

Abstract
Surface fouling is one of the leading causes of infection associated with implants, stents, catheters, and other medical devices. The surface chemistry of medical device coatings is important in controlling and/or preventing fouling. In this study, we have shown that a combination of nitric oxide releasing hydrophobic polymer with a hydrophilic polymer topcoat can significantly reduce protein attachment and subsequently reduce bacterial adhesion as a result of the synergistic effect. Nitric oxide (NO) is a well-known potent antibacterial agent due to its adverse reactions on microbial cell components. Owing to the surface chemistry of hydrophilic polymers, they are suitable as antifouling topcoats. In this study, four biomedical grade polymers were compared for protein adhesion and NO-release behavior: CarboSil 2080A, RTV, SP60D60, and SG80A. SP60D60 was found to resist protein adsorption up to 80% when compared to the other polymers while CarboSil 2080A maintained a steady NO flux even after 24 hours (~0.50 × 10−10 mol cm−2 min−1) of soaking in buffer solution with a loss of less than 3 wt% S-nitroso-N-acetylpenicillamine (SNAP), the NO donor molecule, in the leaching analysis. Therefore, CarboSil 2080A incorporated with SNAP and topcoated with SP60D60, was tested for antibacterial efficacy after exposure to fibrinogen, an abundantly found protein in blood. The NO-releasing CarboSil 2080A with SP60D60 topcoated polymer showed a 96% reduction in Staphylococcus aureus viable cell count compared to the control samples. Hence, the study demonstrated that a hydrophilic polymer topcoat, when applied to a polymer with sustained NO release from underlying SNAP incorporated hydrophobic polymer, can reduce bacterial adhesion and be used as a highly efficient antifouling, antibacterial polymer for biomedical applications.
Keywords: antifouling, hydrophilic, nitric oxide, coating, antibacterial, infection, protein adhesion, biomedical device application, Staphylococcus aureus
Graphical Abstract
1. Introduction
Fouling caused by proteins and bacteria is a very common phenomenon found on interfaces of biomaterials of medical devices and biological environments of the human body.1 According to the Centers for Disease Control and Prevention, fouling is a major problem that causes nosocomial or hospital-associated infections (HAIs) and has led to the increase in medical costs (overall annual direct medical costs of HAIs in U.S. hospitals ranges from $28.4 to $33.8 billion).2 Medical implants and devices like biosensors, drug carriers, soft contact lenses, vascular stents, and urological devices upon contact with the biological milieu can cause subsequent settling of non-specific biomacromolecules on the foreign surfaces 3 and trigger a cascade of events that can bring about device failure. Biomacromolecules like proteins and bacteria settle down on the surface of these medical devices and start forming a layer of extrapolymeric substance (EPS) which is followed by settling of pathogens like bacteria and fungi on these protein layers.4, 5 These layers can develop into large films of a mixture of EPS and bacteria called biofilms. Biofilms are tough to get rid of due to their stable aggregation of bacteria covered with a film of proteins and can thus block pathways for medical devices and cause device failure. This stage of device contamination is infection, and may lead to sepsis and eventually cause patient death. In the recent times, significant research efforts have been directed towards finding efficient antifouling polymers combined with antibacterial properties in the medical industry to combat the problem of biofilms.6–11
Some of the methods that have been used for antifouling mechanisms of polymers are steric repulsion,12, 13 electrostatic repulsion,14, 15 and hydrophilicity.15–17 While steric and electrostatic repulsion are reliable methods for antifouling, both involve major processing changes that is possible in lab scale but not practical for industrial applications. Increasing hydrophilicity of a material for antifouling mechanism is advantageous because most proteins that contaminate surfaces are hydrophobic in nature and the hydration layer formed on the hydrophilic polymer keeps proteins from adhering to the coating surface of the medical device.18, 19 Hydrophilic polymers tend to be smooth and a water layer can easily form on these medical device coatings as they are kept in contact with bodily fluids.20 This hydration layer helps in repulsion of non-specific proteins and acts as an antifouling mechanism.19 Roughness of surfaces also play a critical role in adhesion of proteins.21, 22 It has been found to be an important factor at the nanometer scale for both fibrinogen and bovine serum albumin attachment.21 Increasing random roughness increases the adhesion of proteins to surfaces of biomaterials. Therefore in comparison with smoother polymers that do not allow adherence of proteins, rough polymers tend to act as support for proteins and pathogens to attach to and hence are not considered to be antifouling in nature.23
Antimicrobial polymers can be broadly classified into three types according to their general working principles: polymeric biocides, biocidal polymers, and biocide-releasing polymers.24 Polymeric biocides have repeating units of biocides and in case of biocidal polymers, the whole polymer acts like one antimicrobial agent so no repeating units of biocidal groups are required. Biocide-releasing polymers carry the biocidal agents and release them into the microbes. While passive approaches like polymeric biocides and biocidal polymers are very closely related where the microbe has to come in contact with the polymer to be acted on, the dynamic action of biocide releasing polymers to act from a distance gives them an advantage over the other two classifications. This means that the microbe does not have to be in contact with the polymer to be killed. Another advantage of biocide releasing polymers is that since they discharge antimicrobial agents incorporated within them, the concentration of these biocides can be controlled according to the applications. Biocide releasing polymers also do not let dead microbes bind to the polymers themselves and hence do not allow for accumulation of dead microbes. These advantages of biocide releasing polymers have led to intensive research on antimicrobial agents that can be incorporated within them.
Some commonly studied antimicrobial agents are silver,25–29 antimicrobial peptides,30–34 and nitric oxide (NO).35–37 While silver has been found to be an effective antimicrobial agent, it has also demonstrated concerns for cytotoxicity.38, 39 Antimicrobial peptides are also being studied in great detail but due to the complexity of their interactions,40 they are not a popular class of practically useful antimicrobial agents.
Besides being an endothelium derived relaxing factor which functions as a vasodilator, NO is a well-known antibacterial agent and it has been tested in various polymers along with its production from different sources/donors.35, 41–61 It is a free radical that reacts with superoxides and oxygen to form peroxynitrite and dinitrogen trioxide, respectively.42, 43, 62 Nitric oxide’s mechanisms of action against bacteria include nitrosation of amines and thiols in the extracellular matrix, lipid peroxidation and tyrosine nitration in the cell wall, and DNA cleavage in the cellular matrix.63 It’s antibacterial activity has been found to be successful in various hospital associated infection pathogens such as Pseudomonas aeruginosa,64, 65 Staphylococcus aureus,45, 46, 65 Escherichia coli,45, 46, 56, 65–67 Staphylococcus epidermidis 46, and Acinetobacter baumannii.35, 68
However, even though the antibacterial effects of NO have been studied without any other aids, it’s synergistic effects when combined with an antifouling polymer has not been extensively studied. This is important to study because despite its antibacterial nature, NO by itself cannot prevent the adsorption of proteins. Study of NO releasing materials with the combination of antifouling materials would be essential to eliminate any fouling from proteins that takes place when a medical device first comes in contact with bodily fluids and consequently facilitates bacterial adhesion.
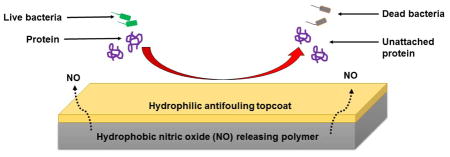
Herein, we combine a hydrophilic antifouling polymer topcoat (SP60D60) on a sustained NO-releasing low water uptake polymer (CarboSil 2080A). Initially, four polymers were chosen to test the different parameters required for the desirable antifouling and antibacterial effects. CarboSil 2080A and Dow Corning® RTV 3140 Silicone Rubber (RTV) were the hydrophobic polymers while SP60D60 and SG80A were the comparatively hydrophilic polymers tested. CarboSil 2080A is a thermoplastic urethane copolymer with a mixed soft segment of poly (dimethyl siloxane) and hydroxyl-terminated polycarbonate and a hard segment of an aromatic diisocyanate.69 RTV is a coating material with a chemical composition of silicone elastomer. SP60D60 is a Tecophilic® solution grade thermoplastic polyurethane resin which is commonly used as a biomedical device coating polymer. SG80A is a Tecoflex® solution processible grade thermoplastic polyurethane resin which, like SP60D60, is also commonly used in the medical industry for coating purposes.
The four polymers were tested for their wetting properties by measurement of static contact angles, water uptake, and surface roughness. Once the wetting nature of the polymers was established, they were tested for their antifouling nature. Protein adhesion test was performed using spectroscopic ellipsometric measurements. Least amount of protein was found attached to SP60D60’s surface which was also supported by its hydrophilic and smooth surface and hence it was chosen as the topcoat material. Following this, NO-release behavior for the polymers were determined with S-nitroso-N-acetylpenicillamine (SNAP) leaching study and nitric oxide release analysis. CarboSil 2080A and RTV both displayed a minimal loss of SNAP (<5% and <7% of total SNAP present in the sample, respectively) during the leaching analysis and hence were chosen for further NO release measurements. Finally, antibacterial efficacy test with gram positive bacteria Staphylococcus aureus was performed to analyze if the hydrophilic coated polymers were more efficient than the control. The increase in killing efficiency of the test samples when compared to the hydrophobic polymer coated controls validated that a SNAP-incorporated CarboSil 2080A with a topcoat of SP60D60 showed the highest reduction in microbial viability.
2. Materials and methods
2.1. Materials
N-acetyl-D-penicillamine (NAP), sodium nitrite, concentrated sulfuric acid (conc. H2SO4), tetrahydrofuran (THF), sodium phosphate monobasic (NaH2PO4), sodium phosphate dibasic (Na2HPO4), potassium chloride, sodium chloride, fibrinogen from bovine plasma, and ethylenediamine tetraacetic acid (EDTA) were obtained from Sigma Aldrich (St. Louis, MO). Luria Agar (LA), Miller and Luria broth (LB), Lennox were purchased from Fischer BioReagents (Fair Lawn, NJ). Concentrated hydrochloric acid (conc. HCl), sodium hydroxide (NaOH), and methanol were purchased from Fisher-Scientific (Hampton, NH). Potassium phosphate monobasic (KH2PO4) was purchased from BDH Chemicals - VWR International (West Chester, PA). Tecophilic SP-60D-60 and Tecoflex SG-80A were products of Lubrizol Advanced Materials Inc. (Cleveland, OH). Dow Corning RTV 3140 Silicone Rubber (SR) was purchased from Ellsworth Adhesives (Germantown, WI). CarboSil™ 2080A was obtained from DSM Biomedical Inc. (Berkeley, CA). Milli-Q filter was used to obtain de-ionized water for the aqueous solution preparations. Staphylococcus aureus (ATCC 5538, S. aureus) was used for all bacterial experiments.
2.2. Synthesis of S-nitroso-N-acetylpenicillamine (SNAP)
S-nitroso-N-acetylpenicillamine was synthesized using methods previously reported with a few modifications.44, 70 1M HCl and 1M H2SO4 was added to an equimolar amount of NAP, methanol and sodium nitrite solution containing DI water. This reaction was stirred for 15 minutes and then cooled in an ice bath for 4 hours. After evaporation of the reaction mixture, precipitated green crystals of SNAP were vacuum filtered, collected and allowed to air dry in dark conditions. Dried crystals of SNAP were used for all experiments.
2.3. Preparation of SNAP-incorporated Polymer Films
Polymers containing 10 wt% SNAP were prepared by solvent evaporation method. The casting solutions were prepared by dissolving 210 mg of the respective polymer (CarboSil 2080A, RTV, SP60D60 or SG80A) in 3 mL of THF. The polymers were allowed to dissolve before the addition of 23.1 mg of SNAP for a final concentration of 10 wt% of SNAP. This mixture was protected from light and stirred until the SNAP crystals dissolved completely. The polymer solutions were then poured into Teflon molds (d = 2.5 cm) and allowed to dry overnight in fume hood. The dried films were then cut into small disks (d = 0.7 cm) and dip coated with a topcoat solution of the polymer without SNAP (40 mg/mL of polymer concentration in THF). The small disks were dried overnight and then dried under vacuum for an additional 24 hours. This was done to remove any residual THF which can hamper the following studies. Weight of each small disk was measured before topcoat. The prepared disks were kept in the freezer (−18°C) in the dark to retain its NO releasing properties and prevent leaching of SNAP. These SNAP-incorporated films were used for NO-release, SNAP leaching and bacterial adhesion studies.
2.4. Preparation of Thin Polymer Films on Silicon wafers
Thin polymer films on silicon wafers were deposited by spin coating the polymer solution using a CHEMAT Technology KW-4A spin coater. Films were spin coated at 2500 rpm for 30 seconds, yielding highly uniform pinhole free layers with a surface thickness of 70–100 nm. These thin films were used for studying protein adhesion measurement (section 2.5.3.) using a M-2000 spectroscopic ellipsometer (J.A. Woollam Co., Inc.).
2.5. Characterization of Topcoat Material with Least Protein Adhesion
2.5.1. Static Contact Angle and Water Uptake Measurement
Static contact angle for the four chosen biomedical grade polymers (two hydrophilic- SP60D60 and SG80A, and two hydrophobic- CarboSil 2080A and RTV, comparatively) were measured using a Krüss DSA100 Drop Shape Analyzer (sessile drop method with deionized water). Spin coated polymers on silicon wafers were used for the measurement.
For water uptake measurement, polymer films prepared with the solvent evaporation method were weighed and soaked in water overnight. Three replicates were used for each measurement. Their mass was checked the next day to measure the water uptake for the polymers.
2.5.2. Surface Roughness Measurement
Surface roughness measurement to check for consistency with protein adhesion study was measured using PeakForce QNM (Bruker Multimode AFM) over a 10 μm2 region. AFM imaging was done on polymer films dried on silicon wafers using solvent evaporation technique. Three images and roughness average measurements (Ra in nm) were collected for each polymer to confirm the measurements.
2.5.3. Protein Adhesion Study
The thicknesses of spin coated films (prepared according to section 2.4.) were measured using a M-2000 spectroscopic ellipsometer (J.A. Woollam Co., Inc.) with a white light source at three angles of incidence (65, 70, and 75°) to the silicon wafer normal. Three replicates were used for each measurement. After thickness of each film was measured, a non-saline phosphate buffer solution was prepared from 1 M sodium phosphate dibasic and 1 M potassium phosphate monobasic. The solution was adjusted to a pH of 7.41 at room temperature (25°C). Samples were kept in the non-saline phosphate buffer for 30 minutes at 37°C. A solution of fibrinogen from bovine plasma and non-saline phosphate buffer was prepared to achieve a concentration of 1mg mL−1 once added to the non-saline phosphate buffer the samples were first placed in. After the protein solution was added to the samples, they were allowed to incubate at 37°C for 90 minutes. After incubation each sample was washed with 5mL of non-saline PBS five consecutive times followed by 5mL of distilled water five consecutive times. The thickness of the wafers before and after submersion in the protein solution was measured by spectroscopic ellipsometry. Care was taken to measure the thickness on the same area of the films as measured before to avoid any inconsistency in data collection.
2.6. Characterization of SNAP-incorporated films for optimized NO-releasing polymer
2.6.1. SNAP leaching Study
The weight percentage of SNAP leached out from the polymers were measured by recording the absorbance of buffer solutions in time intervals of 0.5 h, 1 h, 4 h, and 24 h at 340 nm. Three sample for each type of film was prepared and weighed before topcoating to determine the initial amount of SNAP in each film. These films were then soaked in PBS (with EDTA) at 37°C. A UV-vis spectrophotometer (Thermoscientific Genesys 10S UV-Vis) was used to measure the absorbance of the buffer solutions in the above mentioned time intervals. Absorbance was measured at an optical density of 340 nm which is the maxima in the UV-Vis absorbance spectra for SNAP. The calibration graph of SNAP in PBS (with EDTA) was used to interpolate the absorbance measurements recorded from the study and convert them to concentrations of SNAP in the measured sample. This concentration was converted to weight % of SNAP in the buffer using the initial amount of SNAP present in each sample used. Care was taken to make sure that buffer solution amount for each sample was maintained at the same amount throughout the experiment to avoid any inconsistent readings and three replicates were used for each measurement.
2.6.2. NO-Release Measurements
S-nitroso-N-acetylpenicillamine present in the samples releases NO under physiological conditions (Figure 1) and this was measured and recorded for the study. Real time NO-release from the polymer films was measured using Sievers chemiluminescence NO analyzers® (NOA 280i, GE Analytical, Boulder, CO, USA). The sample holder of the NOA was shielded from light and injected with 4 mL of PBS (containing EDTA). This buffer solution was warmed up to 37°C by a water jacket placed around the sample holder. Once PBS warms up and a baseline of NO flux is established for the sample (prepared according to section 2.3.), the sample is then placed in the sample holder. Nitric oxide released by the sample in the sample holder was swept and purged by a continuous supply of high purity nitrogen maintained at a constant flowrate of 200 mL min−1 through the sweep and bubble flows. At the same time, oxygen produces the ozone required for the reaction that would take place in the reaction chamber. The NO released by the sample is pushed towards the chemiluminescence detection chamber. The voltage signal produced is converted to concentration and displayed on the analyzer’s screen. Using the raw data in ppb form and NOA constant (mol ppb−1 s−1), the data in ppb is normalized and converted to NO flux units (x 10−10 mol cm−2 min−1) according to the surface area of the sample used for analysis. Data is collected in the time intervals mentioned and samples are stored in a PBS (with EDTA) solution at 37°C in dark conditions. The PBS is replaced daily to avoid any accumulation of NO released during the storage time.
Figure 1.

Scheme showing release of nitric oxide from S-nitroso-N-acetylpenicillamine on exposure to heat and/or light.
The samples were always submerged in the buffer solution through the NO-release flux recording. The instrument operating parameters were a cell pressure of 7.4 Torr, a supply pressure of 5.9 psig and a temperature of − 12 °C. Three replicates were used for each measurement.
2.7. Top Coat Stability Study
Static contact angles were measured to make sure that the antifouling hydrophilic polymer, SP60D60, would not delaminate from the hydrophobic polymer, CarboSil 2080A, due to exposure to physiological conditions including soaking in PBS and temperature of 37°C. Spin-coated silicon wafers with CarboSil 2080A and a topcoat of SP60D60 were kept in PBS solution at 37°C. Static contact angle was measured using a Krüss DSA100 Drop Shape Analyzer before and after 24 hours of incubation in the PBS solutions. This environment was used to mimic the physiological environment used for protein adhesion and NO-release study.
2.8. In vitro Analysis of Inhibition of Bacterial Adhesion on Polymer Surface
In this study section, the combined effect of SP60D60 coat on CarboSil 2080A base in terms of bacteria adhesion post protein (fibrinogen from bovine plasma) exposure was assessed using a modified method based on the American Society for Testing and Materials E2180 test protocol. This protocol is effective in testing antimicrobial efficacy of hydrophobic polymers. The multidrug resistant gram positive S. aureus bacteria which is among the most common cause of hospital acquired infections (HAIs) was used as the model organism to test the efficacy of SP60D60 coating in preventing bacteria adhesion.
2.8.1. Bacterial Culture Preparation
Luria Broth (LB) medium was prepared according to the manufacturer’s instructions. This broth was sterilized in an autoclave prior to using it for the study. A bacterial suspension was cultured in LB medium for 14 hours at 37°C and a horizontal rotating speed of 150 rpm in shaker incubator. After 14 hours of culture, the optical density (O.D.) of the culture was measured at a wavelength of 600 nm (O.D.600) using a UV-vis spectrophotometer (Thermoscientific Genesys 10S UV-Vis) to ensure that the bacteria are in actively dividing phase. The bacteria culture was then centrifuged at 3500 rpm for 7.5 mins and the supernatant was discarded. The bacterial cells were washed with fresh sterile phosphate buffer saline (PBS)-pH 7.4, centrifuged at 3500 rpm for 7.5 mins. The supernatant was discarded and fresh PBS was added to resuspend the bacteria. The O.D.600 of the cell suspension in PBS was measured using PBS as blank and adjusted to the CFU in the range of 106–108. In order to verify the consistency of concentration of viable cells between experiments, serial dilutions of S. aureus bacteria were prepared and plated petri dishes containing autoclaved LB agar. LB agar was prepared according manufacturer’s instructions.
2.8.2. Bactericidal Activity Analysis
CarboSil 2080A base with CarboSil 2080A topcoat (CarboSil 2080A/CarboSil 2080A) (n=3) was used as the control to compare the difference in number of viable bacteria on CarboSil 2080A base with SP60D60 topcoat (SP60D60/CarboSil 2080A) and 10 wt.% SNAP films with SP60D60 top coat (SP60D60/SNAP) (n=3). These films were exposed to 1 mg mL−1 of fibrinogen from bovine plasma for 1 hour in the method as described in section 2.5.3. Post 1h of protein exposure, the films were exposed to bacterial cells (106–108 CFU mL−1) at 37°C for 3 hours at a speed of 200 rpm in a shaker incubator. After 3 hours, films were rinsed with sterile PBS to remove any loosely bound bacteria from the film surface and transferred to fresh PBS. The films were homogenized for 45 seconds, vortexed for 20 seconds and S. aureus was plated in the solid LB agar medium after preparing serial dilutions in the range of 10−1–10−5. The LB agar plates with the plated bacteria culture were incubated at 37°C for 20 hours. After 20 hours, the CFUs were counted considering the dilution factor and the number of viable bacteria on SP60D60/SNAP films were compared to the control films. Three replicates were used.
2.9. Statistical analysis of data
All data are expressed as mean ± standard deviation. The results between the control and test films were analyzed by a comparison of means using Student’s t-test. Values of p were obtained for the data analyzed to show significance of results.
3. Results and Discussion
3.1. Characterization of Polymers for an Antifouling Topcoat
The four polymers chosen for this study were at first tested for their wetting properties. These preliminary data would help in predicting their hydrophilic nature and subsequently antifouling properties. At first, static contact angle for the spin coated films was measured. The static contact angle for SP60D60 was the lowest (51.10 ± 2.21°) among the polymers while CarboSil 2080A and RTV both displayed high contact angles (Table 1). This demonstrated that SP60D60 was the most hydrophilic polymer among the four polymers selected for the study. Following this, the results of water uptake study supported the contact angles measurements as predicted. Water uptake was the highest for SP60D60 (57.55 ± 1.80 %) (Table 2) and the least for RTV (0.47 ± 0.25 %).
Table 1.
Static contact angle of the polymers used in the study by a Krüss DSA100 Drop Shape Analyzer. Data represents mean ± SD (n=3).
| Polymer | Static Contact Angle (°) |
|---|---|
| CarboSil | 104.62 ± 0.08 |
| RTV | 111.40 ± 0.30 |
| SP60D60 | 51.10 ± 2.21 |
| SG80A | 93.27 ± 0.66 |
Table 2.
Water uptake of the polymers used in the study measured in weight %. Data represents mean ± SD (n=3).
| Polymer | Water Uptake (wt. %) |
|---|---|
| CarboSil | 0.83 ± 0.27 |
| RTV | 0.47 ± 0.25 |
| SP60D60 | 57.55 ± 1.80 |
| SG80A | 3.37 ± 1.89 |
The four polymers were further studied for their surface roughness (roughness average) by using atomic force microscopy. A 10 μm2 scan was taken for each substrate and as shown in Figure 2, the difference in surface roughness was clearly visible. The hydrophobic polymers, (CarboSil 2080A=0.873 ± 0.048 nm and RTV=2.257 ± 0.458 nm) exhibited higher surface roughness while the hydrophilic polymers (SP60D60=0.360 ± 0.099 nm and SG80A=0.362 ± 0.003 nm) were both smoother. It is understood that this smooth surface of the hydrophilic surface helps in retaining the hydration layer above the polymer and creates a slippery surface on which adhesion by proteins and bacteria is significantly reduced. As previously mentioned, the random roughness of the hydrophobic polymers would also help in protein attachment instead of repulsion. Random roughness increases the amount of surface area available for attachment and hence can increase the amount of adsorbed protein. Random roughness can also increase van der Waals force and electrostatic force, thereby increasing the adsorption of proteins on rough surfaces.21 Therefore, from the results of the wetting properties and surface roughness analysis, the hydrophilic polymers were expected to perform better than the hydrophobic polymers on testing for repulsion of proteins from the polymer surface.
Figure 2.

AFM topography images of the four polymers used in the study to compare surface roughness between the hydrophobic and hydrophilic polymers. The surface roughness (nm) for the polymers were: A) CarboSil = 0.873 ± 0.048 B) RTV = 2.257 ± 0.458 C) SP60D60 = 0.360 ± 0.099 D) SG80A = 0.362 ± 0.003. Data represents mean ± SD. (n=3)
3.2. Protein adhesion test for an Antifouling Topcoat
For the protein adhesion test, all four polymers were soaked in a buffer solution of fibrinogen protein (concentration of 1mg/mL) for 90 minutes because the bulk of protein adhesion to medical device surfaces typically occurs within the first few minutes of exposure to physiological fluids.71 The samples were incubated in the fibrinogen solution at 37°C to mimic physiological conditions.
Fibrinogen (340 kDA) is an anisotropic protein and has shown to increase bacterial adhesion.11, 72–76 It is an adhesive protein that is known to replace proteins like albumin on the surface of materials (Vroman effect) due to its higher surface affinity.72 Fibrinogen molecules act as extracellular matrix (ECM) for bacteria such as S. aureus and the interaction between them has been studied extensively.77 The receptors of ECM molecules bind to adhesins present on bacteria and trigger complex signal transduction cascades in the bacterial cell that can provide impetus to bacterial invasion. In the case of fibrinogen and S. aureus, the fibrinogen binding proteins present on the bacterial cells are called clumping factors A and B.78 The favorable binding interactions between proteins like fibrinogen and bacteria like S. aureus make it an important aspect to control while fabricating implants and medical devices since consequences like biofilm growth can cause device failure.
Any unattached proteins were removed from the polymer surfaces by rinsing the surfaces with non-saline buffer and DI water. The data obtained in Figure 3 shows the change in the thickness for the respective polymers. The nanometer change in thickness of the films were measured using a spectroscopic ellipsometer. Spectroscopic ellipsometry can easily detect changes in the nanometer scale. As studied using electron microscopy and atomic force microscopy, fibrinogen is a highly elongated and anisotropic protein which has dimensions of 5–6.5 nm in diameter and 47.5 nm in length.79, 80 Therefore, in the ellipsometric measurements, any change above 5 nm would be considered adsorption of the fibrinogen protein.
Figure 3.

Graph shows relationship between wetting characteristic and protein adhesion of the surface. It shows thickness of protein layer attached to the polymer after exposure to 1 mg mL−1 of fibrinogen from bovine serum for 90 minutes. Static contact angle represents wetting characteristic of the material with no protein on it. Data represents mean ± SD (n=3) (p<0.004)
From the data, we can infer that protein adhesion was confirmed to be non-existent (p = 0.004) on SP60D60 (2.24 ± 0.68 nm) while the other polymers (CarboSil 2080A = 6.63 ± 0.78 nm, RTV = 5.96 ± 1.66 nm and SG80A = 6.14 ± 1.47 nm) did show a monolayer of fibrinogen adsorption on the surfaces. SP60D60 shows a high resistance to protein adsorption on its surface due to its surface properties. It is hydrophilic in nature which enables it to form a hydration layer with the surrounding environment. This hydration layer in turn protects it from formation of a protein layer on it. The protein molecules are unable to bind to the surface because of the presence of the hydration layer. However, the small increase in thickness observed on SP60D60 can be due to two reasons. It can indicate slight swelling of the SP60D60 surface due to the formation of a hydration layer. It can also indicate the presence of particles on its surface from the environment as spectroscopic ellipsometric measurements are highly sensitive and even despite of avoiding any contact with dust particles, other accidentally settled particles can cause this increase in thickness of only 2.24 nm. Thus, from the mentioned results obtained for characterization of antifouling topcoat polymer, SP60D60 was chosen to be topcoated to the optimal NO-releasing polymer.
3.3. Characterization of Polymers for Optimized NO-release
After selecting SP60D60 for the topcoat, the four commercial biomedical grade polymers were tested for incorporation of SNAP to choose the optimal NO-releasing polymer. SNAP leaching study was performed at first to determine the polymers that retain significant amount of SNAP in them after 24 hours of soaking in PBS (Figure 4). High amount of SNAP retention in the polymers ensures sustained release of NO from the polymers and minimizes the risks (if any) associated with SNAP leaching. After 4 hours of storing the higher water uptake polymer, SP60D60, in the buffer solution at 37°C, a loss of 84.55±1.83 % was recorded. With this initial high rate of leaching from the polymer it was expected that the samples would leach a significant amount of SNAP within 24 h. Overnight storage of the same films of SP60D60 showed a loss of 99.00±0.88 % of SNAP. This was in contrast to the SNAP leaching behavior of lower water uptake polymers, CarboSil 2080A, RTV, and SG80A. At the end of 4 hours, all three had a very low leaching of 2.54±0.18 %, 4.70±1.01 % and 7.25±0.20 % for CarboSil 2080A, RTV, and SG80A, respectively. As expected, these polymers also displayed a minimal leaching of 4.75±0.18 % (CarboSil 2080A),6.96±1.01 % (RTV) and 11.26±0.07 % (SG80A) at the end of 24 h of being soaked in PBS at 37°C. The behavior of high leaching of SNAP from SP60D60 is expected as it is a hydrophilic polymer and hence absorbs water while releasing SNAP molecules from within. However, comparatively hydrophobic polymers, SG80A, CarboSil 2080A, and RTV, leached out <10% of total SNAP content. This characteristic of hydrophobic polymers makes them desirable for incorporating NO donors to maintain a controlled release of NO for a longer period of time.
Figure 4.

SNAP content in PBS buffer as a result of leaching activity in the polymer. Calculated as a percentage of SNAP leached into the PBS buffer from the polymer. Data represents mean ± SD (n=3).
The desirable characteristics of CarboSil 2080A and RTV to retain SNAP within them for with minimal leaching made them ideal polymers to test for NO release. In the first hour, CarboSil 2080A displayed an NO flux of 12.42±3.20 (× 10−10 mol cm−2 min−1) and RTV displayed 5.34±0.91 (x 10−10 mol cm−2 min−1). This trend of higher NO flux from CarboSil 2080A compared to RTV is seen through the 24-hour study (Figure 5). This tendency of higher release of NO from CarboSil 2080A despite lower leaching of SNAP is advantageous because it means the material properties of CarboSil 2080A allow it to release NO without leaching of SNAP. This helped in determining CarboSil 2080A as the polymer for optimal release of NO for this study. This would enable us to carry out further research on the SNAP-incorporated CarboSil 2080A owing to its remoldable nature, high recyclability, chemical resistance, and aesthetic finish.
Figure 5.

NO-release from two hydrophobic (CarboSil and RTV) under physiological conditions (soaked in PBS buffer at 37°C in dark condition). Data represents mean ± SD (n=3)
3.4. Antibacterial Efficacy of SNAP-incorporated Polymer with Antifouling Topcoat
Coating of a hydrophobic polymer with a hydrophilic polymer is sometimes not possible due to the high solubility of the hydrophilic polymer in water. This high solubility can cause delamination of the hydrophilic polymer from the hydrophobic surface that it is coated on. To make sure that the polymers used in the study would complement each other and do not get delaminated, the film of CarboSil 2080A topcoated with SP60D60 was soaked in PBS at 37°C for 24 hours. Contact angle was checked before and after soaking in PBS. Three replicates were checked at three different spots on the samples and found to maintain their static contact angles ~50°. This is the contact angle for SP60D60 and hence it was confirmed that the hydrophilic SP60D60 would not delaminate from the hydrophobic CarboSil 2080A.
Bacteria adhesion, which often results in biofilm formation on the polymer surface, is a very common problem in moist and humid environment, which is found in implanted devices. The basic nutrients important for colony formation may be resourced from the polymer material itself, the fluid’s proteins that adhere to the polymer post-implant or a variety of contaminants that end up on the surface of the material. S. aureus is a major cause of infections associated with wounds, indwelling catheters, and cardiovascular and orthopedic implant devices.77, 81, 82
Owing to the antibacterial properties of NO, active release of NO from the donor molecule incorporated in the hydrophobic polymeric films can reduce the chances of biomedical device related infections or hospital-associated infections. The binding of bacteria to the polymer surface (base) can further be reduced by layering it with a comparatively hydrophilic polymer as the top coat. This hydrophilic layer is antifouling in nature and hence prevents the attachment of non-specific contaminants like proteins and bacteria on the material’s surface. In comparison to CarboSil 2080A (base polymer), SP60D60 (topcoat polymer) is less hydrophobic and hence can form a hydration layer on it. While the hydrophobic nature and low water uptake of CarboSil 2080A provides sustained release of NO, the formation of hydration layer on SP60D60 is expected to prevent protein attachment (as mentioned in the results from section 3.2) and also reduce bacterial adhesion on the material’s surface directly and indirectly (by repulsion of proteins).
Therefore, all samples tested were first exposed to 1mg/ml of fibrinogen protein for 90 minutes (as done in section 2.5.3) and then exposed to the bacterial solution. The polymers used in this study were exposed to 108 CFU mL−1 of S. aureus for three hours. After the three-hour exposure to the bacteria, the SP60D60/CarboSil 2080A hybrid films (first set of test films) with no SNAP showed 78.95 % reduction in viable S. aureus cells when compared to CarboSil 2080A/CarboSil 2080A (control). This reduction is significant and therefore test with SNAP-incorporated material was done to demonstrate the increase in significant reduction. The second set of test films (10 wt. % SNAP in SP60D60/CarboSil 2080A) generated an NO flux of 1.89 ± 0.64 (x 10−10 mol cm−2 min−1) (Figure 6), reducing the bacterial growth on the material’s surface by 95.83 % (p < 0.05) and 80.20 % (p < 0.05), when compared to CarboSil 2080A/CarboSil 2080A (control) and SP60D60/CarboSil 2080A films respectively (Figure 7). This reduction in number of viable bacteria attached to the second set of test films is significant compared to both control and first set of test films. These results are consistent with the theoretical expectations underlying the surface chemistry of SP60D60 and bactericidal properties of NO. To summarize, the combined effect of the tunable NO-release kinetics from CarboSil 2080A’s surface and prevention of protein and/or bacterial adhesion due to SP60D60’s surface chemistry can help reduce undesired clinical consequences post-implantation of a medical device.
Figure 6.

NO flux data before and after the bacterial study. NO flux was 1.89 (x 10−10 mol cm−2 min−1) and 1.33 (x 10−10 mol cm−2 min−1) before and after the bacterial incubation respectively. Data represents mean ± SD (n=3).
Figure 7.

Bacterial adhesion data showing the CFU of S. aureus/cm2 after 24 hours of incubation in the post-protein adhesion treated (1 hour) polymer material. A reduction of 78.95% in viable bacteria is seen on first set of test films (Carbosil topcoated with SP60D60) when compared to control films (CarboSil with CarboSil topcoat) (indicated by *). A reduction of 80.20% is seen on second set of test films when compared to the first set of test films (CarboSil with SP60D60 topcoat and no SNAP) (indicated by #). A reduction of 95.83% in viable bacteria is seen on second set of test films (Carbosil with 10 wt. % SNAP topcoated with SP60D60) when compared to control films (CarboSil with CarboSil topcoat) (indicated by *). Data represents mean ± SD (n=3). * = p≤0.05 for * and *#.
4. Conclusion
In this study we were able to design an efficient antifouling hydrophilic biomedical grade coating for NO releasing hydrophobic polymers which had significant reduction in protein adhesion and microbial growth. This approach was supported by the rigorous testing of the materials in a protein solution found commonly in physiological conditions and analysis of a sustained release of nitric oxide over a period of 24 hours. This study was able to prove two important points: 1) non-NO releasing hydrophobic polymer with a hydrophilic topcoat can reduce bacterial adhesion when compared to hydrophobic polymer topcoats 2) SNAP incorporated in hydrophobic polymer and topcoated with a hydrophilic polymer could significantly reduce microbial growth of a commonly found pathogenic bacteria (S. aureus) compared to polymers with no SNAP. The conclusion supports the theoretical explanation that hydrophilic antifouling polymers can further reduce microbial growth and surface roughness is a very important aspect of surface chemistry while considering it. Therefore, hydrophilic polymers can be a useful strategy of coatings for nitric oxide and other biocide releasing materials which can consequently reduce microbial adhesions while keeping a controlled release of antibacterial agents.
Acknowledgments
The authors declare that this work was supported by the National Institutes of Health, Grant K25HL111213 and Centers for Disease Control and Prevention contract 200-2016-91933.
References
- 1.Harding JL, Reynolds MM. Trends Biotechnol. 2014;32:140–146. doi: 10.1016/j.tibtech.2013.12.004. [DOI] [PubMed] [Google Scholar]
- 2.Douglas Scott R., II . The Direct Medical costs of Healthcare-Associated Infections in U.S. Hospitals and the Benefits of Prevention. Centers for Disease Control and Prevention; 2009. [Google Scholar]
- 3.Dalsin JL, Messersmith PB. Materials Today. 2005;8:38–46. [Google Scholar]
- 4.Donlan RM. Emerging Infectious Diseases. 2001;7:277–281. doi: 10.3201/eid0702.010226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Donlan RM, Costerton JW. Clinical Microbiology Reviews. 2002;15:167–193. doi: 10.1128/CMR.15.2.167-193.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Banerjee I, Pangule RC, Kane RS. Advanced Materials. 2011;23:690–718. doi: 10.1002/adma.201001215. [DOI] [PubMed] [Google Scholar]
- 7.Liu SQ, Yang C, Huang Y, Ding X, Li Y, Fan WM, Hedrick JL, Yang YY. Advanced Materials. 2012;24:6484–6489. doi: 10.1002/adma.201202225. [DOI] [PubMed] [Google Scholar]
- 8.Zhang J, Xu Z, Mai W, Min C, Zhou B, Shan M, Li Y, Yang C, Wang Z, Qian X. Journal of Materials Chemistry A. 2013;1:3101–3111. [Google Scholar]
- 9.Xu C, Hu X, Wang J, Zhang YM, Liu XJ, Xie BB, Yao C, Li Y, Li XS. ACS Appl Mater Interfaces. 2015;7:17337–17345. doi: 10.1021/acsami.5b04520. [DOI] [PubMed] [Google Scholar]
- 10.Mizrahi B, Khoo X, Chiang HH, Sher KJ, Feldman RG, Lee JJ, Irusta S, Kohane DS. Langmuir. 2013;29:10087–10094. doi: 10.1021/la4014575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang H, Chiao M. Journal of Medical and Biological Engineering. 2015;35:143–155. doi: 10.1007/s40846-015-0029-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kang S, Asatekin A, Mayes AM, Elimelech M. Journal of Membrane Science. 2007;296:42–50. [Google Scholar]
- 13.Dong B, Jiang H, Manolache S, Wong ACL, Denes FS. Langmuir. 2007;23:7306–7313. doi: 10.1021/la0633280. [DOI] [PubMed] [Google Scholar]
- 14.Muller M, Rieser T, Lunkwitz K, Meier-Haack J. Macromolecular Rapid Communications. 1999. [Google Scholar]
- 15.Kang G-d, Cao Y-m. Water research. 2012;46:584–600. doi: 10.1016/j.watres.2011.11.041. [DOI] [PubMed] [Google Scholar]
- 16.Tsibouklis J, Stone M, Thorpe AA, Graham P, Peters V, Heerlien R, Smith JR, Green KL, Nevell TG. Biomaterials. 1999;20:1229–1235. doi: 10.1016/s0142-9612(99)00023-x. [DOI] [PubMed] [Google Scholar]
- 17.Callow ME, Fletcher RL. International Biodeterioration & Biodegradation. 1994;34:333–348. [Google Scholar]
- 18.Rana D, Matsuura T. Chem Rev. 2010;110:2448–2471. doi: 10.1021/cr800208y. [DOI] [PubMed] [Google Scholar]
- 19.Chen S, Li L, Zhao C, Zheng J. Polymer. 2010;51:5283–5293. [Google Scholar]
- 20.Louie JS, Pinnau I, Ciobanu I, Ishida KP, Ng A, Reinhard M. Journal of Membrane Science. 2006;280:762–770. [Google Scholar]
- 21.Rechendorff K, Hovgaard MB, Foss M, Zhdanov VP, Besenbacher F. Langmuir. 2006;22:10885–10888. doi: 10.1021/la0621923. [DOI] [PubMed] [Google Scholar]
- 22.Akkas T, Citak C, Sirkecioglu A, Güner FS. Polymer International. 2013;62:1202–1209. [Google Scholar]
- 23.An Q, Li F, Ji Y, Chen H. Journal of Membrane Science. 2011;367:158–165. [Google Scholar]
- 24.Siedenbiedel F, Tiller JC. Polymers. 2012;4:46–71. [Google Scholar]
- 25.Johnson JR, Roberts PL, Olsen RJ, Moyer KA, Stamm WE. Journal of Infectious Diseases. 1990;162:1145–1150. doi: 10.1093/infdis/162.5.1145. [DOI] [PubMed] [Google Scholar]
- 26.Schierholz JM, Lucas LJ, Rump A, Pulverer G. Journal of Hospital Infection. 1998;40:257–262. doi: 10.1016/s0195-6701(98)90301-2. [DOI] [PubMed] [Google Scholar]
- 27.Rupp ME, Fitzgerald T, Marion N, Helget V, Puumala S, Anderson JR, Fey PD. American Journal of Infection Control. 2004;32:445–450. doi: 10.1016/j.ajic.2004.05.002. [DOI] [PubMed] [Google Scholar]
- 28.Kumar R, Münstedt H. Biomaterials. 2005;26:2081–2088. doi: 10.1016/j.biomaterials.2004.05.030. [DOI] [PubMed] [Google Scholar]
- 29.Pickard R, Lam T, Maclennan G, Starr K, Kilonzo M, McPherson G, Gillies K, McDonald A, Walton K, Buckley B, Glazener C, Boachie C, Burr J, Norrie J, Vale L, Grant A, N’Dow J. Health Technol Assess. 2012;16:1–197. doi: 10.3310/hta16470. [DOI] [PubMed] [Google Scholar]
- 30.Gallo RL, Huttner KM. J Investig Dermatol. 1998;111:739–743. doi: 10.1046/j.1523-1747.1998.00361.x. [DOI] [PubMed] [Google Scholar]
- 31.Huttner KM, Bevins CL. Pediatr Res. 1999;45:785–794. doi: 10.1203/00006450-199906000-00001. [DOI] [PubMed] [Google Scholar]
- 32.Shai Y. Peptide Science. 2002;66:236–248. doi: 10.1002/bip.10260. [DOI] [PubMed] [Google Scholar]
- 33.Straus SK, Hancock REW. Biochimica et Biophysica Acta (BBA) - Biomembranes. 2006;1758:1215–1223. doi: 10.1016/j.bbamem.2006.02.009. [DOI] [PubMed] [Google Scholar]
- 34.Marr AK, Gooderham WJ, Hancock REW. Current Opinion in Pharmacology. 2006;6:468–472. doi: 10.1016/j.coph.2006.04.006. [DOI] [PubMed] [Google Scholar]
- 35.Brisbois EJ, Bayliss J, Wu J, Major TC, Xi C, Wang SC, Bartlett RH, Handa H, Meyerhoff ME. Acta Biomaterialia. 2014;10:4136–4142. doi: 10.1016/j.actbio.2014.06.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lutzke A, Neufeld BH, Neufeld MJ, Reynolds MM. Journal of Materials Chemistry B. 2016;4:1987–1998. doi: 10.1039/c6tb00037a. [DOI] [PubMed] [Google Scholar]
- 37.Deppisch C, Herrmann G, Graepler-Mainka U, Wirtz H, Heyder S, Engel C, Marschal M, Miller CC, Riethmüller J. Infection. 2016:1–8. doi: 10.1007/s15010-016-0879-x. [DOI] [PubMed] [Google Scholar]
- 38.Zhang T, Wang L, Chen Q, Chen C. Yonsei Medical Journal. 2014;55:283–291. doi: 10.3349/ymj.2014.55.2.283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.de Lima R, Seabra AB, Duran N. Journal of applied toxicology: JAT. 2012;32:867–879. doi: 10.1002/jat.2780. [DOI] [PubMed] [Google Scholar]
- 40.Brogden KA. Nat Rev Micro. 2005;3:238–250. doi: 10.1038/nrmicro1098. [DOI] [PubMed] [Google Scholar]
- 41.Amoako KA, Sundaram HS, Suhaib A, Jiang S, Cook KE. Advanced Materials Interfaces. 2016 doi: 10.1002/admi.201500646. n/a-n/a. [DOI] [Google Scholar]
- 42.Backlund CJ, Worley BV, Schoenfisch MH. Acta biomaterialia. 2016;29:198–205. doi: 10.1016/j.actbio.2015.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Brisbois EJ, Davis RP, Jones AM, Major TC, Bartlett RH, Meyerhoff ME, Handa H. J Mater Chem B Mater Biol Med. 2015;3:1639–1645. doi: 10.1039/C4TB01839G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Brisbois EJ, Handa H, Major TC, Bartlett RH, Meyerhoff ME. Biomaterials. 2013;34:6957–6966. doi: 10.1016/j.biomaterials.2013.05.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cai W, Wu J, Xi C, Meyerhoff ME. Biomaterials. 2012;33:7933–7944. doi: 10.1016/j.biomaterials.2012.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Charville GW, Hetrick EM, Geer CB, Schoenfisch MH. Biomaterials. 2008;29:4039–4044. doi: 10.1016/j.biomaterials.2008.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fang FC. Nitric oxide: biology and chemistry/official journal of the Nitric Oxide Society. 2012;27(Supplement):S10. [Google Scholar]
- 48.Frost MC, Reynolds MM, Meyerhoff ME. Biomaterials. 2005;26:1685–1693. doi: 10.1016/j.biomaterials.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 49.Gupta S, Amoako KA, Suhaib A, Cook KE. Advanced Materials Interfaces. 2014:1. doi: 10.1002/admi.201400071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Handa H, Brisbois EJ, Major TC, Refahiyat L, Amoako KA, Annich GM, Bartlett RH, Meyerhoff ME. Journal of Materials Chemistry B. 2013;1:3578–3587. doi: 10.1039/C3TB20277A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Handa H, Major TC, Brisbois EJ, Amoako KA, Meyerhoff ME, Bartlett RH. Journal of Materials Chemistry B. 2014;2:1059–1067. doi: 10.1039/C3TB21771J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Harding JL, Reynolds MM. Journal of Materials Chemistry B. 2014;2:2530–2536. doi: 10.1039/c3tb21458c. [DOI] [PubMed] [Google Scholar]
- 53.Hetrick EM, Schoenfisch MH. Chemical Society Reviews. 2006;35:780–789. doi: 10.1039/b515219b. [DOI] [PubMed] [Google Scholar]
- 54.Privett BJ, Broadnax AD, Bauman SJ, Riccio DA, Schoenfisch MH. Nitric oxide: biology and chemistry/official journal of the Nitric Oxide Society. 2012;26:169–173. doi: 10.1016/j.niox.2012.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Regev-Shoshani G, Ko M, Crowe A, Av-Gay Y. Urology. 2011;78:334–339. doi: 10.1016/j.urology.2011.02.063. [DOI] [PubMed] [Google Scholar]
- 56.Regev-Shoshani G, Ko M, Miller C, Av-Gay Y. Antimicrobial Agents and Chemotherapy. 2010;54:273–279. doi: 10.1128/AAC.00511-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Reynolds MM, Frost MC, Meyerhoff ME. Free Radical Biology & Medicine. 2004;37:926–936. doi: 10.1016/j.freeradbiomed.2004.06.019. [DOI] [PubMed] [Google Scholar]
- 58.Taladriz-Blanco P, Pastoriza-Santos V, Pérez-Juste J, Hervés P. Langmuir. 2013;29:8061–8069. doi: 10.1021/la4014762. [DOI] [PubMed] [Google Scholar]
- 59.Wo Y, Li Z, Brisbois EJ, Colletta A, Wu J, Major TC, Xi C, Bartlett RH, Matzger AJ, Meyerhoff ME. ACS Applied Materials & Interfaces. 2015;7:22218–22227. doi: 10.1021/acsami.5b07501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wu B, Gerlitz B, Grinnell BW, Meyerhoff ME. Biomaterials. 2007;28:4047–4055. doi: 10.1016/j.biomaterials.2007.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Goudie MJ, Brisbois EJ, Pant J, Thompson A, Potkay JA, Handa H. International Journal of Polymeric Materials and Polymeric Biomaterials. 2016;65:769–778. doi: 10.1080/00914037.2016.1163570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Brisbois EJ, Major TC, Goudie MJ, Bartlett RH, Meyerhoff ME, Handa H. Acta biomaterialia. 2016;37:111–119. doi: 10.1016/j.actbio.2016.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Fang FC. Journal of Clinical Investigation. 1997;99:2818–2825. doi: 10.1172/JCI119473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hetrick EM, Schoenfisch MH. Biomaterials. 2007;28:1948–1956. doi: 10.1016/j.biomaterials.2007.01.006. [DOI] [PubMed] [Google Scholar]
- 65.Engelsman AF, Krom BP, Busscher HJ, van Dam GM, Ploeg RJ, van der Mei HC. Acta Biomaterialia. 2009;5:1905–1910. doi: 10.1016/j.actbio.2009.01.041. [DOI] [PubMed] [Google Scholar]
- 66.Carlsson S, Weitzberg E, Wiklund P, Lundberg JO. Antimicrobial Agents and Chemotherapy. 2005;49:2352–2355. doi: 10.1128/AAC.49.6.2352-2355.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Regev-Shoshani G, Ko M, Crowe A, Av-Gay Y. Urology. 2011;78:334–339. doi: 10.1016/j.urology.2011.02.063. [DOI] [PubMed] [Google Scholar]
- 68.Mihu MR, Sandkovsky U, Han G, Friedman JM, Nosanchuk JD, Martinez LR. Virulence. 2010;1:62–67. doi: 10.4161/viru.1.2.10038. [DOI] [PubMed] [Google Scholar]
- 69.Yu J, Brisbois E, Handa H, Annich G, Meyerhoff M, Bartlett R, Major T. Journal of Materials Chemistry B. 2016;4:2264–2272. doi: 10.1039/C5TB02419F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chipinda I, Simoyi RH. The Journal of Physical Chemistry B. 2006;110:5052–5061. doi: 10.1021/jp0531107. [DOI] [PubMed] [Google Scholar]
- 71.Statz AR, Barron AE, Messersmith PB. Soft Matter. 2008;4:131–139. doi: 10.1039/B711944E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Martins M, Wang D, Ji J, Feng L, Barbosa M. Biomaterials. 2003;24:2067–2076. doi: 10.1016/s0142-9612(03)00002-4. [DOI] [PubMed] [Google Scholar]
- 73.Shen M, Pan YV, Wagner MS, Hauch KD, Castner DG, Ratner BD, Horbett TA. Journal of biomaterials science Polymer edition. 2001;12:961–978. doi: 10.1163/156856201753252507. [DOI] [PubMed] [Google Scholar]
- 74.Messersmith PB, Textor M. Nature nanotechnology. 2007;2:138–139. doi: 10.1038/nnano.2007.51. [DOI] [PubMed] [Google Scholar]
- 75.Salim M, Mishra G, Fowler GJS, O’Sullivan B, Wright PC, McArthur SL. Lab on a Chip. 2007;7:523–525. doi: 10.1039/b615328c. [DOI] [PubMed] [Google Scholar]
- 76.Zhang Z, Chen S, Chang Y, Jiang S. The Journal of Physical Chemistry B. 2006;110:10799–10804. doi: 10.1021/jp057266i. [DOI] [PubMed] [Google Scholar]
- 77.Boland T, Latour RA, Stutzenberger FJ. Handbook of Bacterial Adhesion. Springer; 2000. pp. 29–41. [Google Scholar]
- 78.Ní Eidhin D, Perkins S, Francois P, Vaudaux P, Höök M, Foster TJ. Molecular Microbiology. 1998;30:245–257. doi: 10.1046/j.1365-2958.1998.01050.x. [DOI] [PubMed] [Google Scholar]
- 79.Jung SY, Lim SM, Albertorio F, Kim G, Gurau MC, Yang RD, Holden MA, Cremer PS. Journal of the American Chemical Society. 2003;125:12782–12786. doi: 10.1021/ja037263o. [DOI] [PubMed] [Google Scholar]
- 80.Toscano A, Santore MM. Langmuir. 2006;22:2588–2597. doi: 10.1021/la051641g. [DOI] [PubMed] [Google Scholar]
- 81.Safdar N, Maki DG. Annals of Internal Medicine. 2002;136:834–844. doi: 10.7326/0003-4819-136-11-200206040-00013. [DOI] [PubMed] [Google Scholar]
- 82.Tong SY, Davis JS, Eichenberger E, Holland TL, Fowler VG. Clinical microbiology reviews. 2015;28:603–661. doi: 10.1128/CMR.00134-14. [DOI] [PMC free article] [PubMed] [Google Scholar]