

Abstract
Background
Compositional differences in bronchial bacterial microbiota have been associated with asthma, but it remains unclear whether the findings are attributable to asthma, to aeroallergen sensitization or to inhaled corticosteroid treatment.
Objectives
To compare the bronchial bacterial microbiota in adults with steroid-naive atopic asthma (AA), with atopy but no asthma (ANA), and non-atopic healthy subjects (HC), and determine relationships of bronchial microbiota to phenotypic features of asthma.
Methods
Bacterial communities in protected bronchial brushings from 42 AA, 21 ANA, and 21 HC subjects were profiled by 16S rRNA gene sequencing. Bacterial composition and community-level functions inferred from sequence profiles were analyzed for between-group differences. Associations with clinical and inflammatory variables were examined, including markers of type 2-related inflammation and change in airway hyperresponsiveness following six weeks of fluticasone treatment.
Results
The bronchial microbiome differed significantly among the three groups. Asthmatic subjects were uniquely enriched in members of the Haemophilus, Neisseria., Fusobacterium, Porphyromonas and Sphingomonodaceae, and depleted in members of the Mogibacteriaceae and Lactobacillales. Asthma-associated differences in predicted bacterial functions included involvement of amino acid and short-chain fatty acid metabolism pathways. Subjects with type 2-high asthma harbored significantly lower bronchial bacterial burden. Distinct changes in specific microbiota members were seen following fluticasone treatment. Steroid-responsiveness was linked to differences in baseline compositional and functional features of the bacterial microbiome.
Conclusion
Even in mild steroid-naive asthma subjects, differences in the bronchial microbiome are associated with immunologic and clinical features of the disease. The specific differences identified suggest possible microbiome targets for future approaches to asthma treatment or prevention.
Keywords: Asthma, atopy, microbiome, corticosteroids, 16S ribosomal RNA, bacteria, Th2 inflammation, Three-gene mean, metabolic pathways, SCFAs
Introduction
Recent culture-independent studies have documented that the composition of commensal lower respiratory tract bacteria (microbiota) differs between asthmatic and healthy adults (1-6). Additionally, phenotypic features of asthma, such as measures of airway hyper-responsiveness, asthma control, and transcriptional response to steroids, correlate with patterns of bronchial microbiota composition (4, 5). Though different studies have reported asthma-associated enrichment (higher relative abundance) of different taxa [i.e. bacterial-derived 16S ribosomal RNA gene sequences that exhibit an operator-defined level of sequence homology (typically 97%)], enrichment in members of the phylum Proteobacteria is a repeating signature. Asthmatic subjects in most previous studies were treated with inhaled corticosteroids (ICS), casting some uncertainty on whether the findings reflect the effects of ICS treatment or of asthma itself. Similarly, many asthmatic patients are atopic (7, 8), raising the question as to whether asthma- associated differences in respiratory microbiota are related to underlying atopy, itself associated with altered mucosal immune function (9, 10). Collectively, these considerations indicate a need to elucidate differences in bronchial microbiota associated with asthma versus atopy, and with important phenotypic features of this disease, such as the level of T2-type inflammation and responsiveness to ICS treatment.
Accordingly, we compared the bronchial bacterial microbiome among adults with mild steroid-naive atopic asthma, with atopy without asthma, and healthy non-atopic nonasthmatic controls. We hypothesized that specific compositional and functional differences in bronchial microbiota are associated with asthma and with distinguishing phenotypic features of the disease, including evidence of Th2 inflammation and responsiveness to ICS treatment.
Methods
Study Population and Sample Collection
This study was conducted at nine sites in the NHLBI AsthmaNet, using a standardized bronchoscopy protocol for sample collection. Of 186 adults screened, 84 subjects were enrolled (Figure S1A-B): 42 atopic asthmatics (AA), 21 atopic non-asthmatics (ANA) and 21 non-atopic healthy control subjects (HC). Atopy was defined by serologic evidence (>0.35 kU/l) of sensitivity to >1 of 12 aeroallergens (specific IgE by ImmunoCap; Thermo-Scientific; Table S1). Asthma was confirmed by airway hyperresponsiveness (methacholine PC20 ≤8 mg/mL or FEV-i improvement >12% postalbuterol). At enrollment, asthmatics had been clinically stable for three months, and had an Asthma Control Questionnaire (ACQ) score of <1.5 (11) without the use of a controller medication. Exclusion criteria included a history of smoking, respiratory infection within six weeks or antibiotic use within 3 months of enrollment (Supplementary Methods).
Samples processed for microbiota analysis included oral wash (OW) (12), a saline flush (10 mL) of the bronchoscope suction channel (“scope-flush”) and protected bronchial brushings (BB; Figure S1C; Supplemental Methods). AA were further randomized in a 2:1 ratio to treatment with inhaled fluticasone propionate (250 mcg, GlaxoSmithKline) or placebo twice daily for six weeks and re-assessed post-treatment. Each subject signed informed consent approved by their center’s IRB; an NHLBI-appointed Data Safety Monitoring Board (DSMB) oversaw the study conduct.
Nucleic acid extraction and quantitation of 16S rRNA gene copy number
Nucleic acids from OW, and 3 BB were extracted as previously described (5, 12) using a modified bead-beating protocol and the AllPrep kit (Qiagen). 16S rRNA gene copy number was assessed by quantitative PCR using universal primers (Supplemental Methods).
16S rRNA-based sequencing and raw data processing
For BB and OW samples, variable region 4 (V4) of the 16S rRNA gene was amplified using the primer combination 515F/806R (13, 14) and sequenced on the Illumina MiSeq platform. Using chimera-checked and quality-filtered sequence reads (Supplemental Methods), a 97% sequence homology cut-off was used to define bacterial taxa (also referred to as operational taxonomic units; OTUs) and classified using the Greengenes database (15). A phylogenetic tree was built using FastTree (16) and used to compute Faith's Phylogenetic Diversity (17) of samples using an OTU table multiply-rarefied to 52,317 sequences per sample (Supplemental Methods).
“Three-gene mean” bronchial epithelial signature of type 2 inflammation
Expression levels of three bronchial epithelial genes (CLCA1, SERPINB2 and POSTN) previously shown to be induced by IL-13 (18, 19), were measured using RNA extracted in parallel with DNA in this study to calculate the “three-gene mean” (TGM) score for each participant (20). Type 2 (T2) - high asthma was defined by TGM scores >1.117 (two standard deviations above the average TGM-score in HC), confirmed by unsupervised cluster and principal component analysis.
Statistical analyses
We applied principal coordinates analyses (PCoA) on an unweighted UniFrac distance matrix and PERMANOVA (21) to identify the determinants of variation in bacterial community beta-diversity. Linear mixed-effects model (LME) (22) was employed to examine relationships between paired OW and BB microbiota; negative binomial (NB) and zero-inflated negative binomial (ZINB) regression models (23-26) corrected for false discovery (Benjamini-Hochberg, q-value <0.1) were used to identify OTUs differentially abundant between subject groups and paired-samples, respectively. We applied Phylogenetic Reconstruction of Unobserved States (PICRUSt) to predict functional capacities of the microbiota (27), and NB regression to compare inferred functional pathway predictions across groups. Procrustes analysis (13) was used to explore the strength of relationships between paired samples in the AA group (Supplemental Methods).
Results
Study group characteristics
AA had mild well-controlled disease, significantly higher serum total IgE and blood and sputum eosinophil cell counts (Table 1, Figure S2A-C) than HC. Compared to the ANA, the asthmatics had significantly higher serum IgE, were sensitized to more of the aeroallergens tested (Table 1), were more likely to be sensitive to cat, dog, and mouse (Table S1), and to report a history of allergic rhinitis and eczema (Table S2). However, they did not exhibit significant differences in environmental exposures as assessed by questionnaire (Table S3).
Table 1. Study cohort characteristics.
| Variable | Atopic asthmatic (AA) (n = 42)** |
Atopic, non-asthmatic (ANA) (n = 21) |
Healthy control (HC) (n = 21) |
p-value# |
|---|---|---|---|---|
| Age (yrs) | 33 (25 - 41) | 28 (24 - 45) | 28 (23 - 47) | NS |
| Age of asthma diagnosis (yrs) | 9 (5 - 22) | - | - | - |
| Duration of Asthma (yrs) | 21 (14 - 27) | - | - | - |
| ACQ Score* | 0.3 (0 - 1.3) | - | - | - |
| % Male | 45% | 48% | 43% | NS€ |
| % White | 60% | 52% | 71% | NS€ |
| BMI (kg/m2) | 26 (23 - 29) | 25 (21 - 28) | 26 (22 - 28) | NS |
| FEV1 % predicted (pre-albuterol) | 89 (76 - 97) | 98 (89 - 109) | 100 (93 - 108) | <0.01& |
| FEV1 % predicted (post-albuterol) | 100 (84 - 106) | 104 (97 - 109) | 102 (98 - 116) | NS& |
| Change in FEV1% | 7.5 (5.0 - 14.3) | 3.0 (-0.5 - 5.5) | 5.0 (2.5 - 5.0) | <0.0001& |
| Methacholine PC20 | 1.2 (0.3 - 3.2) | >32$ | >32$ | - |
| Blood eosinophils (absolute) | 200 (100 - 393) | 100 (87 - 200) | 100 (60 - 200) | <0.01 |
| Blood eosinophils (%) | 3.3 (1.5 - 5.6) | 2.0 (1.4 - 3.0) | 1.4 (1.0 - 3.0) | <0.01¥ |
| Blood neutrophils (%) | 55.5 (49.3 - 62.0) | 56.9 (51.0 - 63.8) | 58.4 (52.7 -63.5) | NS |
| Sputum neutrophils (%) | 54.6 (31.8 - 64.9) | 39.5 (26.5 - 50.0) | 41.8 (29.2 -76.5) | NS |
| Sputum eosinophils (%) | 0.4 (0.0 - 1.1) | 0.0 (0.0 - 0.6) | 0.0 (0.0 - 0.4) | NS¥ |
| Serum IgE (IU/mL) | 169.5 (56.3 - 321.3) | 64.0 (22.0 - 164.5) | 15.0 (5.0 -31.0) | <0.01&¥ |
| Number of positive sIgE¢ | 6 (2-7) | 3 (2-5) | - | <0.05& |
All values are medians (IQR).
ACQ, Asthma Control Questionnaire
Number of exacerbations requiring oral steroids in the past 5 years: zero exacerbations (38 subjects); one exacerbation (3 subjects); two exacerbations (one subject).
Methacholine challenge was stopped at 32 mg/dL and PC20 for these subjects was censored.
For between group statistical comparisons see Supplementary Figure S2A-C.
Number of positive specific IgE (sIgE >0.35 kU/l) from a total of 12 aeroallergens tested by ImmunoCap assay (for breakdown of specific aeroallergens see Supplementary Table S1). Statistical significance was determined using
Kruskal-Wallis or
Chi-square or
Mann-Whitney test for AA vs. ANA comparison.
Bacterial microbiota in bronchial brushings are compositionally distinct from oral wash
To evaluate mucosa-associated bronchial microbiota, we focused on protected BB. Sequence-based bacterial community analysis could be performed in the same proportion (67%) of samples collected in each of the three groups (Figure S1D-E). Samples that could not be sequenced had lower bacterial burden as indicated from 16S rRNA (p<0.0001; Figure S2D), despite recovery of similar mammalian cell burden as assessed by quantifyingβ -actin copy number (Supplemental Methods). Subjects with insufficient 16S rRNA amplicon for microbiota profiling were younger [median 28 (22-37) vs. 34 yrs. (25-44); p=0.03] but did not differ in any other characteristic measured.
Potential oral contamination of BB was evaluated by comparing a random subset of 30 BB-paired scope-flushes. Bacterial burden of scope-flushes for BB with sufficient 16S rRNA for sequencing was indistinguishable from those unable to be sequenced (p>0.1; Figure S2E). Additionally, PCoA analysis of bacterial community composition in paired OW and BB samples showed these two niches to be compositionally distinct (LME β=0.31, p<0.0001; Figure 1A), with BB samples exhibiting lower phylogenetic diversity (Faith's index; p<0.0001; Figure 1B).
Figure 1.
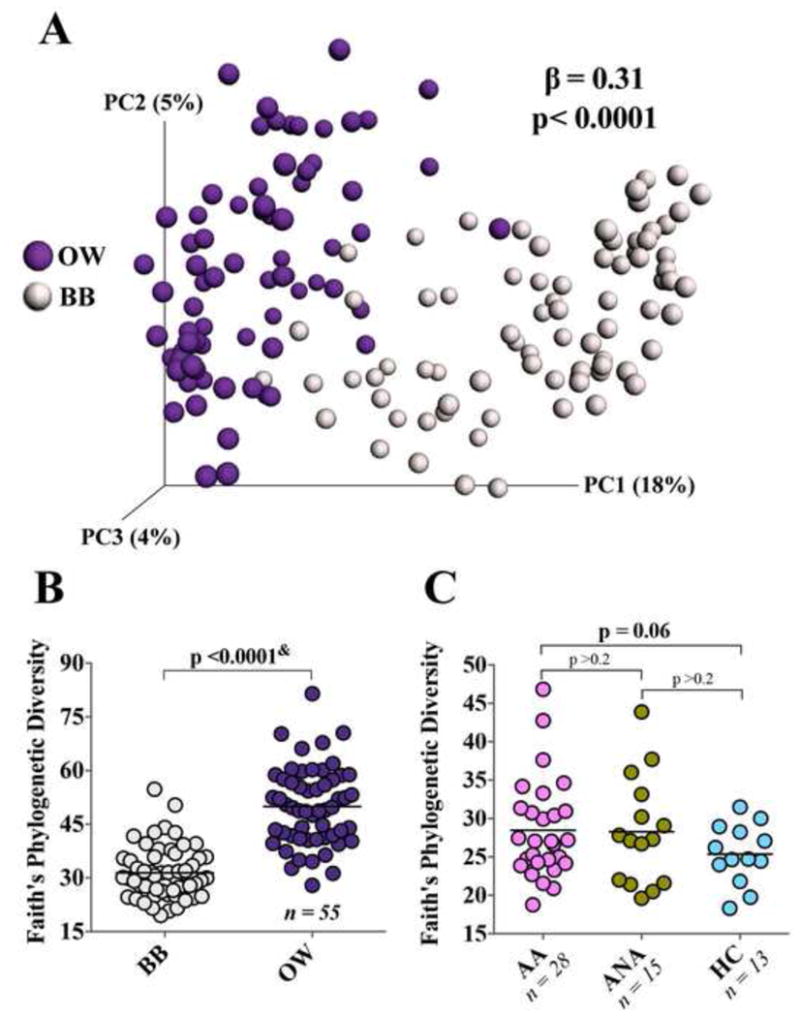
(A) Principal coordinate analysis (unweighted UniFrac) shows compositional dissimilarity between paired BB and OW samples (LME p<0.0001). (B) Phylogenetic diversity (Faith’s index) in BB and paired OW samples (& Wilcoxon matched-pairs signed rank test). (C) Phylogenetic diversity (Faith’s index) in BB samples for the three subject groups (Welch’s corrected t-test).
Features of the bronchial bacterial microbiota and distinct combinations of specific bacterial taxa are associated with atopic asthma or atopy alone
Alpha-diversity indices, such as richness (number of observed taxa), Shannon's diversity, and evenness, as well as bacterial burden did not differ amongst the groups (Figure S2F-I). However, phylogenetic diversity (Faith's index) tended to be higher in AA compared with HC (p=0.06; Figure 1C). Since this index weights phylogenetic relatedness of the bacteria detected, this observation suggests that the bronchial airways of AA harbor more phylogenetically diverse bacterial communities. Although phylogenetic diversity in the ANA also appeared to be greater than in HC (Figure 1C), the difference fell short of significance. Overall, inter-subject bacterial community composition (beta-diversity) was highly heterogeneous across subjects (Figure S3A). This was significantly related to bacterial richness independent of the study group (Figure S3B-E), but was not associated with any of the clinical measures or environmental exposures evaluated in this study (Tables 1 and S1-3). Compositional variability in bronchial bacterial microbiota was significantly greater within the asthmatic group (weighted UniFrac distance F-test p<0.001; Figure S3F), indicating a greater degree of bacterial community heterogeneity in this group.
Following these initial assessments for differences in overall bacterial community composition, we conducted OTU-level analyses to determine if the relative abundance of specific taxa differed significantly across groups; 76 taxa were found to do so in the AA vs HC comparison (Table S4). Phylum-level differences in AA (Figures 2A and S4A) included significant enrichment for members of the Bacteroidetes (Prevotella), Fusobacteria (Fusobacterium), Actinobacteria (Actinomyces) and Proteobacteria (Haemophilus and Neisseria), the latter previously associated with more severe, ICS- requiring asthma (2-6, 28). Conversely, asthmatic subjects showed reduced relative abundance of members of the Fusobacteria (Leptotrichia), Proteobacteria (Actinobacillus), and Firmicutes (Lactobacillus). ANA subjects also demonstrated significant differences in the relative abundance of 100 taxa compared to HC (Table S5B). The taxa most relatively enriched in ANA included members of the Proteobacteria (Aggregatibacter, Haemophilus), Firmicutes (Granulicatella) and Actinobacteria (Corynebacterium) phyla (Figures 2B and S4B); those depleted included members of the phylum Bacteroidetes (Porphyromonas and Prevotella).
Figure 2.
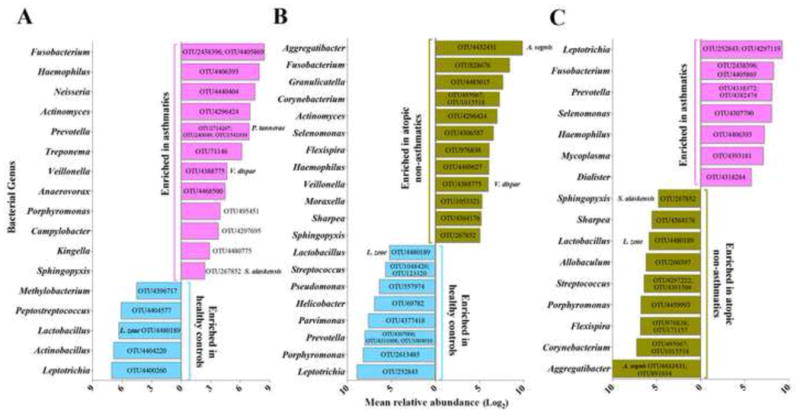
Bacterial taxa, significantly enriched or depleted in relative abundance (at least 2 fold; NB regression, q<0.1) in (A) AA (n=28) compared to HC (n=13); (B) ANA (n=15) compared to HC; (C) AA compared to ANA. The OTUs indicated represent the most abundant representatives within the indicated genus.
We noted that the ANA shared 26% and 29% of taxa that were also relatively enriched or depleted in the AA, when compared to HC. Despite these similarities, OTU-level analysis identified 103 taxa that differed significantly between the AA and ANA groups (Table S6; Figures 2C and S4C). To pinpoint bacteria discretely associated with asthma, we evaluated which taxa among those that distinguished AA from HC, also distinguished AA from ANA (Figure 3). By this approach, asthma-associated taxa included members of the Haemophilus (OTU4406393), Fusobacterium (OTU4405869; OTU2438396), Neisseria (OTU1304), Porphyromonas (OTU495451) and Sphingomonodaceae (OTU8331815); while taxa negatively associated with asthma included members of the Mogibacteriaceae (OTU4335578) and Lactobacillales (OTU4480189; OTU4469032). The same approach also identified taxa specific to atopy- alone, which included enrichment in Aggregatibacter (OTU4432431), Corynebacterium (OTU1015518; OTU495067) and Prevotella (OTU4372058; OTU134265) (Figure 3). Of the taxa identified as specifically associated with either asthma or atopy-alone, a subset exhibited strong, significant correlations with features of atopy (IgE, blood and sputum eosinophil counts); these associations were distinct in the two atopic groups. For example, specific taxa uniquely enriched in AA, belonging to Sphingomonodaceae and Fusobacteria, were positively associated with all three markers of atopy, while of those specifically enriched in ANA, Sharpea and Prevotella (OTU1052181; OTU134265) correlated with IgE and blood eosinophil counts, respectively (Figure 3). These data indicate that while taxonomic overlap exists between AA and ANA subjects, discrete bacterial enrichments characterize these groups, a subset of which are associated with biomarkers of atopic disease.
Figure 3.

Mean difference in specific bacterial taxa between groups. Asthma-specific taxa are similarly abundant in AA compared to HC and ANA (NB regression, q<0.1). Atopy-only taxa are similarly abundant in ANA vs. both HC and AA. Taxa positively correlated (rperson≥0.5, q<0.1) with blood eosinophil counts (BEO), serum IgE (IgE) or sputum eosinophil counts (SEO) in AA (*) or ANA (#) subjects.
We further explored whether predicted functions of the bronchial bacterial microbiome differed among the three groups using PICRUSt (27), an algorithm that predicts bacterial metagenomes in silico from 16S rRNA sequence. This analysis revealed significant differences across groups (Figure 4). Predicted bacterial gene functions enriched in AA included those involved in metabolism of amino acids and carbohydrates, especially of short chain fatty acids (SCFAs) such as butanoate and propanoate (more commonly known as butyrate and propionate, respectively). Also noteworthy was a relative depletion in predicted bacterial functions involved in lipopolysaccharide biosynthesis among asthma-associated bacterial communities.
Figure 4.
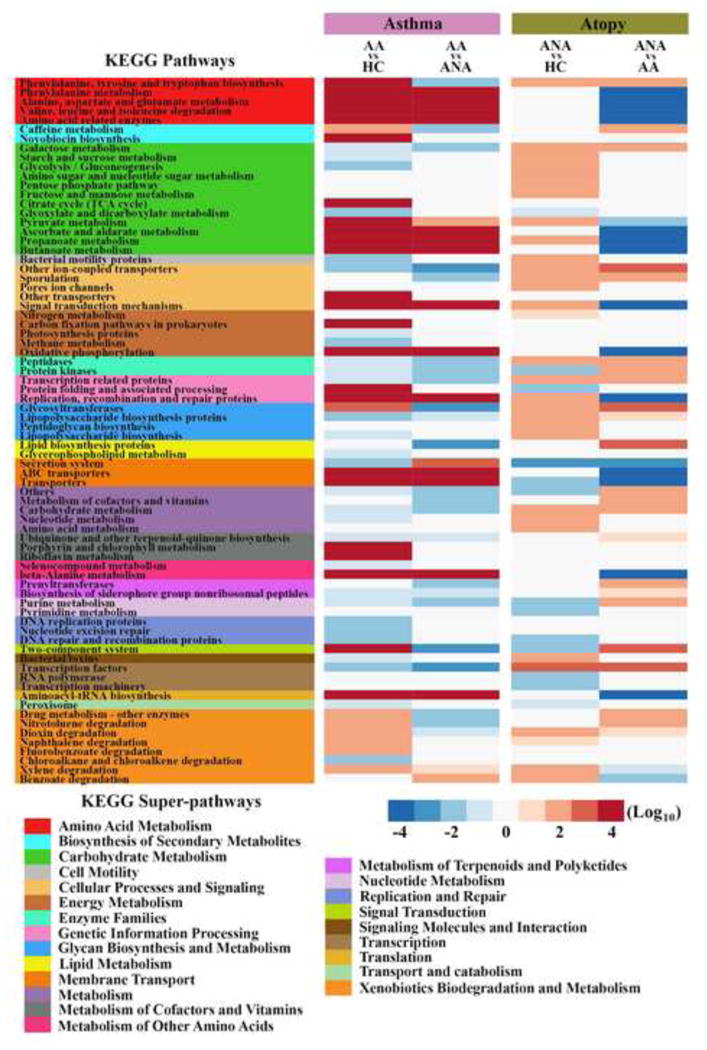
Mean difference in predicted KEGG orthologs (KOs) between groups. Asthma- specific pathways are similarly abundant in AA compared to HC and ANA. Atopy-only pathways are similarly abundant in ANA compared to HC and AA. Statistical significance was determined using NB regression model corrected for false discovery rate (q<0.1).
Type 2-high asthma is associated with reduced bronchial bacterial burden
TGM scores were higher in AA (Figure 5A) and, consistent with prior reports (20, 29), correlated positively with serum IgE (rspearman=0.36, p<0.01), blood and sputum eosinophil counts (rS=0.32 and rS=0.37, respectively, p<0.01), and negatively with FEV-i (rS= -0.45, p<0.005) and PC20 (rS= -0.41, p<0.005). Ten of 40 AA had T2-high asthma and exhibited significantly higher ACQ scores, serum IgE, blood and sputum eosinophil counts, and younger age, than T2-low asthmatics (Table S7). T2-high asthmatics demonstrated significantly lower bronchial bacterial burden than T2-low asthmatics (Figure 5C); bacterial burden also was negatively correlated with TGM scores across all asthmatic subjects (rS= -0.43, p<0.01) independent of age (GLM p=0.03). Because of their low bacterial burden BBs from only four T2-high asthmatics, could be sequenced for profiling, an insufficient sample size to allow meaningful assessment of differences in bacterial microbiota composition between T2-high and T2-low subjects.
Figure 5.
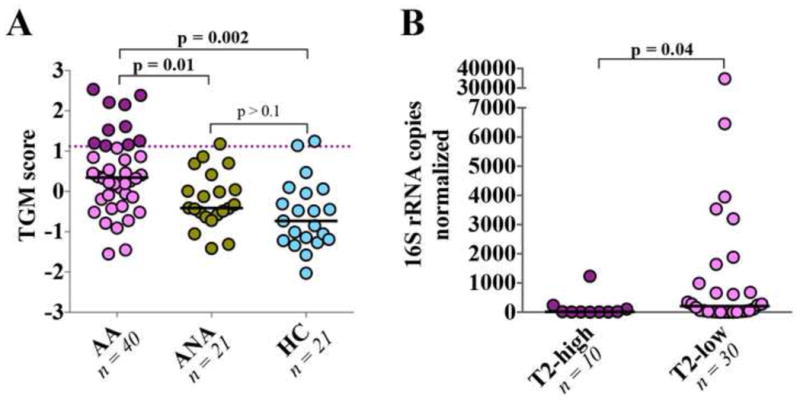
(A) AA displayed greater expression of epithelial genes induced by type 2 cytokines compared to non-asthmatic subjects. T2-high AA, with a TGM >1.117 (cut-off value indicated by a dashed line) are colored in maroon red. (B) Significantly lower bacterial burden was observed among T2-high asthma subjects. Statistical significance was determined using Wilcoxon rank sum test.
Inhaled corticosteroid-responsiveness is associated with distinct features of the bronchial bacterial microbiota present before treatment
We hypothesized that differences in baseline airway microbiota characteristics may be associated with ICS-responsiveness, defined as ≥2-fold increase in PC20Mch after ICS- treatment. Of the asthmatics included, 15 were classified as ICS-responders, 10 as non-responders, and three were excluded as PC20 was not performed (Figure S5A-B). Compared to non-responders, ICS-responders did not differ in PC20, serum IgE, blood and sputum eosinophil counts, or bronchial burden at baseline but did have lower FEV-i and a trend toward a higher TGM score (p<0.08) (Table S8; Figure S5C), which decreased following treatment (p<0.05; Figure S4D).
The baseline composition of profiled bronchial bacterial microbiota in ICS-responders (n=10) differed from that in the non-responders (n=5; unweighted UniFrac PERMANOVA R2=0.13, p=0.01), and was significantly more similar to that of HC (Figure 6A). Bacterial families enriched at baseline in ICS non-responders included Microbacteriaceae, Pasteurellaceae (e.g. asthma-associated Haemophilus OTU4406393) and several others (Figure 6C and Table S9). Conversely, bacterial families enriched at baseline in ICS-responders included Streptococcaceae, Fusobacteriaceae and Sphingomonodaceae. Compared with ICS-responders, the predicted functions of bacterial communities of non-responders were enriched in xenobiotic biodegradation pathways (Figure 6D and Table S10), implicating potential enhanced capacity for synthetic chemical degradation.
Figure 6.
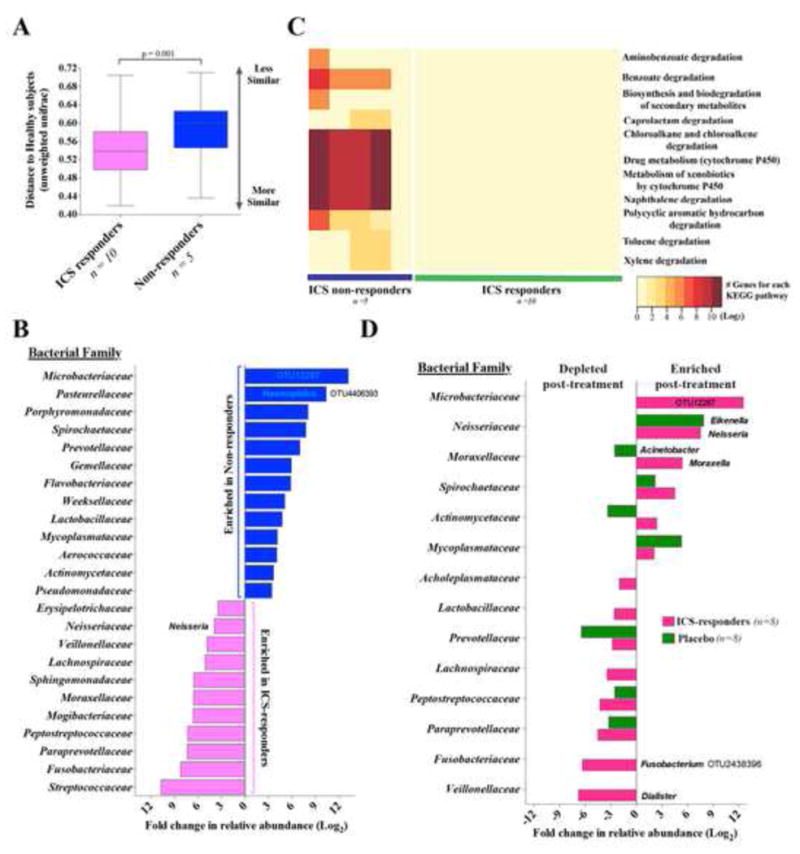
(A) Mean distance to HC (unweighted UniFrac; Bonferroni-corrected t-test). (B) Taxa significantly enriched or depleted in relative abundance (at least 2 fold; NB regression, q<0.1) in ICS-responders vs. non-responders. (C) Predicted KEGG pathways associated with xenobiotic biodegradation and metabolism in ICS-responders and non-responders. (D) Taxa, differentially expressed (at least 2-fold; ZINB regression, q<0.1) in asthmatics following ICS or placebo treatment.
Following six weeks of ICS vs. placebo treatment, we found no significant changes in bacterial burden or phylogenetic diversity in the AA group (Figure S6A-B). Bacterial community analysis in paired samples before and after treatment was limited, as several subjects did not have sufficient 16S rRNA to obtain sequence data at both time points. Nonetheless, we reasoned that ICS exposure might have distinct effects on the microbiotas of non-responders and ICS-responders (2) and explored ICS-induced compositional changes in the latter, more predominant group. No significant differences were seen in the magnitude of changes (beta-diversity assessed by unweighted Unifrac distance) between paired samples (n=8 each in placebo or ICS-treated responders; Figure S6C-E). At the taxon level, however, ICS treatment resulted in increased relative abundance of Microbacteriaceae, Neisseria and Moraxella and depletion of a specific Fusobacterium, which was not observed with the placebo treatment (Figure 6D, Tables S11-12). We unfortunately could not analyze compositional changes in ICS-non- responders, as only 2 subjects in this subgroup had sufficient 16S rRNA in both pre- and post- treatment samples.
Discussion
Our findings show compositional and predicted functional differences in the bronchial bacterial microbiomes of atopic asthmatic, atopic non-asthmatic, and healthy individuals. An important implication of these findings is that control for allergic sensitization is necessary in studies aimed at understanding differences in the respiratory microbiome associated with asthma. Despite overlap in bacterial genera significantly associated with both atopic groups, our study identified specific bacterial taxa whose relative enrichment or depletion were discretely associated with asthma. Analyses based on metagenomic inference further suggest that genes for pathways involved in the metabolism of short- chain fatty acids and amino acids are enriched in the asthmatic bronchial microbiome. We additionally observed that the bronchial microbiome among asthmatics at baseline differed compositionally and functionally according to their responsiveness to treatment with inhaled corticosteroids.
Commonly reported microbial community metrics (e.g., richness, evenness, and burden) did not differ significantly among our three groups. This is not unexpected due to the broad characterization of microbial composition provided by such measures, the inter-individual heterogeneity of microbiota composition found in all human niches, including the lung (30), and the mild disease severity in our subjects. Considering the clinical homogeneity of our asthmatic group, their heterogeneity in bronchial bacterial composition is striking. Moreover, this heterogeneity was predominantly observed among T2-low asthma subjects, in whom bronchial bacterial burden was significantly greater than in T2-high subjects. The trend towards higher bacterial phylogenetic diversity in our mild asthmatic subjects suggests their lower airways are receptive to colonization by a wider variety of bacteria. Although we could not pinpoint any specific clinical features associated with this finding, additional contributing factors might include differences in previous environmental exposures, in clearance of microorganisms, or in other immune function parameters not identified here.
Reasons for the low bronchial bacterial burden in T2-high asthma are unclear, but the finding echoes a recent observation in severe asthma, of an inverse relationship between bronchial bacterial burden and numbers of bronchial biopsy eosinophils (4). A possible explanation is the bactericidal activity of more numerous airway eosinophils (31, 32) in T2-high airway inflammation. Given prior reports of inverse relationships between bacterial and fungal richness (33, 34), the low bacterial burden in T2-high asthma could also reflect increased fungal or bacteriophage burden. Indeed microbial interactions are not limited to bacteria, as bacterial-viral interactions have been associated with asthma exacerbations and risk for asthma development (35, 36). Such inter-kingdom interactions (37) should be considered in future studies.
This study expands the list of bacterial groups previously associated with asthma, further supporting the idea that alterations in microbial composition from a healthy state, is a characteristic of asthma. Enrichment in certain Proteobacteria members (e.g. Haemophilus and Neisseria) among our ICS-naïve asthmatics resembles prior findings in ICS-using patients (2-6, 28). However, we found other taxa discretely associated with asthma including Fusobacterium and Porphyromonas, two oral-associated anaerobes capable of augmenting pathogenic behavior of opportunistic respiratory pathogens, such as Pseudomonas aeruginosa (38). Additionally, both Fusobacterium (39) and Haemophilus (40) induce MUC5AC expression in bronchial epithelial cells. Sphingomonodaceae represented another asthma-specific taxon whose abundance, like that of Fusobacterium, correlated with sputum eosinophilia. This finding echoed the reported activity of glycosphingolipids (present in the cell membrane of Sphingomonodaceae) in activating NKT-cells to produce T2-cytokines (41-43) and the reported association of this bacterial family with bronchial reactivity (5).
Asthma-related alterations in bronchial microbiota composition also involve relative depletion in certain taxa, including members of the Lactobacillales. Numerous studies have demonstrated protective effects of certain Lactobacillus strains against atopy by various mechanisms including alteration of gastrointestinal (44) and respiratory tract permeability (45, 46). Our finding that the predicted functions of asthma-associated microbiota were relatively depleted in machinery for LPS biosynthesis suggests another possibility, for continuous stimulation of airways with LPS has been shown to suppress T2 immune activation (47). We propose, though, that any “pro-asthmatic” activity of a bronchial microbiome is likely dependent on functional effects consequent to interactions among many microbiota members in the airway microenvironment, rather than the activity of any one species. Microbiome-related functions indicated by our analyses as potentially enhanced in asthma include increased capacity for metabolism of butyrate and propionate, SCFAs that maintain epithelial barrier function and immune tolerance in the gut (48-50). We speculate that utilization of anti-inflammatory SCFAs by members of the asthmatic airway microbiota may contribute to atopic asthma by reducing their bioavailability and the consequent capacity to down-regulate host inflammatory responses to aeroallergens and pathogens.
Microbiome-related functions might also affect responsiveness to corticosteroid treatment. We found that pre-treatment enrichment in an asthma-associated Haemophilus, a genus with species previously shown to reduce response of BAL macrophages to corticosteroids (2), was associated with diminished response to six weeks of treatment with inhaled fluticasone. Analysis of the predicted metagenome of pre-treatment bronchial microbiota present in ICS-non-responders also indicated enhancement of xenobiotic degradation capacity, which we hypothesize may contribute to their diminished response. In contrast, ICS-induced changes to the bronchial microbiota in ICS-responsive asthmatics showed enrichment of previously reported asthma-associated taxa such as Neisseria and Moraxella (2, 3, 6). Community shift detected in response to lactose-containing placebo inhaler was not surprising, as an influx of an additional sugar would be expected to alter the composition of microbial communities in the airways, favoring those species with the metabolic capacity to utilize such carbon sources. As importantly, these results emphasize the need to consider the effects of repeated inhalation of particles, especially of an ICS, as a selective pressure and nutritional source for airway microbiome members with the catalytic capacity to degrade such xenobiotics.
Perturbations to the bronchial microbiota associated with atopy-alone is also a novel finding of this study. ANA subjects were less sensitized than the AA subjects and showed primarily low T2 inflammation of bronchial epithelium. While the greater severity of allergy in the AA group cannot be ruled out as contributing to the difference in bacterial signature observed between AA and ANA subjects, distinct taxa associations with different markers of atopy and allergic inflammation were observed, suggesting that distinct microbial interactions with the host immune system occur in these two patient groups. These observations in ANA subjects support an association between airway colonization and atopy-related altered mucosal immune functions (9, 10). Atopy-specific taxa included members of the Pasteurellaceae (specifically Aggregatibacter and Haemophilus). Members of Aggregatibacter are associated with periodontal disease driven by high pro-inflammatory cytokines (e.g., TNF-a, IL1B, IL-6 and IL-8;) and reduced levels of IL-10 (51, 52). It is tempting to speculate that airway enrichment of this genus in ANA subjects could contribute to asthma protection through activation of the T1 arm of the immune system. We also highlight that specific taxa belonging to certain genera (e.g., Prevotella and Haemophilus) were associated with atopy-only or asthma. This underscores the likely importance of species- or strain-level functional differences in microbial interactions related to disease status.
The limitations of our study include exclusive analysis of bacterial communities, the absence of non-atopic asthmatic subjects and the relatively narrow breadth of asthma phenotype captured in this cohort. Moreover, our sample size was small in some comparisons, particularly in assessing changes in the bronchial microbiota in paired samples before and after ICS vs. placebo treatment. However, strengths include the large number of subjects who were characterized and underwent invasive bronchoscopy to collect samples for analysis. Our study protocol also attended carefully to procedural and sampling methods to reduce contamination and analyze for possible contribution of non-bronchial sources of bacterial DNA to the dataset.
Conclusion
Our findings highlight the complexity of bacterial relationships to asthma in a background of atopy, conditions that are associated with distinct alterations in the airway microbiota. To achieve a comprehensive understanding of microbial factors involved in the induction of, management of, or protection against asthma, there is an important need to better understand functions collectively expressed by consortia of airway microbes, which could have a profound influence on asthma.
Supplementary Material
Key messages.
The bronchial bacterial microbiota of both mild atopic-asthma (steroid-naïve) and atopy-alone differ from that of healthy controls and also differ from each other.
Asthma is associated with enrichment in members of the Haemophilus, Neisseria, Fusobacterium, Porphyromonas and Sphingomonodaceae and with depletion of Lactobacillus.
The T2-high asthma phenotype is associated with low bronchial bacterial burden.
ICS-response is linked to a distinct bacterial community composition and functional profile prior to steroid exposure.
Acknowledgments
We additionally would like to thank the following coordinators and staff, and to identify the grants supporting the study: Robert Pedicini, BS1; Kathy Zheng, MPH1; Duanny Alva, MPH2; Assel Biyasheva, PhD2; Jenny Hixon, BS, CCRC2; Lucius Robinson III, BS, CMA, CCRC2; Mary Gill, RN, BSN3; James T. Good, MD3; Christena Kolakowski, MS3; Allen Stevens, CCRC, NREMT3; E. Rand Sutherland, MD3; Julia Bach, RN4; Rich Cornwell, MD4; Holly Eversoll, RN4; Tiffany Huard4; Keith Meyer, MD4; Barbara Miller, RN4; Ann Sexton, MPH4; Michele Wolff, RN4; Merritt Fajt, MD5; Sherri Hill, BS5; Lisa Lane, BS5; Russell Traister, MD5; Cathy Vitari, RN, BSN AE-C5; Vanessa Curtis, RRT6; Brenda Patterson, RN, APN6; Cheryl Shelton, RN, BSN6; Kelly Norsworthy, BA, CPT7; Kelsey Wollen, BA7; Eugene Bleeker, MD8; Christopher Barrios, MD8; Suzan Farris, CCRP8; Jeffrey Krings, FNP8; Victor Ortega, MD8; Cheryl Wilmoth, CCRP8; Matthew Bowman, BS9; Linda Engle, BS9; Jennifer Lucier, BS9; Aimee J. Merchlinski, MS9; Kathryn Trasatt, BS9; Angela Updegrave9; Rachel Weber, BS9; Ronald R. Zimmerman, Jr., MPA9. We also wish to thank Elizabeth F. Juniper of McMaster's University, Canada, for permitting our use of her Asthma Control Questionnaire, and Andrew Manies of UCSF for assistance with manuscript preparation.
Abbreviations
- AA
Atopic asthmatic subjects
- ANA
Atopic non-asthmatic subjects
- HC
Healthy controls
- ICS
Inhaled corticosteroid
- ACQ
Asthma Control Questionnaire
- OW
Oral wash
- BB
Bronchial brush
- OUT
Operational taxonomic unit
- SCFAs
Short chain fatty acids
- TGM
Three-gene mean
- PICRUSt
Phylogenetic Reconstruction of Unobserved States
- KEGG
Kyoto Encyclopedia of Genes and Genomes
References
- 1.Denner DR, Sangwan N, Becker JB, Hogarth DK, Oldham J, Castillo J, Sperling AI, Solway J, Naureckas ET, Gilbert JA, White SR. Corticosteroid therapy and airflow obstruction influence the bronchial microbiome, which is distinct from that of bronchoalveolar lavage in asthmatic airways. Journal of Allergy and Clinical Immunology. 2015 doi: 10.1016/j.jaci.2015.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Goleva E, Jackson LP, Kirk Harris J, Robertson CE, Sutherland ER, Hall CF, Good JTJ, Gelfand EW, Martin RJ, Leung D, Y M. The Effects of Airway Microbiome on Corticosteroid Responsiveness in Asthma. American Journal of Respiratory and Critical Care Medicine. 2013;188:1193–1201. doi: 10.1164/rccm.201304-0775OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hilty M, Burke C, Pedro H, Cardenas P, Bush A, Bossley C, Davies J, Ervine A, Poulter L, Pachter L, Moffatt MF, Cookson WOC. Disordered Microbial Communities in Asthmatic Airways. PLOSone. 2010;5:e8578. doi: 10.1371/journal.pone.0008578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Huang YJ, Nariya S, Harris JM, Lynch SV, Choy DF, Arron JR, Boushey HA. The airway microbiome in patients with severe asthma: Associations with disease features and severity. The Journal of allergy and clinical immunology. 2015;136:874–884. doi: 10.1016/j.jaci.2015.05.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Huang YJ, Nelson CE, Brodie EL, Desantis TZ, Baek MS, Liu J, Woyke T, Allgaier M, Bristow J, Wiener-Kronish JP, Sutherland ER, King TS, Icitovic N, Martin RJ, Calhoun WJ, Castro M, Denlinger LC, Dimango E, Kraft M, Peters SP, Wasserman SI, Wechsler ME, Boushey HA, Lynch SV. Airway microbiota and bronchial hyperresponsiveness in patients with suboptimally controlled asthma. The Journal of allergy and clinical immunology. 2011;127:372–381. doi: 10.1016/j.jaci.2010.10.048. e371-373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Marri PR, Stern DA, Wright AL, Billheimer D, Martinez FD. Asthma-associated differences in microbial composition of induced sputum. The Journal of allergy and clinical immunology. 2013;131:346–352. doi: 10.1016/j.jaci.2012.11.013. e341-343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Burrows B, Martinez FD, Halonen M, Barbee RA, Cline MG. Associations of asthma with serum IgE levels and skin-test reactivity to allergens. N Engl J Med. 1989;320:271–277. doi: 10.1056/NEJM198902023200502. [DOI] [PubMed] [Google Scholar]
- 8.Pearce N, Pekkanen J, Beasley R. How much asthma is really attributable to atopy? Thorax. 1999;54:268–272. doi: 10.1136/thx.54.3.268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kariyawasam HH, Rotiroti G. Allergic rhinitis, chronic rhinosinusitis and asthma: unravelling a complex relationship. Current Opinion in Otolaryngology & Head and Neck Surgery. 2013;21:79–86. doi: 10.1097/MOO.0b013e32835ac640. [DOI] [PubMed] [Google Scholar]
- 10.Tomazic PV, Birner-Gruenberger R, Leitnera A, Obrist B, Spoerkc S, Lang-Loidolt D. Nasal mucus proteomic changes reflect altered immune responses and epithelial permeability in patients with allergic rhinitis. The Journal of allergy and clinical immunology. 2014;133:741–750. doi: 10.1016/j.jaci.2013.09.040. [DOI] [PubMed] [Google Scholar]
- 11.Juniper EF, O'Byrne PM, Guyatt GH, Ferrier PJ, King DR. Development and validation of a questionnaire to measure asthma control. Eur Respir J. 1999;14:902–907. doi: 10.1034/j.1399-3003.1999.14d29.x. [DOI] [PubMed] [Google Scholar]
- 12.Iwai S, Fei M, Huang D, Fong S, Subramanian A, Grieco K, Lynch SV, Huang L. Oral and Airway Microbiota in HIV-Infected Pneumonia Patients. J Clin Microbiol. 2012;50:2995–3002. doi: 10.1128/JCM.00278-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Huntley J, Fierer N, Owens SM, Betley J, Fraser L, Bauer M, Gormley N, Gilbert JA, Smith G, Knight R. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. The ISME Journal. 2012;6:1621–1624. doi: 10.1038/ismej.2012.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Lozupone CA, Turnbaugh PJ, Fierer N, Knight R. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(Suppl 1):4516–4522. doi: 10.1073/pnas.1000080107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.DeSantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, Keller K, Huber T, Dalevi D, Hu P, Andersen GL. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Applied and environmental microbiology. 2006;72:5069–5072. doi: 10.1128/AEM.03006-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Price MN, Dehal PS, Arkin AP. FastTree: Computing Large Minimum Evolution Trees with Profiles instead of a Distance Matrix. Molecular Biology and Evolution. 2009;26:1641–1650. doi: 10.1093/molbev/msp077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Faith DP. Conservation evaluation and phylogenetic diversity. Biological Conservation. 1992;61:1–10. [Google Scholar]
- 18.Woodruff PG, Boushey HA, Dolganov GM, Barker CS, Yang YH, Donnelly S, Ellwanger A, Sidhu SS, Dao-Pick TP, Pantoja C, Erle DJ, Yamamoto KR, Fahy JV. Genome-wide profiling identifies epithelial cell genes associated with asthma and with treatment response to corticosteroids. Proc Natl Acad Sci USA. 2007;104:15858–15868. doi: 10.1073/pnas.0707413104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Woodruff PG, Modrek B, Choy DF, Jia J, Abbas AR, Ellwanger A, Koth LL, Arron JR, Fahy JV. T-helper type 2-driven inflammation defines major subphenotypes of asthma. Am J Respir Crit Care Med. 2009;180:388–395. doi: 10.1164/rccm.200903-0392OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bhakta NR, Solberg OD, Nguyen CP, Nguyen CN, Arron JR, Fahy JV, Woodruff PG. A qPCR-based metric of Th2 airway inflammation in asthma. Clin Transl Allergy. 2013;3:24. doi: 10.1186/2045-7022-3-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Anderson MJ. A new method for non-parametric multivariate analysis of variance. Austral Ecology. 2001;26:32–46. [Google Scholar]
- 22.Pinheiro JC. Encyclopedia of Biostatistics. John Wiley & Sons, Ltd; 2005. Linear Mixed Effects Models for Longitudinal Data. [Google Scholar]
- 23.Gerber GK, Onderdonk AB, Bry L. Inferring dynamic signatures of microbes in complex host ecosystems. PLoS computational biology. 2012;8:e1002624. doi: 10.1371/journal.pcbi.1002624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McMurdie PJ, Holmes S. Waste Not, Want Not: Why Rarefying Microbiome Data Is Inadmissible. PLoS computational biology. 2014;10:e1003531. doi: 10.1371/journal.pcbi.1003531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kassahun W, Neyens T, Molenberghs G, Faes C, Verbeke G. Marginalized multilevel hurdle and zero-inflated models for overdispersed and correlated count data with excess zeros. Statistics in medicine. 2014;33:4402–4419. doi: 10.1002/sim.6237. [DOI] [PubMed] [Google Scholar]
- 26.Min Y, Agresti A. Random effect models for repeated measures of zero-inflated count data. Statistical Modelling. 2005;5:1–19. [Google Scholar]
- 27.Langille MGI, Zaneveld J, Caporaso JG, McDonald D, Knights D, Reyes JA, Clemente JC, Burkepile DE, Vega Thurber RL, Knight R, Beiko RG, Huttenhower C. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat Biotech. 2013;31:814–821. doi: 10.1038/nbt.2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Simpson JL, Daly J, Baines KJ, Yang IA, Upham JW, Reynolds PN, Hodge S, James AL, Hugenholtz P, Willner D, Gibson PG. Airway dysbiosis: Haemophilus influenzae and Tropheryma in poorly controlled asthma. The European respiratory journal. 2015 doi: 10.1183/13993003.00405-2015. [DOI] [PubMed] [Google Scholar]
- 29.Peters MC, Mekonnen ZK, Yuan S, Bhakta NR, Woodruff PG, Fahy JV. Measures of gene expression in sputum cells can identify TH2-high and TH2-low subtypes of asthma. The Journal of allergy and clinical immunology. 2014;133:388–394. doi: 10.1016/j.jaci.2013.07.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ursell LK, Clemente JC, Rideoutb JR, Gevers D, Caporaso JG, Knight R. The interpersonal and intrapersonal diversity of human-associated microbiota in key body sites. J Allergy Clin Immunol. 2012;129:1204–1208. doi: 10.1016/j.jaci.2012.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hogan SP, Waddell A, Fulkerson PC. Eosinophils in Infection and Intestinal Immunity. Current Opinion in Gastroenterology. 2013;29:7–14. doi: 10.1097/MOG.0b013e32835ab29a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Travers J, Rothenberg ME. Eosinophils in mucosal immune responses. Mucosal Immunol. 2015;8:464–475. doi: 10.1038/mi.2015.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dollive S, Chen Y-Y, Grunberg S, Bittinger K, Hoffmann C, Vandivier L, Cuff C, Lewis JD, Wu GD, Bushman FD. Fungi of the Murine Gut: Episodic Variation and Proliferation during Antibiotic Treatment. PLoS ONE. 2013;8:e71806. doi: 10.1371/journal.pone.0071806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Peleg AY, Hogan DA, Mylonakis E. Medically important bacterial-fungal interactions. Nat Rev Micro. 2010;8:340–349. doi: 10.1038/nrmicro2313. [DOI] [PubMed] [Google Scholar]
- 35.Kloepfer KM, Lee WM, Pappas TE, Kang TJ, Vrtis RF, Evans MD, Gangnon RE, Bochkov YA, Jackson DJ, Lemanske RF, Jr, Gern JE. Detection of pathogenic bacteria during rhinovirus infection is associated with increased respiratory symptoms and asthma exacerbations. The Journal of allergy and clinical immunology. 2014;133:1301–1307. doi: 10.1016/j.jaci.2014.02.030. 1307.e1301-1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Teo Shu M, Mok D, Pham K, Kusel M, Serralha M, Troy N, Holt Barbara J, Hales Belinda J, Walker Michael L, Hollams E, Bochkov Yury A, Grindle K, Johnston Sebastian L, Gern James E, Sly Peter D, Holt Patrick G, Holt Kathryn E, Inouye M. The Infant Nasopharyngeal Microbiome Impacts Severity of Lower Respiratory Infection and Risk of Asthma Development. Cell Host & Microbe. 17:704–715. doi: 10.1016/j.chom.2015.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tarkka MT, Sarniguet A, Frey-Klett P. Inter-kingdom encounters: recent advances in molecular bacterium-fungus interactions. Current genetics. 2009;55:233–243. doi: 10.1007/s00294-009-0241-2. [DOI] [PubMed] [Google Scholar]
- 38.Pana Y, Tenga D, Burkea AC, Haasea EM, Scannapieco FA. Oral bacteria modulate invasion and induction of apoptosis in HEp-2 cells by Pseudomonas aeruginosa. Microbial Pathogenesis. 2008;46:73–79. doi: 10.1016/j.micpath.2008.10.012. [DOI] [PubMed] [Google Scholar]
- 39.Nagaoka K, Yanagihara K, Harada Y, Yamada K, Migiyama Y, Morinaga Y, Hasegawa H, Izumikawa K, Kakeya H, Nishimura M, Kohno S. Macrolides inhibit Fusobacterium nucleatum-induced MUC5AC production in human airway epithelial cells. Antimicrobial agents and chemotherapy. 2013;57:1844–1849. doi: 10.1128/AAC.02466-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Araki N, Yanagihara K, Morinaga Y, Yamada K, Nakamura S, Yamada Y, Kohno S, Kamihira S. Azithromycin inhibits nontypeable Haemophilus influenzae-induced MUC5AC expression and secretion via inhibition of activator protein-1 in human airway epithelial cells. European journal of pharmacology. 2010;644:209–214. doi: 10.1016/j.ejphar.2010.06.056. [DOI] [PubMed] [Google Scholar]
- 41.Akbari O, Stock P, Meyer E, Kronenberg M, Sidobre S, Nakayama T, Taniguchi M, Grusby MJ, DeKruyff RH, Umetsu DT. Essential role of NKT cells producing IL-4 and IL-13 in the development of allergen-induced airway hyperreactivity. Nat Med. 2003;9:582–588. doi: 10.1038/nm851. [DOI] [PubMed] [Google Scholar]
- 42.Kinjo Y, Wu D, Kim G, Xing GW, Poles MA, Ho DD, Tsuji M, Kawahara K, Wong CH, Kronenberg M. Recognition of bacterial glycosphingolipids by natural killer T cells. Nature. 2005;434:520–525. doi: 10.1038/nature03407. [DOI] [PubMed] [Google Scholar]
- 43.Zajonc DM, Girardi E. Recognition of Microbial Glycolipids by Natural Killer T Cells. Frontiers in immunology. 2015;6:400. doi: 10.3389/fimmu.2015.00400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Özdemir O. Various effects of different probiotic strains in allergic disorders: an update from laboratory and clinical data. Clin Exp Immunol. 2010;160:295–304. doi: 10.1111/j.1365-2249.2010.04109.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Abreu NA, Nagalingam NA, Song Y, Roediger FC, Pletcher SD, Goldberg AN, Lynch SV. Sinus microbiome diversity depletion and Corynebacterium tuberculostearicum enrichment mediates rhinosinusitis. Sci Transl Med. 2012;12 doi: 10.1126/scitranslmed.3003783. 151ra124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fujimura KE, Demoor T, Rauch M, Faruqi AA, Jang S, Johnson CC, Boushey HA, Zoratti E, Ownby D, Lukacs NW, Lync SV. House dust exposure mediates gut microbiome Lactobacillus enrichment and airway immune defense against allergens and virus infection. Proc Natl Acad Sci USA. 2014;111:805–810. doi: 10.1073/pnas.1310750111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schuijs MJ, Willart MA, Vergote K, Gras D, Deswarte K, Ege MJ, Branco Madeira F, Beyaert R, van Loo G, Bracher F, von Mutius E, Chanez P, Lambrecht BN, Hammad H. Farm dust and endotoxin protect against allergy through A20 induction in lung epithelial cells. Science. 2015;349:1106–1110. doi: 10.1126/science.aac6623. [DOI] [PubMed] [Google Scholar]
- 48.Arrieta MC, Stiemsma LT, Dimitriu PA, Thorson L, Russell S, Yurist-Doutsch S, Kuzeljevic B, Gold MJ, Britton HM, Lefebvre DL, Subbarao P, Mandhane P, Becker A, McNagny KM, Sears MR, Kollmann T, Mohn WW, Turvey SE, Brett Finlay B. Early infancy microbial and metabolic alterations affect risk of childhood asthma. Science translational medicine. 2015;7 doi: 10.1126/scitranslmed.aab2271. 307ra152. [DOI] [PubMed] [Google Scholar]
- 49.Kelly CJ, Zheng L, Campbell EL, Saeedi B, Scholz CC, Bayless AJ, Wilson KE, Glover LE, Kominsky DJ, Magnuson A, Weir TL, Ehrentraut SF, Pickel C, Kuhn KA, Lanis JM, Nguyen V, Taylor CT, Colgan SP. Crosstalk between Microbiota- Derived Short-Chain Fatty Acids and Intestinal Epithelial HIF Augments Tissue Barrier Function. Cell Host Microbe. 2015;17:662–671. doi: 10.1016/j.chom.2015.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Trompette A, Gollwitzer ES, Yadava K, Sichelstiel AK, Sprenger N, Ngom-Bru C, Blanchard C, Junt T, Nicod LP, Harris NL, Marsland BJ. Gut microbiota metabolism of dietary fiber influences allergic airway disease and hematopoiesis. Nat Med. 2014;20:159–166. doi: 10.1038/nm.3444. [DOI] [PubMed] [Google Scholar]
- 51.Bostanci N, Akgül B, Tsakanika V, Allaker RP, Hughes FJ, McKay IJ. Effects of low-dose doxycycline on cytokine secretion in human monocytes stimulated with Aggregatibacter actinomycetemcomitans. Cytokine. 2011;56:656–661. doi: 10.1016/j.cyto.2011.08.039. [DOI] [PubMed] [Google Scholar]
- 52.Hirose M, Ishihara K, Saito A, Nakagawa T, Yamada S, Okuda K. Expression of cytokines and inducible nitric oxide synthase in inflamed gingival tissue. J Periodontol. 2001;72:590–597. doi: 10.1902/jop.2001.72.5.590. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.