

Abstract
It has been proposed that many human cancers are generated by intrinsic mechanisms that produce “Bad Luck” mutations by the proliferation of organ-specific adult stem cells. There have been serious challenges to this interpretation, including multiple extrinsic factors thought to be correlated with mutations found in cancers associated with these exposures. While support for both interpretations provides some validity, both interpretations ignore several concepts of the multistage, multimechanism process of carcinogenesis, namely, (1) mutations can be generated by both “errors of DNA repair” and “errors of DNA replication,” during the “initiation” process of carcinogenesis; (2) “initiated” stem cells must be clonally amplified by nonmutagenic, intrinsic or extrinsic epigenetic mechanisms; (3) organ-specific stem cell numbers can be modified during in utero development, thereby altering the risk to cancer later in life; and (4) epigenetic tumor promoters are characterized by species, individual genetic-, gender-, developmental state-specificities, and threshold levels to be active; sustained and long-term exposures; and exposures in the absence of antioxidant “antipromoters.” Because of the inevitability of some of the stem cells generating “initiating” mutations by either “errors of DNA repair” or “errors of DNA replication,” a tumor is formed depending on the promotion phase of carcinogenesis. While it is possible to reduce our frequencies of mutagenic “initiated” cells, one can never reduce it to zero. Because of the extended period of the promotion phase of carcinogenesis, strategies to reduce the appearance of cancers must involve the interruption of the promotion of these initiated cells.
Keywords: multistage concept of carcinogenesis, errors of DNA repair, errors of DNA replication, adult-organ-specific stem cells, epigenetic mechanisms, tumor promotion
Introduction: Are Mutations the “Drivers” of Human Carcinogenesis?
In the context of understanding the causes of human cancers for the ultimate purpose of prevention and treatment, more and more current paradigms, concepts, and sophisticate technologies are being used to develop practical means for “precision medicine.” In order for this to work for translational clinical trials at the individual level, or for broad public policy to reduce the population burden, it should be safe to say that one should not apply what one does not know. Therefore, it should be prudent to understand, as much as possible, the basic scientific mechanisms of the complex carcinogenic process.
In the context of understanding how exposures to any agent, for example, radiation, chemicals, or microbiological toxins, an organism will manifest a pathological response, the exposure dose, time of exposure, routes of exposure, individual genetic background, gender, developmental state, confounding additive, and synergistic or antagonistic mixtures of endogenous or exogenous agents must be considered. In the case of human carcinogenesis, it is assumed, up front, that cancers are not the result of a “one-hit” process. Given the well-accepted and experimentally demonstrated concept of the multistage, multimechanism process of carcinogenesis, mutagenesis, cytotoxicity, and epigenetic alterations of gene expression occur during the “initiation,” “promotion,” and “progression” stages. The question arises: “Is or are any of these 3 basic toxicological mechanisms involved in the pathogenesis of a cancer dependent on either the linear no-threshold or a threshold dynamic or some hermetic phenomenon?”
With the pressures of today, given the global metabolic disease crises, including cancer, one emerging, though aged, idea is that mutations are the “drivers” of human cancer, and potentially, other diseases. New technological screens to identify several genes, when mutated, are being used to alert a person that they might be “predisposed” to 1 type of cancer or not. With a small percentage of cancers having a major mutated gene that predisposes an organ-specific cancer and the fact that virtually all cells within a tumor contain mutations, it has, unfortunately, led to the conclusion that mutations are, indeed, the “drivers” of human carcinogenesis. With racial, ethnic, and geographical associations of certain kinds of cancer, the emergence of nongenetic, environmental, social, and cultural determinants need to be investigated as to how these factors might influence the role of mutations. That brings to this “Commentary” the idea that “epigenetic” mechanisms should be viewed as the “rate-limiting” step of human carcinogenesis.
In brief, this hypothesis does not negate the roles of mutations in hereditary or somatic human cancers, but rather focuses on the epigenetic role of natural (hormones, cytokines, and growth factors) and synthetic (pollutants, food additives, medications, diet, exercise, stress, etc) chemicals, which can alter the expression of genes, play in the ultimate appearance of a tumor.
Another problem that must be answered is how to explain certain characteristics of cancers, in order to find the most efficacious prevention and treatment strategies. To illustrate this with one example is the observation that human colon cancers that arise from the descending colon are “treatable,” while those colon cancers arising from the right, ascending colon are not treatable.1 Possibly by understanding the role of stem cells, mutations in the initiation phase of the multistep, multimechanisms of human carcinogenesis, the normal physiological roles of the different regions of the colon and the roles of the gut microbiome, one might develop testable hypotheses to see how understanding the roles of mutations and epigenetic mechanisms could influence cancer prevention and treatment.
Role of “Bad Luck” Mutations in Human Cancers
In humans, there are as many different types of cancer as different types of cells in the body— over 200 having diverse risk factors, incidence rates and geographic distribution. Tomasetti and Vogelstein2 proposed that the striking variation in lifetime risk of developing different types of cancer is primarily a consequence of a divergent number of noncancerous stem cell divisions, leading to random mutations during DNA replication. Peculiarly, the authors insinuate that “bad luck” is major determinant over environmental/inherited factors in at least two-thirds of tissue cancer risk variation. Firstly, it ought to be emphasized that, among the 31 tissue types considered by authors, the resulting R-tumors (replicative, having low extra risk score [ERS]) account for a mere 18% to 20% of all human cancers, while the resulting D-tumors (environmental/inherited, with high ERS), along with other major neoplasms (breast, prostate), strongly dependent on lifestyle/environmental factors but not considered in this study, account for 55% to 60% of cancers (National Cancer Institute - Surveillance, Epidemiology and End Results Program). Secondly, although the supplementary material provided by authors indicates the source of data used for their sensitivity analysis, calculations were made largely through arbitrary assumptions based on the variable results in the literature. Furthermore, it is unclear why human tissues such as breast and prostate, despite the characteristics of their stem cell populations have been investigated in a number of studies, have not been included in the analysis. Anyhow, as also admitted by authors, number and doublings of tissue stem cells may well be determined by environmentally dependent epigenetic mechanisms, whose potential impact is impossible to weigh in this study.
As one could expect, there has been substantive criticism3-12 to the conclusions drawn by Tomasetti and Vogelstein. A major challenge to the original idea that differences in inherent cellular processes, namely differential proliferation of organ-specific adult stem cells are the primary reason that some tissues have a higher risk to develop a cancer than others, has been raised recently.
In the history of science where there have been clashes of explanations for a major problem, in this case the cause(s) of human cancer, each side has often used some strong evidence to support its case. In this recent clash to find the “cause” of human cancer, given the important policy implications for the prevention and treatment of cancer, it seems that too many in the cancer field, today, have ignored some classic experimental evidence and concepts that could provide some insights to resolve this recent controversy.
Wu et al,13 while assuming that intrinsic stem cell proliferation rates and extrinsic factors were entirely independent, asked what happens when environmental exposures affect stem cell proliferation rates. This group used epidemiological data showing how migration of people from regions of lower cancer risk to those of higher risk soon develop diseases at rates consistent with their new home. Following up on their original paper and in response to the many criticisms to their original paper, Tomasetti and Volgelstein14 analyzed the different categories of cancer causation (heredity-H, Environmental-E, and mutations of stem cell replication-R) on a large data set of international cancer frequencies. While in agreement with the idea that “errors of DNA replication” of stem cells probably play a major role in human carcinogenesis,15 their analysis still failed to take into account (1) the multistage, multimechanism process of carcinogenesis, especially the nonmutagenic or epigenetic promotion phase of carcinogenesis and (2) the role of modulating the number of organ-specific adult stem cells during development.16-19
Presumptive Mechanisms in the “Initiation,” “Promotion,” “Progression” Model of the Multistep, Multimechanisms Process of Carcinogenesis
In experimental mice, rat, and rabbit animal models,20,21 it has been shown that carcinogenesis is a multistep, multimechanism process.22,23 This led to conceive the carcinogenetic process as an irreversible step that occurred in a single cell (the “initiation” step), after regular, chronic, and threshold levels of exposure to a different class of agents; and in the absence of agents that negate the effect of these agents (“promotion” step), this initiated cell could become clonally amplified to a premalignant lesion, such as a papilloma in the skin, a nodule in the breast, or a polyp in the colon.24 In time, one of those promoted initiated cells could accrue additional genetic and/or epigenetic changes, such that it could invade and metastasize to distal sites (the “progression” phase).
There has been no universal acceptance of the underlying cellular/molecular mechanisms for each of these completely distinct operational steps of carcinogenesis. However, because the initiation process seems to be irreversible, it was only logical to assume initiation came about in single cells by agents that damaged genomic DNA leading to a mutation. The classic case of ultraviolet (UV) light induction of pyrimidine dimers, which were not repaired,25 led to the production of gene mutations as “errors of DNA repair”26,27 in the skin cells of the human hereditary skin cancer-prone, xeroderma pigmentosum.28 The mutations in the oncogene of the skin tumors of these individuals appear to be primarily related to the unrepaired UV-induced pyrimidine dimers.29 Mutations in the genome of cells that can give rise to an initiated cell can be found in another human hereditary cancer-prone syndrome, the Bloom syndrome.30 In this case, mutations can be produced by “errors of DNA replication,”31 Herein lays a major implication of this process of mutation induction. While one can clearly lower the risk of being initiated, such as in Caucasians reducing their exposure to UV light from the sun, human beings cannot reduce their risk to zero of getting an “initiated” cell in one organ or another.
Another series of experimental findings seemed to lead scientists to the conclusion that the cancer originated in a single cell.32-35 However, regardless of how the mutation is formed in a cell to cause it to be “initiated,” that single cell is still not a metastatic cancer cell. That single initiated cell is normally suppressed by surrounding normal cells by either secreted antiproliferative factors or by direct gap junctional intercellular communication, until it is exposed to agents, such as endogenous hormones, growth factors, cytokines, and so on; endogenous agents (pollutants, pesticides, drugs, food additives, solid particles, etc)36; or conditions that stimulate compensatory hyperplasia, such as wound healing or massive cell killing.37 This removal of clonal suppression of the “initiated” cell and its subsequent clonal amplification is the biological process underlying the tumor promotion phase. This promotion process must also prevent or inhibit the apoptotic loss of these stimulated initiated cells.38 Promoters, classified by the manner that stimulates cell proliferation and prevents apoptosis of these initiated cells, act as “epigenetic” agents.39 That is, these “promoting” agents alter the expression of genomic DNA of initiated cells at the transcriptional, translational, and posttranslational levels.
In this respect, convincing evidence is accumulating that many bioactive dietary components may extensively modulate epigenetic mechanisms, eventually leading to a rapid and effective regulation of gene expression and function in response to nutritional changes. In this respect, the term epigenetic diet has been introduced to indicate the consumption of foods, such as soy, grapes, cruciferous vegetables, and green tea, that affect epigenetic mechanisms to protect against cancer and aging.40 It is noteworthy that unbalanced maternal nutrient intake may severely impact on fetal epigenome early during in utero development. There is increasing evidence that nutritionally induced epigenetic alteration of the offspring’s epigenome may be responsible for higher susceptibility to cancer development later in life41 and that several epigenetic marks can be inherited and reshape developmental and cellular features over generations, a phenomenon referred to as epigenetic inheritance.42
Moreover, promoting agents, such as phorbol esters, dichlorodiphenyltrichloroethane, 2,3,7,8-Tetrachlorodibenzo-p-dioxin, phenobarbital, phthalates, and so on, exhibit threshold levels, species, gender, and developmental stage specificities.43 In addition, the initiated cell must be exposed to these agents in a regular and sustained chronic fashion.44,45 Equally as important, these initiated cells must be exposed to these promoting agents and conditions in the absence of “antipromoting” agents that can negate the effects of the promoters.46 Examples of this class of chemicals of antipromoters can be resveratrol,47 green tea components,48 and metformin.49
While most humans die before a tumor is detected, all of us have initiated cells in our bodies in most, if not all, of our organs. Those that develop a tumor in one organ or another before they die had an initiated cell promoted by exposure to some factor, such as specific dietary components, a pollutant, some medication, life-style behavior (smoking, alcoholism), infection, chronic inflammation and, in addition, not being exposed to antipromoters. Even all those who do not develop a cancer can be subjected to these promoting agents and conditions, but their initiated cells might be exposed to these agents at below threshold levels50 or being exposed simultaneously to the presence of antipromoters. This is not to negate genetic or gender factors that can play a role in suppressing the initiated cell from being promoted.
While it is beyond the goal of this Commentary, the question of what is that single cell that can be “initiated” and, subsequently, promoted to become a malignant cell is of outmost importance. The classic “stem cell” hypothesis34,35,51-53 against the “dedifferentiation” or the “reprogrammed” hypothesis54 has been offered up as opposing hypothesis of the origin of single “initiated” cell. Again, acknowledging that the answer to this question is not universally accepted, there is strong evidence that adult organ-specific stem cells exist in the skin, intestine, liver, pancreas, eye, breast, blood, immune system. It is universally recognized that all tumors are composites of heterogeneous cancer and normal cells, and today, most accept that all tumors are sustained by “cancer-initiating stem cells” or “cancer stem cells,” while being surrounded by normal stromal cells, cancer non-stem cells, and other invading cells.55 These “cancer stem cells” seem to be more resistant to toxic physical agents such as radiation and toxic chemicals.56
Finally, to the “clash” between the 2 opposite views as to the “cause” of human cancers, mutations do play a role in the “initiation” of the carcinogenic process. However, with the exception of the mutations caused by exposure to UV light, that are associated with the mutations in oncogenes of skin cancer (“errors of DNA repair”), most mutations are probably induced by “errors of DNA replication” in stem cells that are stimulated to proliferate because of growth, wound healing, massive cell death or because of exposure to endogenous or exogenous agents (epigenetic promoters) that can cause stimulation of these “error of replication”-initiated cells. Recent demonstration of endogenous and exogenous nonmutagenic agents that can stimulate organ-specific stem cells57,58 do provide evidence for the suggestion posed by Wu et al.13
Is there any doubt that nonsmokers can get lung cancers? When looking for any potential “molecular fingerprint” for mutations found in oncogenes in cells of lung cancers of smokers versus nonsmokers, Thilly found the molecular signature of both types of tumors were similar.59 This suggests that the initiated event in both types of lung cancers were caused by “errors in DNA replication” caused by epigenetic promoters that stimulated the proliferation of the adult lung stem cells. In the case of the smokers, chemicals in the cigarette smoke have been shown to be more of a promoter than of being an initiator.60,61 On the other hand, since promoters represent a large class of structurally different endogenous or exogenous epigenetic chemicals, it is unknown what the promoter(s) is (are) in the case of the nonsmoker’s lung cancer. It seems that the clash between the 2 interpretations of differential cancer types had an element of truth in both cases. That which seems to bring the 2 interpretations together is a better understanding of the classic “initiation/promotion/progression” model of carcinogenesis (which, unfortunately, neither many molecular oncologists nor epidemiologists seem to use as their guiding paradigm), together with recent identification of organ-specific stem cells and the mechanism of action of these promoting epigenetic chemicals.
If one assumes for the moment that the organ-specific stem cell is the target cell for the “initiation” of the carcinogenic process, then it follows that by increasing or decreasing the organ-specific stem cell numbers, one should alter the risk for cancers. Consequently, the original question posed by Wu et al,13 “What happens when environmental exposures affect stem cell proliferation rates?,” seems to be supported by both experimental animal and cellular studies showing that endogenous and exogenous epigenetic chemicals can affect organ-specific stem cell behavior. It has been postulated that altering the organ-specific stem cell numbers, especially during fetal development, might explain the Barker hypothesis.62,63 The low frequency of breast cancer in Japanese women, during the Second World War, seen in the study of the atomic bomb survivors,64 might have been the result of the unique diet of those women (caloric restriction, soy-product prevalence, green tea, no smoking, vegetables, etc).65 Since many chemicals have been shown to induce either terminal differentiation or apoptosis of human adult breast stem cells,66 the exposure to dietary components during pregnancy of the female fetus might have reduced the numbers of breast stem cells, such that at puberty the breast tissue would be smaller and the number of adult breast stem cells to be targets for the initiation of breast cancer would be limited. With the current change in the Japanese and the diaspora of Japanese to other countries have changed the patterns of breast cancer frequencies compared to the Japanese women during the Second World War.
The fact the human beings may only partly control DNA mutations by reducing exposure to genotoxic chemicals in the environment or in their diet but cannot set to zero the risk of developing cancer rises the important question as to whether or not it would be more productive to direct most public resources to the design and implementation of primary prevention strategies to avoid or restrain cancer promotion rather than to the cure of cancer. In any case, yet researchers and clinicians need to place common efforts in uniting their own knowledge and abilities to create a perspective large enough to enlighten cancer understanding and to provide effectual solutions to major open questions in both prevention and treatment of human malignancies (see Figure 1).
Figure 1.
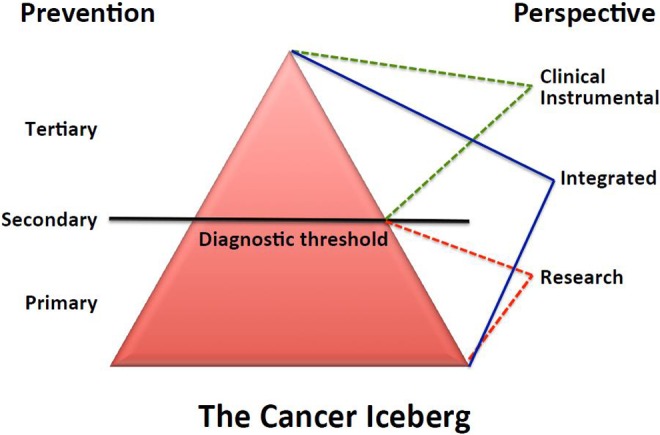
The cancer iceberg. The whole cancer process can be viewed as an iceberg where only the minor emerging part can be perceived from either clinical or instrumental viewpoints. Conversely, the bulk underwater part can be appreciated, though partly, by researchers who in turn miss the iceberg tip. Only an integrated perspective can encompass and provide important inferences for cancer prevention, from primary (control of risk factors) to secondary (early diagnosis) and tertiary (therapy and rehabilitation) type of prevention.
Summary
It is proposed that mutations, as a result of both “errors of DNA repair” and “errors of DNA replication” (The “bad luck” variety), do play a critical role in the initiation step of human carcinogenesis in an organ-specific adult stem cell; the process of clonally amplifying this single initiated step cell during the epigenetic tumor promotion process is the rate-limiting step of the multistage, multimechanism process of human carcinogenesis. This tumor promotion process depends on exposure to threshold levels of the promoting agent, in a regular, chronic sustained fashion, in the absence of antitumor promoters. Since this tumor promotion process takes a long time, it is the step by which efficacious intervention for prevention can take place, since it will never be possible to prevent the initiation step to a zero risk level. Every time a stem cell is forced to proliferate (especially during development), there can always be a finite chance for an “error of DNA replication” or “bad luck” mutation. This initiation step in the adult stem cell probably stops the terminal differentiation of the stem cell, allowing it to accrue more mutations (gene, chromosomal) and epigenetic alterations (during the promotion stage) to take place during its evolution to the “progression,” invasive, metastatic step. So while, strong unequivocal evidence does not exist showing a nonlinear basis of mutagenesis, it seems that both protective mechanisms preventing DNA damage and known DNA repair mechanisms would suggest that the initiation phase must have some “threshold.” On the other hand, the promotion phase, the rate-limiting step of carcinogenesis, clearly exhibits a threshold dynamic.
Footnotes
Declaration of Conflicting Interests: The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The author(s) received no financial support for the research, authorship, and/or publication of this article.
References
- 1. Trosko JE, Lenz HJ. What roles do colon stem cells and gap junctions play in the left and right location of origin of colorectal cancers? J Cell Commun Signal. 2017;11(1):79–87. doi:10.1007/s12079-017-0381-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Tomasetti C, Vogelstein B. Variation in cancer risk among tissues can be explained by the number of stem cell division. Science. 2015;347(6217):78–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Couzin-Frankel J. The bad luck of cancer. Science. 2015;347(6217):12. [DOI] [PubMed] [Google Scholar]
- 4. Couzin-Frankel J. Backlash greets ‘bad luck’ cancer study and coverage. Science. 2015;347(6219):224. [DOI] [PubMed] [Google Scholar]
- 5. Wodarz D, Zauber AG. Risk factors and random chances. Nature. 2015;517(7536):563–564. [DOI] [PubMed] [Google Scholar]
- 6. Ashford NA, Bauman P, Brown HS, et al. Cancer risk: role of the environment. Science. 2015;347(6223):727. [DOI] [PubMed] [Google Scholar]
- 7. Potter JD, Prentise RL. Cancer risk: tumors excluded. Science. 2015;347(6223):727. [DOI] [PubMed] [Google Scholar]
- 8. Wild C, Brennan P, Plummer M, Bray F, Straif K, Zavadil J. Cancer risk: role of chance overstated. Science. 2015;347(6223):728. [DOI] [PubMed] [Google Scholar]
- 9. Gotay C, Dummer T, Spinelli J. Cancer risk: prevention is is crucial. Science. 2015;347(6223):728. [DOI] [PubMed] [Google Scholar]
- 10. Song M, Giovannucci EL. Cancer risk: many factors contribute. Science. 2015;347(6223):728–729. [DOI] [PubMed] [Google Scholar]
- 11. Tomasetti C, Vogelstein B. Cancer risk: role of environment—Response. Science. 2015;347(6223):729–730. [DOI] [PubMed] [Google Scholar]
- 12. Ledford H. Cancer studies clash. Nature. 2015;528(7582):317. [DOI] [PubMed] [Google Scholar]
- 13. Wu S, Powers S, Zhu W, et al. Substantial contribution of extrinsic risk factors to cancer development. Nature. 2016;529(7584):43–47. doi:10.1038/nature16166;2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tomasetti C, Li L, Vogelstein B. Stem cell divisions, somatic mutations, cancer etiology, and cancer prevention. Science. 2017;355(6331):1330–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Trosko JE. Evolution of microbial quorum sensing to human global quorum sensing: an insight to how gap junctional intercellular communication might be linked to the global metabolic disease crisis. Biology. 2016;5(2): doi:10.3390/biology5020029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Trosko JE. A conceptual integration of extra-, intra-, and Gap junctional inter- cellular communication in the evolution of multi-cellularity and stem cells: How disrupted cell-cell communication during development can affect diseases later in life. Int J Stem Cell Res Ther. 2016;3(1):1–6. ISSN: 2469-570X. [Google Scholar]
- 17. Trosko JE. Global Health Crisis Caused by the Collision of Biological and Cultural Evolution: Pre-Natal Influences on Acute and Chronic Diseases in Later Life 2014;2(4):271–280. [Google Scholar]
- 18. Trosko JE. Induction of iPS Cells and of Cancer Stem Cells: The Stem Cell or Reprogramming Hypothesis of Cancer? Anat Rec (Hoboken). 2014;297(1):161–173. [DOI] [PubMed] [Google Scholar]
- 19. Trosko JE. Evolution of energy metabolism, stem cells and cancer stem cells: how the Warburg and Barker hypothesis might be linked. BMC Proc. 2016;7(suppl 2):K8 http://www.biomedcentral.com/1753-6561/7/S2/K8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Berenblum PM. A speculative review: the probable nature of promoting action and its significance in the understanding of the mechanisms of carcinogenesis. Cancer Res. 1954;14(7):471–477. [PubMed] [Google Scholar]
- 21. Yamagiwa K, Ichikawa K. Experimental study of the pathogenesis of carcinoma. CA Cancer J Clin. 1977;27(3):174–181. [DOI] [PubMed] [Google Scholar]
- 22. Weinstein IB, Gattoni CS, Kirschmeier P, et al. Multistage carcinogenesis involves multiple genes and multiple mechanisms. J Cell Physiol. 1984;3(suppl):127–137. [DOI] [PubMed] [Google Scholar]
- 23. Pitot HC, Dragon YP. Facts and theories concerning the mechanism of carcinogenesis. FASEB J. 1991;5(9):2280–2286. [PubMed] [Google Scholar]
- 24. Trosko JE, Tai MH, Sopczynski B, et al. Diet/nutrition, inflammation, cellular senescence, stem cells, diseases of aging and aging In: Rahman I, Bagchi D, eds. Inflammation, Advancing Age and Nutrition. Amsterdam, Netherlands: Elsevier; 2013:125–144. [Google Scholar]
- 25. Cleaver JE, Trosko JE. Absence of excision of ultraviolet-induced cyclobutane dimers in Xeroderma pigmentosum. Photochem Photobiol. 1970;11(6):547–550. [DOI] [PubMed] [Google Scholar]
- 26. Maher VM, McCormick JJ. Effect of DNA repair on the cytotoxicity and mutagenicity of UV irradiation and of chemical carcinogens in normal and xeroderma pigmentosum cells In: Yuhas JM, Tennant RW, Regan JD, eds. Biology of Radiation Carcinogenesis (pp. 129–145). New York, NY: Raven Press; 1976. [Google Scholar]
- 27. Glover TW, Chang CC, Trosko JE, et al. Ultraviolet light induction of diphtheria toxin resistant mutations in normal and xeroderma pigmentosum human fibroblasts. Proc Natl Acad Sci. 1979;76(8):3982–3986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Cleaver JE. Xeroderma pigmentosum: genetic and environmental influences in skin carcinogenesis. J Dermatol. 1978;17(6):435–444. [DOI] [PubMed] [Google Scholar]
- 29. Brash DE, Rudolph JE, Simon JA, et al. A role for sunlight in skin cancer: UV-induced p53 mutations in squamous cell carcinomas. Proc Natl Acad Sci USA. 1991;88(22):10124–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. German J. Bloom syndrome: a mendelian protype: somatic mutations and disease. Medicine. 1993;72(6):393–406. [PubMed] [Google Scholar]
- 31. Warren ST, Schultz RA, Chang CC, Wade MH, Trosko JE. Elevated spontaneous mutation rate in Bloom syndrome fibroblasts. Proc Natl Acad Sci U S A. 1981;78(5):3133–3137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Fialkow PJ, Gartler SM, Yoshida A. Clonal origin of chronic myelopcytic leukemia in man. Proc Natl Acad Sci USA. 1967;58(4):1468–1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Nowell PC. The clonal evolution of tumor cell populations. Science. 1976;194(4260):23–28. [DOI] [PubMed] [Google Scholar]
- 34. Till JE. Stem cells in differentiation and neoplasia. J Cell Physiol Suppl. 1982;1(suppl.S1):3–11. [DOI] [PubMed] [Google Scholar]
- 35. Greaves MF. Differentiation-linked leukemiogenesis in lymphocytes. Science. 1986;234(4777):697–704. [DOI] [PubMed] [Google Scholar]
- 36. Trosko JE, Chang CC. Nongenotoxic mechanisms in carcinogenesis: Role of inhibited intercellular communication In: Hart R, Hoerger FD, eds. Banbury Report 31: New Directions in the Qualitative and Quantitative Aspects of Carcinogen Risk Assessment. New York, NY: Cold Spring Harbor Press, Cold Spring Harbor; 1989:139–170. [Google Scholar]
- 37. Argyris TS. Regeneration and the mechanism of epidermal tumor promotion. CRC Crit Rev Toxicol. 1985;14(3):211–258. [DOI] [PubMed] [Google Scholar]
- 38. Schulte-Hermann R, Grasl-Kraupp B, Bursch W. Dose-response and threshold effects in cytotoxicity and apoptosis. Mutat Res. 2000;464(1):13–18. [DOI] [PubMed] [Google Scholar]
- 39. Upham BL, Trosko JE. Carcinogenic tumor promotion, induced oxidative stress signaling, modulated gap junction function and altered gene expression. Antioxidants Redox Signaling. 2009;11(2):297–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Leone A, Longo C, Trosko JE. The chemopreventive role of dietary phytochemicals through gap junctional intercellular communication. Phytochem Rev. 2012;11:285–307. doi:10.1007/s11101-012- 9235-7. [Google Scholar]
- 41. Thornburg KL, Shannon J, Thuillier P, et al. In utero life and epigenetic predisposition for disease. Adv Genet. 2010;71:57–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bohacek J, Mansuy IM. Epigenetic inheritance of disease and disease risk. Neuropsychopharm Rev. 2013;38(1):220–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Trosko JE. Is the concept of ‘tumor promotion’ a useful paradigm? Mol Carcinogen. 2001;30(3):131–137. [PubMed] [Google Scholar]
- 44. Trosko JE, Ruch RJ. Cell-cell communication in carcinogenesis. Front Biosci. 1998;3:d208–d236. [DOI] [PubMed] [Google Scholar]
- 45. Boutwell RK, Verma AK, Ashendel CL, et al. Mouse skin: a useful model system for studying the mechanism of chemical carcinogenesis. In: Hecker E, Kunz W, Fusenig NE, Marks F, Theilmann HW, eds. Carcinogenesis. vol 7 New York, NY: Raven Press; 1982:1–12. [PubMed] [Google Scholar]
- 46. Trosko JE, Ruch RJ. Gap junctions as targets for cancer chemoprevention and chemotherapy. Curr Drug Targets. 2002;3(6):465–482. [DOI] [PubMed] [Google Scholar]
- 47. Upham B, Guzvic M, Scott J, et al. Inhibition of gap junctional intercellular communication and activation of mitogen-activation protein kinase by tumor-promoting organic peroxides and protection by resveratrol. Nutr Cancer. 2007;57(1):38–47. [DOI] [PubMed] [Google Scholar]
- 48. Sai K, Kanno JR, Hasegawa R, et al. Prevention of the down-regulation of gap junctional intercellular communication by green tea in the liver of mice fed pentachlorophenol. Carcinogenesis. 2007;21(9):1671–1676. [DOI] [PubMed] [Google Scholar]
- 49. Jung JW, Park SB, Lee SJ, et al. Metformin represses self-renewal of the human breast carcinoma stem cells via inhibition of estrogen receptor-mediated OCT4 expression. PLoS One. 2011;6(11):e28068 doi:10.1371/journal.pone.0028068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Williams GM, Latropoulos MJ, Jeffrey AM. Thresholds for the effects of 2-acetyaminofluorene in rat liver. Toxicol Pathol. 2004;32(suppl 2):85–91. [DOI] [PubMed] [Google Scholar]
- 51. Markert CL. Neoplasia: a disease of differentiation. Cancer Res. 1968;28(9):1908–1914. [PubMed] [Google Scholar]
- 52. Pierce B. Neoplasms, differentiation and mutations. Am J Pathol. 1974;77(1):103–118. [PMC free article] [PubMed] [Google Scholar]
- 53. Tai MH, Chang CC, kiupel M, et al. Oct-4 expression in adult human stem cells: evidence in support of the stem cell theory of carcinogenesis. Carcinogenesis. 2005;26(2):495–502. [DOI] [PubMed] [Google Scholar]
- 54. Sell S. Cellular origin of cancer: differentiation or stem cell maturation arrest? Environ Health Perspect. 1993;101(5):15–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. O’Brien CA, Kreso A, Dick JE. Cancer stem cells in solid tumors: an overview. Semin Radiat Oncol. 2009;19(2):71–77. [DOI] [PubMed] [Google Scholar]
- 56. Dean M, Fojo T, Bates S. Tumor stem cells and drug resistance. Nat Rev Cancer. 2005;5(4):275–284. [DOI] [PubMed] [Google Scholar]
- 57. Bessede E, Dubus P, Megraud F, Varon C. Heliobacter pylori infection and stem cells at the origin of gastric cancer. Oncogene. 2014;34(20):2547–2555. [DOI] [PubMed] [Google Scholar]
- 58. Lindemans CA, Calafiore M, Mertelsmann AM, et al. Interleukin-22 promotes intestinal -stem cell-mediated epithelial regeneration. Nature. 2015;528(7583):560–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Thilly WG. Have environmental mutagens caused oncomutations in people? Nat Genet. 2004;34(3):255–259. [DOI] [PubMed] [Google Scholar]
- 60. Upham BL, Weis LM, Trosko JE. Modulated gap junctional intercellular communication as a biomarker of PAH’s epigenetic toxicity in structure/function relationship. Environ Health Perspect. 1998;106(suppl 4):975–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Upham BL, Blaha L, Babica P, et al. Tumor promoting properties of a cigarette smoke prevalent polycyclic aromatic hydrocarbon as indicated by the inhibition of gap junctional intercellular communication via phosphatidylcholine-specific phospholipase C. Cancer Sci. 2008;99(4):696–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Trosko JE. Role of diet and nutrition on the alteration of the quality and quantity of stem cells in human aging and the diseases of aging. Curr Pharm Des. 2008;14(26):2707–2718. [DOI] [PubMed] [Google Scholar]
- 63. Barker DJP. The developmental origins of adult disease. J Am Coll Nutr. 2004;23(suppl 6):588s–595s. [DOI] [PubMed] [Google Scholar]
- 64. Thompson DE, Mabuchi K, Ron E, et al. Cancer incidence in atomic bomb survivors. Part II: Solid tumors 1958-1987. Radiat Res. 1994;137(suppl 2):S17–S67. [PubMed] [Google Scholar]
- 65. Trosko JE, Suzuki K. Adult stem cells, the Barker Hypothesis, epigenetic events and low level radiation effects In: Nakashima M, Takamura N, Tsukasaki K, Nagayama Y, Yamashita S, eds. Radiation Health Risk Sciences. Tokyo, Japan: Springer Publisher; 2009:216–226. [Google Scholar]
- 66. Hsieh CY, Chang CC. Stem cell differentiation and reduction as a potential mechanism for chemoprevention of breast cancer. Chinese Pharm J. 1999;51(1):15–30. [Google Scholar]