonorrhoeae strain FA19 harboring penA35-Ala501 mutants.
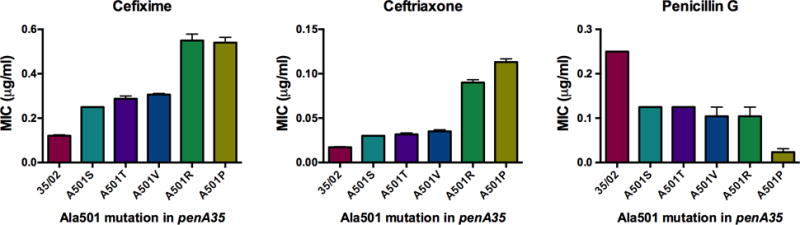
Strains were generated by transformation with the indicated penA allele, and the MICs of the indicated antibiotics were determined as described in Material and Methods. The MICs reported are the averages ± standard deviation determined from a minimum of 3 independent experiments using 3–6 sequence-verified transformants of each strain.
The isolation of only five mutations of Ala501 in the random screen suggested that only a small subset of mutations confers increased resistance to cefixime while retaining essential TPase activity. This result prompted us to investigate why other amino acids were not isolated in the screen. Two reasons seemed likely: 1) the mutant PBP2 proteins are functional as TPases but do not increase the MIC of cefixime to levels high enough to be selected above background, or 2) they are unable to catalyze sufficient levels of TPase activity. To distinguish between these possibilities, we generated four new Ala501 mutations: A501I, A501L, A501K and A501G. Ile and Leu were chosen because they are most similar in size and hydrophobicity to Val, Lys because of its charge similarity to Arg, and Gly because of its smaller size relative to Ala.
FA19 was transformed with pUC18-penA35-A501I, -A501L, -A501K, or –A501G and resistant transformants were selected on 0.04 μg ml−1 cefixime, a concentration ~3-fold lower than the MIC of transformants harboring penA35 without Ala501 mutations, but high enough to ensure that FA19 did not grow. The transformations with penA35-A501L and -A501G yielded no colonies, suggesting that these mutations disrupt essential TPase activity of PBP2. In contrast, transformants were obtained with penA35-A501I and –A501K, and subsequent PCR and sequencing verified that the mutations were present.
When the MICs of cefixime were determined, FA19 penA35-A501K was ~2-fold higher than FA19 penA35, similar to FA19 penA35-A501T, whereas FA19 penA35-A501I was essentially identical to FA19 penA35 (Fig. 2A). The inability of a penA35-A501I mutant allele to increase the MIC of cefixime above that conferred by penA35 is consistent with the absence of such transformants arising from the random screen. It was not evident, however, why A501K mutations were not identified in the screen. One consistent observation from these transformations was that while transformants of FA19 with penA35-A501K could be selected, the number of colonies obtained was markedly lower than that from transformations with other mosaic penA alleles.
Figure 2. MICs and relative transformation efficiencies of Neisseria gonorrhoeae strain FA19 harboring penA35-Ala501 mutants.
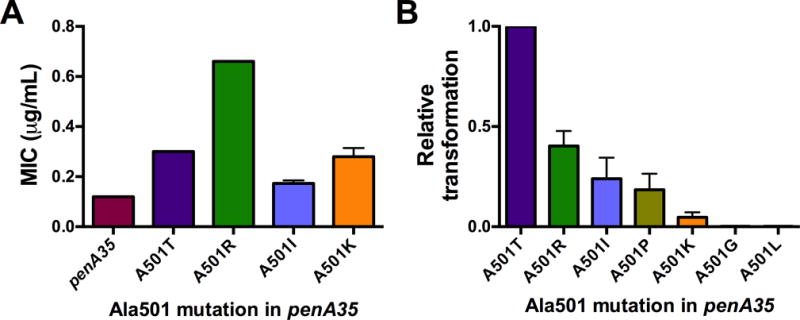
A, MICs of cefixime for the indicated strains; the data were generated and graphed as described in the legend to Fig. 1. B, Normalized transformation frequencies of the indicated penA35-A501 variants. Equal amounts of DNA were used to transform FA19, and transformants were selected on 0.01 μg ml−1 of cefixime. For each experiment, the relative transformation frequency for each strain was determined as a ratio of that obtained with penA35-A501T. The experiment was repeated three times and the ratios were averaged ± standard deviation.
To examine this phenomenon in more detail, we transformed FA19 with equivalent amounts of PCR products of six penA35-Ala501 mutants (penA35-A501T, -A501R, -A501I, -A501K, -A501G, and -A501L) and quantified the number of resistant colonies obtained on plates containing 0.01 μg ml−1 cefixime, which is 10-fold lower than the MIC of FA19 penA35 (Fig. 2B). Compared to penA35-A501T, the number of transformants with penA35-A501R, -A501I and –A501P ranged from 45% to 15% of that obtained with penA35-A501T. In contrast, the transformation efficiency of penA35-A501K was 2.3% of penA35-A501T. Because the six DNAs differed at most by 3 nucleotides (i.e. the mutations at codon-501), our data suggest that the wide variation in the transformation efficiency of the different Ala501 mutants represents the viability of the transformant, and therefore most likely reflects the capacity of each PBP2 mutant to catalyze essential TPase activity. This is an important factor when considering the likelihood that these mutations will emerge in mosaic penA alleles in nature.
In previous work, we have shown that some mutations, while critical for increasing resistance to β-lactam antibiotics, require other mutations (called epistatic mutations) that allow the critical mutations to increase resistance(16). We therefore asked whether a single A501P mutation in the wild-type penA background could confer an increase in the MIC of cefixime. FA19 was transformed with penA-A501P and sequence-verified transformants could be obtained at 0.01 μg/ml cefixime (4-fold higher than its MIC for FA19). Consistent with our MIC data for penicillin G (Fig. 1), no transformants of FA19 penA-A501P were obtained when selected on a concentration of penicillin G 2-fold higher than the MIC for FA19. These data indicate that the A501P mutation is capable of conferring cefixime resistance without any additional mutations present in the penA35 allele.
Acylation rate constants of mutant PBPs
We next assessed the in vitro acylation rates for cefixime, ceftriaxone, and penicillin G. These 2nd order rate constants (defined as k2/KS) describe both the acylation rate constant (k2) and the binding affinity of the antibiotic (KS)(24). As shown in Table 3, the k2/KS constants of the PBP2-35/02-Ala501 mutants identified in the screen were mostly consistent with the MICs of strains containing these variants, with the exception of the rate constants of ceftriaxone, which actually increased in the A501T and A501V mutant constructs compared to PBP-35/02. Consistent with the decreased MICs of penicillin G for FA19 harboring the Ala501 mutants (Fig. 1), the k2/KS values for all of the PBP2 variants were higher than that for PBP235/02. However, the k2/KS values and the MIC did not show a linear or proportional correlation. For example, the k2/KS values for penicillin G of PBP235/02-A501R and –A501P were very similar and ~1.5- to 2-fold lower than the –A501V and -A501T variants, but while FA19 penA35-A501R had a 2-fold lower MIC than FA19 penA35, FA19 penA35-A501P had a 10-fold lower MIC. Likewise, the k2/KS values of PBP235/02-A501P for ceftriaxone and cefixime were ~20-fold lower than PBP235/02-A501R, but the MICs of the two antibiotics for FA19 harboring the different alleles were about the same. As discussed later, these data suggest that, in addition to rates of acylation, other factors can influence the MIC.
Table 3. k2/Ks second order rate constants for acylation.
The values represent the mean ± standard deviation for 3 independent determinations.
| PBP2 Variant | k2/KS (M−1s−1) | ||
|---|---|---|---|
| Cefixime | Ceftriaxone | Penicillin G | |
| PBP2 | 1480000 ± 22000 | 1710000 ± 90000 | 75700 ± 2300 |
| PBP235/02 | 7170 ± 280 | 11300 ± 440 | 510 ± 90 |
| PBP235/02-A501T | 5550 ± 160 | 30400 ± 300 | 1860 ± 130 |
| PBP235/02-A501V | 3150 ± 120 | 20000 ± 390 | 1370 ± 140 |
| PBP235/02-A501R | 820 ± 40 | 6460 ± 70 | 960 ± 100 |
| PBP235/02-A501P | 40 ± 1 | 250 ± 20 | 810 ± 40 |
Effects of Ala501 mutations on bacterial fitness
Antibiotic-resistance determinants, especially those that are altered versions of an essential enzyme, can decrease growth and/or fitness of the bacteria(32). To assess the fitness cost of the Ala501 mutations in the penA35 background, we measured the in vitro growth rate of strains cultured alone or in competition with the parental strain, FA19. The presence of the mutant penA35 allele either with or without Ala501 mutations negatively impacted growth of the bacteria compared to FA19, causing a shorter logarithmic phase as measured by OD600 and a decrease in the number of viable bacteria (CFU ml−1) recovered over time (Fig. 3A, B). When measured in co-culture experiments, all of the resistant strains were out-competed by FA19, with FA19 penA35-A501R and FA19 penA35-A501P showing the smallest competitive index (Fig. 3C). These data indicate that the penA35 allele itself negatively impacts growth of a wild-type strain, particularly at later time points, and that mutation of Ala501 to Arg or Pro, but not Val, Thr, or Ser, further impacts growth.
Figure 3. In vitro fitness of Neisseria gonorrhoeae strain FA19 harboring penA35-Ala501 mutant alleles.
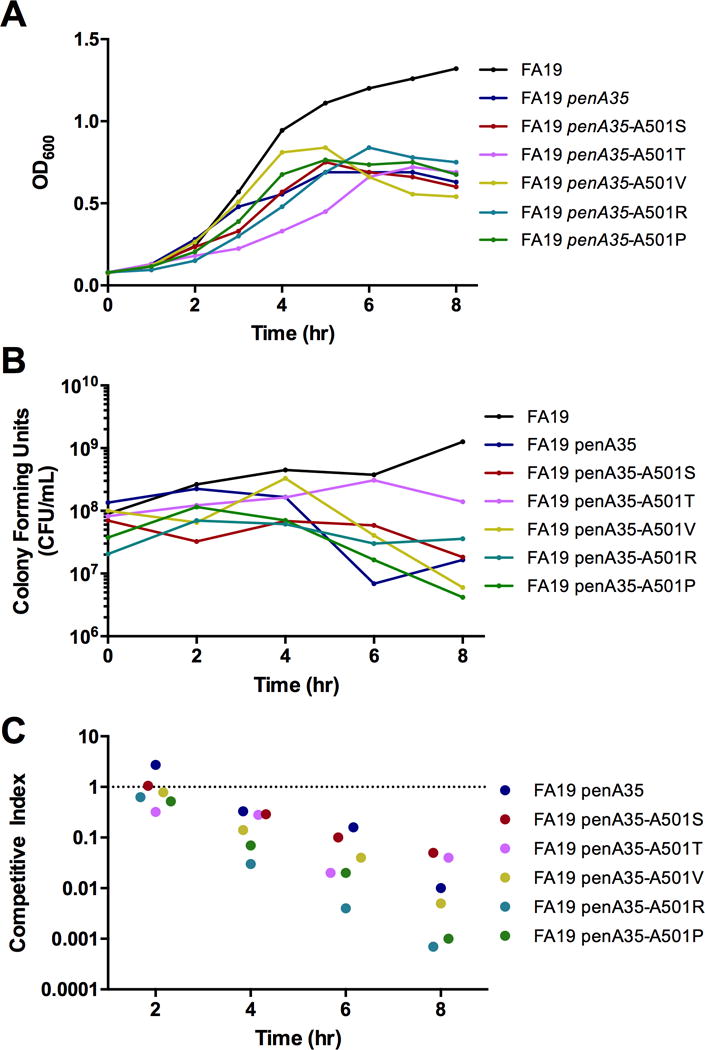
Growth of FA19 harboring wild-type penA, the mosaic penA35, or penA35-Ala501 mutant alleles cultured individually was monitored in liquid GCB medium by quantifying the OD600 (A) and number of colony-forming units (CFU) per ml (B). C, Competitive co-cultures of FA19 versus either FA19 penA35 or individual FA9 penA35-Ala501 mutants. Results are expressed as a Competitive Index (CI) as described in Materials and Methods. CIs less than 1 indicate reduced fitness relative to parent FA19. The experiments were repeated twice with similar results (results from a single experiment are shown).
Effects of Ala501 mutations in a non-mosaic PBP2 background
We next incorporated the Ala501 mutations into PBP2-6140CT (PBP2 containing the four point mutations, but not the Asp346a insertion, found in PBP2 from FA6140;(22)) for structural studies. This was done for several reasons: 1) we had already solved the structure of PBP2-6140CT, which increased the probability of obtaining crystals in this background compared to the PBP235/02 background (as yet, PBP235/02 constructs have not crystallized); 2) Ala501 mutants originally emerged in non-mosaic alleles of penA, making this background clinically relevant(13–15); and 3) interpretation of the effects of the Ala501 mutations would be much less complicated in a non-mosaic PBP2 background.
To ascertain the effects of the Ala501 mutations in a non-mosaic background, we determined the MICs of cefixime and ceftriaxone of FA19 penA-6140CT alone or with one of four Ala501 mutations (Fig. 4). Unlike with penicillin G, the mutations in PBP2-6140CT have no effect on the MICs of cefixime or ceftriaxone. Incorporation of the A501V mutation into PBP2-6140CT conferred a ~2-fold increase in the MIC, whereas incorporation of A501T or A501R increased the MIC ~3-fold. In contrast, incorporation of an A501P mutation into PBP2-6140CT increased the MICs of cefixime and ceftriaxone by 14- and 9-fold, respectively.
Figure 4. MICs of cefixime and ceftriaxone for FA19 alone or FA19 harboring penA6140CT-Ala501 mutant alleles.
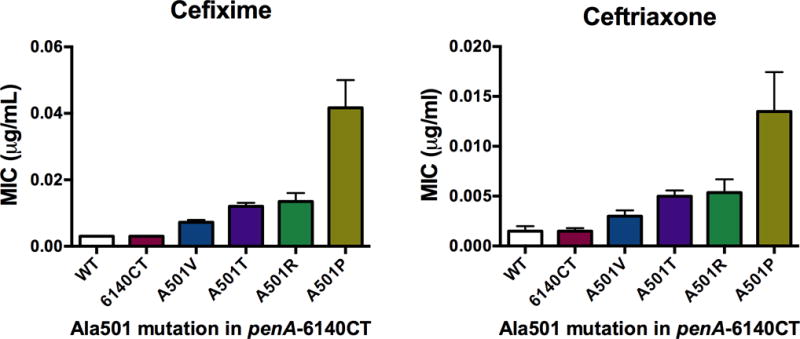
The MICs were determined by agar dilution as described in Materials and Methods and represent the averages (± standard deviation) determined from a minimum of 3 independent experiments using 3–6 sequence-verified transformants of each strain.
Crystal structure description of PBP-6140CT-A501T
The PBP2-6140CT variant containing an A501T mutation crystallized under similar conditions as PBP2 and PBP2-6140CT with two molecules in the asymmetric unit and the structure was solved to 2.4 Å resolution (none of the other PBP2 variants yielded crystals that diffracted to high resolution). Inspection of |Fo|-|Fc| difference electron density maps revealed that the β3-β4 loops of both molecules in the asymmetric unit could be modeled. This is in marked contrast to the structures of PBP2 and PBP2-6140CT, in which residues 501–512 lacked electron density in both molecules of the asymmetric unit. Although the electron density for the side chains in this loop is comparatively weak in places, especially at its apex, that for the main chain is mostly complete, thus establishing the overall structure of the loop.
Two observations suggest that the ordering of the β3-β4 loop is a direct consequence of the A501T mutation and not crystal packing interactions. Firstly, being of the same crystal form, the A501T mutant of PBP2-6140CT has the same packing arrangement as the wild-type structure. Secondly, the structures of the β3-β4 loops in both molecules of the asymmetric unit of PBP2-6140CT-A501T are very similar and yet each has different packing environments. In molecule B, the β3-β4 loop packs against the β2a-β2b loop of a neighboring molecule and against β7n of the N terminal region of another neighboring molecule, whereas the β3-β4 loop of molecule A is exposed to solvent.
The structures of wild-type PBP2 and PBP2-6140CT-A501T superimpose very closely, with an RMS deviation between all common main chain atoms of 0.30 Å for molecule A and 0.36 Å for molecule B. The only significant difference between the structures is the ordering of the β3-β4 loop in the A501T mutant (Fig. 5). Since PBP2 and PBP2-6140CT have essentially the same structures, a superimposition of the A501T mutant with PBP2-6140CT produces the same outcome.
Figure 5. The β3-β4 loop is ordered in the PBP2 A501T mutant.
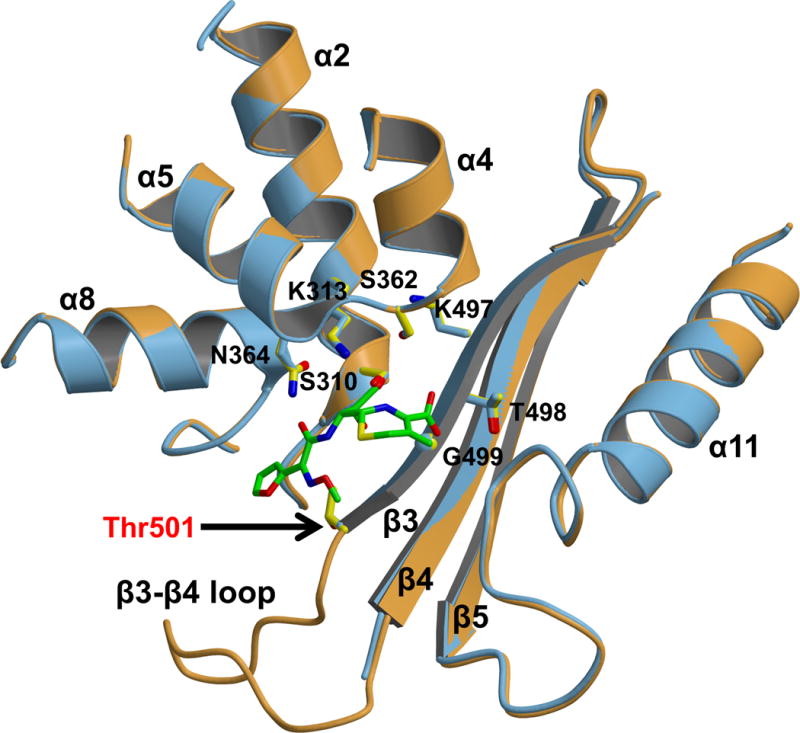
Superimposition of molecule A of wild-type PBP 2 (3EQU; slate blue) with molecule A of PBP2-6140CT-A501T (orange). Cefuroxime has been docked into the wild-type structure based on the structure of the acylated PBP2x from S. pneumoniae (1QMF). Active site residues of the SxxK, SxN, and KTG motifs are shown in stick form.
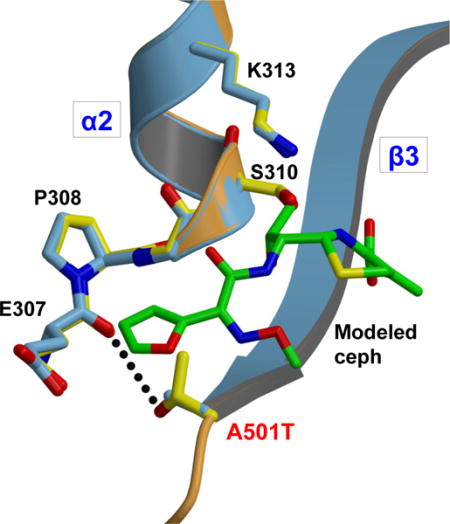
Thr501 forms a hydrogen bond near Ser310
The site of the mutation is at the end of β3, where its threonine side chain packs closely between Val515 on β4 and Tyr422 at the C-terminal end of α8. A further consequence of the A501T mutation is that the side-chain hydroxyl of Thr501 is within hydrogen bonding distance of the main-chain carbonyl of Glu307, thus forming a new connection between β3 and the loop immediately prior to Ser310 (Fig. 6). The nearly exact overlap of wild-type and A501T mutant structures of PBP2 in this region shows that the new hydrogen bond does not alter the architecture, but it may nevertheless be close enough to impact nucleophilic activation of Ser310.
Figure 6. Close-up view of the PBP2 region near the A501T mutation.
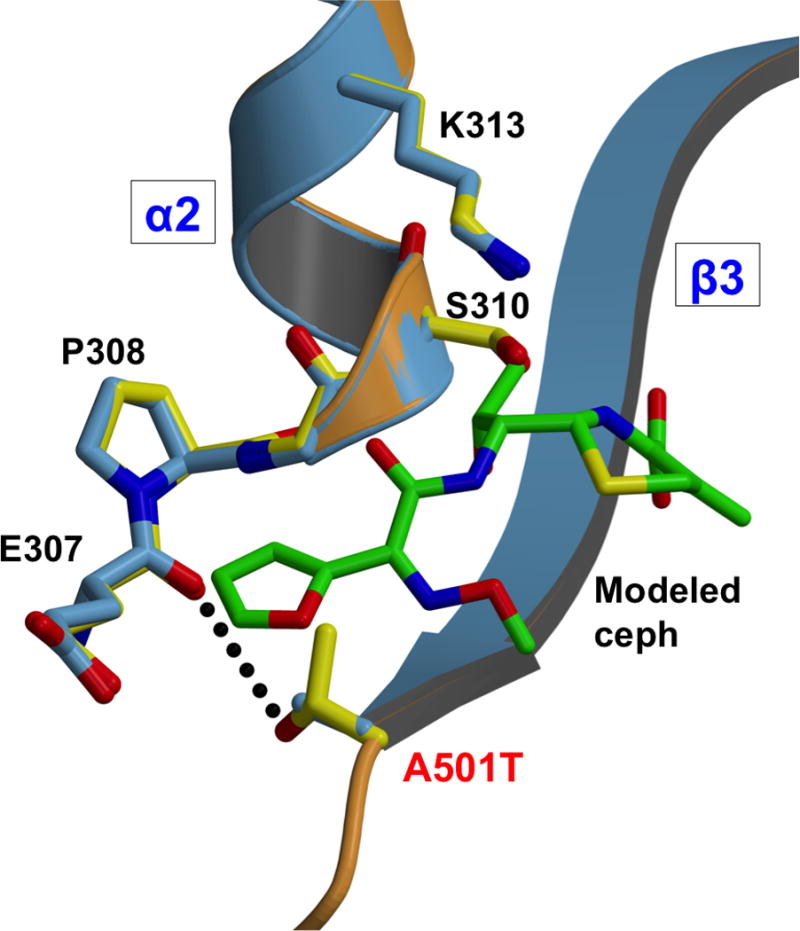
Superimposition of molecule B of wild-type PBP2 (3EQU; blue) and molecule B of PBP2-6140CT-A501T (orange). Cefuroxime has been docked into the wild-type structure based on the structure of the acylated PBP2x from S. pneumoniae (1QMF).
Thermal stability of 501 mutants
The ordering of the β3-β4 loop of PBP2 in the PBP2-6140CT-A501T structure suggests a mechanism of antibiotic resistance involving increased rigidity of the protein. To evaluate the impact of the mutations at position 501 on the stability of the protein, we collected irreversible thermal denaturation curves for the Thr, Val, Pro and Arg mutants of PBP2-6140CT using circular dichroism (CD) spectroscopy. Spectra were also recorded for wild-type PBP2 and for PBP2-6140CT. Surprisingly, the Tm of all the mutants was significantly lower than that of wild-type PBP2, suggesting an overall destabilizing effect of the mutations (Fig. 7). Furthermore, all of the Ala501 mutants exhibit a lower Tm than PBP2-6140CT, with A501R and A501P mutations lowering Tm the most. The contrast between observed ordering of the β3-β4 loop and lowered thermal stability of the mutant proteins is discussed below.
Figure 7. Melting temperatures of PBP2, PBP2-6140CT, and PBP2-6140CT-A501 mutants.
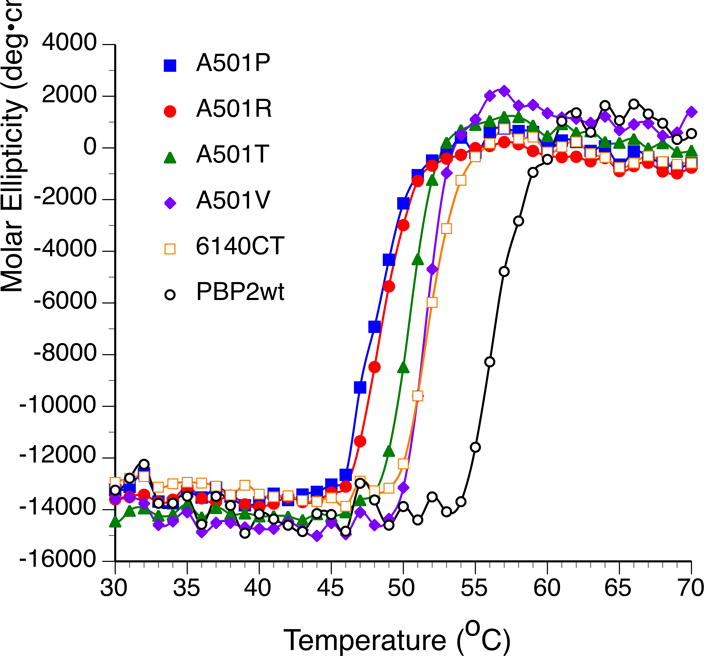
The increase in molar ellipticity at 208 nm of the indicated PBP variants was quantified at different temperatures by circular dichroism.
Discussion
In this study, we identified five amino acids (Thr, Val, Ser, Arg, and Pro) in a random screen for mutations at Ala501 of PBP2, plus an additional amino acid (Lys) by site-directed mutagenesis, that increase resistance of N. gonorrhoeae to expanded-spectrum cephalosporins over that conferred by the parental mosaic PBP, PBP2-35/02. Three of these amino acids, Thr, Val, and Pro, have been identified previously as mutations at Ala501 in penA alleles from gonococcal clinical isolates, while Ser, Arg and Lys have not. Of these, both A501R and A501P mutations have a more pronounced effect on the MIC (~5-fold) and appear to cause a larger fitness deficit compared to the other mutations. The structure of PBP2-6140CT-A501T reveals that the primary impact of the A501T mutation is to promote ordering of the β3-β4 loop.
Antibiotic resistance determinants often have a negative effect on fitness. This is particularly true with mosaic PBP2 variants as these likely have sub-optimal TPase activity. Unfortunately, in the absence of a biochemical assay of TPase activity using purified PBPs (particularly for the class B PBPs such as PBP2)(6), it is not possible to quantify directly the impact of resistance mutations on the TPase function of PBP2. That said, our in vitro growth experiments suggest that bacteria expressing a mosaic penA allele (penA35) grow less well than wild-type bacteria. Moreover, the two Ala501 mutations that increase the MICs of cefixime and ceftriaxone the most (i.e. A501P and A501R) are the least fit in competitive co-culture experiments, which would be expected if these mutations have a more deleterious effect on TPase activity than the other mutations. It seems likely, therefore, that dissemination of any of these alleles containing Ala501 mutations would require compensatory mutations in the strain that increase fitness. Indeed, compensatory mutations that help abrogate the fitness defect of resistance alleles have been identified in other bacteria, and for an mtrA mutant and an mtrR/gyrA/parC triple mutant in N. gonorrhoeae(33, 34).
Surprisingly, nearly 50% of the total transformants sequenced in the random screen harbored an A501R mutation, compared to the identification of a single transformant containing an A501P mutation. The high number of transformants obtained containing A501R mutations compared to A501P mutations was particularly unexpected, because in the clinical setting, three temporally and geographically distinct cefixime-resistant isolates harbored a mosaic penA allele with an A501P mutation(17, 18), but no isolates with an A501R mutation have been observed to date. We investigated this apparent anomaly by comparing the transformation frequencies of identical quantities of penA35 PCR fragments containing different mutations at Ala501, and observed large differences in the capacities of these DNAs to transform competent gonococci. One explanation for these differences is that they reflect the degree to which transpeptidase activity is compromised by the mutation, although this does not explain why only A501P mutations have occurred in nature. Irrespective, these results highlight the fine line that N. gonorrhoeae must navigate to increase resistance to antibiotics by remodeling PBP2 whilst preserving essential TPase activity.
An interesting observation from these studies is that there is not a clear correlation between the acylation rates of purified proteins and the MICs of β-lactam antibiotics for FA19 harboring the corresponding penA alleles. The MIC of a β-lactam antibiotic for N. gonorrhoeae is determined by three main characteristics: i) the acylation rate of PBP2, ii) the capacity to serve as a substrate for efflux pumps, and iii) the rate of influx through porin channels. An additional factor is the degree to which the PBP2 variant needs to be inhibited to cause cell death. For example, the TPase activity of wt PBP2 may need to be inhibited by 80–90% to cause cell death, whereas that of the PBP235/02-A501P variant, which our data suggest is markedly impaired, may only need to be inhibited by 30–40% before the level falls below the threshold that supports cell growth. The lack of correlation observed in this study between the acylation rates and MICs of the different antibiotics is most evident for the A501P mutant, and is consistent with the fitness data and transformation frequencies.
We crystallized PBP2-6140CT containing the A501T mutation and solved its structure to 2.4 Å. The most striking difference compared to the crystal structure of PBP2-6140CT is significant ordering of the β3-β4 loop. This same loop in other PBPs has also been associated with resistance to β-lactams. A Q556E mutation in Streptococcus pneumoniae PBP2x, which is implicated in cephalosporin resistance, lies in a very similar position as Ala501 at the C-terminal end of β3(35). Similarly, for S. pneumoniae PBP1a, there is a clustering of four mutations on the β3-β4 loop that lowers the rates of acylation for cefotaxime and penicillin G(36). Interestingly, whether the β3-β4 loop is ordered or disordered varies in relation to resistance. In the crystal structure of PBP1a from the R6 (susceptible) strain of S. pneumoniae, the β3-β4 loop is ordered, but is disordered in the protein derived from the cephalosporin-resistant strain 5204(37). Opposite to that and in alignment with N. gonorrhoeae PBP2, the crystal structure of PBP2a from S. pneumoniae shows a disordered β3-β4 loop in the protein derived from the susceptible strain and an ordered loop in the protein from the resistant strain(38). These observations have led to the idea that flexibility in this region is a key factor in antibiotic resistance(38).
A key question is how mutations at position 501 confer increased resistance to cephalosporins. One simple mechanism is that larger side chains at position 501 sterically impede the binding of antibiotic. Given the close proximity of position 501 to the core of the active site, direct clashes between side chains at position 501 and the cephalosporin can be envisaged (see Fig. 5). These clashes would block binding of antibiotic (i.e. increase KS) and thereby lower the rate of acylation, while in some way retaining the capacity to recognize the peptide C-terminus of peptidoglycan during transpeptidation. In a similar vein, the negative charge introduced by the Q552E mutation in S. pneumoniae PBP2x is believed to impede binding of antibiotic in a direct manner(35). This mechanism is consistent with the mutations all introducing larger side chains at position 501 and the proportionally higher increase of MICs for the largest side chain, Arg. It does not explain, however, why mutation of Ala501 to Ile, which also introduces a larger side chain, has no effect on resistance, suggesting that steric hindrance alone does not explain the changes in resistance in PBP2.
Perhaps the most compelling mechanism, and consistent with changes in the crystal structure, is that resistance results from increased rigidity of the active site and its immediate environs. As revealed by the structure of PBP2-6140CT-A501T, the β3-β4 loop exhibits significantly increased order, compared to the otherwise identical protein construct lacking the A501T mutation. One possible reason for this is the new hydrogen bond between Thr501 and Glu307 near the SxxK motif that tethers the beginning of β3 with a loop preceding the SxxK motif. The other mutations at position 501 also have the potential to lower flexibility. A serine would likely form the same hydrogen bond with Glu307 as threonine. A valine cannot mediate a hydrogen bond, but could form hydrophobic interactions with Val515 and Tyr422. An arginine could form a new electrostatic interaction with the side chain of Glu307 and introduction of proline would clearly restrict conformational freedom.
At first sight, the idea of increased rigidity causing resistance to cephalosporins appears at odds with the circular dichroism results. These data show that mutations at Ala501 lower the thermal stability of the protein and thus appear more consistent with a mechanism in which resistance mutations destabilize the enzyme. However, the apparent destabilization of the protein detected by a lowered Tm may result from entropic compensation elsewhere in the protein framework. This situation has been described as a rigidity-flexibility equilibrium, where lowered entropy due to a stabilizing mutation at one site is compensated by increased flexibility in remote regions(39). The reverse can also happen when the mutation is destabilizing. Relationships between mutations, protein stability and ligand binding are generally protein specific and specific mutations can reduce ligand affinity by lowering or increasing the stability of proteins(40). In the case of PBP2, it appears that mutations at position 501 reduce reactivity for β-lactams via increasing localized ordering in the active site region and the entropic penalty is paid by increased flexibility elsewhere in the framework that is detected by circular dichroism.
Increased rigidity in the active site could lower rates of acylation by cephalosporins in two ways: by preventing requisite conformational changes necessary for binding of the antibiotic, or by hindering formation of the ideal geometry of the transition state for acylation (i.e. by lowering k2). The first of these mechanisms infers that binding of cephalosporins by PBP2 requires movement of the protein to bind the antibiotic in a productive orientation prior to acylation. Due to the increased rigidity conferred by the mutations at position 501, the energetic barrier against such conformational changes is made higher and thus cephalosporins are less likely to acylate the enzyme successfully. This mechanism may act synergistically with the simple steric hindrance described above. A similar mechanism was proposed for S. aureus PBP2a in which mutations associated with β-lactam resistance were hypothesized to restrict movement of β3(41).
The second mechanism is derived from the mechanistic relationship between PBPs and serine proteases. It is well established that acylation requires the formation of a tetrahedral intermediate in which the negative charge of the intermediate is stabilized by an oxyanion hole, which in PBP2 comprises the amide nitrogens of Ser310 and Thr500. The latter, of course, is immediately adjacent to Ala501. A productive transition state requires precise geometry, and increased rigidity in the structure elicited by mutations at position 501 may create a barrier to successfully achieving this state with the kinetic consequence of lowered rates of acylation.
Of course, in both of these mechanisms, the peptidoglycan substrate of PBP2 must be less affected than antibiotic, and how this is achieved is a matter of significant interest. One possibility is that the larger peptide substrate overcomes the energetic barrier imposed by the Ala501 mutations by making more contacts with the enzyme. That said, it seems inevitable that the efficiency by which the acyl-enzyme complex forms with peptide substrates must be compromised to some degree, and this is consistent with the larger fitness deficit of bacteria harboring penA alleles with Ala501 mutations that increase the MIC the most (Arg and Pro), as revealed in the competitive co-culture experiments.
The opposite effect of the A501P mutation on penicillin G MICs and k2/KS compared to cefixime and ceftriaxone is interesting because the mechanisms that discriminate against cephalosporins must somehow favor reactivity with penicillin. How this happens mechanistically is unclear, but perhaps the precise active-site geometry that is formed by this mutation is ideal for the binding or acylation of penicillin G. There are key chemical differences between cephalosporins and penicillins that can affect binding and/or reactivity with a PBP, including the 6-membered dihydrothiazine ring of cephalosporins rather than 5-membered thiazolidine ring of penicillins, the presence of a C3 substituent in cephalosporins, and the different groups comprising the R1 side chains.
In this study, we have shown that specific mutations of Ala501 in PBP2 can cause cephalosporin resistance in N. gonorrhoeae while retaining sufficient essential transpeptidase function necessary for viability. Substitutions to amino acids with significantly different physicochemical properties compared to Ala confer the biggest increases on MICs. A crystal structure of PBP2 containing an A501T mutation shows dramatic ordering of the β3-β4 loop and suggests that increased rigidity in the active site correlates with resistance. These results are important for predicting future emergence of Ala501 mutations that may threaten the clinical utility of cephalosporins against gonorrhea and for understanding the underlying molecular mechanism of resistance.
Acknowledgments
Use of the Advanced Photon Source was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. W-31-109-ENG-38. Data were collected at Southeast Regional Collaborative Access Team (SER-CAT) 22-ID beam line at the Advanced Photon Source, Argonne National Laboratory. Supporting institutions may be found at www.ser-cat.org/members.html. The X-ray crystallography facility used for this work is supported by the Medical University of South Carolina’s Research Resource Facilities program. The authors acknowledge Dr. Hyun-Sop Choe and Alaa Telchy for their assistance in determining the MICs of penicillin for the various mutants.
Funding Source Statement. This work was supported by grants GM66861 (CD, RAN) and U19 AI11317 (RAN, AEJ) from the National Institutes of Health. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Abbreviations and Textual Footnotes
- PBP
Penicillin-Binding Protein
- TPase
transpeptidase
- MIC
minimum inhibitory concentration
- CephI
cephalosporin-intermediate resistant
- CephR
cephalosporin-resistant
- GCB
GC Medium Base
- LB
lysogeny broth
- CFU
colony forming units
Footnotes
Conflict of Interest
The authors declare that they have no conflicts of interest with the contents of this article.
Author Contributions
RAN, CD, AEJ and MU conceived and coordinated the study, RAN, and CD wrote the manuscript, JT carried out the mutant screen, generated all of the mutant strains, and determined their MICs, AF crystallized PBP2-6140CT-A501T and determined its structure, and LRV conducted the biological fitness experiments. All authors analyzed the results, edited the manuscript, and approved the final version.
References
- 1.Newman L, Rowley J, Vander Hoorn S, Wijesooriya NS, Unemo M, Low N, Stevens G, Gottlieb S, Kiarie J, Temmerman M. Global Estimates of the Prevalence and Incidence of Four Curable Sexually Transmitted Infections in 2012 Based on Systematic Review and Global Reporting. PLoS One. 2015;10:e0143304. doi: 10.1371/journal.pone.0143304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Unemo M, Nicholas RA. Emergence of multidrug-resistant, extensively drug-resistant and untreatable gonorrhea. Future Microbiology. 2012;7:1401–1422. doi: 10.2217/fmb.12.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Unemo M, Shafer WM. Antimicrobial resistance in Neisseria gonorrhoeae in the 21st century: past, evolution, and future. Clin Microbiol Rev. 2014;27:587–613. doi: 10.1128/CMR.00010-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Unemo M. Current and future antimicrobial treatment of gonorrhoea - the rapidly evolving Neisseria gonorrhoeae continues to challenge. BMC Infect Dis. 2015;15:364. doi: 10.1186/s12879-015-1029-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ito M, Deguchi T, Mizutani KS, Yasuda M, Yokoi S, Ito S, Takahashi Y, Ishihara S, Kawamura Y, Ezaki T. Emergence and spread of Neisseria gonorrhoeae clinical isolates harboring mosaic-like structure of penicillin-binding protein 2 in Central Japan. Antimicrob Agents Chemother. 2005;49:137–143. doi: 10.1128/AAC.49.1.137-143.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nicholas RA, Davies C. Structural mechanisms of β-lactam antibiotic resistance in penicillin-binding proteins. In: Dougherty TJ, Pucci MJ, editors. Antibiotic Discovery and Development. Springer; New York: 2012. pp. 397–425. [Google Scholar]
- 7.Spratt BG. Hybrid penicillin-binding proteins in penicillin-resistant strains of Neisseria gonorrhoeae. Nature. 1988;332:173–176. doi: 10.1038/332173a0. [DOI] [PubMed] [Google Scholar]
- 8.Ito M, Yasuda M, Yokoi S, Ito S, Takahashi Y, Ishihara S, Maeda S, Deguchi T. Remarkable increase in central Japan in 2001–2002 of Neisseria gonorrhoeae isolates with decreased susceptibility to penicillin, tetracycline, oral cephalosporins, and fluoroquinolones. Antimicrob Agents Chemother. 2004;48:3185–3187. doi: 10.1128/AAC.48.8.3185-3187.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ochiai S, Sekiguchi S, Hayashi A, Shimadzu M, Ishiko H, Matsushima-Nishiwaki R, Kozawa O, Yasuda M, Deguchi T. Decreased affinity of mosaic-structure recombinant penicillin-binding protein 2 for oral cephalosporins in Neisseria gonorrhoeae. J Antimicrob Chemother. 2007;60:54–60. doi: 10.1093/jac/dkm166. [DOI] [PubMed] [Google Scholar]
- 10.Unemo M, Nicholas RA, Jerse AE, Davies C, Shafer WM. Molecular mechanisms of antibiotic resistance expressed by the pathogenic Neisseria. In: Davies JK, Kahler CM, editors. Pathogenic Neisseria. 2nd. Caister Academic Press; Norfolk, UK: 2014. pp. 161–192. [Google Scholar]
- 11.Zhao S, Duncan M, Tomberg J, Davies C, Unemo M, Nicholas RA. Genetics of chromosomally mediated intermediate resistance to ceftriaxone and cefixime in Neisseria gonorrhoeae. Antimicrob Agents Chemother. 2009;53:3744–3751. doi: 10.1128/AAC.00304-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lindberg R, Fredlund H, Nicholas RA, Unemo M. Neisseria gonorrhoeae isolates with reduced susceptibility to cefixime and ceftriaxone: association with genetic polymorphisms in penA, mtrR, porB1b, and ponA. Antimicrob Agents Chemother. 2007;51:2117–2122. doi: 10.1128/AAC.01604-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Whiley DM, Limnios EA, Ray S, Sloots TP, Tapsall JW. Diversity of penA alterations and subtypes in Neisseria gonorrhoeae strains from Sydney, Australia, that are less susceptible to ceftriaxone. Antimicrob Agents Chemother. 2007;51:3111–3116. doi: 10.1128/AAC.00306-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Osaka K, Takakura T, Narukawa K, Takahata M, Endo K, Kiyota H, Onodera S. Analysis of amino acid sequences of penicillin-binding protein 2 in clinical isolates of Neisseria gonorrhoeae with reduced susceptibility to cefixime and ceftriaxone. J Infect Chemother. 2008;14:195–203. doi: 10.1007/s10156-008-0610-7. [DOI] [PubMed] [Google Scholar]
- 15.Lee SG, Lee H, Jeong SH, Yong D, Chung GT, Lee YS, Chong Y, Lee K. Various penA mutations together with mtrR, porB and ponA mutations in Neisseria gonorrhoeae isolates with reduced susceptibility to cefixime or ceftriaxone. J Antimicrob Chemother. 2010;65:669–675. doi: 10.1093/jac/dkp505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tomberg J, Unemo M, Davies C, Nicholas RA. Molecular and structural analysis of mosaic variants of penicillin-binding protein 2 conferring decreased susceptibility to expanded-spectrum cephalosporins in Neisseria gonorrhoeae: role of epistatic mutations. Biochemistry. 2010;49:8062–8070. doi: 10.1021/bi101167x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Unemo M, Golparian D, Nicholas R, Ohnishi M, Gallay A, Sednaoui P. High-level cefixime- and ceftriaxone-resistant N. gonorrhoeae in France: novel penA mosaic allele in a successful international clone causes treatment failure. Antimicrob Agents Chemother. 2011;56:1273–1280. doi: 10.1128/AAC.05760-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Camara J, Serra J, Ayats J, Bastida T, Carnicer-Pont D, Andreu A, Ardanuy C. Molecular characterization of two high-level ceftriaxone-resistant Neisseria gonorrhoeae isolates detected in Catalonia, Spain. J Antimicrob Chemother. 2012;67:1858–1860. doi: 10.1093/jac/dks162. [DOI] [PubMed] [Google Scholar]
- 19.Kellogg DS, Peacock WL, Deacon WE, Browh L, Perkle CI. Neisseria gonorrhoeae. I. Virulence genetically linked to colonial variation. J Bacteriol. 1963;85:1274–1279. doi: 10.1128/jb.85.6.1274-1279.1963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Elkins C, Thomas CE, Seifert HS, Sparling PF. Species-specific uptake of DNA by gonococci is mediated by a 10-base-pair sequence. J Bacteriol. 1991;173:3911–3913. doi: 10.1128/jb.173.12.3911-3913.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ho SN, Hunt HD, Horton RM, Pullen JK, Pease LR. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene. 1989;77:51–59. doi: 10.1016/0378-1119(89)90358-2. [DOI] [PubMed] [Google Scholar]
- 22.Powell AJ, Tomberg J, Deacon AM, Nicholas RA, Davies C. Crystal structures of penicillin-binding protein 2 from penicillin-susceptible and -resistant strains of Neisseria gonorrhoeae reveal an unexpectedly subtle mechanism for antibiotic resistance. J Biol Chem. 2009;284:1202–1212. doi: 10.1074/jbc.M805761200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stefanova ME, Tomberg J, Olesky M, Holtje JV, Gutheil WG, Nicholas RA. Neisseria gonorrhoeae penicillin-binding protein 3 exhibits exceptionally high carboxypeptidase and β-lactam binding activities. Biochemistry. 2003;42:14614–14625. doi: 10.1021/bi0350607. [DOI] [PubMed] [Google Scholar]
- 24.Frere JM, Nguyen-Disteche M, Coyette J, Joris B. The chemistry of β-lactams. Chapman & Hall; Glasgow: 1992. Mode of action: Interaction with the penicillin binding proteins; pp. 148–196. [Google Scholar]
- 25.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 26.Jones TA, Zhou JY, Cowan SW, Kjeldgaard M. Improved methods for building protein models in electron density maps and the location of errors in those models. Acta Crystallographica A47. 1991:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 27.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 28.Serra-Pladevall J, Barbera MJ, Rodriguez S, Bartolome-Comas R, Roig G, Juve R, Andreu A. Neisseria gonorrhoeae antimicrobial susceptibility in Barcelona: penA, ponA, mtrR, and porB mutations and NG-MAST sequence types associated with decreased susceptibility to cephalosporins. Eur J Clin Microbiol Infect Dis. 2016;35:1549–1556. doi: 10.1007/s10096-016-2696-7. [DOI] [PubMed] [Google Scholar]
- 29.Chen SC, Yin YP, Dai XQ, Unemo M, Chen XS. Antimicrobial resistance, genetic resistance determinants for ceftriaxone and molecular epidemiology of Neisseria gonorrhoeae isolates in Nanjing, China. J Antimicrob Chemother. 2014;69:2959–2965. doi: 10.1093/jac/dku245. [DOI] [PubMed] [Google Scholar]
- 30.Olsen B, Pham TL, Golparian D, Johansson E, Tran HK, Unemo M. Antimicrobial susceptibility and genetic characteristics of Neisseria gonorrhoeae isolates from Vietnam, 2011. BMC Infect Dis. 2013;13:40. doi: 10.1186/1471-2334-13-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Whiley DM, Goire N, Lambert SB, Ray S, Limnios EA, Nissen MD, Sloots TP, Tapsall JW. Reduced susceptibility to ceftriaxone in Neisseria gonorrhoeae is associated with mutations G542S, 551S and 551L in the gonococcal penicillin-binding protein 2. J Antimicrob Chemother. 2010;65:1615–1618. doi: 10.1093/jac/dkq187. [DOI] [PubMed] [Google Scholar]
- 32.Andersson DI. The biological cost of mutational antibiotic resistance: any practical conclusions? Curr Opin Microbiol. 2006;9:461–465. doi: 10.1016/j.mib.2006.07.002. [DOI] [PubMed] [Google Scholar]
- 33.Kunz AN, Begum AA, Wu H, D’Ambrozio JA, Robinson JM, Shafer WM, Bash MC, Jerse AE. Impact of fluoroquinolone resistance mutations on gonococcal fitness and in vivo selection for compensatory mutations. J Infect Dis. 2012;205:1821–1829. doi: 10.1093/infdis/jis277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Warner DM, Folster JP, Shafer WM, Jerse AE. Regulation of the MtrC-MtrD-MtrE efflux-pump system modulates the in vivo fitness of Neisseria gonorrhoeae. J Infect Dis. 2007;196:1804–1812. doi: 10.1086/522964. [DOI] [PubMed] [Google Scholar]
- 35.Mouz N, Di Guilmi AM, Gordon E, Hakenbeck R, Dideberg O, Vernet T. Mutations in the active site of penicillin-binding protein PBP2x from Streptococcus pneumoniae. Role in the specificity for β-lactam antibiotics. J Biol Chem. 1999;274:19175–19180. doi: 10.1074/jbc.274.27.19175. [DOI] [PubMed] [Google Scholar]
- 36.Contreras-Martel C, Job V, Di Guilmi AM, Vernet T, Dideberg O, Dessen A. Crystal structure of penicillin-binding protein 1a (BP1a) reveals a mutational hotspot implicated in β-lactam resistance in Streptococcus pneumoniae. J Mol Biol. 2006;355:684–696. doi: 10.1016/j.jmb.2005.10.030. [DOI] [PubMed] [Google Scholar]
- 37.Job V, Carapito R, Vernet T, Dessen A, Zapun A. Common alterations in BP1a from resistant Streptococcus pneumoniae decrease its reactivity toward β-lactams: structural insights. J Biol Chem. 2008;283:4886–4894. doi: 10.1074/jbc.M706181200. [DOI] [PubMed] [Google Scholar]
- 38.Contreras-Martel C, Dahout-Gonzalez C, Martins Ados S, Kotnik M, Dessen A. PBP active site flexibility as the key mechanism for beta-lactam resistance in pneumococci. J Mol Biol. 2009;387:899–909. doi: 10.1016/j.jmb.2009.02.024. [DOI] [PubMed] [Google Scholar]
- 39.Li T, Tracka MB, Uddin S, Casas-Finet J, Jacobs DJ, Livesay DR. Redistribution of flexibility in stabilizing antibody fragment mutants follows Le Chatelier’s principle. PLoS One. 2014;9:e92870. doi: 10.1371/journal.pone.0092870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Teilum K, Olsen JG, Kragelund BB. Protein stability, flexibility and function. Biochim Biophys Acta. 2011;1814:969–976. doi: 10.1016/j.bbapap.2010.11.005. [DOI] [PubMed] [Google Scholar]
- 41.Lim D, Strynadka NC. Structural basis for the β-lactam resistance of BP2a from methicillin-resistant Staphylococcus aureus. Nat Struct Biol. 2002;9:870–876. doi: 10.1038/nsb858. [DOI] [PubMed] [Google Scholar]