

Abstract
By applying a multitude of experimental techniques including 1H, 14N, 207Pb NMR and 127I NMR/NQR, tracer diffusion, reaction cell and doping experiments, as well as stoichiometric variation, conductivity, and polarization experiments, iodine ions are unambiguously shown to be the mobile species in CH3NH3PbI3, with iodine vacancies shown to represent the mechanistic centers under equilibrium conditions. Pb2+ and CH3NH3 + ions do not significantly contribute to the long range transport (upper limits for their contributions are given), whereby the latter exhibit substantial local motion. The decisive electronic contribution to the mixed conductivity in the experimental window stems from electron holes. As holes can be associated with iodine orbitals, local variations of the iodine stoichiometry may be fast and enable light effects on ion transport.
Keywords: charge carriers, halide perovskite, ion migration, methylammonium lead iodide, perovskite solar cells
Rarely have solid compounds gained such a sudden attention as the methylammonium lead halide (X=Cl, Br, I) perovskites. Their application as light‐harvesters in dye‐sensitized solar cells has allowed for the fabrication of all‐solid‐state devices with very high cell efficiencies (>22 %).1, 2, 3, 4 However, stability of these chemically weak compounds has proven to be a tough obstacle to overcome,5 and considerable polarization phenomena affecting charge transport have been observed at long timescales/low frequencies in devices during operation.6, 7, 8 For all these phenomena, ion conduction is a key process, and while its influence on degradation kinetics9, 10 and electrode reactions11 is rather evident, its role in polarization processes deserves some comments. When a material exhibiting both electronic and significant ionic conductivity is charged by a current while placed between neighboring phases that block ion transfer, stoichiometric gradients throughout the bulk of the material occur, necessarily involving both ionic and electronic charge carriers.12, 13 This situation can be expected to take place in all perovskite solar cell devices due to the ion‐blocking nature of the neighboring phases.14 In addition to this phenomenon, which comprises the entire bulk, space charge polarization occurs at the interface between perovskite and neighboring phases but on a completely different length scale (for generalized treatments the reader is referred to literature15, 16). Mobile ionic point defects are of great significance for such space charge potentials at the contacts, be they present already in equilibrium or generated under load,17 and hence they are important for electronic carrier extraction and thus for device performance.18 Even though the halide ion has been known to be conductive in similar compounds,19, 20 mixed conduction in hybrid halide perovskites (specifically in CH3NH3PbI3, MAPbI3 in short) has been measured directly only recently.14 In that study, predominant I− motion is reported, in agreement with recent modeling results21, 22 and experimental observations.23, 24, 25, 26, 27 In parallel, dominant ion conduction through methylammonium (MA) ions has been claimed in a number of other contributions.28, 29, 30, 31 Some reports even state the possibility of both ions being mobile.32, 33 Furthermore, as various lead compounds34, 35 show significant motion of the highly polarizable Pb2+ cation, this ion might also be a potential candidate for diffusion in MAPbI3. The ongoing controversy around this issue is reflected by recent publications.36, 37
There is an urgent need to clarify this point, not only regarding device performance and understanding but also because conditioning of these compounds through stoichiometry control or doping depends crucially on the nature and effective charge of the relevant ionic charge carriers. In this respect, understanding equilibrium conditions is a prerequisite to analyze the situation under illumination, therefore in this paper we refer to dark conditions. Interestingly, as already mentioned in Refs. 14, 38, ionic conductivity in these compounds seems heavily influenced by light, an issue that deserves more detailed studies.39 Herein, by applying electrochemical measurements (a.c. impedance, d.c. polarization, variation of stoichiometry, doping, faradaic reaction cells, emf measurements), tracer experiments, and NMR techniques (207Pb1/2, 1H1/2, 13C1/2, 14N1 NMR and 127I5/2 NQR) on MAPbI3, we give compelling evidence for iodine vacancies dominating the substantial ionic conductivity in this material. Our experiments also clearly establish electron holes as dominant electronic carriers in MAPbI3, if the iodine partial pressure is not extremely small. We focus on long‐range motion; a detailed discussion about short‐range dynamics (rotational/vibrational motion), as addressed in earlier studies,40, 41, 42, 43, 44 is outside the scope of this paper.
In addition to applying d.c. galvanostatic polarization and a.c impedance measurements (Supporting Information, Figure S1) to separate ionic and electronic conductivity in MAPbI3,14 we determine the ratio of the two contributions by measuring the emf of an iodine concentration cell, addressing the same thermodynamic conditions (Figure 1 a). The open circuit voltage is found to be close to the Nernst voltage indicating predominant ionic conductivity under the measurement conditions, in agreement with d.c. polarization studies.
Figure 1.
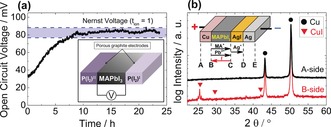
a) Equilibration of the emf voltage across a MAPbI3 pellet when the two faces are exposed to different iodine partial pressures (1.7×10−7 bar and 1.5×10−5 bar), measured at 403 K. Inset: Schematic of the experiment. According to polarization studies, under such conditions ionic conductivity should dominate (ionic transport number, t ion≈1), therefore the emf voltage is expected to approach the thermodynamic Nernst voltage of 77 mV (details in Supporting Information Section 7). Error from P(I2) control was estimated as ±10 mV. b) XRD patterns of surfaces A and B of Cu foil, taken after the Faradaic reaction cell experiment, showing formation of CuI on B side. Inset: Schematic of the reaction cell (+Cu|MAPbI3|AgI|Ag−). 25 nA cm−2 current was applied for one week under Ar atmosphere. Note that iodine is supplied to MAPbI3 from the AgI contact.
As a second step, the major mobile ionic species is identified by applying a current for a given time through a Faradaic reaction cell of type +Cu | MAPbI3 | AgI | Ag−. The only phase change observed is the formation of CuI at the Cu‐contact (B‐interface, Figure 1 b and the Supporting Information, Figure S2), which indicates only I− transport and excludes significant Pb2+ or MA+ conductivity (details in the Supporting Information, Section 5). Consistent conclusions were obtained by replacing Cu with Pb electrodes. Indeed, as shown in the Supporting Information (Section 6) and in our previous publication,14 a substantial formation of PbI2 is detectable for a Pb reaction cell at the expense of the Pb contact (B‐side). No changes are observable at the AgI contact (C‐side, Figure S4).
Interestingly, as shown in the Supporting Information (Section 2), ionic transport in MAPbI3 does not only persist under illumination, it appears to be enhanced (Figure S1 c,d). This peculiar feature was already briefly remarked on in Ref. 14 and confirmed by a recent publication.38 At the moment, however, we do not have conclusive evidence on the nature of the conductive species under illumination.
Having measured ionic conductivity and having shown that it is due to iodide ions, we demonstrate that this conductivity is mechanistically due to iodine vacancies and not to iodine interstitials, as claimed in Refs. 45, 46. A firm indication is provided by analyzing the iodine partial pressure [P(I2)] dependence of the ionic conductivity. Iodine partial pressure variation, analogously to P(O2) in oxides, leads to a change in stoichiometry and, as a consequence, of charge carrier concentrations (Supporting Information, Section 4). As shown in Figure 2 a, in MAPbI3 ionic conductivity clearly decreases as a function of P(I2), while electronic conductivity increases. This behavior identifies the mobile ionic defect as vacancies of iodine, the concentration of which is decreased due to incorporation of I2 from the gas phase. More detailed considerations show that also the slope is in the expected range (slightly below 0, details in the Supporting Information, Section 4). Note that an ionic conduction due to iodine interstitials (or MA vacancies) would increase with P(I2). Additionally, the increment of electronic conductivity shown in Figure 2 a is indicative of p‐type electronic conduction, with the stronger increase being also theoretically expected (slope slightly below 0.5, details in the Supporting Information, Section 4).
Figure 2.

a) Ionic (σion, red) and electronic (σeon, black) conductivity of a pure MAPbI3 pellet vs. P(I2), obtained from d.c. galvanostatic polarisation at 373 K. Closed (▪, ▪) and open (○, ▵) symbols represent points taken from low‐to‐high and high‐to‐low P(I2) respectively, to observe reversibility. b) Comparison between d.c. galvanostatic polarization curves of pure and Na‐doped (0.5 % at.) samples, when applying 150 nA cm−2 at 378 K, with P(I2)=2×10−7 bar. c) Schematic of the tracer diffusion experiment. d) ToF‐SIMS depth profile of a pure MAPbI3 pellet after diffusion annealing in contact with a pellet of 13CH3 15NH3PbI3 (20 % enriched) for 133 hours at 378 K. Thick lines are to guide the eye. The ratio of 13C/Ctot (in black) is almost constant through the entire depth (ca. 1.1 %, that is, natural abundance).
Doping experiments with sodium provide further independent and very clear evidence. Doping materials with aliovalent ions is a well‐established technique to tune their electrical (ionic and electronic) conductivities. According to the ionic radius, Na is expected to replace Pb, leading to an aliovalent substitutional doping and hence to an enhancement of the concentration of iodine vacancies and electron holes (details in the Supporting Information, Section 4). Na substitution on the Pb site of MAPbI3 was also recently referred to in Ref. 47. Note that substitution of MA by Na would not lead to aliovalent doping and thus not to significant conductivity variations. As shown in Figure 2 b, doping with 0.5 % at. Na (Supporting Information, Figure S5) results in a large and similarly great increase of both ionic and electronic conductivity (>1 order of magnitude) compared to pure MAPbI3. This behavior is in complete agreement with the proposed defect chemistry and specifically with an ionic conductivity due to iodine vacancies. Iodine interstitial or MA vacancy concentrations would be instead decreased by such an acceptor doping (details in the Supporting Information, Section 4). As the electron holes as electronic carriers can be attributed to the iodine orbitals, changes in iodine stoichiometry may be locally very fast, which may explain light effects not only on electronic but also on ionic transport (Supporting Information, Section 2).
To provide even more evidence for MA cations not being mobile, a tracer exchange experiment was performed, in which two samples of MAPbI3, one of which was 13C‐ and 15N‐enriched, were put in contact with each other (Figure 2 c). The secondary ion mass spectroscopy (SIMS) depth profile of a long‐time annealed sample in Figure 2 d shows absence of significant MA+ diffusion. Note that the surface roughness of the sample (Supporting Information, Figure S6) is well below the probed depth. If present at all, we may recognize a very weak diffusion profile in the first 2–3 μm, yielding an upper limit for the MA+ tracer diffusion coefficient of 9×10−14 cm2 s−1 at 378 K.
Additional information can be gained from the NMR and NQR data (typical 1H, 13C spectra are reported in Figure S7).
14N NMR spectra in Figure 3 a show, below phase transition, a clear quadrupolar splitting, that can exist only in the absence of isotropic motion of MA cations, that would otherwise result in a collapse of the signals. From this we conclude that, in the tetragonal phase, no long‐range MA diffusion can be present and that the rotational motion must take place without full reorientation of the molecule (tilted mode). Since above the phase transition only a single isotropic signal is expected,40 its evaluation does not allow such a conclusion. Instead, direct evidence for the absence of long‐range MA+ motion also in the cubic phase stems from the temperature dependence of the 1H NMR signal, displayed in Figure 3 c. Indeed, the signal shows an almost constant linewidth between 193 and 503 K, indicating that the interactions responsible for such width are nearly constant in this range. As explained in detail in the Supporting Information (Section 13), the linewidth of 1H signal in MAPbI3 is comprised of several contributions (for example, 1H–127I dipole–dipole interactions), that are differently averaged by the motion (rotation) of the MA cation. The presence of fast isotropic translational motion of the MA cation (for example, due to the sample melting or to the occurrence of fast solid‐state diffusion) would lead to complete averaging of these interactions, causing the linewidth of the 1H signal to decrease 1000‐fold to a liquid‐like signal. The measurements up to 503 K (Figure 3 c) show no evidence of such behavior, ruling out perceptible long‐range MA+ diffusion in the temperature range probed. From these considerations we can estimate an upper limit for the MA+ diffusion coefficient of 4×10−15 cm2 s−1 at 343 K (and 6×10−13 cm2 s−1 at 378 K), which is consistent with the upper limit obtained from tracer diffusion. We can state that the values are 2–3 orders of magnitude too low to account for the ion conductivity measured. By assuming Schottky disorder we can also extract an upper limit for the methylammonium vacancy diffusivity of 3×10−12 cm2 s−1 at 343 K. Note that the lower limit for the diffusivity of the dominant ionic defect at the same temperature is 2×10−9 cm2 s−1, derived from conductivity measurements of Na‐doped samples (Supporting Information, Section 10). A similarly low upper limit for the diffusion coefficient of Pb (4×10−15 cm2 s−1 at 343 K) is derived from 207Pb NMR spectra. The position of the 207Pb signal in MAPbI3 at room temperature is in good agreement with what expected for Pb2+ ions coordinated to iodine48 and shifted downfield with respect to pure PbI2 (Figure 4 a). The position of this peak prior to the measurements was estimated by DFT calculations (Supporting Information, Section 14). The signal position depends strongly on the temperature due to lattice expansion (Figure 4 b) but shows no sign of the phase transition at 327 K.
Figure 3.
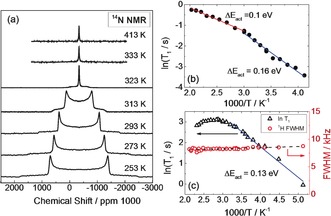
a) 14N NMR spectra as function of temperature across the phase transition. Below 327 K the 14N spectra demonstrate a characteristic quadrupolar splitting with the asymmetry parameter of η=0. Temperature dependences of b) 14N spin‐lattice relaxation time T 1 and c) linewidth and spin‐lattice relaxation time T 1 in 1H NMR spectrum of MAPbI3. All the experimental points shown in this figure were obtained in several runs with both increasing and decreasing temperature. No indications of degradation have been detected.
Figure 4.
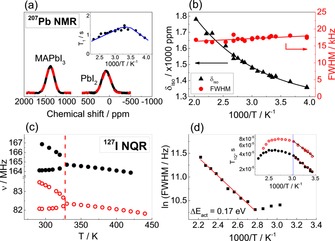
a) Experimental 207Pb NMR spectra of MAPbI3 and PbI2 for comparison. Inset: Spin‐lattice relaxation T 1 as a function of temperature. The dependence is rather weak and is similar to what was reported for pure PbI2.48 b) Temperature dependences of the 207Pb chemical shift and FWHM. Solid lines are a guide to the eye. A very short spin‐spin relaxation time T 2 (ca. 2×10−5 s) is detected for this signal, which is consistent with strong dipolar coupling and particularly cross‐relaxation to quadrupolar 127I.49 c) Temperature dependence of 127I NQR transition frequencies (•=ν±3/2↔±5/2, ○=ν±1/2↔±3/2) in MAPbI3, demonstrating the phase transition at 327 K. d) Temperature dependence of the linewidth in ν1 NQR transition of MAPbI3. Inset: Temperature dependence of the spin‐spin relaxation time in the two 127I NQR transitions of MAPbI3 (•=ν±3/2↔±5/2, ○=ν±1/2↔±3/2). Blue line indicates phase transition.
The chemical shift anisotropy is either small or completely absent, as expected from a strong and symmetrical coordination of Pb2+ to I−. The fact that the signal, as shown in Figure 4 b, maintains a nearly constant linewidth with temperature indicates that no appreciable long‐range motion is present in the range probed (250–493 K). Along with the insensitivity to the phase transition and the strong dipolar coupling with 127I, this behavior strongly asserts the absence of long‐ and/or short‐range Pb2+ motion in MAPbI3. Concerning iodine, in spite of its large quadrupolar coupling constant (>500 MHz), we were able to detect a reliable 127I NMR signal in MAPbI3 (Supporting Information, Figure S8). However, 127I NQR (Figure S9) turned out to be a more meaningful and suitable technique for this material. Even though these spectra do not give insights into long‐range motion of iodine, the results provide useful information on short‐range (rotational/vibrational) dynamics. Information on short‐range motions can be extracted also from 14N, 1H, and 207Pb spectra. As this is not the major focus of the present paper, we restrict ourselves to a few key points. The character of changes in the quadrupolar frequency with temperature in 127I NQR shown in Figure 4 c can be associated with planar torsional oscillation of I atoms about Pb centers, in a correlated fashion.50, 51 The FWHM of the 127I signal massively increases with temperature (Figure 4 d) indicating highly active short‐range dynamics of iodine atoms, which can either happen as a pre‐step or in parallel to long‐range motion. The aforementioned results revealed also fast rotational motion for MA+ (in agreement with Refs. 40, 42, 52), which however appears decoupled from short‐range I− dynamics (compare activation energies in Figures 3 b and 4 d, also Ref. 53). Pb2+ cations finally appear immobile even at short length scale.
In summary, using a set of complementary techniques we revealed the nature of the decisive charge carriers in MAPbI3 under equilibrium conditions. Using d.c. polarization, a.c. impedance spectroscopy, and emf measurements, we have shown that the dark conductivity in MAPbI3 is to a substantial degree ionic. Faradaic reaction experiments demonstrated that this ion conduction is primarily due to iodine transport, whereby significant Pb2+ and MA+ diffusion can be excluded. The latter was also directly demonstrated by tracer experiments. NMR measurements are completely consistent in showing absence of significant Pb2+ and MA+ long‐range transport. Irrespective of long‐range motion, these results gave worthwhile information on the short‐range processes, in which highly active iodine dynamics were clearly observed. On the short‐range scale, and only in this length scale, MA cations show fast rotational dynamics as opposed to Pb cations that appear immobile on all length scales. Having elucidated the nature of the mobile ion, the next step was to identify the mechanism, that is, whether iodide motion occurs interstitially or through vacancies. This was unambiguously shown by studying ionic (and electronic) conductivity as a function of iodine partial pressure as well as by doping experiments. Both independently reveal iodine vacancies to be the responsible carriers for ion conduction (and electron holes for electronic conduction) in the parameter window under consideration. Having clarified the nature of charge carriers in MAPbI3 in equilibrium, a firm basis is now available on which results under illumination, in particular the indications of enhanced ionic transport, can rely on.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
The authors are grateful to Tolga Acarturk for ToF‐SIMS measurements, Yvonne Stuhlhofer for AFM picture and surface profiling, Dr. Rotraut Merkle for many suggestions and discussions and Dr. Helga Hoier for XRD measurements. The authors are also thankful to V. Terskikh (University of Ottawa, Canada) for help with the UHF NMR experiments. Access to the 900 MHz NMR spectrometer was provided by the National Ultrahigh Field NMR Facility for Solids managed by University of Ottawa, Ottawa, Canada (http://www.nmr900.ca). This work was performed within the framework of the Max Planck‐EPFL Center for Molecular Nanoscience and Technology.
A. Senocrate, I. Moudrakovski, G. Y. Kim, T.-Y. Yang, G. Gregori, M. Grätzel, J. Maier, Angew. Chem. Int. Ed. 2017, 56, 7755.
Contributor Information
Dr. Igor Moudrakovski, Email: i.moudrakovski@fkf.mpg.de
Prof. Joachim Maier, Email: s.weiglein@fkf.mpg.de
References
- 1. Kojima A., Teshima K., Shirai Y., Miyasaka T., J. Am. Chem. Soc. 2009, 131, 6050–6051. [DOI] [PubMed] [Google Scholar]
- 2. Kim H. S., Lee C. R., Im J. H., Lee K. B., Moehl T., Marchioro A., Moon S. J., Humphry-Baker R., Yum J. H., Moser J. E., et al., Sci. Rep. 2012, 2, 591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Burschka J., Pellet N., Moon S.-J., Humphry-Baker R., Gao P., Nazeeruddin M. K., Gratzel M., Nature 2013, 499, 316–319. [DOI] [PubMed] [Google Scholar]
- 4. Grätzel M., Nat. Mater. 2014, 13, 838–842. [DOI] [PubMed] [Google Scholar]
- 5. Berhe T. A., Su W.-N., Chen C.-H., Pan C.-J., Cheng J.-H., Chen H.-M., Tsai M.-C., Chen L.-Y., Dubale A. A., Hwang B.-J., Energy Environ. Sci. 2016, 9, 323–356. [Google Scholar]
- 6. Xiao Z., Yuan Y., Shao Y., Wang Q., Dong Q., Bi C., Sharma P., Gruverman A., Huang J., Nat. Mater. 2015, 14, 193–198. [DOI] [PubMed] [Google Scholar]
- 7. Juarez-Perez E. J., Sanchez R. S., Badia L., Garcia-Belmonte G., Kang Y. S., Mora-Sero I., Bisquert J., J. Phys. Chem. Lett. 2014, 5, 2390–2394. [DOI] [PubMed] [Google Scholar]
- 8. Snaith H. J., Abate A., Ball J. M., Eperon G. E., Leijtens T., Noel N. K., Stranks S. D., Wang J. T.-W., Wojciechowski K., Zhang W., J. Phys. Chem. Lett. 2014, 5, 1511–1515. [DOI] [PubMed] [Google Scholar]
- 9. Cheng Y., Li H.-W., Qing J., Yang Q.-D., Guan Z., Liu C., Cheung S. H., So S. K., Lee C.-S., Tsang S.-W., J. Mater. Chem. A 2016, 4, 12748–12755. [Google Scholar]
- 10. Bag M., Renna L. A., Adhikari R. Y., Karak S., Liu F., Lahti P. M., Russell T. P., Tuominen M. T., Venkataraman D., J. Am. Chem. Soc. 2015, 137, 13130–13137. [DOI] [PubMed] [Google Scholar]
- 11. Carrillo J., Guerrero A., Rahimnejad S., Almora O., Zarazua I., Mas-Marza E., Bisquert J., Garcia-Belmonte G., Adv. Energy Mater. 2016, 6, 1502246. [Google Scholar]
- 12. Wagner C., Z. Elektrochem. 1956, 60, 4–7. [Google Scholar]
- 13. Yokota I., J. Phys. Soc. Jpn. 1961, 16, 2213–2223. [Google Scholar]
- 14. Yang T.-Y., Gregori G., Pellet N., Grätzel M., Maier J., Angew. Chem. Int. Ed. 2015, 54, 7905–7910; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 8016–8021. [Google Scholar]
- 15. Jamnik J., Maier J., Ber. Bunsen-Ges. 1997, 101, 23–40. [Google Scholar]
- 16. Riess I., Leshem A., Solid State Ionics 2012, 225, 161–165. [Google Scholar]
- 17. Maier J., Prog. Solid State Chem. 1995, 23, 171–263. [Google Scholar]
- 18. Chen B., Yang M., Zheng X., Wu C., Li W., Yan Y., Bisquert J., Garcia-Belmonte G., Zhu K., Priya S., J. Phys. Chem. Lett. 2015, 6, 4693–4700. [DOI] [PubMed] [Google Scholar]
- 19. Yamada K., Isobe K., Tsuyama E., Okuda T., Furukawa Y., Solid State Ionics 1995, 79, 152–157. [Google Scholar]
- 20. Mizusaki J., Arai K., Fueki K., Solid State Ionics 1983, 11, 203–211. [Google Scholar]
- 21. Eames C., Frost J. M., Barnes P. R. F., O'Regan B. C., Walsh A., Islam M. S., Nat. Commun. 2015, 6, 7497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Haruyama J., Sodeyama K., Han L., Tateyama Y., J. Am. Chem. Soc. 2015, 137, 10048–10051. [DOI] [PubMed] [Google Scholar]
- 23. Li C., Tscheuschner S., Paulus F., Hopkinson P. E., Kießling J., Köhler A., Vaynzof Y., Huettner S., Adv. Mater. 2016, 28, 2446–2454. [DOI] [PubMed] [Google Scholar]
- 24. Besleaga C., Abramiuc L. E., Stancu V., Tomulescu A. G., Sima M., Trinca L., Plugaru N., Pintilie L., Nemnes G. A., Iliescu M., et al., J. Phys. Chem. Lett. 2016, 7, 5168–5175. [DOI] [PubMed] [Google Scholar]
- 25. De Bastiani M., Dell′Erba G., Gandini M., D′Innocenzo V., Neutzner S., Kandada A. R. S., Grancini G., Binda M., Prato M., Ball J. M., et al., Adv. Energy Mater. 2016, 6, 1501453. [Google Scholar]
- 26. Li J., Dong Q., Li N., Wang L., Adv. Energy Mater. 2017, 1602922. [Google Scholar]
- 27. Zhang T., Meng X., Bai Y., Xiao S., Hu C., Yang Y., Chen H., Yang S., Ginger D. S., Stranks S. D., et al., J. Mater. Chem. A 2017, 5, 1103–1111. [Google Scholar]
- 28. Yuan Y., Chae J., Shao Y., Wang Q., Xiao Z., Centrone A., Huang J., Adv. Energy Mater. 2015, 5, 1500615. [Google Scholar]
- 29. Leijtens T., Hoke E. T., Grancini G., Slotcavage D. J., Eperon G. E., Ball J. M., De Bastiani M., Bowring A. R., Martino N., Wojciechowski K., et al., Adv. Energy Mater. 2015, 5, 1500962. [Google Scholar]
- 30. Jacobs D. A., Wu Y., Shen H., Barugkin C., Beck F. J., White T. P., Weber K., Catchpole K. R., Snaith H. J., Hodes G., et al., Phys. Chem. Chem. Phys. 2017, 19, 3094–3103. [DOI] [PubMed] [Google Scholar]
- 31. Contreras L., Idígoras J. A., Todinova A., Salado M., Kazim S., Ahmad S., Anta J., Phys. Chem. Chem. Phys. 2016, 18, 31033–31042. [DOI] [PubMed] [Google Scholar]
- 32. Domanski K., Roose B., Matsui T., Saliba M., Turren-Cruz S.-H., Correa-Baena J.-P., Carmona C. R., Richardson G., Foster J. M., De Angelis F., et al., Energy Environ. Sci. 2017, 10, 604–613. [Google Scholar]
- 33. Yuan Y., Wang Q., Shao Y., Lu H., Li T., Gruverman A., Huang J., Adv. Energy Mater. 2016, 6, 1501803. [Google Scholar]
- 34. Lingras A., Simkovich G., Solid State Commun. 1978, 27, iii–iv. [Google Scholar]
- 35. Dunn B., Ostrom R. M., Seevers R., Farrington G. C., Solid State Ionics 1981, 5, 203–204. [Google Scholar]
- 36. Organic-Inorganic Halide Perovskite Photovoltaics: From Fundamentals to Device Architectures (Eds.: N.-G. Park, M. Graetzel, T. Miyasaka), Springer, Heidelberg, 2016. [Google Scholar]
- 37. Berry J., Buonassisi T., Egger D. A., Hodes G., Kronik L., Loo Y.-L., Lubomirsky I., Marder S. R., Mastai Y., Miller J. S., et al., Adv. Mater. 2015, 27, 5102–5112. [DOI] [PubMed] [Google Scholar]
- 38.Y. Zhao, W. Zhou, Z. Zhou, K. Liu, D. Yu, Q. Zhao, Light Sci. Appl 2017, https://doi.org/10.1038/lsa.2016.243. [DOI] [PMC free article] [PubMed]
- 39.G. Y. Kim, A. Senocrate, J. Maier, unpublished results.
- 40. Wasylishen R. E., Knop O., Macdonald J. B., Solid State Commun. 1985, 56, 581–582. [Google Scholar]
- 41. Knop O., Wasylishen R. E., White M. A., Cameron T. S., Van Oort M. J. M., Can. J. Chem. 1990, 68, 412–422. [Google Scholar]
- 42. Poglitsch A., Weber D., J. Chem. Phys. 1987, 87, 6373. [Google Scholar]
- 43. Leguy A. M. A., Goñi A. R., Frost J. M., Skelton J., Brivio F., Rodríguez-Martínez X., Weber O. J., Pallipurath A., Alonso M. I., Campoy-Quiles M., et al., Phys. Chem. Chem. Phys. 2016, 18, 27051–27066. [DOI] [PubMed] [Google Scholar]
- 44. Franssen W. M. J., van Es S. G. D., Dervişoğlu R., de Wijs G. A., Kentgens A. P. M., J. Phys. Chem. Lett. 2017, 8, 61–66. [DOI] [PubMed] [Google Scholar]
- 45. Yin W.-J., Shi T., Yan Y., Appl. Phys. Lett. 2014, 104, 063903. [Google Scholar]
- 46. Du M. H., J. Mater. Chem. A 2014, 2, 9091–9098. [Google Scholar]
- 47. Abdi-Jalebi M., Dar M. I., Sadhanala A., Senanayak S. P., Grätzel M., Friend R. H., J. Vis. Exp. 2017, e55307–e55307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Taylor R. E., Beckmann P. A., Bai S., Dybowski C., J. Phys. Chem. C 2014, 118, 9143–9153. [Google Scholar]
- 49. Shmyreva A. A., Safdari M., Furó I., Dvinskikh S. V., J. Chem. Phys. 2016, 144, 224201. [DOI] [PubMed] [Google Scholar]
- 50. Bayer H., Z. Phys. 1951, 130, 227–238. [Google Scholar]
- 51. Jeffrey K. R., Armstrong R. L., Phys. Rev. 1968, 174, 359–369. [Google Scholar]
- 52. Leguy A. M. A., Frost J. M., McMahon A. P., Sakai V. G., Kochelmann W., Law C., Li X., Foglia F., Walsh A., O'Regan B. C., et al., Nat. Commun. 2015, 6, 7124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Xu Q., Eguchi T., Nakayama H., Nakamura N., Kishita M., Z. Naturforsch. A 1991, 46, 240–246. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary