

Introduction
Ovotesticular disorder of sexual development (otDSD) is characterized by the presence of testicular and ovarian tissue in the same individual.1 These patients generally present at birth with ambiguous genitalia.1–4 Karyotypes vary including 46,XX, 46,XY, and several mosaic and aneuploidic forms.1, 2, 5 Here we report on two phenotypic males who presented late during adolescence and were found to have otDSD.
Patient presentations
Case Presentation 1
A 15-year-old phenotypic male presented late with concern for testicular rupture following a bike straddle injury. He reported initial scrotal swelling and pain but waited for self-resolution. He denied dysuria, gross hematuria, or fever. Notably, he had a history of penile surgery during infancy.
On exam, he was a well-appearing, non-dysmorphic adolescent without gynecomastia. Examination showed circumcised phallus with orthotopic meatus and well-healed ventral shaft scar, Tanner V development, normal left palpable testis, and moderately enlarged, tender right hemiscrotum.
Ultrasound revealed possible right testicular rupture with hematocele (Figure 1A). A 1.7 cm circular structure abutting the cleavage plane of the right testis demonstrated increased flow on both color Doppler and contrast enhanced ultrasound (Figure 1B). Tumor markers were normal.
Figure 1.
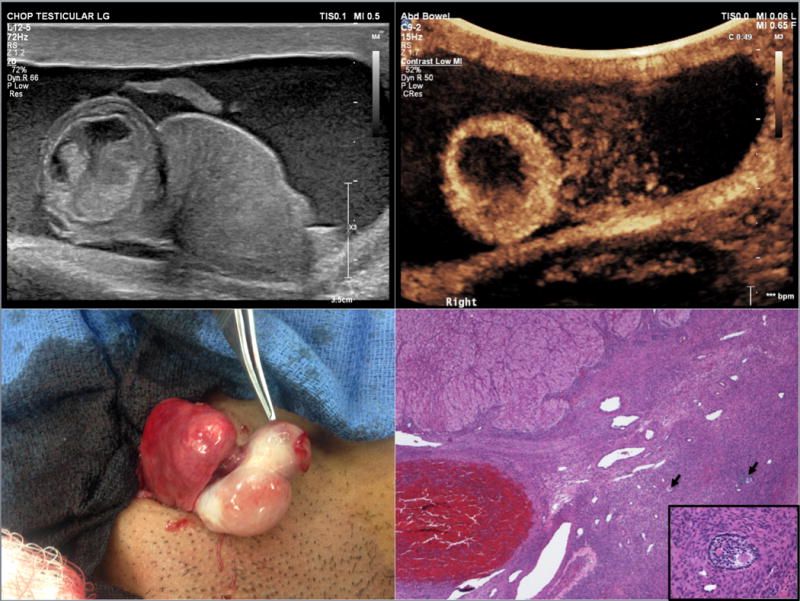
15-year-old phenotypic male who presented with testicular rupture. (A) Right testicular rupture with upper pole mass and hematocele. (B) After intravenous administration of ultrasound contrast there is marked peripheral enhancement of the right upper pole mass. The lower half of the testis does not demonstrate homogenous enhancement. (C) Intraoperative gross pathology demonstrating the abnormal round mass at the superior pole of the right testis (indicated by forceps). (D) Ovarian stroma with primary and secondary follicles (→), corpus luteum in upper left corner, and corpora albicans in the lower center, (4× magnification; Hematoxylin and eosin stain); inset: secondary follicle, (40× magnification; Hematoxylin and eosin stain)
The patient underwent inguinal exploration. In addition to findings of testicular rupture, a mass was seen at the superior pole of the testis consistent with ultrasound findings (Figure 1C). This was excised. Frozen section showed ovarian tissue (luteinized cells and follicular cells) with peripheral seminiferous tubules. Karyotype, testosterone (T), luteinizing hormone (LH), and follicle stimulating hormone (FSH) were sent. A testicular biopsy was performed. Remaining testicular tissue was preserved.
Permanent pathology sections showed ovarian stroma with corpora albicantia, follicles, and a hemorrhagic luteinized cyst (Figure 1D). Hyalinized seminiferous tubules were identified in the testis biopsy. T and LH were decreased, FSH was normal, and karyotype returned 46,XX.
Closer questioning revealed proximal hypospadias at birth but always bilateral palpable descended gonads. Genetic conditions or familial DSD were denied. Fluorescence-in-situ-hybridization (FISH) for SRY demonstrated 46,XX and no SRY gene. Chromosomal single nucleotide polymorphism (SNP) microarray revealed the same karyotype and no copy number changes with SRY, SOX3, SOX9, or NR0B1. The diagnosis was thus SRY-negative 46,XX otDSD. The patient’s phenotype indicated he had either an undetected mosaicism with a Y-bearing cell line or a single-gene mutation that allowed differentiation downstream of SRY. Patient has not yet returned for follow-up.
Case Presentation 2
A 15-year-old phenotypic male presented with worsening asymptomatic gynecomastia since age 12. He denied medication or marijuana use. Examination showed a well-appearing adolescent with Tanner IV breast tissue, Tanner V pubic hair, and 3 cm mobile mass anterosuperior to the left gonad. He had noted the scrotal mass one year prior but thought it was normal. He denied any pain or changes in mass size. T was low, LH and FSH were normal, and prolactin and estradiol were elevated.
Ultrasound showed a focal cystic mass with fluid level contained within an intratesticular cyst on the right (Figure 2A). No normal appearing left testis was demonstrated; there was a mass-like heterogeneous structure (5.0 × 1.0 × 1.4 cm) with multiple small cysts and echogenic linear structure (Figure 2B).
Figure 2.
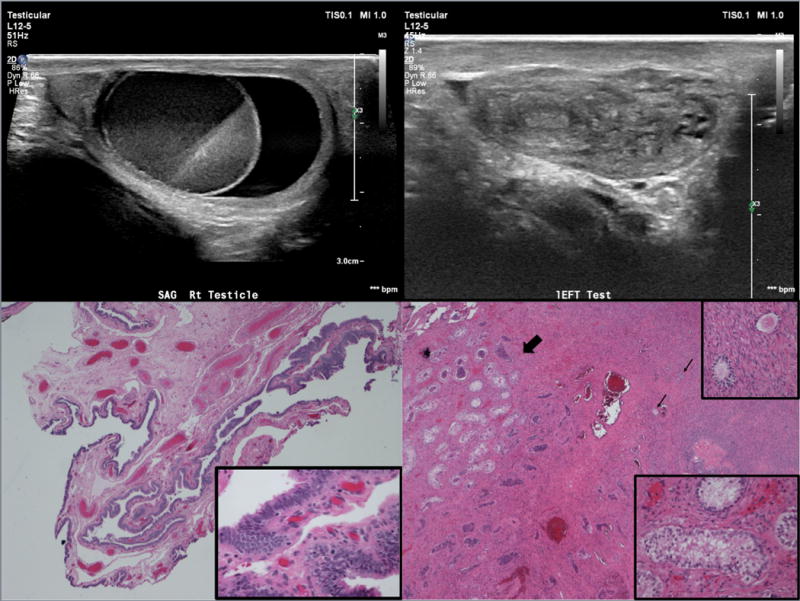
15-year-old phenotypic male who presented with gynecomastia. (A) Longitudinal scan of the right testis demonstrates a cystic mass with fluid-fluid level contained within a cystic testis surrounded by a thin rim of normal appearing testicular parenchyma. (B) The left hemiscrotum is filled with a heterogenous mass. There are echogenic structures as well as small cysts. No normal testicular tissue is identified. (C) Fibrovascular stroma predominantly lined by ciliated columnar cells consistent with fallopian tube tissue, (4× magnification; Hematoxylin and eosin stain); inset: Higher power of ciliated columnar cell lining, (40× magnification; Hematoxylin and eosin stain) (D) Fibrous tissue with Sertoli-only seminiferous tubules (➜) and ovarian stroma with corpora albicans, primary follicles (→), (4× magnification; Hematoxylin and eosin stain); right upper inset: Higher power of primary follicles, (40× magnification; Hematoxylin and eosin stain); right lower inset: Higher power of Sertoli-only seminiferous tubules, (40× magnification; Hematoxlyin and eosin stain)
Chest X-ray and abdomen/pelvis CT were normal. Tumor markers were normal.
The patient underwent bilateral inguinal exploration. The left scrotal mass resembled an ovary with fallopian tube and hemi-uterus; respective frozen section biopsies showed a corpus luteum cyst and endometrial tissue. All structures were removed en bloc. The right scrotal mass contained both testicular tissue and abnormal mass. Frozen section of the mass revealed focal oocytes, focal rete, and focal Sertoli cell-only seminiferous tubules. The mass was removed. A testicular biopsy frozen section revealed Sertoli cell-only tissue. Remaining testicular tissue was preserved.
Permanent pathology sections showed a left ovary with hemorrhagic corpus luteum cyst (3.8 cm) and cystic ovarian follicles; a fallopian tube with a small paratubal cyst (0.6 cm) (Figure 2C); and a hemi-uterus with benign endometrium and myometrium. The right side revealed an ovotestis with cystic ovarian follicles and Sertoli-cell only seminiferous tubules separated by ovarian and fibrous stroma (Figure 2D).
The patient’s blood karyotype was 46,XX[26]/47,XXY[4] consistent with mosaic Klinefelter Syndrome (KS) with 46,XX predominance, suggesting loss of Y chromosome over time or an undetected predominance of the 47,XXY cell line in the genitourinary region. Prolactin normalized postoperatively but T remained low. He is being followed for androgen replacement therapy.
Discussion
Here we present two patients with otDSD on histopathological exam but with very different initial clinical presentations, imaging, and surgical findings. This is consistent with the current model of gonad development, which acknowledges that directionality towards testicular versus ovarian development is exquisitely sensitive. Perturbation, from a variety of underlying diagnoses, can manifest with ovotesticular gonads, along with a variety of other malformations as determined by the overarching diagnosis. This heterogeneity complicates studying the otDSD population. Published reports have varying outcome measures, ages of diagnosis, years of follow-up, and mixed populations. Additionally, gender assignment surgery at a young age further complicates analyses.
Reports of delayed otDSD diagnosis are rare (Table) and often come from developing countries.1, 6–9 Clinical features often depend on the underlying karyotype. The most common karyotype in otDSD is 46,XX,1, 2, 4, 5 as in our first patient. However, findings of 46,XX/47,XXY (mosaic KS) in the setting of otDSD are much more rare. A large series of 20 otDSD Brazilian patients had a mean age of diagnosis of 11 years.1 Clinical characteristics included bilateral gynecomastia and cyclical hematuria. Karyotype was 46,XX in 18 and 46,XX/46,XY in 2 patients. In a case report from India, a 16-year-old phenotypic male presented with cyclic hematuria.6 He had bilateral gynecomastia, which is common in patients with KS and other DSDs. Karyotype was 46,XX/47,XXY. He had sparse axillary hair, Tanner III feminine-type pubic hair distribution, and left cryptorchidism. Work-up revealed a mass behind the bladder; surgical exploration found an ovary, uterus, and fallopian tube. Another case report from Turkey described a 14-year-old phenotypic female who presented with masculinization at puberty.9 Karyotype was 46,XX, SRY(-). Other reports of delayed otDSD diagnosis are shown in the Table. Of note, the KS population is at increased risk of developing malignancy, especially extragonadal germ cell tumors with a reported risk of 66.7%.10
Table.
Reports of delayed diagnosis or presentation of ovotesticular disorder of sexual differentiation.
| Reference | Country | No. patients | Mean age at diagnosis or presentation | Karyotype characteristics | Clinical characteristics |
|---|---|---|---|---|---|
| Sircili1 | Brazil | 20 | 11 years | 46,XX (n=18) 46,XX/46,XY (n=2) |
Gynecomastia, cyclical hematuria |
| Talreja6 | India | 1 | 16 years | 46,XX/47,XXY Klinefelter Syndrome |
Cyclical hematuria, gynecomastia, left cryptorchidism |
| Isguven7 | Turkey | 1 | 14 years | 46,XX/47,XXY Klinefelter Syndrome |
Gynecomastia, cyclical hematuria, eunuchoid body habitus, mild mental retardation |
| Eberenz8 | United States | 1 | 15 years | 46,XX | Gynecomastia, intermittent scrotal pain/swelling |
| Selver Eklioglu9 | Turkey | 1 | 14 years | 46,XX, SRY(-) | Pubertal masculinization, deafness, visual impairment, short stature |
The imaging diagnosis of ovotestis can be challenging in the absence of appropriate history and focused search. The presence of ovotestis in the scrotum can mimic testicular or paratesticular tumors as well as a hematoma. Ovotestis can be suspected if multiple cysts are present.8 In their absence the diagnosis becomes difficult.
Conclusion
OtDSD is a rare finding that is heterogenous in its genetic etiology, clinical presentation, and surgical findings. While many patients are diagnosed in infancy or childhood, we presented two cases of otDSD diagnosed in adolescence including a rare case of ovotestis in a patient with Klinefelter mosaicism and a second one in combination with testicular rupture. Both patients likely presented in a delayed fashion due to normal phenotypic male development over time and will require long-term follow-up to assess for hormonal status and sexual function. These cases highlight the need for high index of suspicion for DSD in the adolescent with abnormal pubertal development or unusual genital findings.
Acknowledgments
Support/Financial Disclosures:
Jyoti D. Chouhan- none
David I. Chu- Source of extra-institutional funding: T32 DK00778514 from the National Institute of Diabetes and Digestive and Kidney Diseases. The NIDDK had no role in the design and conduct of the study; collection, management, analysis, and interpretation of the data; and preparation, review, or approval of the manuscript. The views expressed in this article are those of the authors and do not necessarily represent the official view of the NIDDK. Funded by the National Institutes of Health (NIH).
Antoinette Birs- none
Louise C. Pyle- Source of extra-institutional funding: T32 GM00863820 from the National Institute of General Medical Sciences. The NIGMS had no role in the design and conduct of the study; collection, management, analysis, and interpretation of the data; and preparation, review, or approval of the manuscript. The views expressed in this article are those of the authors and do not necessarily represent the official view of the NIGMS. Funded by the National Institutes of Health (NIH).
Jason P. Van Batavia- none
Rebecca L. Linn- none
Susan J. Back- none
Pierre Russo- none
Kassa Darge- none
Thomas F. Kolon- none
Arun K. Srinivasan- none
Footnotes
Conflicts of Interest: None
References
- 1.Sircili MH, Denes FT, Costa EM, et al. Long-term followup of a large cohort of patients with ovotesticular disorder of sex development. J Urol. 2014;191:1532–1536. doi: 10.1016/j.juro.2013.10.037. [DOI] [PubMed] [Google Scholar]
- 2.Matsui F, Shimada K, Matsumoto F, et al. Long-term outcome of ovotesticular disorder of sex development: a single center experience. Int J Urol. 2011;18:231–236. doi: 10.1111/j.1442-2042.2010.02700.x. [DOI] [PubMed] [Google Scholar]
- 3.Kojima Y, Mizuno K, Nakane A, Kato T, Kohri K, Hayashi Y. Long-term physical, hormonal, and sexual outcome of males with disorders of sex development. J Pediatr Surg. 2009;44:1491–1496. doi: 10.1016/j.jpedsurg.2008.10.111. [DOI] [PubMed] [Google Scholar]
- 4.Khadilkar KS, Budyal SR, Kasaliwal R, et al. Ovotesticular Disorder of Sex Development: A Single-Center Experience. Endocr Pract. 2015;21:770–776. doi: 10.4158/EP15606.OR. [DOI] [PubMed] [Google Scholar]
- 5.Wiersma R, Ramdial PK. The gonads of 111 South African patients with ovotesticular disorder of sex differentiation. J Pediatr Surg. 2009;44:556–560. doi: 10.1016/j.jpedsurg.2008.08.013. [DOI] [PubMed] [Google Scholar]
- 6.Talreja SM, Banerjee I, Yadav SS, Tomar V. A rare case of lateral ovotesticular disorder with Klinefelter syndrome mosaicism 46, XX/47, XXY: An unusual presentation. Urol Ann. 2015;7:520–523. doi: 10.4103/0974-7796.164855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Isguven P, Yildiz M, Arslanoglu I, Adal E, Erguven M, Tuzlali S. True hermaphroditism with characteristics of Klinefelter’s syndrome: a rare presentation. J Pediatr Endocrinol Metab. 2005;18:603–606. doi: 10.1515/jpem.2005.18.6.603. [DOI] [PubMed] [Google Scholar]
- 8.Eberenz W, Rosenberg HK, Moshang T, Chatten J, Keating MA. True hermaphroditism: sonographic demonstration of ovotestes. Radiology. 1991;179:429–431. doi: 10.1148/radiology.179.2.2014286. [DOI] [PubMed] [Google Scholar]
- 9.Selver Eklioglu B, Atabek ME, Akyurek N, Ari Yuca S, Piskin M. The 46XX Ovotesticular Disorders of Sexual Development with Dismorphic Features. J Pediatr Adolesc Gynecol. 2015;28:e157–159. doi: 10.1016/j.jpag.2015.02.112. [DOI] [PubMed] [Google Scholar]
- 10.Song JS, Lee SH, Jin DK, Kim SH. A case report of rare XXY/XX mosaicism in a phenotypic male with Klinefelter syndrome and mediastinal germ cell tumor. Genet Couns. 2014;25:215–220. [PubMed] [Google Scholar]