

Abstract
We report a multi‐component asymmetric Brønsted acid‐catalyzed aza‐Darzens reaction which is not limited to specific aromatic or heterocyclic aldehydes. Incorporating alkyl diazoacetates and, important for high ee's, ortho‐tert‐butoxyaniline our optimized reaction (i.e. solvent, temperature and catalyst study) affords excellent yields (61–98 %) and mostly >90 % optically active cis‐aziridines. (+)‐Chloramphenicol was generated in 4 steps from commercial starting materials. A tentative mechanism is outlined.
Keywords: asymmetric catalysis, aziridine, Brønsted acids, multicomponent reactions
Such is the versatility of organocatalysis and its ability to mediate a plethora of diverse reaction types1 it is, now, an indispensable “tool” in the synthetic chemists “toolbox”.2 Indeed, improving atom‐ and reaction‐efficiency is a key driver to developing new reactions and protocols; in this context organocatalysis has demonstrated its importance by efficiently mediating many different convergent reactions or multi‐component syntheses. The work here supports these aspects by generating structure and function‐diverse motifs via fewer synthetic, isolation and purification steps.
Optically active aziridines have many diverse uses, especially as key intermediates3 “on route” to important “secondary” products for example, α‐/β‐amino acids, polymers, azasugars, auxilaries, oxazolidinones, imidazolidines, β‐lactams and pyrrolidines. Further applications include synthesis of non‐aziridine containing bioactive compounds for example, kainoids, (−)‐mesembrine, (−)‐platynesine, actinomycin and feldamycin, in addition to synthetic bioactive aziridines for example, NSC676892 as well as natural products for example, azinomycin and maduropeptide.4
Using a BINOL N‐triflylphosphoramide Brønsted acid a 61–98 % yielding asymmetric aza‐Darzens reaction affords N‐aryl‐cis‐aziridines in, mostly, 90–99 % ee. The reaction is straightforward to set up and has minimal requirements for strictly anhydrous or anaerobic conditions, furthermore it does not require organocatalyst pre‐generation or activation, or an “activated” arylglyoxal starting material. Exploiting the protocol synthesis of aziridines based on 4 uses readily generated or commercially available aldehydes (1), amines (2) and alkyl diazoacetates (3, Scheme 1).
Scheme 1.
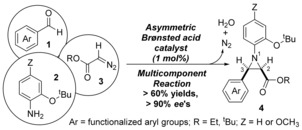
Multicomponent asymmetric synthesis of N‐aryl‐cis‐aziridines 4.
Activating the C=N bond of an imine with a BINOL phosphoric acid5 lowers its LUMO energy and generates an imminium ion pair that can, but not always will, react with a nucleophile. Seminal work by Akiyama et al. established chiral BINOL phosphoric acids [pK a≈13 (CH3CN)6] activate aldimines (derived from, specifically, arylglyoxals and p‐anisidine) and react with ethyl diazoacetate (EDA) affording cis‐aziridines in 92–97 % ee.7 Similarly, other Brønsted acids8 and pyridinium triflate activate a diverse array of imines, including for example, 2‐pyridyl derived 5, enabling the presumed imminium ion‐pair (not shown) to react with EDA and afford cis‐rac‐aziridine (8, 83 % yield) (Scheme 2).9 With these racemic studies complete our focus shifted to developing a substrate enhanced and diverse, multi‐component asymmetric aza‐Darzens reaction. Inspired by the work of Akiyama et al.7 and the Mannich reaction reported by Yamanaka et al. we considered the inclusion of 5 may generate a constrained hydrogen‐bonded and activated complex similar to 11; we were drawn to the use of 5 to generate 11 due to similarities in the chiral non‐racemic rigid environment proposed by Yamanaka (using a N‐(2‐hydroxyphenyl)imine starting material).10 Screening chiral non‐racemic BINOL and VANOL phosphoric acids, as well as a H‐QUIN‐BAM triflate salt11 we were disappointed no reactions were observed. We attribute the failure using 5, as well as other alternative imines, to the low pK a’s of the Brønsted acids and their inability to generate a sufficiently “activated” form of 10 or 11.
Scheme 2.
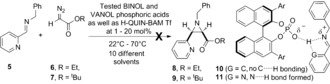
Failed attempts at synthesising cis‐8 and cis‐9.
Switching to the more acidic BINOL N‐triflylphosphoramides for example, pK a 14 ≈6 (CH3CN)6 (Figure 1) the synthesis of (S)‐3,3′‐bis(phenyl)‐14, (S)‐3,3′‐bis(4‐methylphenyl)‐15 and sterically encumbered (S)‐3,3′‐bis(4‐tert‐butylphenyl)‐16 was straightforward.12 By using 10 mol % all three catalysts, independently, at room temperature mediated the synthesis of cis‐aziridine 8 in 73 %, 85 % and 87 % yields, respectively. 1H‐NMR of the unpurified reactions confirmed no enamide5 i.e. Z‐12 or Z‐13 (Figure 1) or trans‐8 (J 2,3≈2 Hz, not shown) had formed. Disappointingly, chiral column HPLC analysis established cis‐8 was racemic when generated using 14 or 15; in contrast, 16 afforded non‐racemic cis‐8 but in a poor 16 % ee (Table 1, Entries 1–3 respectively). Clearly, the bulky 4‐tert‐butyl group had a positive stereochemical advantage over 14 and 15. Increasing 3,3′‐steric congestion at the 2‐ and 6‐ positions using (S)‐3,3′‐bis(2,4,6‐triisopropyl)phenyl‐17 returned cis‐8 in excellent yield and increased 23 % ee (entry 4).
Figure 1.
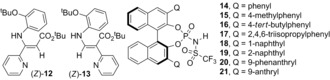
Enamides and 3,3′‐bis(aryl) (S)‐BINOL N‐triflylphosphoramides.
Table 1.
Probing the asymmetric synthesis of cis‐8 and cis‐9 using 14–21.
| Entry | Catalyst | 8 (R=Et, ee) | Entry | Catalyst | 9 (R=tBu, ee) | |
|---|---|---|---|---|---|---|
| 1 | 14 | racemic | 9 | 14 | racemic | |
| 2 | 15 | racemic | 10 | 15 | 18 % | |
| 3 | 16 | 16 % | 11 | 16 | 13 % | |
| 4 | 17 | 23 % | 12 | 17 | racemic | |
| 5 | 18 | 28 % | 13 | 18 | 20 % | |
| 6 | 19 | 26 % | 14 | 19 | 20 % | |
| 7 | 20 | 35 % | 15 | 20 | 22 % | |
| 8 | 21 | 47 % | 16 | 21 | 31 % |
The 69 % increase in ee when 2‐ and 6‐isopropyl groups were incorporated (17) suggests these “lateral” positions have key roles in reaction stereoselectivity. Probing this, multicyclic 1‐naphthyl (18), 2‐naphthyl (19), 9‐phenanthryl (20) and 9‐anthryl (21) were incorporated (10 mol %) into our “test” reaction (Scheme 2). All afforded excellent yields of cis‐8. A gradual increase in ee was observed as the “lateral” groups were added. Thus catalyst 14 afforded rac‐8, whereas 1‐naphthyl‐18 offered cis‐8 with a 28 % ee. An almost identical 26 % ee was provided by 2‐naphthyl‐19 and 9‐phenanthryl‐20 gave an improved 35 % ee, finally, 9‐anthryl‐21 generated cis‐8 in a respectable 47 % ee.
Encouraged by the results with 6, sterically encumbered tert‐butyl ester 7 was investigated. A gradual increase in ee was observed but, overall, the levels of stereoinduction were, generally, inferior. So, N‐benzyl 5 was substituted for a rotationally less flexible N‐4‐(methoxyphenyl) or N‐PMP group. Reacting the corresponding imine (not shown) with 7 mediated by 21 afforded the cis‐aziridine in an 81 % yield (J 2,3 6.8 Hz). Further verifying the importance of including the, presumed, rotationally less flexible N‐PMP the product was afforded with a significantly improved 67 % ee. Exchanging the N‐PMP for the regioisomeric N‐2‐methoxyphenyl imine 22 (Scheme 3) its activation (21) and reaction with tert‐butyl ester‐7 afforded cis‐25 with a 72 % ee. Evidently, the 2‐methoxyphenyl had a positive influence on the stereochemical outcome of the aza‐Darzens reaction. The steric effect was probed using 2‐isopropoxyphenyl‐23, 2‐n‐butoxyphenyl (not shown) and 2‐tert‐butoxyphenyl‐24 (Scheme 3) each reacted, independently, with 7 and 21. In this series and at ambient temperature the tert‐butoxy group on 24 afforded cis‐27 with a 74 % ee.
Scheme 3.
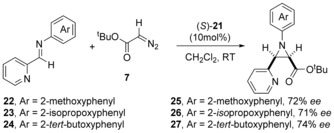
Asymmetric synthesis of N‐(alkoxyphenyl)‐cis‐aziridines 25–27.
A solvent and temperature study using 1 mol % of 21 established chloroform at −60 °C was the optimum combination for transforming 2‐(tert‐butoxyphenyl)‐24 into cis‐27 with an excellent 98 % ee and 95 % yield. Probing the catalytic activity of 21 at 0.5 and 0.25 mol % loadings the reaction times increased to 48 and 62 hours. In both examples cis‐27 was afforded in very similar 87 %/86 % ee and 98 %/95 % yield, respectively.
The synthesis of 38–47 (Scheme 4) was examined using 21 (1 mol %) in CHCl3 at −60 °C. Incorporating (E)‐2‐(tert‐butoxyphenyl)‐28 cis‐38 was afforded in an excellent 91 % ee and 90 % yield. Confirming reaction versatility electron‐withdrawing 4‐cyano imine‐30 and 4‐nitrophenyl imine‐31 were transformed into cis‐40 and cis‐41 with excellent optical purities both 98 % and yields that is, 98 % and 97 % respectively (Scheme 4). Similarly, electron‐rich 4‐hydroxybenzaldehyde (O‐Fmoc protected) afforded cis‐42 in a 90 % ee and 91 % yield. Cis‐43 to cis‐47 were synthesized in excellent yields and ee’s; 4‐bromophenyl‐cis‐45 (93 % ee) and 4‐iodophenyl‐cis‐46 (92 % ee) appear readily amenable to further elaboration via transition‐metal mediated transformations.
Scheme 4.
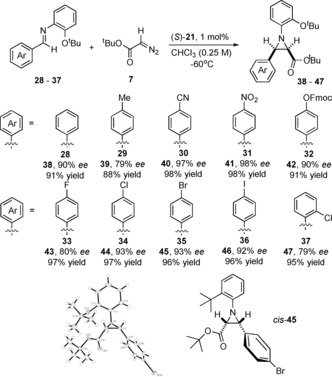
Asymmetric synthesis of structure and function diverse N‐(2‐tert‐butoxyphenyl)‐cis‐aziridines 38–47 and the X‐ray structure of cis‐45.
The magnitude of an aziridine coupling constant (J 2,3) indicates the relative stereochemical assignment of the C2,3‐substituents that is, J 2,3 5–9 Hz=cis and 2–6 Hz=trans. For 38–47 we tentatively assigned a cis‐stereochemical relationship; confirming this was essential. Recrystallising 45 [J 2,3 6.7(4) Hz] afforded colorless orthorhombic plates. X‐ray diffraction established the cis‐stereochemical relationship between the 4‐bromophenyl and the tert‐butyl carboxylate ester (Scheme 4).13
Generating aziridines via multicomponent asymmetric syntheses is advantageous, they are however, still, rare.14 It was crucial to verify 21 mediated the multicomponent synthesis of cis‐aziridines. A three‐component, two‐step, one‐pot protocol generated 2‐pyridyl‐27, 4‐cyanophenyl‐40 and 4‐nitrophenyl‐41 in excellent yields that is, 74–96 % and ee’s that is, 27 (96 %) 40, (99 %) and 41 (96 %, Scheme 5). These ee’s are, within experimental error, identical to those generated via the pre‐synthesis imine route (Scheme 4). Incorporating 4‐thiomethyl‐, 4‐trifluoromethyl‐ and pentafluorobenzaldehyde afforded cis‐49 to cis‐51. The efficient synthesis of thioether cis‐49 is worthy of note; Davis et al. exploited similar (S)‐N‐(4‐toluenesulfinyl)‐derived aziridines transforming them into thiamphenicol and florfenicol.15
Scheme 5.
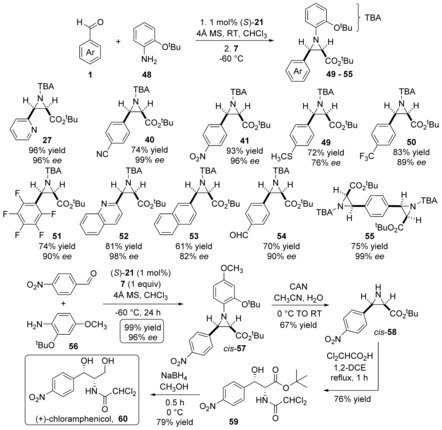
Asymmetric synthesis of cis‐aziridines and (+)‐chloramphenicol.
4‐Trifluoromethylbenzaldehyde and pentafluorobenzaldehyde afforded cis‐50 and cis‐51 in excellent 89 % and 90 % ee’s, respectively. Integrating bicyclic quinoline‐2‐carboxyaldehyde was also straightforward; optically active cis‐52 was afforded in a 98 % ee. Interestingly, the formation of cis‐27 and cis‐52 was faster than for example 40, 41, 49–50; the rapid evolution of, presumably, N2 was attributed to formation of a more reactive intramolecular chelated hydrogen bond (cf. 11). Combining benzene‐1,4‐dicarboxyaldehyde and 48 (1 equiv) a one‐pot, single asymmetric aziridination afforded mono‐aziridine cis‐54. Alternatively, 2 equiv of 48 generated bis‐aziridine cis‐55. Both reactions worked very well, cis‐54 was afforded in a 70 % yield and 90 % ee and bis‐aziridine cis‐55 with a 99 % ee. Seemingly, the installation of the second aziridine on optically active cis‐54 to generate cis‐55 was not negatively influenced by the first optically active cis‐aziridine. (−)‐Chloramphenicol is an important natural product with antibiotic properties. A one‐pot multicomponent aziridination using 4‐nitrobenzaldehyde, amine 56 and tert‐butyl diazoester 7 afforded cis‐57 in near quantitative yield and an excellent 96 % ee. By using cerium(IV) ammonium nitrate in aqueous acetonitrile an important objective was to establish the cleavage “potential” of the 2‐tert‐butoxy‐4‐methoxyphenyl on cis‐57; NH‐cis‐58 was afforded in an unoptimized 67 % yield, this was ring‐opened to amide 59 with dichloroacetic acid, finally reducing the tert‐butyl ester generated primary alcohol 60 (79 % yield). Physicochemical analysis and comparison with the literature confirmed (+)‐chloramphenicol 60 had been synthesized using (S)‐21 in 4 steps and an overall 40 % yield.16
Scheme 6 outlines a tentative mechanism for cis‐aziridine diastereoselectivity. Initial N‐protonation of 31 via Brønsted acid (S)‐21 [pK a≈6 (CH3CN)],6 affords imminium‐phosphoramide anion 60 (Path A, Scheme 6) whilst the weaker triflate salts and phosphoric acids (see Supporting Information, page 3) do not form sufficiently reactive imminium‐triflate/phosphate anions.
Scheme 6.
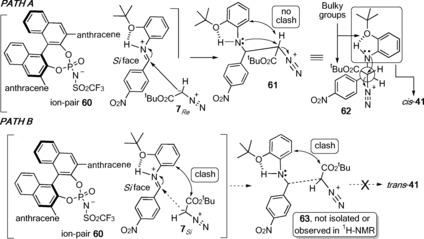
Mechanistic rational for the synthesis of cis‐41 and not trans‐41.
Supporting protonation, not hydrogen‐bond activation,17 Houk et al. described a mechanism and origins of catalysis DFT and experimental study in which a similarly N‐protonated, to 60, reactive hydrazonium‐phosphoramide18 anion (not shown) was formed from a BINOL N‐triflylphosphoramide and a hydrazone. Activation of 31 is crucial; the widely accepted aza‐Darzens mechanism19 invokes attack of a diazo nucleophile (i.e. 7) on an imminium cation (i.e. 60) generating an α‐diazonium β‐amino ester (i.e. 61, see Path A). The importance of the latter, from a reaction kinetics and enantioselectivity point of view has been established by the reluctance of these intermediates to undergo a retro‐Mannich reaction.20 Generating, presumed, kinetic product 61 with excellent enantioselectivity is possible only if 7, with its heterotopic faces that is, 7 Re and 7 Si, efficiently discriminates between the Si and Re faces of optically active 60. Path A outlines how anti‐diazonium intermediate 62 (Scheme 6) forms when the sterically encumbered heterotopic 7 Re face approaches the Si face of imine 60 minimising the steric interactions between the intramolecularly hydrogen bonded bulky ortho‐(tert‐butoxy)phenyl imminium and the tert‐butyl ester on 7 Re. Although we have no direct evidence (1H‐NMR) for the backbone rigidifying hydrogen bond in 60 similar intramolecular hydrogen bonds in ortho‐substituted Schiff base's are known.21 Newman projection 62 affords a detailed depiction of the minimized steric interactions between the tert‐butyl ester and ortho‐tert‐butylphenyl ether. An intramolecular SN2 cyclization (release of N2) between the anti‐periplanar amine and diazonium groups affords cis‐41. Path B proceeds via ion‐pair 60, however approach of 7 Si onto the imine Si face is, now, inhibited by the two sterically bulky groups. Thus, formation of α‐diazonium β‐amino ester 63 and trans‐41 is disfavoured. The crude 1H‐NMRs of our reactions afforded no evidence of trans‐41 or α‐diazonium β‐amino ester 63.
Experimental Section
A flame dried Radleys tube and stirrer bar was charged with 4‐cyanobenzaldehyde (34 mg, 0.26 mmol) and 2‐tert‐butoxy‐phenylamine (43 mg, 0.26 mmol). Anhydrous chloroform (1 mL) and (S)‐21 (2 mg, 0.0025 mmol, 1 mol %) were added followed by 40 mg of freshly powdered 4 Å molecular sieves. The reaction was stirred for 6 hours. Cooling the tube to −60 °C, 7 (40 μL, 0.29 mmol) was added via syringe. The reaction was stirred at −60 °C and monitored via TLC (hexane/ether:80/20) until the starting materials had been consumed. In vacuo removal of solvent allowed flash purification on silica gel (hexane/ether:80/20). Physicochemical analysis confirmed the identity of the solid as cis‐40. Chiral column analytical HPLC established cis‐40 had an ee of 99 %.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This work was supported via an EPSRC GSK CASE award.
S. P. Bew, J. Liddle, D. L. Hughes, P. Pesce, S. M. Thurston, Angew. Chem. Int. Ed. 2017, 56, 5322.
References
- 1. List B., Maruoka K., Science of Synthesis Asymmetric Organocatalysis, Thieme Stuttgart, 2012. [Google Scholar]
- 2. MacMillan D. W. C., Nature 2008, 455, 304–308. [DOI] [PubMed] [Google Scholar]
- 3. Lu P., Tetrahedron 2010, 66, 2549–2560. [Google Scholar]
- 4. Botuha C., Chemla F., Ferreira F., Perez-Luna A., Majumdar (Eds.: K. C., Chattopadhyay) S. K., Heterocycles in Natural Product Synthesis, Wiley, Hoboken: 2011, pp. 3–39. [Google Scholar]
- 5.
- 5a. Akiyama T., Chem. Rev. 2007, 107, 5744–5758; [DOI] [PubMed] [Google Scholar]
- 5b. Mori K., Akiyama T., Chem. Rev. 2015, 115, 9277–9306; [DOI] [PubMed] [Google Scholar]
- 5c. Kampen D., Reisinger C. M., List B., Top. Curr. Chem. 2010, 291, 395–456; [DOI] [PubMed] [Google Scholar]
- 5d. Akiyama T., Itoh J., Fuchibe K., Angew. Chem. Int. Ed. 2004, 43, 1566–1568; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2004, 116, 1592–1594; [Google Scholar]
- 5e. Akiyama T., Morita H., Fuchibe K., J. Am. Chem. Soc. 2006, 128, 13070–13071; [DOI] [PubMed] [Google Scholar]
- 5f. Hashimoto T., Maruoka K., J. Am. Chem. Soc. 2007, 129, 10054–10055; [DOI] [PubMed] [Google Scholar]
- 5g. Terada M., Machioka K., Sorimachi K., Angew. Chem. Int. Ed. 2006, 45, 2254–2257; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2006, 118, 2312–2315; [Google Scholar]
- 5h. Rueping M., Antonchick A. P., Theissmann T., Angew. Chem. Int. Ed. 2006, 45, 6751–6755; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2006, 118, 6903–6907; [Google Scholar]
- 5i. Bew S. P., Bachera D. U., Coles S. J., Hiatt-Gipson G. D., Pesce P., Pitak M., Thurston S. M., Zdorichenko V., Chem 2016, 1, 921–945. [Google Scholar]
- 6. Rueping M., Parmar D., Sugiono E., Raja S., Chem. Rev. 2014, 114, 9047–9153. [DOI] [PubMed] [Google Scholar]
- 7.
- 7a. Mori K., Suzuki T., Akiyama T., Org. Lett. 2009, 11, 2445–2447; [DOI] [PubMed] [Google Scholar]
- 7b. Akiyama T., Chem. Rev. 2007, 107, 5744–5758; [DOI] [PubMed] [Google Scholar]
- 7c. Terada M., Synthesis 2010, 1929–1982. [Google Scholar]
- 8.
- 8a. Desai A. A., Wulff W. D., J. Am. Chem. Soc. 2010, 132, 13100–13103; [DOI] [PubMed] [Google Scholar]
- 8b. Hashimoto N., Nakatsu H., Watanabe S., Maruoka K., Org. Lett. 2010, 12, 1668–1671; [DOI] [PubMed] [Google Scholar]
- 8c. Zeng X., Zeng X., Xu Z., Lu M., Zhong G., Org. Lett. 2009, 11, 3036–3039; [DOI] [PubMed] [Google Scholar]
- 8d. Hashimoto T., Nakatsu H., Yamamoto K., Maruoka K., J. Am. Chem. Soc. 2011, 133, 9730–9733; [DOI] [PubMed] [Google Scholar]
- 8e. Rowland E. B., Rowland G. B., Rivera-Otero E., Antilla J. C., J. Am. Chem. Soc. 2007, 129, 12084–12085; [DOI] [PubMed] [Google Scholar]
- 8f. Mahoney J. M., Smith C. R., Johnston J. N., J. Am. Chem. Soc. 2005, 127, 1354–1355; [DOI] [PubMed] [Google Scholar]
- 8g. Williams A. L., Johnston J. N., J. Am. Chem. Soc. 2004, 126, 1612–1613. [DOI] [PubMed] [Google Scholar]
- 9. Bew S. P., Carrington R., Hughes D. L., Liddle J., Pesce P., Adv. Synth. Catal. 2009, 351, 2579–2588. [Google Scholar]
- 10. Yamanaka M., Itoh J., Fuchibe K., Akiyama T., J. Am. Chem. Soc. 2007, 129, 6756–6764. [DOI] [PubMed] [Google Scholar]
- 11.See the Supporting Information for the structures of the catalysts used.
- 12. Nakashima D., Yamamoto H., J. Am. Chem. Soc. 2006, 128, 9626–9627. [DOI] [PubMed] [Google Scholar]
- 13.CCDC 1519076 (cis-45) contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre.
- 14. Albrecht K., Jiang H., Jørgensen K. A., Angew. Chem. Int. Ed. 2011, 50, 8492–8509; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 8642–8660. [Google Scholar]
- 15. Davis F. A., Zhou P., Tetrahedron Lett. 1994, 41, 7525–7528. [Google Scholar]
- 16. Franchino A., Jakubec P., Dixon D. J., Org. Biomol. Chem. 2016, 14, 93–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Merino P., Delso I., Tejero T., Roca-López D., Isasi A., Matute R., Curr. Org. Chem. 2011, 15, 2184–2209. [Google Scholar]
- 18. Hong X., Küçük H. B., Maji M. S., Yang Y.-F., Rueping M., Houk K. N., J. Am. Chem. Soc. 2014, 136, 13769–13780. [DOI] [PubMed] [Google Scholar]
- 19.
- 19a. Vetticatt M. J., Desai A. A., Wulff W. D., J. Am. Chem. Soc. 2010, 132, 13104–13107; [DOI] [PubMed] [Google Scholar]
- 19b. Johnston J. J., Muchalski H., Troyer T. L., Angew. Chem. Int. Ed. 2010, 49, 2290–2298; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 2340–2349. [Google Scholar]
- 20. Trover T. L., Muchalski H., Hong K. B., Johnston J. N., Org. Lett. 2011, 13, 1790–1792. [DOI] [PubMed] [Google Scholar]
- 21.
- 21a. Zheng J., Kwak K., Chen X., Asbury J. B., Fayer M. D., J. Am. Chem. Soc. 2006, 128, 2977–2987; [DOI] [PubMed] [Google Scholar]
- 21b. Kabak K., Elmali A., Elerman Y., Durlu T. N., J. Mol. Struct. 2000, 553, 187–192. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary