

Abstract
Nipah virus (NiV, Henipavirus) is a highly lethal emergent zoonotic paramyxovirus responsible for repeated human outbreaks of encephalitis in South East Asia. There are no approved vaccines or treatments, thus improved understanding of NiV biology is imperative. NiV matrix protein recruits a plethora of cellular machinery to scaffold and coordinate virion budding. Intriguingly, matrix also hijacks cellular trafficking and ubiquitination pathways to facilitate transient nuclear localization. While the biological significance of matrix nuclear localization for an otherwise cytoplasmic virus remains enigmatic, the molecular details have begun to be characterized, and are conserved among matrix proteins from divergent paramyxoviruses. Matrix protein appropriation of cellular machinery will be discussed in terms of its early nuclear targeting and later role in virion assembly.
Keywords: Henipavirus, matrix, paramyxovirus
Nipah virus: a highly lethal emergent paramyxovirus
Nipah virus (NiV) is a highly pathogenic emerging zoonotic virus, first identified as the causative agent of an outbreak of acute encephalitis in Malaysia and Singapore that started in 1998. This outbreak continued into 1999, with over 265 reported cases, and a fatality rate of approximately 40% [1]. Since 2001, repeated sporadic NiV outbreaks have occurred almost every year in Bangladesh, and occasionally in India, collectively causing hundreds of cases with lethality rates sometimes in excess of 70% [2,3]. Nipah virus isolates from Malaysia and subsequent Bangladesh and India outbreaks show a relatively high degree of conservation at the amino acid level, with over 99% identity in the matrix (M) protein, and at least 92% identity across the other structural proteins [1,4,5]. There are currently no approved vaccines or therapeutics for NiV, and given its remarkably high case fatality rate, it is classified as a biosafety level 4 (BSL4) pathogen. However, prophylactic and therapeutic administration of monoclonal antibodies, as well as several vaccine strategies, have shown efficacy in animal challenge models, indicating progress toward the development of an effective vaccine (Reviewed [6]).
The natural reservoir for NiV is pteropid fruit bats [7], and direct bat to human transmission can occur, frequently as a result of consumption of date palm sap contaminated with saliva or urine from infected bats [3]. Alternatively, transmission to the human population can proceed via an amplifying host, as during the initial NiV outbreak in Malaysia, in which transmission was from close contact with infected domesticated swine [1]. Transmission is primarily via the oronasopharyngeal route, with initial infection in the respiratory mucosa, followed by viral dissemination and high levels of viral replication in the endothelial cells of the central nervous system vasculature, causing the often-fatal encephalitis [8,9]. In most outbreaks, limited chains of direct human-to-human transmission have been documented, usually from an infected patient with respiratory symptoms to a direct care-giver, both in community and hospital settings [3,10–12].
NiV is classified in the Henipavirus (HNV) genus, within the Paramyxoviridae family. The only other HNV known to cause human disease is Hendra virus (HeV), a zoonotic pathogen identified following the deaths of several individuals who had contact with infected horses in Australia in 1994 [2]. More recently, Mojiang paramyxovirus (MojV), another HNV, was circumstantially associated with three cases of fatal pneumonia in mine workers in Yunnan Province in China [13,14]. While the current geographical locations of all HNV spillover events causing human disease have been restricted to South East Asia (Fig. 1), surveillance efforts have additionally identified evidence of HNVs in sera collected from bats in Subsaharan Africa, greatly expanding our knowledge of both the geographical and bat species restriction of these viruses (Fig. 1) [15–19]. Moreover, a recent study looking at bat and human serum samples from Cameroon found that 3–4% of human samples were HNV seropositive, and that this was almost exclusively among individuals who reported butchering bat meat, providing the first evidence of human HNV spillover infections in Africa [20].
Fig. 1.
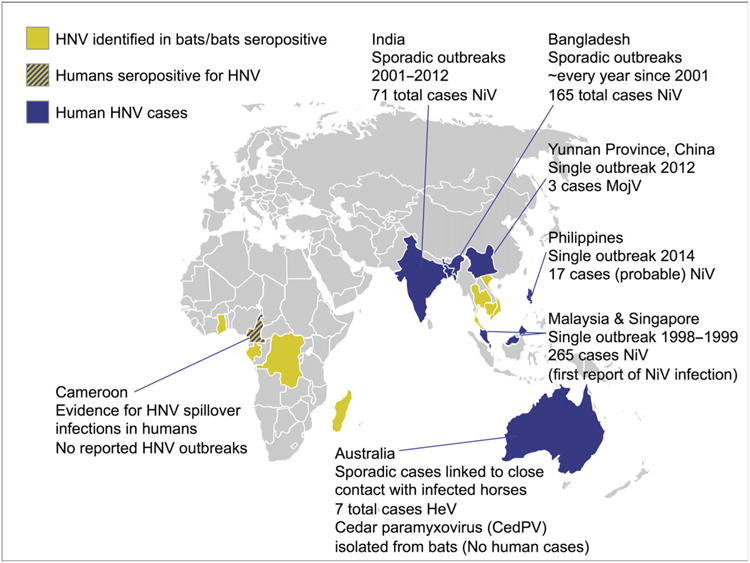
Global distribution of HNVs in human and bat populations. The map depicts countries where human HNV outbreaks have been reported (dark blue) [1–3,10,13,127,128], where human seropositivity to HNV has been identified (dark yellow) [20], and where there is evidence of HNV circulating in bat populations (light yellow) [15–19,129–132].
At the molecular level, paramyxoviruses are pleomorphic enveloped viruses, with nonsegmented, negative-strand RNA genomes, generally encoding six structural proteins and three accessory proteins [21]. Paramyxovirus cellular entry is mediated by the attachment (G, H or HN) and fusion (F) transmembrane glycoproteins. In the case of HNVs, the G attachment protein engages ephrinB2, or to a lesser extent, ephrinB3, as a cellular receptor [22–24]. Following fusion, viral gene expression and replication occurs in the cytoplasm, mediated by the large protein (L), the catalytic subunit of the RNA-dependent RNA polymerase, in complex with the nucleocapsid protein (N), and the phosphoprotein (P). Assembly and budding is orchestrated by the matrix protein (M), which associates with the inner leaflet of the plasma membrane and recruits the ribonucleoprotein complex and the transmembrane glycoproteins to nascent virions [21].
NiV matrix protein
Although the tertiary structure of NiV matrix protein (NiV-M) remains unknown, there are crystal structures of M proteins from a different paramyxovirus, Newcastle disease virus (NDV, Avulavirus) [25]; as well as the more distantly related respiratory syncytial virus (RSV, Pneumovirus) [26]. Despite relatively low primary sequence homology (˜ 20%), NDV and RSV matrix (RSV-M) share a remarkably common architecture, comprising two similarly folded β-sandwich domains, connected by a relatively unstructured linker, and decorated by several surface-exposed α-helices. Homology modeling suggests other M proteins also have similar structures, with most differences mapping to surface-exposed loops [27]. The existing structures show that paramyxovirus M proteins contain large continuous areas of positive charge and numerous surface-exposed hydrophobic residues on the membrane-binding face; features which together drive membrane association [25,27], and are conserved in homology-modeled NiV-M (Fig. 2A). M proteins assemble into stable dimers, which then associate into pseudotetrameric arrays underlying the plasma membrane. These dimer–dimer interactions generate the membrane curvature required for budding of viral particles [25]. For some paramyxoviruses, including NiV, ectopic expression of M alone is sufficient for budding of viral-like particles (VLPs) [28–31]. However, in the context of infection, M also mediates efficient recruitment and incorporation of the other viral structural proteins. The critical role of NiV-M in coordinating budding is highlighted by the recent characterization of recombinant NiV engineered to lack the M gene: Compared to wild-type virus, viral titers were dramatically reduced, virions were substantially less stable, had less regular morphology and frequent defects in the integrity of the viral envelope [32].
Fig. 2.
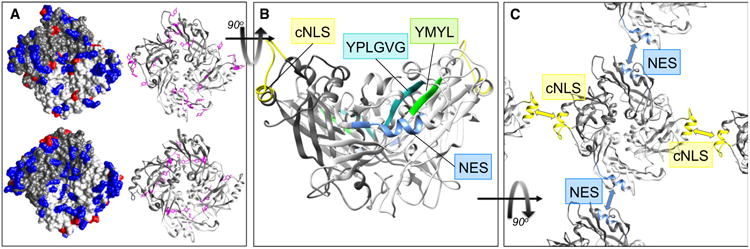
Homology-modeled structure and oligomerization of NiV matrix. (A) Surface features of M proteins that enable membrane association. Homology-modeled structure of dimeric NiV-M protein (top) generated using SWISS-MODEL [133–135] based on the existing X-ray crystal structure of NDV-M protein (bottom), PDB ID code 4G1G [25]. The two monomers are shaded light gray and dark gray in both the space-filling models (left), and the ribbon diagrams (right). The structures are oriented to show the surface that interacts with the cellular plasma membrane, with features thought to contribute to membrane association highlighted: There is an enrichment of surface-exposed positively charged residues (blue, left) and a planar distribution of tyrosine residues (fushia, right). (B) Sequences involved in NiV-M nuclear-cytoplasmic trafficking. Homology-modeled NiV-M (as in A) viewed from the side, with the membrane-binding surface at the top. The cNLS (yellow) and NES (blue) [33] are both located in surface-exposed α-helices. The putative late domain sequences ‘YMYL’ [74] (green) and ‘YPLGVG’ [107] (cyan) form a contiguous motif in the predicted tertiary structure, and together comprise two strands of the β-sheet that underlies the NES helix. (C) The cNLS and NES sequences form the two predicted dimer–dimer interfaces in NiV-M. Homology-modeled NiV-M (as A) shaded as (B), oriented to show the membrane-interacting surface. The blue and yellow arrows indicate the two dimer–dimer interfaces as identified for NDV-M [25], which are contributed by the NES (blue) and cNLS (yellow) α-helices, respectively.
Interestingly, at early timepoints during infection, the M protein of NiV [33,34], and other paramyxoviruses [28,29], traffics through the nucleus and nucleolus. Although the functional relevance of this trafficking of M proteins to paramyxovirus biology remains elusive, it is tightly regulated, requiring co-option of multiple host pathways, and is a prerequisite for budding [34]. These features imply biological importance, and strongly suggest as-yet-unidentified nonstructural roles for M proteins prior to budding of progeny virions.
Viral hijacking of cellular machinery
RNA viruses exhibit the highest mutation rates known among viruses and organisms across all kingdoms of life, and as a result, exist at the brink of error catastrophe, a phenomenon in which a small increase in the rate of mutations causes a dramatic loss of fitness, and population collapse [35,36]. This relatively low fidelity of genome replication strongly constrains RNA virus genome size, but also generates sequence diversity, providing vast potential for rapid adaptive evolution [37]. Together, these features conspire to make RNA viruses master molecular mimics: exploiting binding interfaces to co-opt, disrupt, and subvert host cell pathways to achieve efficient replication in spite of a minimal coding capacity. NiV-M is no exception, and this review will focus on its known exploitation of host cell machinery, as well as discussing the open questions as to how the M protein fulfills both its structural and putative nonstructural roles during NiV infection.
Matrix nuclear and subnuclear targeting early during infection
As discussed in the introduction, the best-characterized role of NiV-M is in assembly and budding of progeny virions at the plasma membrane. However, at early timepoints–approximately the first 16–20 h post infection–NiV-M has been observed to localize to the nucleus and nucleolus [33,34]. This phenotype is counterintuitive given that nuclear localization of M is obviously incompatible with its known structural function, and that paramyxoviruses replicate in the cytoplasm. Indeed, the only other NiV protein known to localize to the nucleus is the accessory protein, W, which inhibits induction of type I interferon (IFN) gene expression by sequestering the transcription factor STAT1 in the nucleus in an inactive form [38–41].
However, the nuclear localization of paramyxovirus M proteins appears to be broadly conserved among even distantly related paramyxoviruses [28,34,42], and was documented as early as 1979 in the case of sendai virus (SeV, Respiroviridae) [43]. Despite this long-standing observation, the role(s) of M in the nucleus remain opaque, although increased nuclear localization of SeV matrix (SeV-M) has been associated with the ability of the virus to establish persistent infection in cell culture systems [44]. Our understanding of the interaction between paramyxoviruses and the host cell nucleus may be accelerated by the very recent study by Deffrasnes et al. [45], who conducted the first genome-wide siRNA screen to identify host factors required for paramyxovirus infection. Interestingly, almost half of the highest confidence hits for genes required for HeV infection encode nucleolar proteins, and more generally, approximately a third of all significant hits encode nuclear proteins [45]. Follow-up studies characterizing the requirements for the identified genes should shed light on the currently enigmatic interactions of paramyxovirus M proteins with the nucleus and nucleolus.
While the biological significance of the nuclear localization of NiV-M is currently unclear, this phenotype is conserved among M proteins from closely related (e.g. HeV, HNV, > 90% amino acid identity to NiV-M) and more distantly related (e.g. Mumps virus, Rubulavirus ˜ 25% amino acid identity to NiV-M) paramyxoviruses. The molecular details governing the transient nuclear and nucleolar residence of M proteins have begun to be elucidated, and have revealed an exquisite system of regulation that relies on exploitation of multiple host pathways, as well as an apparent requirement for this nuclear sojourn in order for M to mediate subsequent budding [33,34]. The interactions of NiV and other paramyxovirus M proteins with host machinery for subcellular trafficking and for putative nuclear functions will be discussed below, and are summarized in Fig. 3.
Fig. 3.
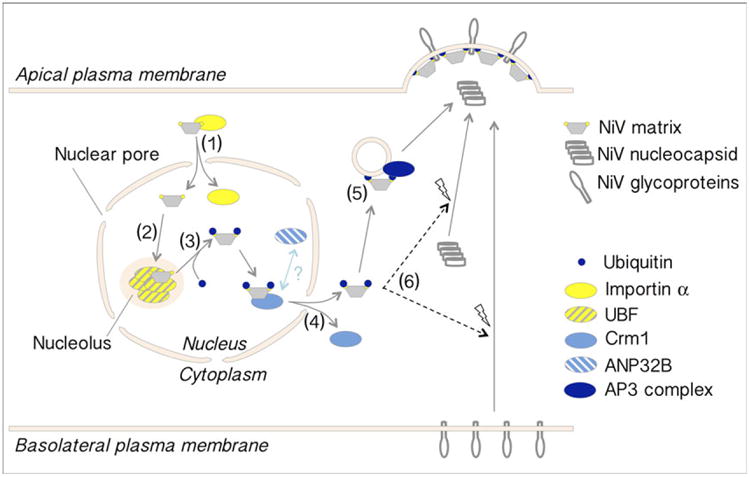
Proposed model for NiV-M trafficking and host protein interactions. Schematic showing a proposed model summarizing NiV-M interactions with host proteins and pathways including (1) cNLS-dependent nuclear import via interaction with importin α [33,34], (2) nucleolar targeting, putatively via interaction with UBF [34], (3) ubiquitin modification within the cNLS motif (by a currently unknown E3 ligase) [33,34], (4) NES-dependent nuclear export involving interaction with Crm1 and ANP32B [33,66], (5) membrane association and trafficking to the plasma membrane via interaction with the AP3 complex [79], and (6) recruitment and assembly of the viral nucleocapsid and glycoproteins to drive budding from the plasma membrane (by a currently unknown mechanism) [84].
Nuclear import of NiV-M
Monomeric NiV-M has a calculated molecular weight of 39 kD, which is approximately at the upper size limit (˜40 kD) for free diffusion of proteins through the nuclear pore complex (NPC) [46]. However, based on structural characterization of related paramyxovirus M proteins [25,47], NiV-M is likely to exist in the cell predominantly as homodimers (or higher order oligomers), indicating that active use of transport machinery is almost certainly required for nuclear import. Active host protein nuclear import is mediated primarily by nuclear transport receptors belonging to the karyopherin-β superfamily. These receptors interact with cargo protein (either directly or via an importin-α (impα) adaptor protein), with structural components of the NPC, and with RanGTPase, which regulates cargo binding and release in a mechanism dependent on GTP hydrolysis (reviewed in [46]). Cargo protein engagement of transport receptors or adaptors is usually dependent on the presence of a linear amino acid motif that conforms to one of the several known classes of nuclear localization signal (NLS) [48,49]. Lee and colleagues identified two putative NLS motifs within NiV and other paramyxovirus M proteins, and demonstrated by mutagenesis that both motifs contribute somewhat to nuclear localization of NiV-M, but that the bipartite classical NLS (cNLS) is absolutely required [33,34]. A bipartite cNLS motif is defined by two clusters of basic amino acid residues, separated by a 10–12 residue linker, and is recognized by one of the six human impα adaptor proteins, which form a ternary complex with importin-β (impβ) and GTP-bound-RanGTPase to mediate nuclear import of the cargo protein [46,50]. Certain cargoes show a strong preference for binding to a specific impα, but for the majority of cargoes, there appears to be at least some redundancy, and specificity is perhaps determined by relative abundance and moderate differences in affinity in the context of the crowded intracellular environment [48,50,51]. Consistent with this, NiV-M efficiently pulled down all members of the impα family when each was individually coexpressed with M [34]. However, when the NiV-M interactome was interrogated using an unbiased mass spectrometry-based approach for endogenous level host proteins, four of the six impα transport receptors were still identified as interaction partners, along with a strong enrichment of NPC proteins [34]. It is tempting to speculate that in addition to interacting with components of the nuclear import pathway for its transport, NiV-M could also compete with host proteins for transport, or disrupt the transport machinery, in order to antagonize the host innate immune response, as has been demonstrated for numerous other viral proteins, such as Ebola virus VP24 [52] and Polio virus proteases [53], and reviewed in detail by Yarbrough et al. [54].
Mutation of a single lysine residue within the NiV-M bipartite cNLS (K258A) significantly inhibited nuclear localization, demonstrating the importance of this motif for transport [33]. Conversely, mutation of this residue to arginine (K258R), which retains the positive charge important for the cNLS motif, was imported to the nucleus as anticipated, but unexpectedly accumulated, and was retained in the nucleolus [33]. These phenotypes were consistent when the corresponding mutations were introduced to other paramyxovirus M proteins from viruses spanning several genera including HeV, SeV, and mumps virus (MuV, Rubulavirus) [34]. Further experiments revealed that as well as forming part of the cNLS, K258 is an important site of regulatory ubiquitination, without which, M proteins become trapped in the nucleolus [33,34]. Therefore, M not only hijacks the cellular nuclear-cytoplasmic transport machinery, but also exploits the ubiquitin conjugation pathway. Although the E3 ubiquitin ligase(s) involved are yet to be identified, regulatory mono-ubiquitination within, or proximal to, an NLS motif by Mdm2 and UBE2O has previously been shown to inhibit the nuclear import of the tumor suppressor proteins p53 [55] and BAP1 [56], respectively.
As might be expected, given its nuclear-retention phenotype, the K258R mutation completely inhibited budding of NiV VLPs [33]. Similarly, engineering the equivalent mutation into recombinant SeV using a reverse genetics system abrogated production of infectious virions, despite efficient viral rescue [34]. These results raise the possibility that this absolute dependence on ubiquitin modification of M for budding may represent a useful therapeutic target. Indeed, treatment of cells with proteasome inhibitors–which deplete pools of free ubiquitin, so globally inhibit ubiquitin conjugation–potently inhibited replication of NiV [33].
While the nuclear retention of K258R NiV-M suggests an obvious explanation for the inhibition of budding, unexpectedly, budding was also abrogated by the K258A mutation, which results in diffuse cytoplasmic localization of M [33]. A similar phenotype was observed when a K258R NiV-M-ubiquitin fusion protein was expressed, which restored nuclear export (or inhibited nuclear import), but failed to target to the plasma membrane or induce VLP budding [33]. It is currently unclear why ubiquitination at this residue is so critical for subsequent budding–it could be that both the ubiquitin fusion and K258A block a necessary transit through the nuclear compartment, and so prevent a required interaction with additional host proteins, perhaps that mediate further post-translational modifications. An alternative hypothesis is that ubiquitin modification at this site is required for oligomerization of M, which is critical to its function in budding. Modeling the predicted tertiary structure of NiV-M (based on the existing crystal structure [25] of NDV matrix (NDV-M)) reveals that the cNLS motif is likely to comprise one of the two exposed α-helices in dimeric NDV-M that each form antiparallel helix–helix interactions at the two dimer–dimer interfaces that assemble the pseudotetrameric M protein array (Fig. 2B,C and [25]). The structural model also indicates that this positively charged bipartite cNLS motif is located at the membrane-proximal face of M, and therefore is likely to contribute to membrane association via electrostatic interactions with the negatively charged phospholipid head groups (Fig. 2A,B). Given that this motif is likely critical to both membrane association and dimer–dimer homo-oligomerization, it is possible that ubiquitination regulates these functions of NiV-M, in addition to its nuclear-cytoplasmic trafficking. This reflects a remarkably efficient use of coding capacity, as a single motif, and perhaps even a single residue, engages the host nuclear import machinery and ubiquitination machinery, while also contributing a critical interface for a viral protein–protein interaction.
Nucleolar targeting of NiV-M
Following nuclear import, NiV, HeV, and NDV-M are transiently targeted to specific subnuclear structures [29,33,57], and depletion of free ubiquitin, or engineering in the K258R (or equivalent) mutation, results in nucleolar retention of ectopically expressed M protein from NiV, HeV, SeV, and MuV. Together these results indicate that M proteins co-opt the host ubiquintation machinery for dynamic regulation of nucleolar targeting [34].
The nucleolus was first described as a structurally distinct compartment within the nucleus in the mid-1800s, and is a nuclear body formed around chromosomal features called nucleolar organizing regions (NORs) as reviewed by Pederson [58]. These NORs contain arrays of ribosomal DNA sequences and binding sites for the transcription factor upstream binding factor (UBF) [59], and have a critical role in ribosome biogenesis, processing and assembly [58]. Nucleolar localization signals (NoLS) in host proteins are less well defined than NLS motifs, and appear to be relatively divergent in both primary sequence and length [60,61]. It has been suggested that targeting to the nucleolus, which lacks a limiting membrane, is achieved by target protein interactions with nucleolar ‘hub’ proteins, including UBF [62]. Although it is currently unknown which sequence(s) within NiV-M are required for targeting to the nucleolus, UBF was identified as a NiV-M interaction partner, and overexpression of UBF-enhanced nucleolar localization of ectopically expressed NiV-M [34].
Other paramyxovirus M proteins have also been shown to interact with nucleolar proteins. In particular, nucleophosmin (Npm, B23), a protein with chaper-one activity that is involved in shuttling ribosome components from the nucleolus to the cytoplasm, has been identified as an interaction partner of both RSV-M [63] and NDV-M [57]. In the case of NDV, this initially mediates localization of NDV-M to the nucleolus, but at later stages of infection, both M and B23 are relocalized to the cytoplasm. In addition, depletion of B23 inhibited NDV replication, although the mechanistic details mediating this were not examined [57]. It remains an open but important question as to whether NiV-M localization to the nucleolus similarly induces redistribution of nucleolar proteins, and thus disrupts normal nucleolar structure and/or function.
In addition to roles in ribosome biogenesis, the nucleolus is implicated in cellular responses to stress [64]. For example, following heat shock or acidotic stress, the nucleolus undergoes a dramatic structural reorganization, and becomes the site of sequestration of numerous otherwise cytosolic or nuclear proteins in a mechanism dependent on expression of stress-induced long noncoding RNA [65]. While the cellular consequences of stress-induced nucleolar detention remain opaque, it is possible that infection may provoke similar reorganization, with potential relevance both for viral replication and cell survival.
The first functional link between NiV and the nucleolus comes from the recent finding that fibrillarin (FBL), a nucleolar methyltransferase involved in ribo-some biogenesis, is critical for an early postentry step in NiV replication [45]. The requirement for FBL was identified in a genome-wide siRNA screen for host factors affecting HeV infection, and was subsequently found to be required for infection by diverse paramyxoviruses including NiV, MuV, and measles virus (MeV, Morbillivirus); but dispensable for infection by the unrelated influenza virus A (IAV) [45]. While the mechanistic role of FBL in paramyxovirus infection is not yet fully understood, it is dependent on FBL methyltransferase activity, and HeV-M interacts with FBL, although depletion of FBL did not prevent the previously documented [34] targeting of HeV-M to the nucleolus [45]. An interesting question is whether the role of this nucleolar protein in paramyxovirus infection is dependent on its interaction with M proteins, and therefore whether this could explain, at least in part, the strong requirement, tight regulation, and remarkable conservation of nuclear and nucleolar targeting of paramyxovirus M proteins.
NiV-M nuclear export
The motifs that facilitate nuclear export of NiV-M have also been defined by comparison to known host nuclear export signals (NESs), followed by mutation and experimental confirmation of their role in trafficking [33]. Disruption of either of the two putative classical leucine-rich NES sequences in NiV-M increased nuclear localization of M, but only the N-terminal NES-mediated nuclear export when fused to a heterologous protein [33]. As expected for a protein bearing a leucine-rich NES, nuclear export of M is sensitive to inhibition with leptomycin B (LMB) [66], indicating transport requires chromosome region maintenance 1 (Crm1, XPO1), one of the seven karyo-pherin-β superfamily nuclear export proteins (XPO1-7) [67,68].
This NES is functionally conserved among several paramyxovirus M proteins, and is required for budding, both in the context of NiV VLP production, and in budding of infectious SeV virions in a reverse genetics system [33,34]. Strikingly, looking at the predicated tertiary structure of NiV-M, this NES forms the second antiparallel helix–helix dimer–dimer oligomerization interface–as discussed above, the other is contributed by the cNLS helix–again highlighting the dual importance of conserved residues both in M protein trafficking and in the oligomerization required for virion assembly (Fig. 2B,C).
Interestingly, a proteomics-based interaction study identified acidic leucine-rich nuclear phosphoprotein 32 member B (ANP32B), a Crm1 transport adaptor for mRNA [69], as a HNV matrix (HNV-M)-interacting protein [66]. Overexpression of ANP32B resulted in nuclear retention of both NiV and HeV-M, suggesting a possible role in M protein trafficking [66]. However, ANP32B overexpression did not affect VLP budding [66], and depletion of ANP32B using siRNA did not significantly affect HeV infection [45], thus both the mechanistic and biological consequences of this interaction are currently unclear.
Matrix protein co-option of host machinery for virion assembly and budding
As discussed above, it appears that paramyxovirus M proteins may have important nonstructural roles early in the viral replication cycle, but the primary, and best characterized, role of M proteins is in orchestrating assembly and budding of progeny virions at the plasma membrane. The first evidence for the structural function of M proteins came from characterization of temperature-sensitive mutant paramyxoviruses that yielded critical insights into paramyxovirus biology in the era prior to availability of reverse genetics systems to engineer mutations to interrogate gene function. In 1978, Yoshida et al. correlated a change in the SeV-M protein apparent molecular weight at the permissive versus nonpermissive temperature, with a defect in viral budding. This deficit was accompanied by a more diffuse distribution of the viral glycoproteins throughout the plasma membrane, and a failure to recruit the viral nucleoprotein to the plasma membrane fraction, suggesting a critical role for M in assembly [43]. Decades later, the same group were able to confirm that this phenotype is due to instability and degradation of M at the nonpermissive temperature [70]. Further evidence that M scaffold assembly came from studies demonstrating that SeV-M can self-assemble in vitro into highly ordered tubes and sheets with curvature and spacing remarkably similar to the dense protein arrays detected underlying the plasma membrane in SeV-infected cells [71,72]; and that ectopic expression of SeV-M results in budding of VLPs [30]. In concordance with the established role of M in SeV budding, overexpression of NiV-M can also drive budding of VLPs independently of other viral proteins [73,74]. Moreover, genetic ablation of the M gene in recombinant NiV markedly reduces viral titers, as well as resulting in production of particles with reduced stability, more irregular morphology, and less efficient incorporation of viral proteins, together contributing to a dramatic decrease in the infectivity-to-particle ratio [32].
NiV-M is heavily reliant on host factors in order to accomplish each aspect of infectious virion budding, from viral protein recruitment to the plasma membrane, to the final membrane fission of particle release. The known, and putative, hijacking of cellular pathways by NiV-M, as well as other paramyxovirus M proteins, will be discussed in the following section.
Matrix protein membrane association
Many cellular and viral nonintegral membrane proteins are targeted to membranes by conjugation of fatty acyl chains, by post-translational myristoylation of glycine residues or palmitoylation of cysteine residues, as reviewed in detail by Resh [75]. However, there is no evidence of acylation of paramyxovirus M proteins, suggesting an alternate mode of membrane interaction. In fact, recent biophysical characterization of IAV and HIV-type 1 M proteins suggests that although both are acylated, membrane association is primarily driven by electrostatic and hydrophobic interactions between surface-exposed amino acids and membrane lipids, with acylation surprisingly dispensable for membrane targeting [76,77]. Structural analysis of the M proteins from NDV [25] and RSV [27] has revealed that the membrane-binding surface has a high proportion of positively charged residues, important for interaction with negatively charged phospholipid head groups; and a planar arrangement of tyrosine residues thought to mediate hydrophobic interactions with the lipid bilayer. These, and other studies [78], have also identified a remarkable conservation in the overall structure of diverse M proteins from across the entire Mononegavirales order, thus the existing NDV-M structure can be used to generate a homology model of the NiV-M structure, which indicates that these surface features are conserved (Fig. 2A), suggesting a similar mode of membrane interaction.
Although membrane association per se is mediated by intrinsic properties of the surface features of paramyxovirus M proteins, several lines of evidence indicate that this is not a passive process, but rather, it requires utilization of multiple cellular pathways. Firstly, introduction of a single lysine to alanine point mutation within the NiV-M NLS (K258A) almost entirely abrogates membrane association of M and results in diffuse, cytoplasmic, localization. This is unlikely to be due to loss of positive charge required for membrane binding, as mutation of a nearby lysine residue (K263A) had no impact on membrane association [33]. As discussed earlier, detailed mutagenesis studies investigating both the subcellular trafficking and budding capacity of NiV have revealed that a positively charged residue is required at amino acid position 258 for nuclear import of NiV-M, and ubiquitin modification is required for subsequent nuclear export and membrane association. However, it is interesting to note that while ubiquitin fusion to the otherwise nucleolar-retained K258R mutant restored nuclear export (or perhaps inhibited nuclear import), it failed to mediate membrane association [33,34]. This suggests that this motif is necessary for interaction with host factors that facilitate membrane association, in addition to its known role in nuclear-cytoplasmic trafficking of M.
Further insights into the exploitation of host pathways to mediate NiV-M membrane targeting come from the identification of the beta subunit (AP3B1) of the adaptor protein 3 (AP-3) complex as a cellular interaction partner of HNV-M proteins by two independent proteomics studies [34,79]. Disruption of M interaction with the AP-3 complex caused a severe defect in M VLP budding, which appeared to be attributable to failure in sorting of M from internal cellular membranes to the plasma membrane [79]. This hints at a requirement for the AP-3 complex in trafficking of M-bound vesicles, consistent with its known role in vesicular sorting from tubular sorting endosomes [80]. Ectopic expression of a polypeptide derived from the AP3B1 hinge region, which was identified as the M-interacting domain, severely impaired budding of VLPs, suggesting that this protein–protein interface could be a viable drug target [79]. It is interesting to note that the homologous hinge domain of the delta subunit of the AP-3 complex has previously been shown to interact with HIV-1 Gag, and similarly promote its association with the plasma membrane and assembly of HIV-1 particles [81], suggesting the function of this host protein complex may be similarly commandeered by divergent viruses.
Sorting of matrix and the viral glycoproteins to the apical membrane
Epithelial cells are specialized in that they have a polarized distribution of membrane proteins and lipids between the apical (luminal) and basolateral plasma membrane domains. This is of important consequence for the pathobiology of many paramyxoviruses, including NiV, as they are primarily transmitted via the oronasopharyngeal route, and the initial site of replication is the airway mucosa [82]. In particular, vectorial budding of progeny virions from the apical membrane can result in infection restricted to the respiratory epithelia, which may favor viral shedding and transmission; whereas, budding from the basolateral domain can rapidly disseminate virus, causing systemic infection with obvious implications for pathology, as discussed by Schmitt and Lamb [83]. Membrane asymmetry in a monolayer of polarized epithelial cells is maintained by the separation of the two membrane domains by tight junctions, combined with specialized sorting machinery that differentially recycles and segregates lipids and membrane proteins between the apical and basolateral domains. In order to achieve directional budding, viral proteins must hack into these sorting pathways to concentrate the structural proteins at either the apical or basolateral face. Recent interrogation of NiV infection in polarized epithelial cell cultures demonstrated that budding of NiV occurs almost exclusively from the apical surface [84]. However, there is a bipolar distribution of the surface glycoproteins, which drives cell–cell fusion and syncytia formation, disrupting the epithelial monolayer, therefore facilitating dissemination of virions from the respiratory epithelia as infection progresses [84].
Interestingly, in spite of the observed bipolar membrane distribution of NiV glycoproteins [84], both NiV F and G possess intrinsic basolateral sorting signals, contributed by residues within the transmembrane domains (TMDs) and tyrosine-based motifs in the cytoplasmic tails of each protein [85]. Consistent with this, when expressed in the absence of other viral proteins [85], as well as at early timepoints during infection [84], F and G localize exclusively to the basolateral membrane. In contrast, ectopically expressed NiV-M is efficiently targeted to the apical membrane, and at later timepoints during infection, plasma membrane localization of M coincides with redistribution of the glycoproteins to the apical membrane domain [84]. Therefore, although it was previously assumed that viral glycoproteins govern the membrane domain from which budding occurs, in the case of NiV, it is rather driven by M.
Similar dependence on M for directional budding in epithelial cells has been observed for MeV: ectopically expressed glycoproteins are targeted to the basolateral membrane, but there is a more equal membrane distribution of the glycoproteins during infection, with exclusively apical budding [86]. Further evidence that the site of budding is determined by M comes from experiments with recombinant MeV, which demonstrated that in the absence of M, the glycoproteins fail to relocalize to the apical membrane domain during infection [87]. It is worth noting that this is perhaps an emerging theme given that the M proteins of various more distantly related viruses including IAV (Orthomyxoviridae), Marburg virus (MARV, Filoviridae), and Vesicular Stomatitis virus (VSV, Rhabdoviridae) also appear to determine directional budding, as reviewed by El Najjar et al. [88].
These observations raise important mechanistic questions as to how M proteins achieve specific apical localization, and redistribution of the viral transmembrane glycoproteins. It is possible that NiV-M contains as-yet uncharacterized intrinsic apical sorting motifs that are recognized by cellular membrane trafficking machinery, generating its asymmetric distribution. On the other hand, M may become concentrated at the apical membrane more passively, via a stabilizing interaction with a protein component of the apical cell cortex, as postulated by Lamp et al. [84]. An alternative hypothesis is that apical localization of M results from its preferential segregation into lipid raft membrane microdomains, which are enriched in cholesterol and sphingolipids have markedly different protein contents from the surrounding lipid bilayer, and are largely restricted to the apical membrane of epithelial cells [89,90]. As reviewed [83], many, but notably not all, RNA viruses are known to bud from lipid raft domains, although this has not been thoroughly investigated in the case of NiV. Interestingly, expressing cytoplasmic NiV-M fused to membrane-targeting motifs from other proteins to artificially target NiV-M to either lipid raft, or nonlipid raft, membrane domains resulted in equally efficient VLP production [33]. While this suggests that NiV budding is not dependent on lipid raft domains, it should be noted that the requirements for efficient budding of VLPs in nonpolarized cells may be markedly different from the in vivo determinants of infectious NiV particle budding, and could also vary between different cell types. It therefore remains an open question as to how M drives budding specifically from the apical membrane domain, and the molecular mechanisms by which M induces redistribution of the NiV glycoproteins from the basolateral to apical surface are currently similarly obscure [84].
M engagement with the actin cytoskeleton
Many viruses are known to usurp and reorganize the actin cytoskeleton to facilitate or enhance virtually every stage of replication, from initial cell entry, to assembly and virion egress, as reviewed by Taylor et al. [91]. Paramyxoviruses are no exception, and the importance of their engagement with the actin cytoskeleton has been postulated ever since large quantities of cellular actin were identified in purified MeV preparations in the 1970s [92]. More direct evidence of paramyxovirus hijacking of actin structures was subsequently provided by electron microscopic studies that revealed that MeV buds from microvillus-like structures containing specifically negative-end oriented actin filaments, that are tightly associated with budding virions [93]. Furthermore, MeV budding is inhibited by cytochalasin B, a fungal metabolite that blocks polymerization and elongation of actin filaments [94]. Manipulation of the actin cytoskeleton is not a mechanism specific to MeV, in fact, infection of cultured fibroblasts with paramyxoviruses spanning four genera (MeV, MuV, SeV, NDV) all induced gross reorganization of the cytoskeleton, with a dramatic reduction in detectable actin bundles [95].
The M proteins of at least SeV and NDV are known to interact directly with purified actin [96]. In the case of SeV-M, a putative actin-binding motif was identified by sequence comparison with other actin-binding proteins, and mutation of these residues inhibited VLP production without loss of M membrane association, suggesting interaction with actin is required specifically for egress [30]. In addition, suppression of M gene expression during SeV infection almost completely prevented the extensive actin remodeling otherwise observed, providing further evidence that M is central to these changes in actin dynamics [97]. While it is currently unconfirmed whether NiV-M similarly interacts either directly with actin, with regulatory proteins to subvert actin dynamics, or indeed both; a proteomics-based interaction study identified several important regulators of actin cytoskeleton remodeling–cofilin and the ARP2/3 complex–as cellular binding partners of NiV-M [34].
Virion budding and ESCRT recruitment
The process of virion membrane envelopment involves two key processes: firstly, the induction of membrane curvature around the viral structural proteins and genome; and secondly, the membrane scission that releases the virion from the host membrane. For many enveloped viruses, including paramyxoviruses, induction of membrane curvature is driven, at least in part, by the oligomerization of membrane-bound M protein. Structural analysis of NDV-M indicates that dimer–dimer interactions between M homodimers result in an angle of approximately 6–20° between neighboring dimers [25], and in vitro studies have demonstrated that purified paramyxovirus M proteins added to lipid bilayers induce membrane deformation and the formation of tubular structures, although they do not spontaneously induce budding [71,72]. The fission of the viral envelope and host membrane is an energetically demanding event, and most viruses are either known, or thought, to hijack the cellular vesiculation machinery in order to achieve this final stage of the infectious cycle [98].
In classical endocytic vesicle formation, the endo-some forms into the cell, and this process is mediated by recruitment of cytoplasmic factors such as clathrin and dynamin, which surround the outside of the vesicle and generate membrane curvature and the force needed for membrane scission, as reviewed by Doherty and McMahon [99]. In contrast, virion budding from cellular membranes is topologically opposite to this process, thus cytoplasmic factors have access only to the inside of the virion. However, the requirement for vesiculation in this orientation is not a challenge unique to viruses–in fact, the membrane topology in this process is analogous to that in vesicle budding into the lumen of maturing endosomes in multivesicular body (MVB) formation, as well as the membrane constriction and scission during the final step of cytokinesis in cell division. Mammalian cells have an endosomal sorting complex required for transport (ESCRT) pathway that mediates these processes, and it is therefore no surprise that many enveloped viruses have evolved strategies to hijack the cellular ESCRT machinery [100].
The mammalian ESCRT machinery is a complex pathway composed of over 30 proteins, and has been reviewed in detail elsewhere [100–102]. Briefly, adaptor proteins serve to recruit ESCRT I/ESCRT II complex proteins or Bro1 domain proteins to cellular membranes, and this stabilizes membrane curvature, and promotes recruitment of the late-acting ESCRT III complex proteins, and vacuolar protein sorting-associated protein 4 (VPS4) ATPase, which together catalyze membrane fission. ESCRT III proteins are thought to oligomerize to form tight filamentous and spiral structures around the neck of the budding membrane, and VPS4 ATP hydrolysis somehow provides the force necessary for pinching off and disassembly of the ESCRT III oligomers to allow subsequent rounds of vesicle formation. Viral exploitation of the ESCRT machinery for budding is well characterized for various retroviral Gag proteins, and a remarkable conservation in viral strategy for ESCRT recruitment has emerged: Many diverse viruses, including Retroviruses, Arenaviruses, Filoviruses, Flaviviruses, Rhabdoviruses, and Herpesviruses all employ one of three short linear amino acid motifs (P(T/S)AP, PPxY, YP(x)nL) that each mimic a cellular protein–protein interaction motif and serve to recruit components of the ESCRT machinery [100]. These motifs are known as late (or ‘L’) domains, are relatively position-independent within viral proteins, and have been found to be generally interchangeable. For example, the motif from Ebola virus VP40 M protein will restore full budding function to HIV-1 Gag in which the late domain has been deleted [103]. Interestingly, paramyxovirus M proteins generally lack motifs that conform to these well-defined classes of viral late domains. As such, it remains unclear in most cases whether paramyxoviruses employ a distinct, ESCRT-independent, mechanism for budding, or whether they merely achieve co-option of ESCRT machinery components by an alternate mechanism [104]. Efficient budding of NDV, MuV, and parainfluenza virus 5 (PIV5, Rubulavirus) all require the catalytic activity of VPS4, and their budding is dependent on a conserved ‘FP(I/V)(I/V)’ motif, in the M protein, which was identified based on more distant sequence similarity to known viral late domains [31,105,106]. This motif has been suggested to function as a nonclassical late domain in these viruses, although interaction between M and the ESCRT machinery has not yet been demonstrated [31,105,106], and the motif is not conserved in other paramyxoviruses.
Several studies have attempted to identify putative noncanonical late domains in NiV-M, by similarly identifying amino acid motifs that show more distant homology to known late domains: In both cases, deletion of these motifs abrogated budding of ectopically expressed NiV-M, but also resulted in nuclear retention of M protein, a phenotype never previously observed with deletion of a bona fide late domain [74,107]. Given that nuclear localization alone is clearly incompatible with M budding from the plasma membrane, regardless of recruitment of cellular machinery, it is difficult to interpret the role of these results in terms of possible dependence on the ESCRT machinery. Interestingly, inspection of a homology-modeled structure of NiV-M indicates that these two putative late domains (which are noncontinuous in the primary sequence) each contribute to adjacent strands of an antiparallel β-sheet in the NTD of M, and this underlies the NES α-helix, offering an alternative interpretation as to why deletion of these motifs would result in nuclear localization and therefore disrupt budding (Fig. 2B).
As VPS4 is the only known enzyme in the ESCRT pathway, an alternative approach that has been employed to interrogate whether various viruses usurp the ESCRT machinery is to express a dominant negative version of the two mammalian VPS4 isoforms, and determine whether this inhibits budding [100]. Patch et al. [107] found that VPS4 activity is dispensable for budding of NiV VLPs generated from ectopic expression of NiV-M, consistent with the absence of a canonical late domain in NiV-M. However, a recent study found that budding during live NiV infection is strongly dependent on VPS4 activity, while also confirming previous results that budding of VLPs from expression of NiV-M alone is independent [108]. This apparent conflict was resolved by the finding that NiV C protein–an accessory protein previously only known to mediate antagonism of the innate immune response–enhances M-dependent budding via bridging interaction between NiV-M and Tsg101, a core component of the cellular ESCRT I complex [101]. This strategy for ESCRT recruitment is unusual both in that it is mediated by a nonstructural protein, and that it is dependent on a motif in the C protein that lacks homology to any previously defined late domains. However, the NiV C protein does show significant homology to Vps28, a component of the ESCRT I complex heterotetramer that interacts with Tsg101, suggesting that this also conforms to the classical late domain mechanism of viral mimicry of a host proteinprotein interaction motif. Interestingly, this mechanism was found to be conserved among all known HNVs. These results are also somewhat similar to reports that SeV C protein enhances budding of SeV-M VLPs via recruitment of ALIX, an ESCRT complex accessory protein, and that this enhancement is dependent on VPS4 catalytic activity [109,110]. However, a different study identified a YLDL motif in SeV-M also responsible for interaction with ALIX, and required for budding independently of the C protein [111]. These apparently conflicting results have been further confounded by experiments demonstrating that live SeV budding is neither inhibited by depletion of ALIX nor inhibition of the VPS4 ATPase [112], highlighting the often critical differences between determinants of budding of ectopically expressed M proteins compared to budding of live virus, and leaving the requirement for the ESCRT machinery in SeV budding currently unclear.
Conclusions and perspectives
Key open questions
In order to achieve tightly regulated and essential trafficking through the nucleus and nucleolus, NiV-M makes extensive use of host pathways, including the nuclear import and export machinery, nucleolar-targeting pathway(s) and ubiquitin conjugation system (Fig. 3) [33,34]. While originally identified for NiV-M, this is also remarkably conserved among M proteins from relatively diverse paramyxoviruses [34]. Although many of the molecular details governing M trafficking have been characterized, further elucidation of both the host and M protein requirements for the nuclear transit of M will be important. For example, while it is known that ubiquitin modification of M at residue K258 regulates trafficking [33], the host E3 ubiquitin ligase responsible has not yet been identified, and the possible contribution of modification at other residues has generally not been assessed. Similarly, the signals within M that mediate subnuclear targeting to the nucleolus are currently unknown, as are the cognate host components that facilitate this.
Importantly, nuclear localization is incompatible with the currently known function of paramyxovirus M proteins–driving virion assembly and budding at the plasma membrane–thus the major unanswered question posed by these observations concerns the evolutionary advantage of targeting the M protein to the nucleus at early timepoints during infection. An important aspect of this question is whether M is targeted to the nucleus to sequester it and prevent it disrupting the efficiency of the early stages of viral replication, or to mediate an active function that requires nuclear localization, or indeed both. Given that M protein can drive budding of VLPs in the absence of the genome and nucleocapsid, it is possible that it is advantageous to sequester M in the nucleus until sufficient replication has occurred to allow efficient building of infectious virions, as M budding prior to this could be associated with a fitness cost to the virus both in terms of the waste of cellular membrane and protein resources, and of release of noninfectious–but potentially immunogenic–particles. Alternatively, temporary nuclear and nucleolar sequestration of NiV-M could have evolved because M may regulate viral RNA synthesis by inhibiting the activity of the viral RNA-dependent-RNA-polymerase (RdRP) complex, as has been demonstrated for the M protein of MeV [113]. On the other hand, further investigation may identify active functions of NiV-M within the nucleus, perhaps hacking into cellular pathways to modify the cellular environment for optimal viral replication, for example by antagonizing the host innate immune response, or modulating the cell cycle, as proteins from numerous diverse viruses are known to do [114,115].
Conversely, while the functional importance of M in budding is well defined, the molecular details and necessary host interactions for the role of NiV-M in virion assembly and budding are perhaps less well characterized than those required for nuclear trafficking of M, leaving many open questions as to how NiV-M is able to exploit host cell pathways to achieve each of the major functions associated with budding of infectious virions and dependent on M.
The potential–and need–for antiviral interventions
Although the current disease burden of NiV is relatively small (approximately 500 cases spanning the past 18 years) the extremely high case fatality rate, capacity for human-to-human transmission, and cross-continental HNV distribution in bat populations, together make NiV a pathogen of global health concern with pandemic potential [2,10,19]. Indeed, NiV was recently named one of the top eight priority emerging pathogens by the world health organization (WHO) [116]. While efforts have been made to introduce public health measures, such as educating at-risk populations in Bangladesh about the dangers of consuming raw date palm sap that may be contaminated with saliva and urine from infected bats, repeated outbreaks continue to occur [3]. Moreover, the increasing deforestation and bat bushmeat hunting in many regions across western and central Africa [117,118], where HNVs are frequently detected in bat populations [15,17,19,20,119,120] strongly suggest that the geographical distribution of NiV and other HNV outbreaks is likely to increase dramatically in the future.
Detailed characterization of essential NiV-M interactions with host proteins has already identified a number of host pathways that represent potential therapeutic targets, including the ubiquitination machinery, nuclear export apparatus, and vesicle sorting and trafficking components. Excitingly, these studies have also raised the possibility of repurposing FDA-approved proteasome inhibitors to treat NiV infection [33]. An additional strategy for inhibition of NiV replication by targeting host pathways M is reliant on, is to selectively inhibit nuclear export with SINE compounds. Experiments using a relatively toxic SINE (LMB) have demonstrated proof-of-concept for inhibition of NiV-M function [66], paving the way for possible repurposing of the numerous novel SINE compounds with vastly improved toxicity profiles that are currently showing promise at various stages of cancer clinical trials [121]. Interestingly, repurposing of SINE compounds as antiviral drugs has already been investigated for IAV [122] and HIV-1 [123].
In addition to identification of host pathways that can be disrupted to block essential functions of NiV-M, characterization of NiV-M-host interactions, such as the interaction between M and the hinge domain of AP3B1 [79], may generate additional therapeutic targets. While protein–protein interactions have generally proved a particularly challenging class of therapeutic target [124], there has been substantial recent progress in this field, driven both by improved in silico rational drug design, and by advances in the bio-engineering of peptide- and protein-based therapeutics [124,125]. Indeed, proof-of-principle experiments have demonstrated relatively broad-spectrum antiviral activity by efficiently blocking the conserved interaction between the PPxY viral late domain and the host ESCRT machinery [126].
Further progress in elucidating the molecular details of the myriad ways in which NiV-M hijacks cellular pathways will certainly continue to identify potential weak spots in NiV biology, and perhaps paramyxovirus biology more generally, that that can be exploited with antiviral therapeutics.
Abbreviations
- ANP32B
leucine-rich nuclear phosphoprotein 32 member B
- AP3B1
adaptor protein 3 beta subunit
- BSL4
biosafety level 4
- cNLS
classical nuclear localization signal
- Crm1
chromosome region maintenance 1
- ESCRT
endosomal sorting complex required for transport
- FBL
fibrillarin
- HeV
Hendra virus
- HIV-1
human immunodeficiency virus type 1
- HNV
Henipavirus
- IAV
influenza A virus
- imp-α
importin-α
- impβ
importin-β
- LMB
leptomycin B
- M
matrix
- MARV
Marburg virus
- MeV
Measles virus
- MojV
Mojiang paramyxovirus
- MuV
mumps virus
- NDV
Newcastle disease virus
- NES
nuclear export signal
- NiV
Nipah virus
- NiV-M
NiV matrix protein
- NOR
nucleolar organizing region
- NPC
nuclear pore complex
- Npm
B23, nucleophosmin
- NTD
N-terminal domain
- PIV5
parainfluenza virus 5
- RSV
respiratory syncytial virus
- SeV
Sendai virus
- SINE
selective inhibitor of nuclear export
- UBF
upstream binding factor
- VLP
viral-like particle
- VPS4
vacuolar protein sorting-associated protein 4
- VSV
Vesicular Stomatitis virus
- WHO
World Health Organization
Footnotes
Author contributions: REW and BL wrote the manuscript.
References
- 1.Chua AKB, Bellini WJ, Rota PA, Harcourt BH, Tamin A, Lam SK, Rollin PE, Zaki SR, Shieh W, Goldsmith CS, et al. Nipah virus: a recently emergent deadly paramyxovirus. Science. 2000;288:1432–1435. doi: 10.1126/science.288.5470.1432. [DOI] [PubMed] [Google Scholar]
- 2.Eaton BT, Broder CC, Middleton D, Wang LF. Hendra and Nipah viruses: different and dangerous. Nat Rev Microbiol. 2006;4:23–35. doi: 10.1038/nrmicro1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Islam MS, Sazzad HMS, Satter SM, Sultana S, Hossain MJ, Hasan M, Rahman M, Campbell S, Cannon DL, Stroher U, et al. Nipah virus transmission from bats to humans associated with drinking traditional liquor made from date palm sap, Bangladesh, 2011–2014. Emerg Infect Dis. 2016;22:664–670. doi: 10.3201/eid2204.151747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Harcourt BH, Lowe L, Tamin A, Liu X, Bankamp B, Bowden N, Rollin PE, Comer JA, Ksiazek TG, Hossain MJ, et al. Genetic characterization of Nipah virus, Bangladesh, 2004. Emerg Infect Dis. 2005;11:1594–1597. doi: 10.3201/eid1110.050513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Arankalle VA, Bandyopadhyay BT, Ramdasi AY, Jadi R, Patil DR, Rahman M, Majumdar M, Banerjee PS, Hati AK, Goswami RP, et al. Genomic characterization of nipah virus, West Bengal, India. Emerg Infect Dis. 2011;17:907–909. doi: 10.3201/eid1705.100968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Satterfield BA, Dawes BE, Milligan GN. Status of vaccine research and development of vaccines for Nipah virus. Vaccine. 2016;34:2971–2975. doi: 10.1016/j.vaccine.2015.12.075. [DOI] [PubMed] [Google Scholar]
- 7.Halpin K, Hyatt AD, Fogarty R, Middleton D, Bingham J, Epstein JH, Rahman SA, Hughes T, Smith C, Field HE, et al. Pteropid bats are confirmed as the reservoir hosts of henipaviruses: a comprehensive experimental study of virus transmission. Am J Trop Med Hyg. 2011;85:946–951. doi: 10.4269/ajtmh.2011.10-0567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wong KT, Shieh WJ, Kumar S, Norain K, Abdullah W, Guarner J, Goldsmith CS, Chua KB, Lam SK, Tan CT, et al. Nipah virus infection. Am J Pathol. 2002;161:2153–2167. doi: 10.1016/S0002-9440(10)64493-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.De Wit E, Munster VJ. Animal models of disease shed light on Nipah virus pathogenesis and transmission. J Pathol. 2015;235:196–205. doi: 10.1002/path.4444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Luby SP. The pandemic potential of Nipah virus. Antiviral Res. 2013;100:38–43. doi: 10.1016/j.antiviral.2013.07.011. [DOI] [PubMed] [Google Scholar]
- 11.Homaira N, Rahman M, Hossain MJ, Epstein JH, Sultana R, Khan MSU, Podder G, Nahar K, Ahmed B, Gurley ES, et al. Nipah virus outbreak with person-to-person transmission in a district of Bangladesh, 2007. Epidemiol Infect. 2010;138:1630–1636. doi: 10.1017/S0950268810000695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sazzad HMS, Hossain MJ, Gurley ES, Ameen KMH, Parveen S, Islam MS, Faruque LI, Podder G, Banu SS, Lo MK, et al. Nipah virus infection outbreak with nosocomial and corpse-to-human transmission, Bangladesh. Emerg Infect Dis. 2013;19:210–217. doi: 10.3201/eid1902.120971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fernández-Aguilar X, Pujols J, Velarde R, Rosell R, López-Olvera JR, Marco I, Pumarola M. Novel Henipa-like virus, Mojiang Paramyxovirus, in rats, China, 2012. Emerg Infect Dis. 2014;20:1064–1066. doi: 10.3201/eid2006.131022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zeltina A, Bowden TA, Lee B. Emerging paramyxoviruses: receptor tropism and zoonotic potential. PLoS Pathog. 2016;12:e1005390. doi: 10.1371/journal.ppat.1005390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Drexler JF, Corman VM, Gloza-Rausch F, Seebens A, Annan A, Ipsen A, Kruppa T, Müller MA, Kalko EKV, Adu-Sarkodie Y, et al. Henipavirus RNA in African bats. PLoS One. 2009;4:e6367. doi: 10.1371/journal.pone.0006367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hayman DTS, Wang LF, Barr J, Baker KS, Suu-Ire R, Broder CC, Cunningham AA, Wood JLN. Antibodies to henipavirus or henipa-like viruses in domestic pigs in Ghana, West Africa. PLoS One. 2011;6:e25256. doi: 10.1371/journal.pone.0025256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hayman DTS, Suu-Ire R, Breed AC, McEachern JA, Wang L, Wood JLN, Cunningham AA. Evidence of henipavirus infection in West African fruit bats. PLoS One. 2008;3:e2739. doi: 10.1371/journal.pone.0002739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Iehlé C, Razafitrimo G, Razainirina J, Andriaholinirina N, Goodman SM, Faure C, Georges-Courbot MC, Rousset D, Reynes JM. Henipavirus and tioman virus antibodies in pteropodid bats, Madagascar. Emerg Infect Dis. 2007;13:159–161. doi: 10.3201/eid1301.060791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Drexler JF, Corman VM, Müller MA, Maganga GD, Vallo P, Binger T, Gloza-Rausch F, Cottontail VM, Rasche A, Yordanov S, et al. Bats host major mammalian paramyxoviruses. Nat Commun. 2012;3:796. doi: 10.1038/ncomms1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pernet O, Schneider BS, Beaty SM, LeBreton M, Yun TE, Park A, Zachariah TT, Bowden TA, Hitchens P, Ramirez CM, et al. Evidence for henipavirus spillover into human populations in Africa. Nat Commun. 2014;5:5342. doi: 10.1038/ncomms6342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lamb RA, Parks GD. Paramyxoviridae: the viruses and their replication. In: Knipe DM, Howley PM, editors. In Fields Virology. 5th. Lippincott, Williams and Wilkins; Philadelphia, PA: 2006. pp. 1449–1496. [Google Scholar]
- 22.Negrete OA, Levroney EL, Aguilar HC, Bertolotti-Ciarlet A, Nazarian R, Tajyar S, Lee B. EphrinB2 is the entry receptor for Nipah virus, an emergent deadly paramyxovirus. Nature. 2005;436:401–405. doi: 10.1038/nature03838. [DOI] [PubMed] [Google Scholar]
- 23.Negrete OA, Wolf MC, Aguilar HC, Enterlein S, Wang W, Muhlberger E, Su SV, Bertolotti-Ciarlet A, Flick R, Lee B. Two key residues in EphrinB3 are critical for its use as an alternative receptor for Nipah virus. PLoS Pathog. 2006;2:0078–0086. doi: 10.1371/journal.ppat.0020007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bonaparte MI, Dimitrov AS, Bossart KN, Crameri G, Mungall BA, Bishop KA, Choudhry V, Dimitrov DS, Wang LF, Eaton BT, et al. Ephrin-B2 ligand is a functional receptor for Hendra virus and Nipah virus. Proc Natl Acad Sci USA. 2005;102:10652–10657. doi: 10.1073/pnas.0504887102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Battisti AJ, Meng G, Winkler DC, McGinnes LW, Plevka P, Steven AC, Morrison TG, Rossmann MG. Structure and assembly of a paramyxovirus matrix protein. Proc Natl Acad Sci USA. 2012;109:13996–14000. doi: 10.1073/pnas.1210275109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Money VA, Mcphee HK, Mosely JA, Sanderson JM, Yeo RP. Surface features of a Mononegavirales matrix protein indicate sites of membrane interaction. Proc Natl Acad Sci USA. 2009;106:4441–4446. doi: 10.1073/pnas.0805740106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Leyrat C, Renner M, Harlos K, Huiskonen JT, Grimes JM. Structure and self-assembly of the calcium binding matrix protein of human metapneumovirus. Structure. 2014;22:136–148. doi: 10.1016/j.str.2013.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Peeples ME, Wang C, Gupta KC, Coleman N. Nuclear entry and nucleolar localization of the Newcastle disease virus (NDV) matrix protein occur early in infection and do not require other NDV proteins. J Virol. 1992;66:3263–3269. doi: 10.1128/jvi.66.5.3263-3269.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Monaghan P, Green D, Pallister J, Klein R, White J, Williams C, McMillan P, Tilley L, Lampe M, Hawes P, et al. Detailed morphological characterisation of Hendra virus infection of different cell types using super-resolution and conventional imaging. Virol J. 2014;11:200. doi: 10.1186/s12985-014-0200-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Takimoto T, Murti KG, Bousse T, Scroggs RA, Portner A. Role of matrix and fusion proteins in budding of sendai virus. J Virol. 2001;75:11384–11391. doi: 10.1128/JVI.75.23.11384-11391.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li M, Schmitt PT, Li Z, McCrory TS, He B, Schmitt AP. Mumps virus matrix, fusion, and nucleocapsid proteins cooperate for efficient production of virus-like particles. J Virol. 2009;83:7261–7272. doi: 10.1128/JVI.00421-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dietzel E, Kolesnikova L, Sawatsky B, Heiner A, Weis M, Kobinger GP, Becker S, von Messling V, Maisner A. Nipah virus matrix protein influences fusogenicity and is essential for particle infectivity and stability. J Virol. 2015;90:2514–2522. doi: 10.1128/JVI.02920-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang YE, Park A, Lake M, Pentecost M, Torres B, Yun TE, Wolf MC, Holbrook MR, Freiberg AN, Lee B. Ubiquitin-regulated nuclear-cytoplasmic trafficking of the Nipah virus matrix protein is important for viral budding. PLoS Pathog. 2010;6:e1001186. doi: 10.1371/journal.ppat.1001186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pentecost M, Vashisht AA, Lester T, Voros T, Beaty SM, Park A, Wang YE, Yun TE, Freiberg AN, Wohlschlegel JA, et al. Evidence for ubiquitin-regulated nuclear and subnuclear trafficking among paramyxovirinae matrix proteins. PLoS Pathog. 2015;11:e1004739. doi: 10.1371/journal.ppat.1004739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Drake JW, Holland JJ. Mutation rates among RNA viruses. Proc Natl Acad Sci USA. 1999;96:13910–13913. doi: 10.1073/pnas.96.24.13910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Crotty S, Cameron CE, Andino R. RNA virus error catastrophe: direct molecular test by using ribavirin. Proc Natl Acad Sci USA. 2001;98:6895–6900. doi: 10.1073/pnas.111085598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Makeyev E, Makeyev E, Bamford D, Bamford D. Evolutionary potential of an RNA virus. J Virol. 2004;78:2114–2120. doi: 10.1128/JVI.78.4.2114-2120.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shaw ML, Cardenas WB, Zamarin D, Palese P, Basler CF. Nuclear localization of the Nipah virus W protein allows for inhibition of both virus-and toll-like receptor 3-triggered signaling pathways. J Virol. 2005;79:6078–6088. doi: 10.1128/JVI.79.10.6078-6088.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shaw ML, García-Sastre A, Palese P, Basler CF. Nipah virus V and W proteins have a common STAT1-binding domain yet inhibit STAT1 activation from the cytoplasmic and nuclear compartments, respectively. J Virol. 2004;78:5633–5641. doi: 10.1128/JVI.78.11.5633-5641.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kulkarni S, Volchkova V, Basler CF, Palese P, Volchkov VE, Shaw ML. Nipah virus edits its P gene at high frequency to express the V and W proteins. J Virol. 2009;83:3982–3987. doi: 10.1128/JVI.02599-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ciancanelli MJ, Volchkova VA, Shaw ML, Volchkov VE, Basler CF. Nipah virus sequesters inactive STAT1 in the nucleus via a P gene-encoded mechanism. J Virol. 2009;83:7828–7841. doi: 10.1128/JVI.02610-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ghildyal R, Baulch-Brown C, Mills J, Meanger J. The matrix protein of Human respiratory syncytial virus localises to the nucleus of infected cells and inhibits transcription. Arch Virol. 2003;148:1419–1429. doi: 10.1007/s00705-003-0112-y. [DOI] [PubMed] [Google Scholar]
- 43.Yoshida T, Nagai Y, Maeno K, Iinuma M, Hamaguchi M, Matsumoto T, Nagayoshi S, Hoshino M. Studies on the role of M protein in virus assembly using a ts mutant of HVJ (Sendai virus) Virology. 1979;92:139–154. doi: 10.1016/0042-6822(79)90220-4. [DOI] [PubMed] [Google Scholar]
- 44.Ito M, Takeuchi T, Nishio M, Kawano M, Komada H, Tsurudome M, Ito Y. Early stage of establishment of persistent Sendai virus infection: unstable dynamic phase and then selection of viruses which are tightly cell associated, temperature sensitive, and capable of establishing persistent infection. J Virol. 2004;78:11939–11951. doi: 10.1128/JVI.78.21.11939-11951.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Deffrasnes C, Marsh GA, Foo CH, Rootes CL, Gould CM, Grusovin J, Monaghan P, Lo MK, Tompkins SM, Adams TE, et al. Genome-wide siRNA screening at biosafety level 4 reveals a crucial role for fibrillarin in henipavirus infection. PLoS Pathog. 2016;12:e1005478. doi: 10.1371/journal.ppat.1005478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Terry LJ, Shows EB, Wente SR. Crossing the nuclear envelope: hierarchical regulation of nucleocytoplasmic transport. Science. 2007;318:1412–1416. doi: 10.1126/science.1142204. [DOI] [PubMed] [Google Scholar]
- 47.Förster A, Maertens GN, Farrell PJ, Bajorek M. Dimerization of matrix protein is required for budding of respiratory syncytial virus. J Virol. 2015;89:4624–4635. doi: 10.1128/JVI.03500-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kosugi S, Hasebe M, Matsumura N, Takashima H, Miyamoto-Sato E, Tomita M, Yanagawa H. Six classes of nuclear localization signals specific to different binding grooves of importin alpha. J Biol Chem. 2009;284:478–485. doi: 10.1074/jbc.M807017200. [DOI] [PubMed] [Google Scholar]
- 49.Marfori M, Mynott A, Ellis JJ, Mehdi AM, Saunders NFW, Curmi PM, Forwood JK, Boden M, Kobe B. Molecular basis for specificity of nuclear import and prediction of nuclear localization. Biochim Biophys Acta. 2011;1813:1562–1577. doi: 10.1016/j.bbamcr.2010.10.013. [DOI] [PubMed] [Google Scholar]
- 50.Marfori M, Lonhienne TG, Forwood JK, Kobe B. Structural basis of high-affinity nuclear localization signal interactions with importin alpha. Traffic. 2012;13:532–548. doi: 10.1111/j.1600-0854.2012.01329.x. [DOI] [PubMed] [Google Scholar]
- 51.Kohler M, Speck C, Christiansen M, Bischoff FR, Prehn S, Haller H, Gorlich D, Hartmann E. Evidence for distinct substrate specificities of importin alpha family members in nuclear protein import. Mol Cell Biol. 1999;19:7782–7791. doi: 10.1128/mcb.19.11.7782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Xu W, Edwards MR, Borek DM, Feagins AR, Mittal A, Alinger JB, Berry KN, Yen B, Hamilton J, Brett TJ, et al. Ebola virus VP24 targets a unique NLS binding site on karyopherin alpha 5 to selectively compete with nuclear import of phosphorylated STAT1. Cell Host Microbe. 2014;16:187–200. doi: 10.1016/j.chom.2014.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gustin KE, Sarnow P. Effects of poliovirus infection on nucleo-cytoplasmic trafficking and nuclear pore complex composition. EMBO J. 2001;20:240–249. doi: 10.1093/emboj/20.1.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yarbrough ML, Mata MA, Sakthivel R, Fontoura BMA. Viral subversion of nucleocytoplasmic trafficking. Traffic. 2014;15:127–140. doi: 10.1111/tra.12137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li M, Brooks CL, Wu-Baer F, Chen D, Baer R, Gu W. Mono- versus polyubiquitination: differential control of p53 fate by Mdm2. Science. 2003;302:1972–1975. doi: 10.1126/science.1091362. [DOI] [PubMed] [Google Scholar]
- 56.Mashtalir N, Daou S, Barbour H, Sen NN, Gagnon J, Hammond-Martel I, Dar HH, Therrien M, Affar EB. Autodeubiquitination protects the tumor suppressor BAP1 from cytoplasmic sequestration mediated by the atypical ubiquitin ligase UBE2O. Mol Cell. 2014;54:392–406. doi: 10.1016/j.molcel.2014.03.002. [DOI] [PubMed] [Google Scholar]
- 57.Duan Z, Chen J, Xu H, Zhu J, Li Q, He L, Liu H, Hu S, Liu X. The nucleolar phosphoprotein B23 targets Newcastle disease virus matrix protein to the nucleoli and facilitates viral replication. Virology. 2014;452-453:212–222. doi: 10.1016/j.virol.2014.01.011. [DOI] [PubMed] [Google Scholar]
- 58.Pederson T. The nucleolus. Cold Spring Harb Perspect Biol. 2011;3:a000638. doi: 10.1101/cshperspect.a000638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Grob A, Colleran C, McStay B. Construction of synthetic nucleoli in human cells reveals how a major functional nuclear domain is formed and propagated through cell division. Genes Dev. 2014;28:220–230. doi: 10.1101/gad.234591.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Scott MS, Boisvert FM, McDowall MD, Lamond AI, Barton GJ. Characterization and prediction of protein nucleolar localization sequences. Nucleic Acids Res. 2010;38:7388–7399. doi: 10.1093/nar/gkq653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mekhail M, Rivero-Lopez L, Al-Masri A, Brandon C, Khacho M, Lee S. Identification of a common subnuclear localization signal. Mol Biol Cell. 2007;18:3966–3977. doi: 10.1091/mbc.E07-03-0295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Emmott E, Hiscox JA. Nucleolar targeting: the hub of the matter. EMBO Rep. 2009;10:231–238. doi: 10.1038/embor.2009.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Oliveira AP, Simabuco FM, Tamura RE, Guerrero MC, Ribeiro PGG, Libermann TA, Zerbini LF, Ventura AM. Human respiratory syncytial virus N, P and M protein interactions in HEK-293T cells. Virus Res. 2013;177:108–112. doi: 10.1016/j.virusres.2013.07.010. [DOI] [PubMed] [Google Scholar]
- 64.Jacob MD, Audas TE, Uniacke J, Trinkle-Mulcahy L, Lee S. Environmental cues induce a long noncoding RNA-dependent remodeling of the nucleolus. Mol Biol Cell. 2013;24:2943–2953. doi: 10.1091/mbc.E13-04-0223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Audas TE, Jacob MD, Lee S. Immobilization of proteins in the nucleolus by ribosomal intergenic spacer noncoding RNA. Mol Cell. 2012;45:147–157. doi: 10.1016/j.molcel.2011.12.012. [DOI] [PubMed] [Google Scholar]
- 66.Bauer A, Neumann S, Karger A, Henning AK, Maisner A, Lamp B, Dietzel E, Kwasnitschka L, Balkema-Buschmann A, Keil GM, et al. ANP32B is a nuclear target of henipavirus M proteins. PLoS One. 2014;9:e97233. doi: 10.1371/journal.pone.0097233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wolff B, Sanglier JJ, Wang Y. Leptomycin B is an inhibitor of nuclear export: inhibition of nucleo- cytoplasmic translocation of the human immunodeficiency virus type 1 (HIV-1) Rev protein and Rev-dependent mRNA. Chem Biol. 1997;4:139–147. doi: 10.1016/s1074-5521(97)90257-x. [DOI] [PubMed] [Google Scholar]
- 68.Kudo N, Matsumori N, Taoka H, Fujiwara D, Schreiner EP, Wolff B, Yoshida M, Horinouchi S. Leptomycin B inactivates CRM1/exportin 1 by covalent modification at a cysteine residue in the central conserved region. Proc Natl Acad Sci USA. 1999;96:9112–9117. doi: 10.1073/pnas.96.16.9112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Natalizio BJ, Wente SR. Postage for the messenger: designating routes for nuclear mRNA export. Trends Cell Biol. 2013;23:365–373. doi: 10.1016/j.tcb.2013.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kondo T, Yoshidas T, Miura N, Nakanishis M. Temperature-sensitive phenotype of a mutant Sendai virus strain is caused by its insufficient accumulation of the M protein. J Biol Chem. 1993;268:21924–21930. [PubMed] [Google Scholar]
- 71.Bachi T. Intramembrane structural differentiation in Sendai virus maturation. Virology. 1980;106:41–49. doi: 10.1016/0042-6822(80)90219-6. [DOI] [PubMed] [Google Scholar]
- 72.Heggeness MH, Smith PR, Choppin PW. In vitro assembly of the nonglycosylated membrane protein (M) of Sendai virus. Proc Natl Acad Sci USA. 1982;79:6232–6236. doi: 10.1073/pnas.79.20.6232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Patch JR, Crameri G, Wang LF, Eaton BT, Broder CC. Quantitative analysis of Nipah virus proteins released as virus-like particles reveals central role for the matrix protein. Virol J. 2007;4 doi: 10.1186/1743-422X-4-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ciancanelli MJ, Basler CF. Mutation of YMYL in the Nipah virus matrix protein abrogates budding and alters subcellular localization. J Virol. 2006;80:12070–12078. doi: 10.1128/JVI.01743-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Resh MD. Fatty acylation of proteins: new insights into membrane targeting of myristylated and palmitoylated proteins. Biochim Biophys Acta. 1999;1451:1–16. doi: 10.1016/s0167-4889(99)00075-0. [DOI] [PubMed] [Google Scholar]
- 76.Thaa B, Siche S, Herrmann A, Veit M. Acylation and cholesterol binding are not required for targeting of influenza A virus M2 protein to the hemagglutinin-defined budozone. FEBS Lett. 2014;588:1031–1036. doi: 10.1016/j.febslet.2014.02.014. [DOI] [PubMed] [Google Scholar]
- 77.Mercredi PY, Bucca N, Loeliger B, Gaines CR, Mehta M, Bhargava P, Tedbury PR, Charlier L, Floquet N, Muriaux D, et al. Structural and molecular determinants of membrane binding by the HIV-1 matrix protein. J Mol Biol. 2016;24:1637–1655. doi: 10.1016/j.jmb.2016.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Latiff K, Meanger J, Mills J, Ghildyal R. Sequence and structure relatedness of matrix protein of human respiratory syncytial virus with matrix proteins of other negative-sense RNA viruses. Clin Microbiol Infect. 2004;10:945–948. doi: 10.1111/j.1469-0691.2004.00980.x. [DOI] [PubMed] [Google Scholar]
- 79.Sun W, McCrory TS, Khaw WY, Petzing S, Myers T, Schmitt AP. Matrix proteins of Nipah and Hendra viruses interact with beta subunits of AP-3 complexes. J Virol. 2014;88:13099–13110. doi: 10.1128/JVI.02103-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Dell'Angelica EC. AP-3-dependent trafficking and disease: the first decade. Curr Opin Cell Biol. 2009;21:552–559. doi: 10.1016/j.ceb.2009.04.014. [DOI] [PubMed] [Google Scholar]
- 81.Dong X, Li H, Derdowski A, Ding L, Burnett A, Chen X, Peters TR, Dermody TS, Woodruff E, Wang JJ, et al. AP-3 directs the intracellular trafficking of HIV-1 gag and plays a key role in particle assembly. Cell. 2005;120:663–674. doi: 10.1016/j.cell.2004.12.023. [DOI] [PubMed] [Google Scholar]
- 82.Maisner A, Neufeld J, Weingartl H. Organ-and endotheliotropism of Nipah virus infections in vivo and in vitro. Thromb Haemost. 2009;102:1014–1023. doi: 10.1160/TH09-05-0310. [DOI] [PubMed] [Google Scholar]
- 83.Schmitt AP, Lamb RA. Escaping from the cell: assembly and budding of negative-strand RNA viruses. Curr Top Microbiol Immunol. 2004;283:145–196. doi: 10.1007/978-3-662-06099-5_5. [DOI] [PubMed] [Google Scholar]
- 84.Lamp B, Dietzel E, Kolesnikova L, Sauerhering L, Erbar S, Weingartl H, Maisner A. Nipah virus entry and egress from polarized epithelial cells. J Virol. 2013;87:3143–3154. doi: 10.1128/JVI.02696-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Weise C, Erbar S, Lamp B, Vogt C, Diederich S, Maisner A. Tyrosine residues in the cytoplasmic domains affect sorting and fusion activity of the Nipah virus glycoproteins in polarized epithelial cells. J Virol. 2010;84:7634–7641. doi: 10.1128/JVI.02576-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Maisner A, Klenk H, Herrler G. Polarized budding of measles virus is not determined by viral surface glycoproteins. J Virol. 1998;72:5276–5278. doi: 10.1128/jvi.72.6.5276-5278.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Naim HY, Ehler E, Billeter MA. Measles virus matrix protein specifies apical virus release and glycoprotein sorting in epithelial cells. EMBO J. 2000;19:3576–3585. doi: 10.1093/emboj/19.14.3576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.El Najjar F, Schmitt AP, Dutch RE. Paramyxovirus glycoprotein incorporation, assembly and budding: a three way dance for infectious particle production. Viruses. 2014;6:3019–3054. doi: 10.3390/v6083019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Diaz-Rohrer B, Levental KR, Levental I. Rafting through traffic: membrane domains in cellular logistics. Biochim Biophys Acta. 2014;1838:3003–3013. doi: 10.1016/j.bbamem.2014.07.029. [DOI] [PubMed] [Google Scholar]
- 90.Cao X, Surma MA, Simons K. Polarized sorting and trafficking in epithelial cells. Cell Res. 2012;22:793–805. doi: 10.1038/cr.2012.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Taylor MP, Koyuncu OO, Enquist LW. Subversion of the actin cytoskeleton during viral infection. Nat Rev Microbiol. 2011;9:427–439. doi: 10.1038/nrmicro2574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Tyrrell DLJ, Norrby E. Structural polypeptides of measles virus. J Gen Virol. 1978;39:219–229. doi: 10.1099/0022-1317-39-2-219. [DOI] [PubMed] [Google Scholar]
- 93.Bohn W, Rutter G, Hohenberg H, Mannweiler K, Nobis P. Involvement of actin filaments in budding of measles virus: studies on cytoskeletons of infected cells. Virology. 1986;149:91–106. doi: 10.1016/0042-6822(86)90090-5. [DOI] [PubMed] [Google Scholar]
- 94.Stallcup KC, Raine CS, Fields BN. Cytochalasin B inhibits the maturation of measles virus. Virology. 1983;124:59–74. doi: 10.1016/0042-6822(83)90290-8. [DOI] [PubMed] [Google Scholar]
- 95.Fagraeus A, Tyrrell DLJ, Norberg R, Norrby E. Actin filaments in paramyxovirus-infected human fibroblasts studied by indirect immunofluoreseence. Arch Virol. 1978;296:291–296. doi: 10.1007/BF01320068. [DOI] [PubMed] [Google Scholar]
- 96.Giuffre RM, Tovell DR, Kay CM, Tyrrell DL. Evidence for an interaction between the membrane protein of a paramyxovirus and actin. J Virol. 1982;42:963–968. doi: 10.1128/jvi.42.3.963-968.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Miazza V, Mottet-Osman G, Startchick S, Chaponnier C, Roux L. Sendai virus induced cytoplasmic actin remodeling correlates with efficient virus particle production. Virology. 2011;410:7–16. doi: 10.1016/j.virol.2010.10.003. [DOI] [PubMed] [Google Scholar]
- 98.Chen BJ, Lamb RA. Mechanisms for enveloped virus budding: can some viruses do without an ESCRT? Changes. 2012;29:997–1003. doi: 10.1016/j.virol.2007.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Doherty GJ, McMahon HT. Mechanisms of endocytosis. Annu Rev Biochem. 2009;78:857–902. doi: 10.1146/annurev.biochem.78.081307.110540. [DOI] [PubMed] [Google Scholar]
- 100.Votteler J, Sundquist WI. Virus budding and the ESCRT pathway. Cell Host Microbe. 2013;14:232–241. doi: 10.1016/j.chom.2013.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wollert T, Yang D, Ren X, Lee HH, Im YJ, Hurley JH. The ESCRT machinery at a glance. J Cell Sci. 2009;122:2163–2166. doi: 10.1242/jcs.029884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Shields SB, Piper RC. How ubiquitin functions with ESCRTs. Traffic. 2011;12:1306–1317. doi: 10.1111/j.1600-0854.2011.01242.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Martin-Serrano J, Zang T, Bieniasz PD. HIV-1 and Ebola virus encode small peptide motifs that recruit Tsg101 to sites of particle assembly to facilitate egress. Nat Med. 2001;7:1313–1319. doi: 10.1038/nm1201-1313. [DOI] [PubMed] [Google Scholar]
- 104.Harrison MS, Sakaguchi T, Schmitt AP. Paramyxovirus assembly and budding: building particles that transmit infections. Int J Biochem Cell Biol. 2010;42:1416–1429. doi: 10.1016/j.biocel.2010.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Duan Z, Hu Z, Zhu J, Xu H, Chen J, Liu H, Hu S, Liu X. Mutations in the FPIV motif of Newcastle disease virus matrix protein attenuate virus replication and reduce virus budding. Arch Virol. 2014;159:1813–1819. doi: 10.1007/s00705-014-1998-2. [DOI] [PubMed] [Google Scholar]
- 106.Schmitt AP, Leser GP, Morita E, Sundquist WI, Lamb RA. Evidence for a new viral late-domain core sequence, FPIV, necessary for budding of a paramyxovirus. J Virol. 2005;79:2988–2997. doi: 10.1128/JVI.79.5.2988-2997.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Patch JR, Han Z, McCarthy SE, Yan L, Wang LF, Harty RN, Broder CC. The YPLGVG sequence of the Nipah virus matrix protein is required for budding. Virol J. 2008;5:137. doi: 10.1186/1743-422X-5-137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Park A, Yun T, Vigant F, Pernet O, Won ST, Dawes BE, Bartkowski W, Freiberg AN, Lee B. Nipah virus C protein recruits Tsg101 to promote the efficient release of virus in an ESCRT-dependent pathway. PLoS Pathog. 2016;12:e1005659. doi: 10.1371/journal.ppat.1005659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Irie T, Nagata N, Yoshida T, Sakaguchi T. Recruitment of Alix/AIP1 to the plasma membrane by Sendai virus C protein facilitates budding of virus-like particles. Virology. 2008;371:108–120. doi: 10.1016/j.virol.2007.09.020. [DOI] [PubMed] [Google Scholar]
- 110.Sugahara F, Uchiyama T, Watanabe H, Shimazu Y, Kuwayama M, Fujii Y, Kiyotani K, Adachi A, Kohno N, Yoshida T, et al. Paramyxovirus Sendai virus-like particle formation by expression of multiple viral proteins and acceleration of its release by C protein. Virology. 2004;325:1–10. doi: 10.1016/j.virol.2004.04.019. [DOI] [PubMed] [Google Scholar]
- 111.Irie T, Shimazu Y, Yoshida T, Sakaguchi T. The YLDL sequence within Sendai virus M protein is critical for budding of virus-like particles and interacts with Alix/AIP1 independently of C protein. J Virol. 2007;81:2263–2273. doi: 10.1128/JVI.02218-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Gosselin-Grenet AS, Marq JB, Abrami L, Garcin D, Roux L. Sendai virus budding in the course of an infection does not require Alix and VPS4A host factors. Virology. 2007;365:101–112. doi: 10.1016/j.virol.2007.03.039. [DOI] [PubMed] [Google Scholar]
- 113.Iwasaki M, Takeda M, Shirogane Y, Nakatsu Y, Nakamura T, Yanagi Y. The matrix protein of measles virus regulates viral RNA synthesis and assembly by interacting with the nucleocapsid protein. J Virol. 2009;83:10374–10383. doi: 10.1128/JVI.01056-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Chaurushiya MS, Weitzman MD. Viral manipulation of DNA repair and cell cycle checkpoints. DNA Repair (Amst) 2009;8:1166–1176. doi: 10.1016/j.dnarep.2009.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Levy DE, García-Sastre A. The virus battles: IFN induction of the antiviral state and mechanisms of viral evasion. Cytokine Growth Factor Rev. 2001;12:143–156. doi: 10.1016/s1359-6101(00)00027-7. [DOI] [PubMed] [Google Scholar]
- 116.WHO. Blueprint for R&D preparedness and response to public health emergencies due to highly infectious pathogens. WHO Work. prioritization Pathog. Rep. 2015:1–7. [Google Scholar]
- 117.Wolfe ND, Daszak P, Kilpatrick AM, Burke DS. Bushmeat hunting, deforesation, and prediction of zoonotic disease emergence. Emerg Infect Dis. 2005;11:1822–1827. doi: 10.3201/eid1112.040789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Murray KA, Daszak P. Human ecology in pathogenic landscapes: two hypotheses on how land use change drives viral emergence. Curr Opin Virol. 2011;72:181–204. doi: 10.1016/j.coviro.2013.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Peel AJ, Baker KS, Crameri G, Barr JA, Hayman DTS, Wright E, Broder CC, Fernandez-Loras A, Fooks AR, Wang LF, et al. Henipavirus neutralising antibodies in an isolated island population of African fruit bats. PLoS One. 2012;7:e30346. doi: 10.1371/journal.pone.0030346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Weiss S, Nowak K, Fahr J, Wibbelt G, Mombouli JV, Parra HJ, Wolfe ND, Schneider BS, Leendertz FH. Henipavirus-related sequences in fruit bat bushmeat, Republic of Congo. Emerg Infect Dis. 2012;18:1536–1537. doi: 10.3201/eid1809.111607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Parikh K, Cang S, Sekhri A, Liu D. Selective inhibitors of nuclear export (SINE)–a novel class of anti-cancer agents. J Hematol Oncol. 2014;7:78. doi: 10.1186/s13045-014-0078-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Perwitasari O, Johnson S, Yan X, Howerth E, Shacham S, Landesman Y, Baloglu E, McCauley D, Tamir S, Tompkins SM, et al. Verdinexor, a novel selective inhibitor of nuclear export, reduces influenza A virus replication in vitro and in vivo. J Virol. 2014;88:10228–10243. doi: 10.1128/JVI.01774-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Boons E, Vanstreels E, Jacquemyn M, Nogueira TC, Neggers JE, Vercruysse T, van den Oord J, Tamir S, Shacham S, Landesman Y, et al. Human exportin-1 is a target for combined therapy of HIV and AIDS related lymphoma. EBioMedicine. 2015;2:1102–1113. doi: 10.1016/j.ebiom.2015.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Corbi-Verge C, Kim PM. Motif mediated protein-protein interactions as drug targets. Cell Commun Signal. 2016;14:8. doi: 10.1186/s12964-016-0131-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Petta I, Lievens S, Libert C, Tavernier J, De Bosscher K. Modulation of protein-protein interactions for the development of novel therapeutics. Mol Ther. 2015;2:1–12. doi: 10.1038/mt.2015.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Han Z, Lu J, Liu Y, Davis B, Lee MS, Olson MA, Ruthel G, Freedman BD, Schnell MJ, Wrobel JE, et al. Small molecule probes targeting the Viral PPxY-host Nedd4 interface block egress of a broad range of RNA viruses. J Virol. 2014;88:7294–7306. doi: 10.1128/JVI.00591-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Aljofan M. Hendra and Nipah infection: emerging paramyxoviruses. Virus Res. 2013;177:119–126. doi: 10.1016/j.virusres.2013.08.002. [DOI] [PubMed] [Google Scholar]
- 128.Ching PKG, de Los Reyes VC, Sucaldito MN, Tayag E, Columna-Vingno AB, Malbas FF, Bolo GC, Sejvar JJ, Eagles D, Playford G, et al. Outbreak of henipavirus infection, Philippines, 2014. Emerg Infect Dis. 2015;21:328–331. doi: 10.3201/eid2102.141433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Marsh GA, de Jong C, Barr JA, Tachedjian M, Smith C, Middleton D, Yu M, Todd S, Foord AJ, Haring V, et al. Cedar virus: a novel Henipavirus isolated from Australian bats. PLoS Pathog. 2012;8:e1002836. doi: 10.1371/journal.ppat.1002836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Reynes JM, Counor D, Ong S, Faure C, Seng V, Molia S, Walston J, Georges-Courbot MC, Deubel V, Sarthou JL. Nipah virus in Lyle's flying foxes, Cambodia. Emerg Infect Dis. 2005;11:1042–1047. doi: 10.3201/eid1107.041350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Wacharapluesadee S, Boongird K, Wanghongsa S, Ratanasetyuth N, Supavonwong P, Saengsen D, Gongal GN, Hemachudha T. A longitudinal study of the prevalence of Nipah virus in Pteropus lylei bats in Thailand: evidence for seasonal preference in disease transmission. Vector Borne Zoonotic Dis. 2010;10:183–190. doi: 10.1089/vbz.2008.0105. [DOI] [PubMed] [Google Scholar]
- 132.Hasebe F, Thuy NTT, Inoue S, Yu F, Kaku Y, Watanabe S, Akashi H, Dat DT, Mai LTQ, Morita K. Serologic evidence of Nipah virus infection in bats, Vietnam. Emerg Infect Dis. 2012;18:536–537. doi: 10.3201/eid1803.111121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Biasini M, Bienert S, Waterhouse A, Arnold K, Studer G, Schmidt T, Kiefer F, Cassarino TG, Bertoni M, Bordoli L, et al. SWISS-MODEL: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res. 2014;42:252–258. doi: 10.1093/nar/gku340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Kiefer F, Arnold K, Künzli M, Bordoli L, Schwede T. The SWISS-MODEL Repository and associated resources. Nucleic Acids Res. 2009;37:387–392. doi: 10.1093/nar/gkn750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Arnold K, Bordoli L, Kopp J, Schwede T. The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics. 2006;22:195–201. doi: 10.1093/bioinformatics/bti770. [DOI] [PubMed] [Google Scholar]