

Abstract
Craniosynostosis is a relatively common birth defect characterized by the premature fusion of one or more cranial sutures. Examples of craniosynostosis syndromes include Crouzon (CS), Pfeiffer (PS) and Apert (AS) syndrome, with clinical characteristics such as midface hypoplasia, hypertelorism and in some cases, limb defects. Mutations in Fibroblast Growth Factor Receptor-2 comprise the majority of known mutations in syndromic forms of craniosynostosis. A number of clinical reports of FGFR-associated craniosynostosis patients and mouse mutants have been linked to gastrointestinal tract (GIT) disorders, leading to the hypothesis of a direct link between FGFR-associated craniosynostosis syndromes and GIT malformations. We conducted an investigation to determine GIT symptoms in a sample of FGFR-associated craniosynostosis syndrome patients and a mouse model of CS containing a mutation (W290R) in Fgfr2. We found that, compared to the general population, the incidence of intestinal/bowel malrotation (IM) was present at a higher level in our sample population of patients with FGFR-associated craniosynostosis syndromes. We also showed that the mouse model of CS had an increased incidence of cecal displacement, suggestive of IM. These findings suggest a direct relationship between FGFR-related craniosynostosis syndromes and GIT malformations. Our study may shed further light on the potential widespread impact FGFR mutations on different developmental systems. Based on reports of GIT malformations in children with craniosynostosis syndromes and substantiation with our animal model, GIT malformations should be considered in any child with an FGFR2-associated craniosynostosis syndrome.
Keywords: Crouzon syndrome, Pfeiffer syndrome, Apert syndrome, intestinal malrotation, FGFR2 mutations
INTRODUCTION
Craniosynostosis, characterized by the premature fusion of one or more cranial sutures, has an overall incidence estimated at 1:2100 to 1:3000 live births [Lajeunie et al., 1995]. While most single suture craniosynostosis is isolated, multi-suture craniosynostosis exists, often with other associated anomalies in syndromes such as Crouzon (CS), Pfeiffer (PS) and Apert (AS) [Cohen, 2002; Cornejo-Roldan et al., 1999]. These syndromes share characteristics such as midface hypoplasia, hypertelorism, and exorbitism. Although similar in many respects, each syndrome differs, in particular in the extent of involvement of the limbs and other associated abnormalities. CS is the most common and phenotypically the mildest syndrome of the three, with absence of any limb defects [Johnson and Wilkie, 2011]. PS is characterized by multi-suture craniosynostosis, broad radially deviated thumbs and toes, partial cutaneous syndactyly of the hands and feet [Johnson and Wilkie, 2011; Lajeunie et al., 2006] and central nervous system malformations in some cases [Kabbani and Raghuveer, 2004; Ranger et al., 2010]. In addition to craniosynostosis, patients with AS present with severe bilateral and symmetrical complex syndactyly of the hands and feet, and cleft palate in some [Johnson and Wilkie, 2011; Kaplan, 1991; Wilkie et al., 1995].
Over 60 different mutations have been identified in syndromic forms of craniosynostosis. The majority of these mutations occur in Fibroblast Growth Factor Receptor-2, FGFR2 [Johnson and Wilkie, 2011]. All four receptors, FGFR1-4, share the same general protein structure with an amino-terminal signal sequence, three extracellular immunoglobulin (Ig) domains (IgI-III), a single membrane-spanning region, and an intracellular split tyrosine kinase domain [recent review, Carter et al., 2015]. Two highly conserved cysteine residues each form an intramolecular disulfide bridge stabilizing the loop structure of the three Ig-like domains. Ligand binding to the FGFR, in conjunction with heparan sulfate proteoglycan, causes receptor dimerization, transphosphorylation and activation of an intracellular tyrosine kinase domain [Eswarakumar et al., 2005; Jaye et al., 1992]. The extracellular region of the protein has important roles in regulating the affinity and specificity of ligand binding. The base FGF binding site is mediated by the IgII domain, the IgII/III linker region, and the N-terminus of IgIII, with further specificity of binding provided by the exclusive alternate splicing of two exons (IIIb or IIIc) coding for the C-terminal half of IgIII [McKeehan and Kan, 1994; Wang et al., 1999].
In FGFR-associated craniosynostosis syndromes, missense or splice-site mutations account for the majority of all observed nucleotide changes with some small inframe insertions and deletions [Cohen 2002]. Approximately 95% of the more than 50 FGFR mutations in patients with CS are located in the extracellular IgIII domain of FGFR2 [Johnson and Wilkie, 2011]. Interestingly, there is considerable overlap of mutations associated with CS and PS; such mutational “hot spots” have been identified in codons 278, 290 and 342 [Kress et al., 2000; Schaefer et al., 1998]. For example, two FGFR2 mutations creating cysteine residues (p.Trp290Cys and p.Tyr340Cys) cause severe forms of PS whereas conversion of the same residues into another amino acid (p.Trp290Gly, p.Trp290Arg, p.Tyr340His) results exclusively in the CS phenotype [Lajeunie et al., 2006]. For AS, two missense mutations in the linker between the IgII and IgIII domains of FGFR2, codons p.Ser252Trp (66%) or p.Pro253Arg (32%), account for over 98% [Johnson and Wilkie, 2011]. Mutations causing AS result in the loss of ligand binding specificity [Ibrahimi et al., 2001; Mangasarian et al., 1997; Marie et al., 2012; Plotnikov et al., 2000] whereas FGFR2 mutations in CS and PS frequently lead to constitutive activation of the receptor [Neilson and Friesel, 1996].
Recently, we demonstrated that a mouse model of human CS containing a mutation in codon 290 of Fgfr2 (Fgfr2W290R) presented with numerous gastrointestinal tract (GIT) malformations [Dab et al., 2013; Mai et al., 2010]. Duodenal atresia was found with a 35% penetrance in Fgfr2IIIb mutant embryos [Fairbanks et al., 2004]. Clinically, mutations in FGFR2 have been linked to several reports of GIT disorders. For example, GIT malformations (e.g., intestinal malrotation and atresia) were found in 22% of 23 PS patients [Koga et al., 2012]. Autopsy examinations in 12 of a sample population of 135 AS showed that 9 (of these 12 patients) had several visceral anomalies of the GIT, although only 1.5% of these 135 AS patients presented with GIT clinical symptoms [Cohen and Kreiborg, 1993]. Other isolated examples are present in the literature: Intestinal malrotation in a patient with PS [Eaton et al., 1975]; bowel obstruction and intestinal malrotation in two patients with PS [Barone et al., 1993]; a mother and son both with CS and epithelial-derived anal atresia [Park et al., 1995]; and gastro-esophageal reflux in a 16-month old male with craniosynostosis [Zarate et al., 2010]. These reports suggest the possibility that GIT malformations could be a direct effect of gene mutations associated with craniosynostosis. We set out to test the hypothesis of a direct link between FGFR-associated craniosynostosis syndromes and the presence of GIT malformation. We first investigated the presence of GIT symptoms in FGFR-associated craniosynostosis syndrome patients treated at a major provincial hospital. We found that, compared to the general population, the incidence of intestinal/bowel malrotation was higher in our sample population of patients with FGFR-associated craniosynostosis syndromes. Increased incidence of intestinal malrotations was also confirmed in a mouse model of CS with a specific mutation in Fgfr2.
MATERIALS AND METHODS
Patient study
Patient Recruitment
A retrospective chart review of patients diagnosed with CS, PS or AS at the Hospital for Sick Children (SickKids) between 1990 and 2011 was performed. Approval was obtained from the Research Ethics Board at SickKids (#1000029124).
Inclusion criteria of charts to be included for review included:
Record of genetic counseling, testing or discussion.
Diagnosis of a craniosynostosis syndrome (CS, PS or AS)
Identification of a suspected mutation in FGFR1, 2 or 3
Documentation of a GIT malformation according to the EUROCAT classification system and common congenital GIT malformations seen at SickKids
Patients with incomplete records and lack of documented evidence of mutations in FGFR1, 2 or 3 were excluded.
Three databases were searched for patients meeting the study inclusion criterion:
Division of Clinical and Metabolic Genetics clinical database (SHIRE systems)
Department of Paediatric Laboratory Medicine molecular genetics database (SHIRE systems), and
Division of Plastic Surgery craniofacial database.
The EUROCAT classification system, a network of population-based registries for the epidemiologic surveillance of congenital anomalies, was used. This database is useful for providing incidence data for rare congenital anomalies, including those of the digestive system and abdominal wall defects. This registry has documented 1.7 million births in 21 countries of Europe (http://www.eurocat-network.eu/) under separate categories of the EUROCAT heading of Digestive System.
Medical records of the selected patients treated at SickKids included review of molecular genetic test reports, general clinic letters, consultations, diagnostic imaging reports, clinic notes for plastic surgery, genetics and GI visits and emergency records. These charts were reviewed using a systematic approach that included a step-by-step analysis of all the records contained in each patient folder to assess for a definitive diagnosis of craniosynostosis, presence of mutations in FGFR1, 2 or 3, and any record of GIT abnormalities. If GIT abnormalities were noted, they were classified using the EUROCAT classification system (http://www.eurocat-network.eu/) for the following GIT anomalies: esophageal atresia with or without tracheo-esophageal fistula, intestinal malrotation, stenosis or atresia, ano-rectal atresia or stenosis, Hirschsprung disease, atresia of the bile ducts, annular pancreas, diaphragmatic hernia, gastroschisis, and omphalocele. Findings were documented in a coded spreadsheet to preserve patient anonymity.
Statistical analysis
Statistical analysis was carried out by an independent statistician. A power calculation and a binomial test was conducted to establish the statistical significance of the deviation in the proportion of patients with GIT malformations in this sample compared to that of the general population, at the 0.05 significance level.
Animal Study
Animals, GI phenotyping, histological analyses
The Fgfr2W290R mouse model of CS were bred and genotyped according to Mai et al. [2010] and approved for use by the University of California, San Francisco, Institutional Animal Care and Use Committee (IACUC). Both male and female wildtype (WT) and heterozygous (HET) mice were sacrificed at 3 to 33 weeks postnatally. The total number of animals studied were WT=22, HET = 26.
For each animal, an incision was made across the abdominal cavity through the skin and underlying peritoneum, taking care not to disturb the organs in the cavity. After fixing the mice in 4% formalin for at least 1 day, wider incisions were made through the abdominal walls to allow the gentle removal of the contents of the cavity in its entirety. The heart, liver and diaphragm were next carefully separated and removed; the remaining GIT was examined individually to assess overall size and arrangements of the individual parts. The presence of malrotation was determined by taking advantage of the size and positioning of the cecum relative to other parts of the GIT. Malrotation refers to the abnormal or incomplete rotation of the intestine during embryogenesis whereby the upper and lower intestines are positioned in abnormal location. The cecum represents the first part of the large intestines, connecting the ileum of the small intestines to the proximal colon of the large intestines. In contrast to humans, mice possess a large cecum (and a missing appendix) that is highly visible in the abdominal cavity. The arrangement of the cecum, especially with regard to its position and where the end of the cecum faced (right/left) was used to determine the absence or presence of malrotation. Photographs were obtained of the GI from the ventral and dorsal views.
RESULTS
Patient sample – types of syndromes and mutations and GIT anomalies
A total of 32 patient charts were identified that met the inclusion criteria of the study. An initial review of the three databases of patients treated at SickKids between 1990 and 2011 identified a total of 50 charts of patients with clinical diagnoses of CS, PS and AS. As SickKids is a hospital for children, all patients were 18 years of age or younger. Of the 50 charts identified initially, 18 patients were excluded from the study: 5 due to inadequate health records, 8 for a lack of documentation of genetic testing for FGFR1, 2 or 3, and 5 due to the documented absence of a detectable FGFR mutation. The 32 patient charts were reviewed for the congenital GIT malformations as described in the EUROCAT classification system.
Of 32 patients that met the inclusion criteria, 10 were diagnosed with CS, 7 with PS and 15 with AS (Table I and II). All 32 patients had documented FGFR2 mutations, with no mutations in FGFR1 or FGFR3. Information on specific FGFR2 mutations was only available for 25 patients. In total, 16 different mutations were identified in the 25 patients (Table I).
Table I.
Number and type of mutations identified in a sample of 32 FGFR2-associated craniosynostosis syndromic patients at the Hospital for Sick Children (Toronto, Ontario) between 1990 and 2011. IM - intestinal (bowel) malrotation.
| Mutation in FGFR2 | # of Patients | GI Symptoms | |
|---|---|---|---|
|
| |||
| Crouzon (10 patients) | p.Ser282Cys | 1 | — |
| p.Trp290Gly | 1 | — | |
| p.Trp290Arg | 2 | — | |
| p.Tyr328Cys | 1 | — | |
| Missense mutations in IgIII domain of FGFR2 | p.Cys342Ser | 2 | — |
| p.Cys342Phe | 1 | — | |
| p.Ala344Ala | 1 | — | |
| Specific mutation not identified | 1 | — | |
|
| |||
| Pfeiffer (7 patients) | p.Ser252Trp | 1 | — |
| p.Cys278Phe | 1 | — | |
| Mutations in IgIII domain and S252 of FGFR2 | c.940-1G>A | 1 | — |
| p.Cys342Arg | 2 | IM | |
| Specific mutation not identified | 2 | — | |
|
| |||
| Apert (15 patients) | p.Ser252Trp | 1 | IM |
| p.Ser252Trp | 6 | — | |
| Deletion and missense mutations in S252 and P253 of FGFR2 | p.Pro253Arg | 3 | — |
| g.79706_81077del1372* | 1 | — | |
| Specific mutation not identified | 4 | — | |
Previously reported in Fenwick et al., [2011]
Table II.
Frequency of intestinal/bowel malrotation (IM) in the sample population of CS, PS and AS patients.
| Malrotation | |||
|---|---|---|---|
| Sample Size | Frequency | (% of total) | |
| AS | 15 | 1 | 3.13 |
| PS | 7 | 2 | 6.25 |
| CS | 10 | 0 | 0 |
| Total | 32 | 3 | 9.38 |
GIT anomalies in craniosynostosis patients
The review of the charts for GIT abnormalities according to the EUROCAT classification system revealed a relative absence of documentation of most GIT problems. The most common abnormalities observed were intestinal/bowel malrotation (IM) (Table I). Three of the 32 patients had IM (Tables I and II). Of the three patients with IM, two were diagnosed with PS, with the p.Cys342Arg mutation. The third patient was diagnosed with AS with a p.Ser252Trp mutation. The two PS patients with IM had the same p.Cys342Arg mutation, whereas the AS patient with IM had the p.Ser252Trp mutation. Of the two PS with IM, one was diagnosed with IM during her second year of life and presented with a jejunum located to the right of the midline with abnormal location of the cecum and ascending colon. The second PS patient was an infant who presented with a small bowel obstruction secondary to volvulus and a duodenal web. Fortunately, prompt diagnosis prevented further morbidity. Lastly, the AS patient with IM was diagnosed later in life as a result of recurrent vomiting and abdominal pain.
The incidence in the general population of IM is 1 in 500 (pgeneral=0.002) [Cassart et al., 2006; Nehra & Goldstein, 2011; Pickhardt & Bhalla, 2002]. Statistical analysis comparing the incidence of IM in this sample population revealed a significantly higher incidence of this GI abnormality compared to the general population (p < 0.05). Thus, it can be concluded that the proportion with IM in our sample was greater than that of the general population.
Animal studies
In order to characterize possible GIT problems in FGFR-associated craniosynostosis syndrome, the arrangement and the histological features of the GIT of a previously characterized mouse model of CS were analyzed. This mutant mouse model of CS contains a mutation in codon 290 (Trp to Arg) of Fgfr2. Fgfr2W290R heterozygotes exhibit many of the features found in humans with CS such as coronal craniosynostosis and retruded midface and are considered the human model of CS [Mai et al., 2010].
Gross anatomic findings on the GIT of Fgfr2W290R heterozygous mice
Because of the finding of IM in our patient cohort with craniosynostosis syndromes, we characterized the presence of malrotation in the Fgfr2W290R CS mouse model. We took advantage of the size and positioning of the highly visible cecum in mice to classify malrotation. In WT mice, the cecum lay almost horizontally in the abdominal cavity (from the ventral view), with the distal end of this blind J-shaped pouch facing towards the right (n=22; Figs. 1, 2a–c). Of the 26 heterozygous Fgfr2W290R mice examined, 13 (50%) presented with an arrangement of the cecum that closely resembled that of WT mice, cecum present ventrally with its cecal end facing right. In contrast, in the “malrotation” group, the cecum of 5 of the remaining 13 heterozygous mice appeared to come from the right side of the abdomen with the end of the sac facing left (Fig. 2d–f). The remaining 8 heterozygous mice presented with what was described as “partial rotation” where the bulk of the cecum appeared to be hidden dorsally and only a small portion of the cecum was visible from the ventral views and terminating below the stomach (see ventral and dorsal views, Fig. 2g–h). Therefore, 50% of HET in this sample was considered to have malrotation, either non- or partial rotation, whereas the remaining 50% were of the normal phenotype with regard to the positioning of the cecum (Fig. 1).
Figure 1. Phenotypic analysis of position of cecum in wildtype (WT) and heterozygous (HET) Fgfr2W290R mice.
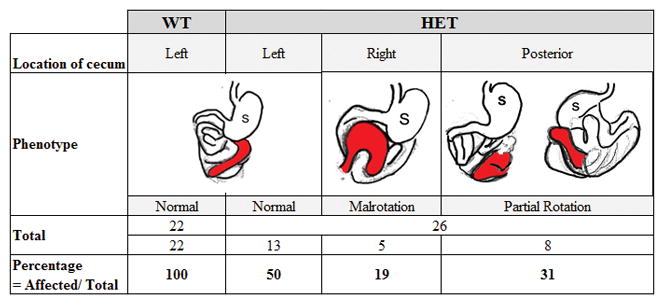
The position of the cecum in the abdominal cavity is colored in red, as viewed ventrally. In wildtype (WT) and 50% of mutant mice, the cecum lies across the abdominal cavity, stretching from left to right (denoted under “left” column). In 19% of the mutant mice, the cecum appears from right to left (“Right” column). In the remaining 31% of the mutants, the cecum is located towards the posterior aspect of the contents of the abdominal cavity (“Posterior” column where figures on left and right represent ventral and dorsal views, respectively).
Figure 2. Position of cecum in the abdominal cavity of Fgfr2W290R mice.

Abdominal contents were removed en masse from mutant mice and photographed after removing the overlying liver (a – i: ventral views; j – l: dorsal views of same guts as shown in g–i). All wildtype (WT) mice and 50% of the Fgfr2W290R heterozygous (HET) mice presented with the cecum lying ventrally across from the left side, with the ends of the pouched-like cecum facing right (a – c are examples from three mice). In the partial rotation group (g & j, h & k, and i & l are guts from the same embryos, photographed from the ventral and dorsal views, respectively), the cecum is barely visible on the ventral surface and the bulk remains dorsally. C- colon; D- duodenum; I- ileum; J-jejunum; S-stomach; stippled line- outline of cecum.
DISCUSSION
In line with many previous reports, mutational hotspots in codons 342 and 290 of FGFR2 [Kress et al., 2000; Schaefer et al., 1998] were identified among the 16 different genetic mutations in the present study. Two patients with IM had the Cys342Arg mutation in the FGFR2 that, along with Ser351Cys and Trp290Cys, has been detected in the more severe phenotype of PS [Johnson and Wilkie, 2011]. In addition to the missense mutations noted, one PS patient in our cohort had a splice site mutation at 952-1G>A, similar to the mutation previously described by Teebi and colleagues [2002]. With AS, a similar trend was observed with regard to the presence of the Ser252Trp and Pro253Arg mutations of AS correlating with the specific phenotypic changes [Jadico et al., 2006; Johnson and Wilkie, 2011]: Six of seven AS individuals with Ser252Trp mutation in our sample had cleft palate (data not shown).
Our results showed a statistically significant difference in the number of FGFR-associated craniosynostosis patients with an IM compared to the normal population. That is, our cohort was approximately 47 times more likely to have bowel malrotation. Our findings of displaced cecal positioning in the abdominal cavity of 50% of a CS mouse model provided an additional line of evidence, at the very least, that some of the important development events during the formation of the GIT can be altered due to mutations in FGFR. Our findings are not surprising given what is known about FGFR involvement in normal growth and development. At a molecular level, studies have demonstrated that the FGFRs and the FGF ligands are expressed along the entire GIT and are known to play a critical role in the development of GI structures and other organ systems. High levels of FGFR2 expression were found throughout the GIT in adult human tissues, specifically throughout the epithelium and submucosal macro- and microvasculature [Hughes, 1997]. Intestinal rotation is important to ensure the proper positioning of the different parts, including the intestines [Martin and Shaw-Smith, 2010]. In humans, the embryonic GIT undergoes two rounds of rotation, an initial 90° followed by a subsequent 180° anti-clockwise turns in the 4th and 8–10th gestational weeks, respectively. The final outcome of the rotations is the placement of the duodenal-jejunal junction lying left of the midline and the cecum in the right iliac fossa [Adams and Stanton, 2014]. In malrotation, the herniated midgut loop undergoes the first rotation normally but fails to undergo its later 180° rotation, such that the colon and cecum return to lie on the left side of the abdominal cavity. In a partial rotation, the cranial limb of midgut alone undergoes the 90° rotation and only the caudal limb undergoes the 180° rotation. The duodenum comes to lie on the right side of the abdominal cavity and the cecum comes to lie just inferior to the stomach [Strouse, 2004]. We used the information of the rotational events during normal GIT development to derive a classification system for IM in the mouse model. Based on cecal position, and by comparing WT with HET mice, we were able to determine that 50% of the mice had normal rotational morphology, whereas the rest had either malrotation or partial rotation. Our findings of the 50% change in the HETs appear to be supported by the findings in our patient cohort of the incidence of IM in 3/32 patients with craniosynostosis syndromes. The less than 50% of IM in our craniosynostosis patients could be related to the fact that many patients with non-rotation and reverse rotation are mostly asymptomatic [Adams and Stanton, 2014]. It is quite possible that our sample may represent an under ascertainment of the true incidence of rotation in this craniosynostosis population. Further research will need to be conducted to confirm this relationship and may involve a multi-center approach to look at a larger number of FGFR-associated syndromic patients.
IM is a serious and potentially fatal developmental anomaly. The small and large intestines can be involved, affecting both the position and peritoneal attachments leading to malrotation [Cassart et al., 2006]. In the majority, diagnosis is made during the first year of life [Pickhardt and Bhalla, 2002]. Diagnostic symptoms include abdominal pain, bile stained vomit secondary to bowel obstruction, bowel dilatation, ascites or meconium peritonitis [Nehra and Goldstein, 2011]. Some reports illustrate the importance of early recognition of IM as a possible comorbidity associated with FGFR-related craniosynostosis syndromes. In our study, the diagnosis of IM was associated with the more severe craniosynostosis syndromes, AS and PS, and not CS, the mildest of the three. For example, a patient with the amino acid substitution in codon 290 to Gly (p.Trp290Gly; Table I), a mutation commonly associated with a milder form of CS [Lajeunie et al., 2006], did not present with GIT symptoms. The amino acid substitution in codon 290 to Arg in the Fgfr2W290R mutants is associated with a more severe form of CS in humans. Based on the findings of IM in 50% of the mutants in our mouse model, it is possible that GIT problems may be asymptomatic in humans with CS.
The findings of this study suggest a correlation between FGFR-related craniosynostosis syndromes and GIT malformations. Our findings corroborate the numerous case reports and mouse studies that revealed GIT anomalies in the syndromic craniosynostosis populations and animal models. Our study may shed further light on the potential widespread impact an FGFR mutation on different developmental systems such as the GIT to provide a sound scientific rationale for the possible implementation of changes, e.g., additional testing and screening procedures for GIT problems, in the clinical management of patients with FGFR-associated craniosynostosis.
Acknowledgments
The authors declare no conflicts of interest in the study. This work was supported in part by National Sciences and Engineering Research Council of Canada-RGPIN 371539-10 (S-G Gong), Canadian Foundation for Innovation (S-G Gong) and the National Institutes of Health (NIH R01-DE018234 to RM).
References
- Adams SD, Stanton MP. Malrotation and intestinal atresias. Early Hum Dev. 2014;90(12):921–925. doi: 10.1016/j.earlhumdev.2014.09.017. [DOI] [PubMed] [Google Scholar]
- Barone CM, Marion R, Shanske A, Argamaso RV, Shprintzen RJ. Craniofacial, limb, and abdominal anomalies in a distinct syndrome: relation to the spectrum of Pfeiffer syndrome type 3. Am J Med Genet. 1993;45(6):745–750. doi: 10.1002/ajmg.1320450616. [DOI] [PubMed] [Google Scholar]
- Carter EP, Fearon AE, Grose RP. Careless talk costs lives: fibroblast growth factor receptor signalling and the consequences of pathway malfunction. Trends Cell Biol. 2015;25(4):221–233. doi: 10.1016/j.tcb.2014.11.003. [DOI] [PubMed] [Google Scholar]
- Cassart M, Massez A, Lingier P, Absil AS, Donner C, Avni F. Sonographic prenatal diagnosis of malpositioned stomach as a feature of uncomplicated intestinal malrotation. Pediatr Radiol. 2006;36(4):358–360. doi: 10.1007/s00247-005-0074-1. [DOI] [PubMed] [Google Scholar]
- Cohen MJ. Malformations of the craniofacial region: Evolutionary, embryonic, genetic, and clinical perspectives. Am J Med Genet. 2002;115(4):245–268. doi: 10.1002/ajmg.10982. [DOI] [PubMed] [Google Scholar]
- Cohen MM, Kreiborg S. Visceral anomalies in the Apert syndrome. Am J Med Genet. 1993;45(6):758–760. doi: 10.1002/ajmg.1320450618. [DOI] [PubMed] [Google Scholar]
- Cornejo-Roldan LR, Roessler E, Muenke M. Analysis of the mutational spectrum of the FGFR2 gene in Pfeiffer syndrome. Hum Genet. 1999;104(5):425–431. doi: 10.1007/s004390050979. [DOI] [PubMed] [Google Scholar]
- Dab S, Sokhi R, Lee JC, Sessle BJ, Aubin JE, Gong SG. Characterization of esophageal defects in the Crouzon mouse model. Birth Defects Res A Clin Mol Teratol. 2013;97(9):578–586. doi: 10.1002/bdra.23172. [DOI] [PubMed] [Google Scholar]
- Eaton AP, Sommer A, Sayers MP. The Kleeblattschädel anomaly. Birth Defects Orig Artic Ser. 1975;11(2):238–246. [PubMed] [Google Scholar]
- Eswarakumar VP, Lax I, Schlessinger J. Cellular signaling by fibroblast growth factor receptors. Cytokine Growth Factor Rev. 2005;16(2):139. doi: 10.1016/j.cytogfr.2005.01.001. [DOI] [PubMed] [Google Scholar]
- Fairbanks TJ, Kanard R, Del Moral PM, Sala FG, De Langhe S, Warburton D, Anderson KD, Bellusci S, Burns RC. Fibroblast growth factor receptor 2 IIIb invalidation--a potential cause of familial duodenal atresia. J Pediatr Surg. 2004;39(6):872–874. doi: 10.1016/j.jpedsurg.2004.02.026. [DOI] [PubMed] [Google Scholar]
- Fenwick AL, Bowdin SC, Klatt RE, Wilkie AO. A deletion of FGFR2 creating a chimeric IIIb/IIIc exon in a child with Apert syndrome. BMC Med Genet. 2011;12:122. doi: 10.1186/1471-2350-12-122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes SE. Differential expression of the fibroblast growth factor receptor (FGFR) multigene family in normal human adult tissues. J Histochem Cytochem. 1997;45(7):1005–1019. doi: 10.1177/002215549704500710. [DOI] [PubMed] [Google Scholar]
- Ibrahimi OA, Eliseenkova AV, Plotnikov AN, Yu K, Ornitz DM, Mohammadi M. Structural basis for fibroblast growth factor receptor 2 activation in Apert syndrome. Proc Natl Acad Sci USA. 2001;98(13):7182–7187. doi: 10.1073/pnas.121183798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jadico SK, Young DA, Huebner A, Edmond JC, Pollock AN, McDonald-McGinn DM, Li YJ, Zackai EH, Young TL. Ocular abnormalities in Apert syndrome: genotype/phenotype correlations with fibroblast growth factor receptor type 2 mutations. J AAPOS. 2006;10(6):521–527. doi: 10.1016/j.jaapos.2006.07.012. [DOI] [PubMed] [Google Scholar]
- Jaye M, Schlessinger J, Dionne CA. Fibroblast growth factor receptor tyrosine kinases: molecular analysis and signal transduction. Biochim Biophys Acta. 1992;1135(2):185–199. doi: 10.1016/0167-4889(92)90136-y. [DOI] [PubMed] [Google Scholar]
- Johnson D, Wilkie AO. Craniosynostosis. Eur J Hum Genet. 2011;19(4):369–376. doi: 10.1038/ejhg.2010.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabbani H, Raghuveer TS. Craniosynostosis. Am Fam Physician. 2004;69(12):2863–2870. [PubMed] [Google Scholar]
- Kaplan LC. Clinical assessment and multispecialty management of Apert syndrome. Clin Plast Surg. 1991;18(2):217–225. [PubMed] [Google Scholar]
- Koga H, Suga N, Nakamoto T, Tanaka K, Takahashi N. Clinical expression in Pfeiffer syndrome type 2 and 3: surveillance in Japan. Am J Med Genet A. 2012;158A(10):2506–2510. doi: 10.1002/ajmg.a.35590. [DOI] [PubMed] [Google Scholar]
- Kress W, Collmann H, Büsse M, Halliger-Keller B, Mueller C. Clustering of FGFR2 gene mutations inpatients with Pfeiffer and Crouzon syndromes (FGFR2-associated craniosynostoses) Cytogenet Cell Genet. 2000;91(1–4):134–137. doi: 10.1159/000056833. [DOI] [PubMed] [Google Scholar]
- Lajeunie E, Heuertz S, El Ghouzzi V, Martinovic J, Renier D, Le Merrer M, Bonaventure J. Mutation screening in patients with syndromic craniosynostoses indicates that a limited number of recurrent FGFR2 mutations accounts for severe forms of Pfeiffer syndrome. Eur J Hum Genet. 2006;14(3):289–298. doi: 10.1038/sj.ejhg.5201558. [DOI] [PubMed] [Google Scholar]
- Lajeunie E, Ma HW, Bonaventure J, Munnich A, Le Merrer M, Renier D. FGFR2 mutations in Pfeiffer syndrome. Nat Genet. 1995;9(2):108–108. doi: 10.1038/ng0295-108. [DOI] [PubMed] [Google Scholar]
- Mai S, Wei K, Flenniken A, Adamson SL, Rossant J, Aubin JE, Gong SG. The missense mutation W290R in Fgfr2 causes developmental defects from aberrant IIIb and IIIc signaling. Dev Dyn. 2010;239(6):1888–1900. doi: 10.1002/dvdy.22314. [DOI] [PubMed] [Google Scholar]
- Mangasarian K, Li Y, Mansukhani A, Basilico C. Mutation associated with Crouzon syndrome causes ligand-independent dimerization and activation of FGF receptor-2. J Cell Physiol. 1997;172(1):117–125. doi: 10.1002/(SICI)1097-4652(199707)172:1<117::AID-JCP13>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- Marie PJ, Miraoui H, Sévère N. FGF/FGFR signaling in bone formation: progress and perspectives. Growth Factors. 2012;30(2):117–123. doi: 10.3109/08977194.2012.656761. [DOI] [PubMed] [Google Scholar]
- Martin V, Shaw-Smith C. Review of genetic factors in intestinal malrotation. Pediatr Surg Int. 2010;26(8):769–781. doi: 10.1007/s00383-010-2622-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKeehan WL, Kan M. Heparan sulfate fibroblast growth factor receptor complex: structure-function relationships. Mol Reprod Dev. 1994;39(1):69–81. doi: 10.1002/mrd.1080390112. discusison 81–62. [DOI] [PubMed] [Google Scholar]
- Nehra D, Goldstein AM. Intestinal malrotation: varied clinical presentation from infancy through adulthood. Surgery. 2011;149(3):386–393. doi: 10.1016/j.surg.2010.07.004. [DOI] [PubMed] [Google Scholar]
- Neilson KM, Friesel R. Ligand-independent activation of fibroblast growth factor receptors by point mutations in the extracellular, transmembrane, and kinase domains. J Biol Chem. 1996;271(40):25049–25057. doi: 10.1074/jbc.271.40.25049. [DOI] [PubMed] [Google Scholar]
- Park W-J, Meyers GA, Li X, Theda C, Day D, Oriow SJ, Jones MC, Jabs EW. Novel FGFR2 mutations in Crouzon and Jackson-Weiss syndromes show allelic heterogeneity and phenotypic variability. Hum Mol Genet. 1995;4(7):1229–1233. doi: 10.1093/hmg/4.7.1229. [DOI] [PubMed] [Google Scholar]
- Pickhardt PJ, Bhalla S. Intestinal malrotation in adolescents and adults: spectrum of clinical and imaging features. AJR Am J Roentgenol. 2002;179(6):1429–1435. doi: 10.2214/ajr.179.6.1791429. [DOI] [PubMed] [Google Scholar]
- Plotnikov AN, Hubbard SR, Schlessinger J, Mohammadi M. Crystal Structures of Two FGF-FGFR Complexes Reveal the Determinants of Ligand-Receptor Specificity. Cell. 2000;101(4):413–424. doi: 10.1016/s0092-8674(00)80851-x. [DOI] [PubMed] [Google Scholar]
- Ranger A, Al-Hayek A, Matic D. Chiari type 1 malformation in an infant with type 2 Pfeiffer syndrome: further evidence of acquired pathogenesis. J Craniofac Surg. 2010;21(2):427–431. doi: 10.1097/SCS.0b013e3181cfa792. [DOI] [PubMed] [Google Scholar]
- Schaefer F, Anderson C, Can B, Say B. Novel mutation in the FGFR2 gene at the same codon as the Crouzon syndrome mutations in a severe Pfeiffer syndrome type 2 case. Am J Med Genet. 1998;75(3):252–255. doi: 10.1002/(sici)1096-8628(19980123)75:3<252::aid-ajmg4>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- Strouse PJ. Disorders of intestinal rotation and fixation (“malrotation”) Pediatr Radiol. 2004;34(11):837–851. doi: 10.1007/s00247-004-1279-4. [DOI] [PubMed] [Google Scholar]
- Teebi AS, Kennedy S, Chun K, Ray PN. Severe and mild phenotypes in Pfeiffer syndrome with splice acceptor mutations in exon IIIc of FGFR2. Am J Med Genet. 2002;107(1):43–47. doi: 10.1002/ajmg.10125. [DOI] [PubMed] [Google Scholar]
- Wang F, Lu W, McKeehan K, Mohamedali K, Gabriel JL, Kan M, McKeehan WL. Common and Specific Determinants for Fibroblast Growth Factors in the Ectodomain of the Receptor Kinase Complex. Biochemistry. 1999;38(1):160–171. doi: 10.1021/bi981758m. [DOI] [PubMed] [Google Scholar]
- Wilkie AOM, Slaney SF, Oldridge M, Poole MD, Ashworth GJ, Hockley AD, Hayward RD, David DJ, Pulleyn LJ, Rutland P, Malcolm S, Winter RM, Reardon W. Apert syndrome results from localized mutations of FGFR2 and is allelic with Crouzon syndrome. Nat Genet. 1995;9(2):165–172. doi: 10.1038/ng0295-165. [DOI] [PubMed] [Google Scholar]
- Zarate YA, Putnam PE, Saal HM. Intestinal malrotation in a patient with Pfeiffer syndrome type 2. Cleft Palate Craniofac J. 2010;47(6):638–641. doi: 10.1597/09-115. [DOI] [PubMed] [Google Scholar]