

Abstract
Objective:
To assist the interpretation of genomic data for common epilepsies, we asked whether variants implicated in mild epilepsies in autosomal dominant families are present in the general population.
Methods:
We studied 12 genes for the milder epilepsies and identified published variants with strong segregation support (de novo germline mutation or ≥4 affected family members). These variants were checked in the Exome Aggregation Consortium (ExAC), a database of genetic variation in over 60,000 individuals. We subsequently evaluated variants in these epilepsy genes that lacked strong segregation support. To determine whether the findings in epilepsies were representative of other diseases, we also assessed the presence of variants in other dominant neurologic disorders (e.g., CADASIL).
Results:
Published epilepsy variants with strong segregation support (n = 65) were absent (n = 61) or present once (n = 4) in ExAC. By contrast, of 46 published epilepsy variants without strong segregation support, 8 occurred recurrently (2–186 times). Similarly, none of the 45 disease-associated variants from other neurologic disorders with strong segregation support occurred more than once in ExAC. Reanalysis using the larger ExAC V2 plus gnomAD reference cohort showed consistent results.
Conclusions:
Variants causing autosomal dominant epilepsies are ultra-rare in the general population. Variants observed more than once in ExAC were only found among reports without strong segregation support, suggesting that they may be benign. Clinicians are increasingly faced with the interpretation of genetic variants of unknown significance. These data illustrate that variants present more than once in ExAC are less likely to be pathogenic, reinforcing the valuable clinical role of ExAC.
Recent advances in DNA technologies have increased the influence that genomic data have in epilepsy clinical practice. Finding a genetic cause can have important implications for diagnosis and prognosis, treatment and genetic counseling, as well as the psychological and financial benefits associated with ending the diagnostic journey.1,2
Clinical laboratories are understandably conservative in ascribing pathogenicity; however, the elevated reporting of variants of unknown significance (VOUS) makes interpretation complex and renders follow-up efforts, such as segregation analyses and functional studies, impractical at a large scale. Novel missense variants are particularly challenging. Establishing the frequency of a VOUS in control populations is currently among the most reliable considerations in variant interpretation.
The Exome Aggregation Consortium (ExAC) database contains exome data for more than 60,000 individuals compiled at the Broad Institute.3 For de novo mutations affecting established epilepsy genes, absence from ExAC is widely accepted as one supportive argument for pathogenicity and this is particularly true for epileptic encephalopathies and most neurodevelopmental disorders.4 The question is whether this utility extends to the milder epilepsies where negative selection may not have been as strong.
The milder presentations and the observation of reduced penetrance within dominant families raise the possibility that some causative variants might be present in the general population at low frequencies. Here, to assess how informative ExAC might be to ascribing pathogenicity to inherited variants among milder epilepsies, we evaluated the frequency in ExAC of published epilepsy variants, where de novo status or segregation in large families supported pathogenicity.
METHODS
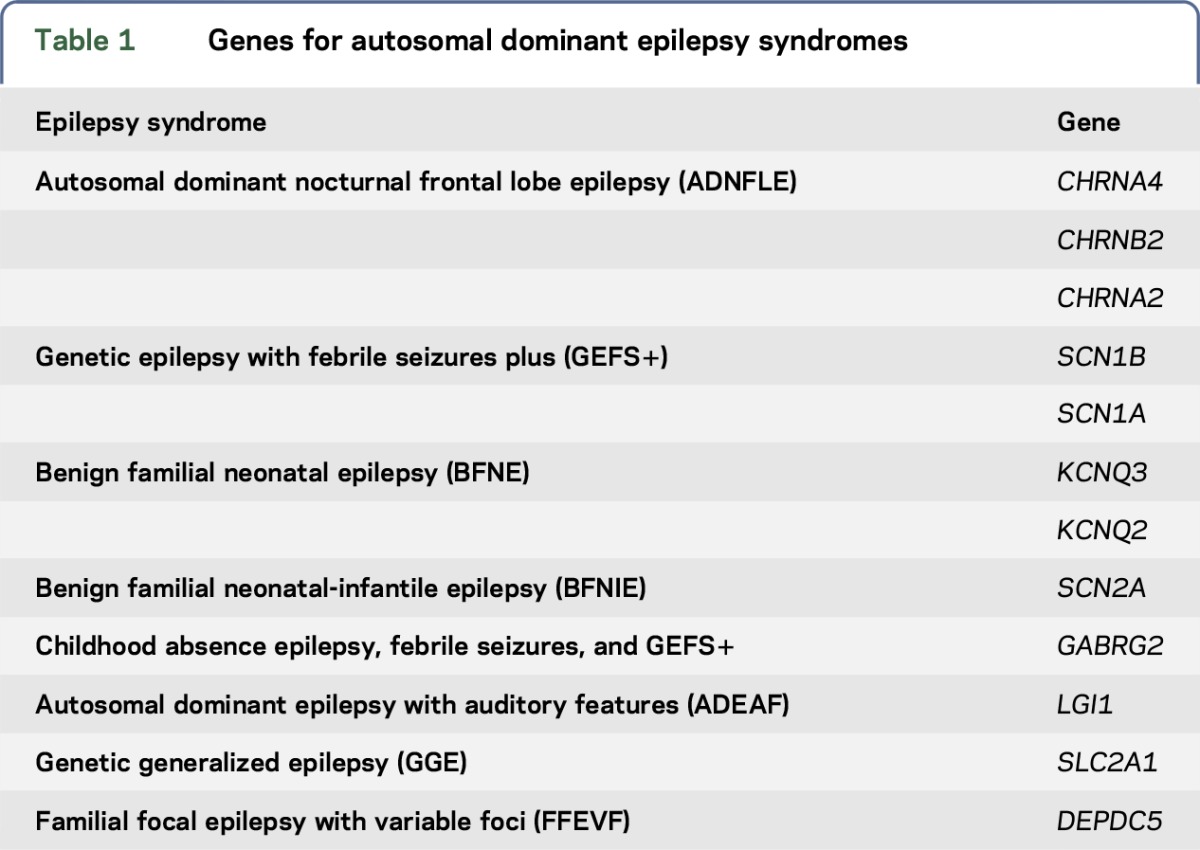
A list of 12 genes was generated for the “milder” epilepsies (table 1). We excluded PRRT2, as it is prone to alignment errors in next-generation sequencing.
Table 1.
Genes for autosomal dominant epilepsy syndromes
Literature search.
We attempted an exhaustive PubMed search for missense and splice variants deemed causative for dominant familial epilepsies, published in English, on the basis of strong segregation support, which we defined as prior linkage analysis or familial segregation in at least 4 affected family members. We note that segregation in 4 individuals is inadequate to support gene discovery but, in the context of an established gene for an appropriate phenotype, such segregation provides considerable support for the role of the variant in the particular family. Sanger validated de novo variants found in our list of 12 epilepsy genes, among sporadic cases of “mild” epilepsies, were also included. We deliberately limited our search to the literature published prior to 2015, as this literature can realistically be considered to precede the launch of the ExAC database. A separate review was conducted for each of the 12 genes by searching the gene name and “epilepsy.” For genes associated with both milder and more severe phenotypes, such as genetic epilepsy with febrile seizures plus (GEFS+) and Dravet syndrome with SCN1A, only variants found among families or individuals with the milder phenotype were included. Variants reported exclusively among the epileptic encephalopathies were excluded.
A second list of variants without strong segregation support was generated. Lack of strong segregation support was defined as segregation data based on less than 4 affected individuals or not performed at all. We hypothesized that this group of variants might be more enriched for background genetic variants in epilepsy genes.
To determine whether findings in mild epilepsies were reflective of other autosomal dominant neurologic diseases, we evaluated variants with strong segregation support from 45 published families with familial hemiplegic migraine, CADASIL, autosomal dominant Alzheimer disease or frontotemporal dementia. The genes examined were ATP1A2, CACNA1A, SCN1A, NOTCH3, APP, PSEN1, PSEN2, GRN, MAPT, and TARDBP1.
Finally, after the study was completed, the expanded ExAC V2 plus gnomAD was released and the analysis was rerun. This effectively doubled the size of the ExAC control cohort, with whole-exome sequencing (WES) and whole-genome sequencing data available on 123,136 individuals and 15,496 individuals, respectively.3
RESULTS
Sixty-five missense or predicted splice variants for the milder familial epilepsies with strong segregation support were identified. Of these, 61 (94%) were absent from ExAC and 4 (6%) were present in ExAC once (a single allele was reported) (figure; table e-1 at Neurology.org/ng).
Figure. Frequency distribution of allele counts in ExAC for variants in epilepsy families.
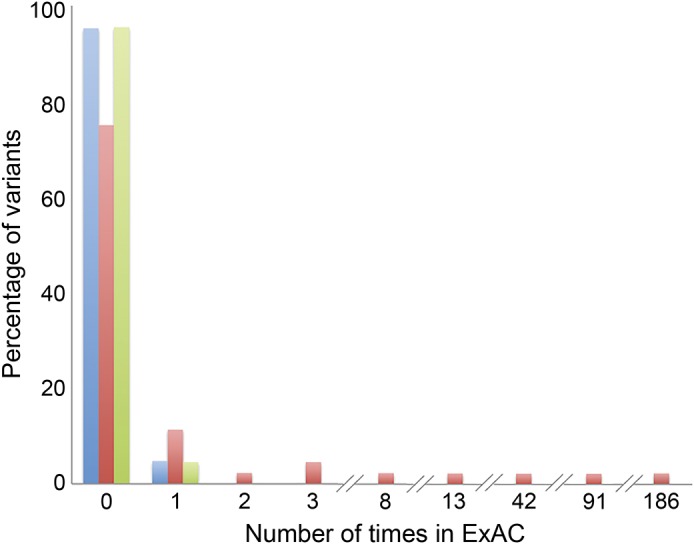
Variants with strong segregation support, including confirmed de novo mutations among sporadic common epilepsies, are shown in blue (n = 65); variants without strong segregation support are shown in red (n = 46); and variants from other neurologic disorders are shown in green (n = 45). ExAC = Exome Aggregation Consortium.
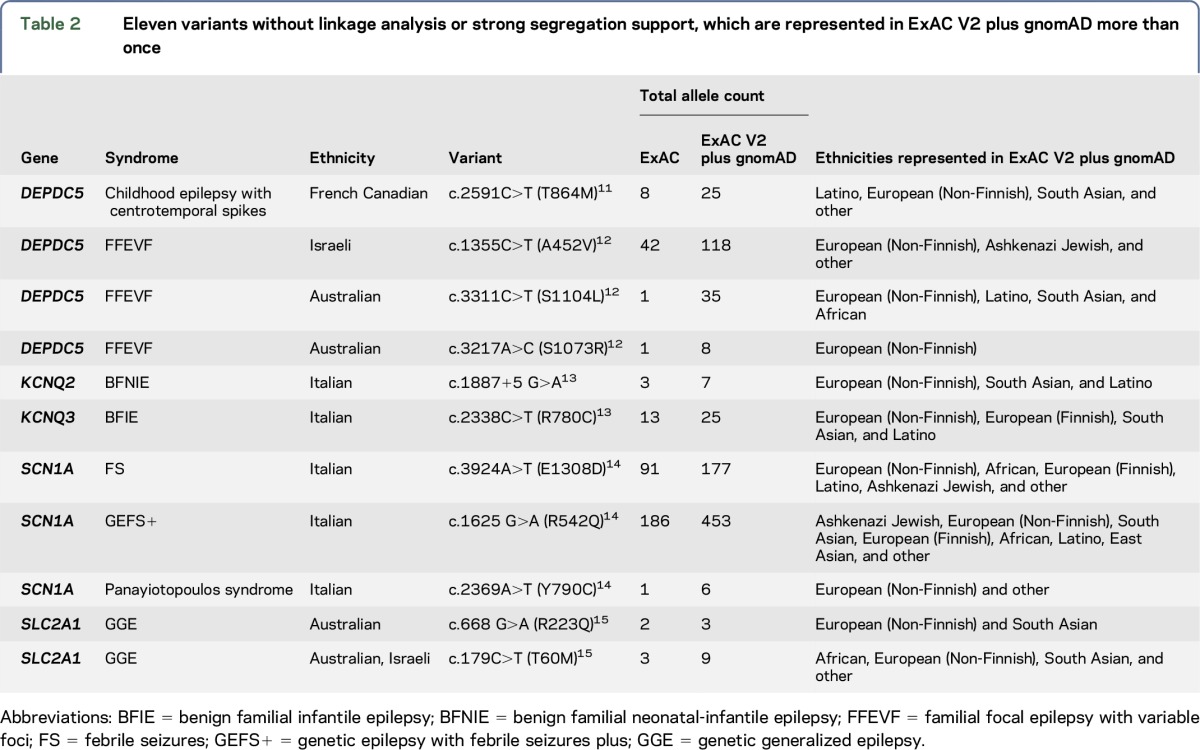
By contrast, of the 46 missense or splice variants contained in the same publications but lacking the strong segregation support, 13 (28%) were present in ExAC. Of these, 8 variants were observed among 2–186 carriers (figure; table e-1). These 8 variants are listed in table 2.
Table 2.
Eleven variants without linkage analysis or strong segregation support, which are represented in ExAC V2 plus gnomAD more than once
Of the 45 missense variants with strong segregation support for other neurologic disorders, 43 (96%) were absent from ExAC and 2 (4%) were present once (figure; table e-1).
Reanalysis with ExAC V2 and gnomAD datafiles showed that the epilepsy variants in our current study with strong segregation support are present no more than once, with the exception of 2 variants in SCN1B associated with febrile seizures and GEFS+ which are present 3 and 4 times (table e-1).
For variants without strong segregation support, their frequency in ExAC V2 approximately doubled, as expected, compared with the initial analysis and 3 new variants were observed multiple times in ExAC V2 (table 2).
DISCUSSION
The interpretation of putative epilepsy genetic variants remains complex for researchers and clinicians alike. Here, we assessed the allele count of literature-reported epilepsy variants in the ExAC database and found that variants with strong segregation evidence for pathogenicity were present no more than once in the ExAC collection of over 60,000 individuals, illustrating that these variants are ultra-rare in the general population. This pattern was also seen in the variants found in other neurologic disorders that were reported based on strong segregation support. However, the literature-reported epilepsy variants without strong segregation support were more likely to be present in more than 1 carrier among the ExAC collection (figure), suggesting that many of these variants (table 2) are unlikely to be penetrant disease-causing alleles, but rather background variation in established epilepsy genes or possibly risk alleles contributing to a more complex genetic architecture that is not currently well understood.
WES in patients with epilepsy can be a powerful diagnostic tool, but interpretation of the data remains complex. In the field of epilepsy genetics, this complexity is highlighted by the likely false attribution of pathogenicity to 2 genes, EFHC1 and SRPX2. SRPX2 was deemed a pathogenic gene in 2006 in 2 families with epilepsy, speech dyspraxia, and intellectual impairment, including one from our centre.5 The key family was subsequently shown to have a GRIN2A variant, and in fact the SRPX2 variant from this family appears in ExAC 47 times. Similarly, the pathogenicity of EFHC1 as a gene for juvenile myoclonic epilepsy has been disputed.6
Even for widely accepted epilepsy genes, the interpretation of VOUS is complex. A large contribution to false-positive epilepsy variants comes from variants identified through “secondary” studies, wherein pathogenicity is largely inferred based on the knowledge of prior disease-causing variants within the gene. These variants are often not subject to the same rigorous evaluation of pathogenicity as variants from the original discovery,6 resulting in more permissive pathogenicity assignments. Indeed, we know that epilepsy genes carry background variation in the general population. For example, the variants in table 2, which are in established epilepsy genes, were published as pathogenic and some of these are present in ExAC at relatively high frequencies, emphasizing the caution necessary when assessing pathogenicity solely on the basis of a known epilepsy gene. This is one of the most important considerations in the interpretation of results from clinical gene panels.
The issue of variant misclassification extends well beyond the field of epilepsy to other monogenic diseases. It has recently been demonstrated that each ExAC participant carries, on average, ∼54 variants that have previously been reported as disease causing.3 Of these, ∼41 variants have a minor allele frequency of >1%.3 It is therefore likely that many of these variants are either benign variants or contributing to complex inheritance.
The frequency of VOUS in control samples is thus an invaluable line of evidence in interpretation, and the ExAC database offers a control cohort of unprecedented size. ExAC is not enriched for pathogenic variants in genes that are commonly tested in a clinical diagnostics setting, thus supporting its utility in classifying variants.7 In a recent study, the ExAC database was found to have an excess of ostensibly pathogenic variants for prion disease but found that the variants with the strongest independent evidence of pathogenicity were absent from ExAC, which is consistent with our findings.8 The authors attributed the excess allele frequency for variants with weak evidence for pathogenicity to the incorrect attribution of pathogenicity for certain variants or reduced penetrance of these alleles, an interpretation shared here.
Thus, the interrogation of ExAC provides a highly useful way of assessing the relevance of VOUS for patients with epilepsy and is likely to be widely applicable to other neurologic disorders. We have shown that disease-causing variants in epilepsy are very rare, and thus variants that are present more than once in the current ExAC cohort are unlikely to contribute to disease in a large way. It is important that the inverse is definitely not true—we cannot infer that a variant's absence in ExAC is sufficient evidence of pathogenicity.
The use of large reference cohorts as a clinical tool is not without limitations. As the minor allele frequency resolution improves with additional samples added to large reference cohorts (like ExAC), it is possible that clinically relevant variants for the milder epilepsies might begin to appear more than once. Indeed, our results with ExAC were supported with ExAC V2 plus gnomAD where the sample size was doubled. The only exception was 2 variants in SCN1B associated with febrile seizures and GEFS+ which are present 3 and 4 times (table e-1). A possible explanation for this slightly elevated frequency is that variants causing febrile seizures (a mild phenotype often resolving with age) are expected to have a subtler impact on reproductive fitness than variants causing other common genetic generalized and nonacquired focal epilepsies, and this is also coupled with the higher rates of febrile seizures in the general population than other common epilepsies. Thus, it is unlikely that variants with a minor allele frequency >0.0005% (approximate current ExAC V2 resolution) have a major contribution to autosomal dominant forms of mild epilepsies.
Our study and the message presented here are independently supported by our recent Epi4K study, where we found that when comparing to a control population, the epilepsy risk signals identified in epilepsy genes among large collections of epilepsy samples were driven by ultra-rare variants absent in ExAC.9 Our current study takes an alternative approach, comparing the frequency in ExAC of variants with and without strong segregation support, to reach the same conclusion. It is important to note that although European and some other genetic ancestries are well represented in ExAC, many ethnic minorities are underrepresented or not represented at all, posing additional issues in interpretation when assessing data from a patient from an ethnic background not well represented in these databases.10
Supplementary Material
GLOSSARY
- ExAC
Exome Aggregation Consortium
- GEFS+
genetic epilepsy with febrile seizures plus
- VOUS
variants of unknown significance
- WES
whole-exome sequencing
Footnotes
Supplemental data at Neurology.org/ng
AUTHOR CONTRIBUTIONS
Caitlin A. Bennett: acquisition and analysis of data and drafted the manuscript. Slavé Petrovski: study concept and design; interpretation of data; and revised the manuscript. Karen L. Oliver: interpretation of data and revised the manuscript. Samuel F. Berkovic: study concept and design; study supervision; and revised the manuscript.
STUDY FUNDING
Study funded by NHMRC Program Grant 1091593.
DISCLOSURE
C.A. Bennett reports no disclosures. S. Petrovski serves on the scientific advisory board and has interest in Pairnomix and serves on the editorial board of Epilepsia. K.L. Oliver reports no disclosures. S.F. Berkovic has served on the scientific advisory boards for UCB Pharma Eisai and Janssen-Cilag; serves on the editorial boards of Lancet Neurology and Epileptic Disorders and the Advisory Board of Brain; and may accrue future revenue on pending patent WO61/010176: Therapeutic compound that relates to discovery of PCDH19 gene as the cause of familial epilepsy with mental retardation limited to females; is one of the inventors listed on a patent held by Bionomics Inc. on diagnostic testing of using the SCN1A gene, WO2006/133508, and on pending patent WO61/010176: Therapeutic compound that relates to discovery of PCDH19 gene as the cause of familial epilepsy with mental retardation limited to females; has received speaker honoraria from UCB; has received unrestricted educational grants from UCB, Janssen-Cilag, and Sanofi-Aventis; and receives/has received research support from the National Health and Medical Research Council of Australia and NINDS. Go to Neurology.org/ng for full disclosure forms.
REFERENCES
- 1.EpiPM Consortium. A roadmap for precision medicine in the epilepsies. Lancet Neurol 2015;14:1219–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Scheffer IE. Genetic testing in epilepsy: what should you be doing? Epilepsy Curr 2011;11:107–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lek M, Karczewski KJ, Minikel EV, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016;536:285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kosmicki J, Samocha K, Howrigan D, et al. Refining the role of de novo protein truncating variants in neurodevelopmental disorders using population reference samples. Nat Genet 2017;49:504–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Roll P, Rudolf G, Pereira S, et al. SRPX2 mutations in disorders of language cortex and cognition. Hum Mol Genet 2006;15:1195–1207. [DOI] [PubMed] [Google Scholar]
- 6.Pal D, Helbig I. Commentary: pathogenic EFHC1 mutations are tolerated in healthy individuals dependent on reported ancestry. Epilepsia 2015;56:195–196. [DOI] [PubMed] [Google Scholar]
- 7.Song W, Gardner SA, Hovhannisyan H, et al. Exploring the landscape of pathogenic genetic variation in the ExAC population database: insights of relevance to variant classification. Genet Med 2015;18:850–854. [DOI] [PubMed] [Google Scholar]
- 8.Minikel EV, Vallabh SM, Lek M, et al. Quantifying prion disease penetrance using large population control cohorts. Sci Transl Med 2016;8:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Consortium Epi4K, Epilepsy Phenome/Genome Project. Ultra-rare genetic variation in common epilepsies: a case-control sequencing study. Lancet Neurol 2017;16:135–143. [DOI] [PubMed] [Google Scholar]
- 10.Petrovski S, Goldstein DB. Unequal representation of genetic variation across ancestry groups creates healthcare inequality in the application of precision medicine. Genome Biol 2016;17:157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Martin C, Meloche C, Rioux MF, et al. A recurrent mutation in DEPDC5 predisposes to focal epilepsies in the French-Canadian population. Clin Genet 2014;86:570–574. [DOI] [PubMed] [Google Scholar]
- 12.Dibbens LM, de Vries B, Donatello S, et al. Mutations in DEPDC5 cause familial focal epilepsy with variable foci. Nat Genet 2013;45:546–551. [DOI] [PubMed] [Google Scholar]
- 13.Zara F, Specchio N, Striano P, et al. Genetic testing in benign familial epilepsies of the first year of life: clinical and diagnostic significance. Epilepsia 2013;54:425–436. [DOI] [PubMed] [Google Scholar]
- 14.Orrico A, Galli L, Grosso S, et al. Mutational analysis of the SCN1A, SCN1B and GABRG2 genes in 150 Italian patients with idiopathic childhood epilepsies. Clin Genet 2009;75:579–581. [DOI] [PubMed] [Google Scholar]
- 15.Arsov T, Mullen S, Rogers S, et al. Glucose transporter 1 deficiency in the idiopathic generalized epilepsies. Ann Neurol 2012;72:807–815. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.