

Abstract
Non-random chromosomal conformations, including promoter--enhancer loopings that bypass kilo- or megabases of linear genome, provide a critical layer of transcriptional regulation, and move vast amounts of non-coding sequence into the physical proximity of genes that are important for neurodevelopment, cognition and behavior. Activity-regulated changes of the neuronal ‘3D genome’ could govern transcriptional mechanisms associated with learning and plasticity, and loop-bound intergenic and intronic non-coding sequences have been implicated in psychiatric and adult-onset neurodegenerative disease. Recent studies have begun to clarify the roles of spatial genome organization in normal and abnormal cognition.
INTRODUCTION
Comprehensive exploration of the functional organization of the human genome has to go far beyond linear sequencing of 6 billion base pairs. Although high resolution mapping of the various constituents of the epigenome, including DNA methylation and histone modifications, has enabled the ‘linear’ genome to be understood in two dimensions, this does not account for the spatial configuration and packaging of interphase chromosomes. Despite the importance of the spatial organization of chromosomal material in the regulation of gene expression, the maintenance of genome integrity and stability, and the control of growth and differentiation, surprisingly little is known.
According to some estimates,, up to 40% of the human genome is epigenomically decorated with various types of histone modifications and DNA methylation, localized transcription factor complexes or enrichment with chromatin scaffolding proteins1. By contrast, only ~1.5% of genome sequence encodes protein. Therefore, in addition to knowledge of the genome and epigenome, mapping the 3D genome in neurons and glia is essential for a full understanding of how genes are regulated and expressed. Such an understanding could enable identification of novel distal regulatory elements and in turn, to develop an understand of how these elements assemble in 3D to ‘bypass’ the linear genome and regulate gene expression.
Early findings from a select set of candidate gene loci indicate that chromosomal contacts and ‘loopings’ could be heavily regulated by neuronal activity, suggesting that the 3D genome plays a part in activity-dependent regulation of gene expression in brain cells. In addition, studies on a small number of candidate genes indicate that loop-bound non-coding DNA contributes to the genetic risk architecture of cognitive disease with onset in early childhood or young adulthood, including autism2 and schizophrenia3. Of note, deleterious mutations in genes encoding regulators of chromosomal scaffolding severely impact brain development and function, further underscoring that proper packaging and organization of the genomic material inside the nuclei of brain cells is of pivotal importance.
Advances in epigenomic editing techniques are now being developed that enable neuronal or glial control of transcriptional units, including genes, to be manipulated artificially by placing transcriptional activators at the side of regulatory sequences that are separated from their target genes by many thousands of base pairs. Therefore, loop-bound regulatory sequences could be harnessed to modulate expression of disease relevant genes without interfering with basal transcription. In this review, we will briefly introduce the key concepts of the spatial genome and the experimental approaches used to study it. We then discuss recent developments that have fueled the growing interest in exploring the spatial organization of chromatin fibers and chromosomes in brain cells.
The 3D genome and its Constituents
Eukaryote nuclei, separated by a nuclear membrane from the cytoplasm, contain the genome packaged into chromatin fibers as nucleosomal arrays (Figure 1). Nucleosomes are comprised of 146bp DNA wrapped around a core histone octamer, and interconnected by linker DNA and linker histones. Chromatin can exist in different ‘states’, including ‘open’ (eu-) and condensed (hetero-) chromatin. These are differentially defined by three characteristics: (1) loose or dense nucleosomal packaging euchromatin or heterochromatin, respectively), (2) specific types of post-translational histone modifications (such as acetylation), and (3) presence or absence of various chromatin regulatory proteins that either facilitate or repress transcription. For example, actively expressed genes in open chromatin show high levels of histone acetylation, with nucleosome-free intervals occupied by activator proteins (transcription factors) and the RNA polymerase II initiation complex (Figure 1). Superimposed upon these types of nucleosomal organization is the 3-dimensional conformation of chromatin fibers and entire chromosomes, often described in terms such as ‘loopings’ or ‘globules’ and in toto referred to as the ‘3D genome’. This includes the ‘clustering’ of euchromatic and heterochromatic sequences that tend to assemble into alternating regions of approximately ~5 megabases (Mb). These ‘compartments’, positioned along the same chromosome, are able to interact with compartments from different chromosomes4. Euchromatic regions are termed‘ A compartments’ and are enriched with open/decondensed chromatin and correspond to much higher overall levels of transcription, whereas ‘B compartments’ harbor inactive and heterochromatic sequences5 (Figure 1). In most cell types, large clusters of heterochromatin are enriched at the nuclear periphery, in multiple pericentromeric foci in the nuclear interior and around nucleolar membranes6.
Figure 1. The 3-dimensional genome, from nucleosome to nucleus.

a. Chromatin fibers that surround a DNA inside the nucleus are organized of arrays of the elementary unit, the nucleosome (146 bp of DNA wrapped in 2.5 loops around the core histone octamer). In ‘A compartments’, chromatin is in an open or active conformation and is permissive for transcription. This state is defined by high occupancy of RNA polymerase II complex and transcription factors and increased portions of nucleosome free DNA. In ‘B compartments’, chromatin is condensed and RNA polymerase and transcription factor occupancy is decreased. Each compartment can contain several megabases of 3D genome sequences. Superimposed upon this generalized model of nucleosomal organization is the 3D genome. This includes topological-associated domains (TADs) which extend on average (median size) across 185 kb and that can exist within both A compartments and in B compartments that primarily harbor repressive and condensed chromatin54. The constituent loci within TADs come into contact with each other much more frequently than with loci from outside domains5. The 3D genome also includes lamina-associated domains (LAD), which are condensed heterochromatin enriched around the nuclear periphery and physically bound to lamin proteins at the inner nuclear lamina.b. In ‘B compartments’, chromatin is condensed and is enriched with a different set of proteins to A compartments, which include, among others, HP1 (heterochromatin-associated protein 1) and (not shown) repressive histone methylation markings (H3K9me3, H4K20me3). c. TAD boundaries, but also specific chromosomal loop formations, including promoter-enhancer loopings, are often demarcated by CTCF and cohesins, and additional proteins which serve to regulate the formation of 3D genome structures118. Formation of promoter—enhancer loopings in A compartments, for example, involves a sequence of steps:. (1) transcription factors (TF) bind to promoter and enhancer sequences of DNA; (2) the co-activator Mediator is recruited which in turn recruits the cohesin complex (3), which as ring structure creates or stabilizes the loop. CTCF operates in parallel or synergistically to Mediator119. This structure allows activation and modulation of the RNA polymerase II core transcription machinery
Scaffolding and other regulatory proteins play a crucial role in the chromosomal spatial conformations, and a prime example of this is the cohesin complex and the CCCTC-binding factor/zinc finger protein CTCF, where accessory proteins load or release the cohesion complex onto chromosomes7. In humans, cohesins are comprised of five core subunits SMC1, SMC3, RAD21/REC8, and STAG1-37. Cohesins were initially explored in the context of sister chromatid cohesion and segregation during cell division (mitosis) 7. However, these proteins continue to be present at high levels in the nuclear proteome of postmitotic cells including neurons8. Cohesins form ring-like structures, literally entrapping distant chromatin fibers into chromosomal loopings7 (Figure 1). Cohesins are highly enriched at actively expressed genes in a tissue- and cell-type specific manner7. By contrast, CTCF, although dispensable for cohesin loading onto DNA, orchestrates cohesin enrichment at select binding sites8. As a result, chromosomal loopings co-occupied by cohesins and CTCF at both ends often associate with broader stretches of regulatory domains at other parts of the chromosome, marking the co-regulated repression or expression of groups of genes in a cell-type specific manner9. The CTCF binding sites often are positioned in inward and/or convergent orientation, and to a lesser degree, tandem orientation, at the two contact sites of the loop5,10. CTCF directionally recognizes binding sites via an 11 zinc finger array. Cohesin, in turn, is assembled from the CTCF’s C-terminal end, resulting in loop-bound head-to-head CTCF configurations11.
Importantly, powerful sequencing technologies in conjunction with chromosome conformation capture assays (Textbox 1, with Figure 4 in textbox) make it possible to map chromosomal contacts on a genome-wide scale, and have provided vast amounts of information on intra- and interchromosomal interactions5. The functional implications thereof, however, remain to be explored.
Textbox 1. Charting the 3D genome–methods and challenges.
The most widely used approach to chart contacts and physical proximity of non-contiguous DNA is chromosome conformation capture (termed 3C). Intact chromatin undergoes enzyme-based restriction digest followed by enzymatic fusion of the cut DNA end with DNA ligase. This results in chimeric reads if the DNA molecule is a fusion product of (previously) two DNA molecules that each map to a different locations in the genome with different coordinates either on the same or on a different chromosome. The 3C technique is scalable, up to comprehensive mapping on a genome-wide scale (‘Hi-C’) 54 (Figure). The most advanced protocols can be applied to intact nuclei (‘in situ Hi-C’), and allow for an incredibly high resolution (~1kb) mapping of chromosomal contacts. However, this requires cost-intensive DNA libraries sequencing at significant depth, or at least five billion reads (number of DNA molecules sequenced) per assay5. Not all interactions are functionally relevant and polymer features of chromatin fibers could influence to some degree the interactions between loci, in the absence of biological regulation115,116. Moreover, systematic comparison of 3C derivatives with DNA fluorescence in situ hybridization (DNA FISH), a traditional low throughput method measuring proximity of candidate genomic loci at single cell resolution, has uncovered additional confounds117. This is because seemingly subtle differences in chromosome conformation capture protocols and downstream bioinformatical analyses of the Hi-C data have a major effect on the quantity and quality of chromosomal contacts observed. As a result, there is still debate whether the number of ‘loopings’ (distinct contacts between non-contiguous DNA) in a typical vertebrate cell is in the order of 1×104 5, or perhaps as many as 1×106 21. These challenges, while not diminishing the potential of chromosome conformation capture assays to fundamentally advance knowledge in virtually all areas of genomics, clearly emphasize the importance of additional analyses and functional validation, after investigators have charted the 3D genome by chromosome conformation capture.
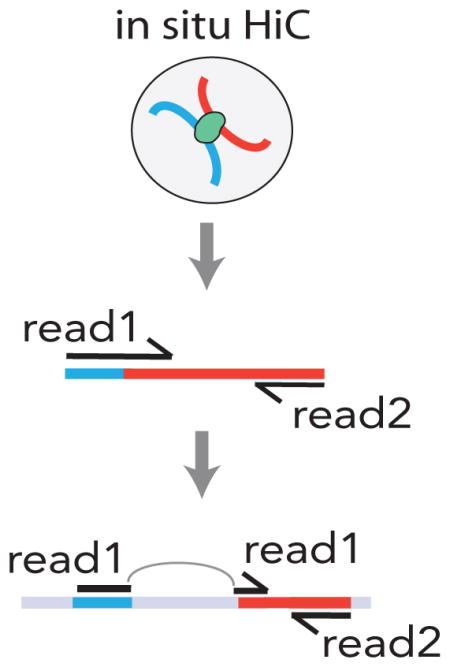
Promoter-enhancer loopings
Promoter-enhancer loopings, considered to be of fundamental importance for transcriptional regulation in eukaryotes, are one type of chromosomal interaction that is becoming increasingly well understood. Promoters are commonly defined as cis-regulatory sequences within 1000 base pairs from the next gene transcription start site. By contrast, enhancers are a type of cis-regulatory sequence positioned >1kb from the nearest transcription start site12. Promoters (but not enhancers) typically include a core promoter as docking site for general transcription factors (TFIIA/B/D/E/F/H) and components of RNA polymerase II holoenzyme as part of the preinitiation complex12. These core promoters drive low levels of basal transcription. However, gene expression is heavily stimulated by ‘activators’ or transcription factors that bind, in sequence-specific fashion, at the site of promoters and enhancers12. The transcription factors bind to promoters and enhancer sequences in nucleosome-free intervals in open chromatin and recruit co-activator complexes, such as Mediator and CREB-binding protein (CPB)/p30012. Promoters, in contrast to enhancers, are often CpG rich12. Furthermore, on a genome-wide scale. promoters and enhancers show robust differences in their histone modification landscapes. These include, for example, sharp peaks of trimethyl-histone H3-lysine 4 and enrichment of mono-methyl-histone H3-lysine 4 at enhancers13. Each form of H3-lysine 4 methylation is thought to bind a different set of methyl-reader proteins and transcriptional regulators14. However, enhancers are separated from their target gene by many kilo- or even megabases of interspersed linear genome12. Various mechanisms have been proposed by which enhancer chromatin could regulate the expression of target genes from distant chromosomal locations. Such types of mechanisms include sliding along the chromosome to ‘track’ promoters15, or alternatively, a physical ‘bridge’ built via protein-protein interactions16. Presently, however, most studies implicate the promoter-enhancer (chromosomal) loop model which involves physical interaction, or at least spatial proximity, between the enhancer chromatin and the promoter target17.
Recently, an important role in the shaping of promoter-enhancer loopings has been ascribed to Mediator, a multi-subunit complex acting as co-factor for many transcription factors18. Mediator loads cohesins onto chromatin fibers, potentially promoting loop formations in self-organizing fashion18. According to this model, transcription factors that recognize specific DNA motifs bind to specific genome sequences, in turn attracting the Mediator complex. This recruitment subsequently promotes the build-up of the ring-shaped cohesin complex at the site. This protein ring structure thus could promote and stabilize promoter-enhancer loopings (Figure 1).
Activity-regulated Chromosomal Contacts
Dynamic remodeling of neuronal connectivity, both in terms of structural plasticity (e.g. dendritic spines and axonal branching) and functional plasticity (e.g. changes in synaptic strength) is fundamentally important for normal brain development, learning, and memory. In the nucleus, promoter-enhancer interactions are likely to play an important role for these types of plasticity. To mention just one example, postnatal differentiation and functional maturation of cerebellar granule neurons is associated with dynamic changes in chromatin accessibility and levels of histone acetylation at thousands of putative enhancer sequences19. It is noteworthy that in differentiated tissues, including the brain, perhaps as many as 60% of all promoters and transcription start sites could be under control by multiple enhancer sequences20–22. Indeed, pharmacologically induced membrane depolarization produces transcriptional changes in neurons that are associated with highly dynamic changes in chromosomal loopings at selected loci23–25, which supports a role for these mechanisms in neuronal plasticity. However, in the absence of genome-wide chromosome conformation studies, it remains unknown to what extent these mechanisms operate more broadly across the genome. Not all developmental stimuli trigger reorganization of the 3D genome because human fibroblasts exposed to Tumor Necrosis Factor alpha (TNFα), a cytokine with profound effects on growth and proliferation, do not show widespread changes in their chromosomal contact map21.
Promoter—enhancer interactions and plasticity
Based on recent studies, four models have been proposed for how promoter-enhancer interactions might be involved in neuronal plasticity. In the first model (Figure 2A), distal sequences carrying a cargo of transcription factors are moved into close spatial proximity to the target gene promoter. This mechanism has been proposed for enhancer loopings targeting the transcription start site of NMDA receptor subunits23 or GABA synthesis genes24. In this model, expression of a subset of immediate early gene (IEG) transcription factors is under tight control of enhancer elements, which rapidly assemble into chromosomal loopings upon synaptic stimulation25. This process is combined with localized transcription from enhancer elements -- ‘enhancer RNAs -- (eRNAs) and is required for activity-induced expression of the c-fos IEG25. According to these studies, promoter-enhancer loopings mediate the ‘re-location’ of enhancer-bound transcription factor protein (the ‘cargo’) towards activity-regulated neuronal gene promoters, thereby facilitating the transcriptional process24,25 (Figure 2A).
Figure 2. Enhancer-mediated regulation of neuronal gene expression.

Four models for enhancer-dependent up- and down-regulation of transcription have been described in neurons (see text). The models are not mutually exclusive. (A, top) Protein ‘cargo’ model, which has been demonstrated for the GAD1 gene: Distal enhancer sequences bind transcription factors. Promoter and enhancer sequences are separated and interaction between transcription factors, enhancer and promoter sequences cannot occur, and the rate of transcription is reduced (dashed arrow). Loop formations that enable physical proximity between the enhancer’s target (such as be a gene transcription start site), transcription factors and the proximal promoter region upregulate transcription (bottom). (B, top) enhancer RNA (eRNA) decoy model, which has been demonstrated for the neuronal gene Arc: When the Arc gene promoter is occupied by negative elongation factor (NELF), the action of RNA polymerase II complex is stalled and Arc transcription is low. However, the distal enhancer sequences of the gene produce enhancer RNAs (eRNAs), and when the enhancer region moves via specific loop formations into physical proximity to the Arc promoter, these short non-coding eRNAs bind NELF, which reduces NELF occupancy at the target gene (Arc), thereby liberating RNA polymerase II complex and promoting the transcriptional process (Bottom). (C) Loop competition model, shown for the gene GRIN2B: Two or more non-contiguous (separated by interspersed sequence) cis-regulatory sequences, potentially with opposing effects on transcription (i.e. a promoter sequence and a repressor sequence), are competing to access the GRIN2B gene promoter. Top: When the GRIN2B promoter interacts with a silencer protein occupying loop-bound repressor DNA elements, and transcriptional activity is reduced. Bottom: When the same promoter interacts with an active enhancer element that has bound transcription factors present, GRIN2B expression is increased. (D) Strand break mobilization at the c-fos immediate early gene (IEG) promoter: Promoter activity is low at baseline when the genome is in linear form (bottom). To activate transcription, topoisomerase IIβ enzyme induces DNA strand breaks. This results in the mobilization of promoter sequences into physical proximity with enhancer elements that were previously separated by several kb of interspersed linear genome (bottom). The physical proximity of these promoter and enhancer DNA then leads to synergistic activation of gene expression via enhancer-bound transcription factor proteins.
An entirely different mechanistic model ascribes a decoy function to the eRNAs, in order to liberate the target promoter from negative regulators of transcription. This model emerged from the study of loop-bound enhancer elements governing the transcription of Activity-regulated cytoskeletal (Arc), which encodes a small protein that is important for numerous aspects of synaptic plasticity26. In this system, loop-bound enhancer DNA produces short-lived non-coding RNAs that bind the negative elongation factor (NELF) complex away from Arc promoter sequences27. Because NELF functions as an inhibitor of transcription, the small RNAs produced by the Arc enhancer essentially act as a ‘sponge’ for NELF. As a result, NELF occupancy decreases at the Arc gene and paused RNA polymerase II complex, previously stalled by NELF, now becomes unlocked and is rapidly ramping up production of Arc RNA27. Therefore, enhancer-mediated localized production of an RNA decoy for negative regulators of RNA polymerase II could temporarily disinhibit target gene expression27 (Figure 2B).
Additional types of promoter-enhancer interactions are variations of the transcription factor protein cargo model put forth in Figure 2A. One example is competition between loop-bound enhancer and repressor sequences (Figure 2C). Furthermore, Figure 2D summarizes a recently described mechanism for shorter-range promoter-enhancer loopings extending a few kilobases from the transcription start site of the IEG and transcriptional regulators Fos, FosB, Npas4, Egr1, Nr4a1 and Nr4a328. These IEG promoters, which show increased expression within minutes after neuronal stimulation, are positioned close to regulated DNA strand breaks induced by type II DNA topoisomerase28. This effectively increases the mobility of the local cis-regulatory sequences, including shorter-range chromosomal loop formations enriched with CTCF protein. As a result, there is an increased interaction of promoters with neighboring enhancers 28 (Figure 2D).
With an estimated total of ~12,000 neuronal activity-regulated enhancers, and eRNA expression at ~2,000 extra- and ~1,000 intra-genic activity-regulated enhancers in cortical neurons29, it is likely that neurons use a wide array of chromosomal conformations and loop structures to orchestrate gene expression programs in response to changes in synaptic activity.
The 3D Genome in neurodevelopmental disorders
Mutations in chromosomal scaffolding proteins
It seems beyond doubt that genome folding and packaging are crucial for normal brain development and function. To this end, the nuclear lamina, a protein meshwork located at the inner layer of the nuclear membrane and primarily comprised of three types of filamentous protein (lamin A/B/C) could play an important role by interacting with a wide range of repressive chromatin regulators and thereby tethering heterochromatic sequences to the nuclear periphery6,30. Such types of lamina-associated heterochromatic chromosomal domains become surprisingly mobile during the course of differentiation of neural precursor cells into astrocytes31. As a result, many hundreds of genes undergo regulated repositioning during the course of astrocyte differentiation. By moving into or out of the nuclear lamina, these genes are thought to participate in transcriptional programs associated with astrocyte cell identity31. Similar phenomena have been observed during neuronal differentiation. For example, a subtype among the olfactory receptor gene family, the trace amine-associated receptors, undergo mono-allelic dissociation from lamin proteins and the nuclear periphery during the course of olfactory sensory neuron differentiation32. Early studies suggest that by relocating to inner compartments of the nucleus, olfactory receptor genes could encounter localized accumulations of transcription factors and an open chromatin environment that facilitates promoter-enhancer interactions and gene expression32, 33.
In this context, it is noteworthy that deleterious mutations in genes encoding scaffolding proteins for the 3D genome have been linked to disease. These include not only neurodevelopmental disorders such as Cornelia de Lange Syndrome (CdLS) 7, 34, 35, but also adult-onset progressive demyelination syndromes36 (Table 1). Neurodevelopmental disease phenotypes in CdLS include intellectual disability, psychosis and other psychiatric maladies. The underlying genetic defect includes microdeletions and copy number variations affecting core members of the cohesin complex including SMC1A and SMC3, and the accessory subunit NIPBL 34 (Table 1). The neurological manifestations could be due to 3D genome disorganization in brain cells and de-compaction of chromatin, however, the precise molecular mechanisms remain to be elucidated37.
Table 1.
Monogenic neurological disease associated with altered function of 3D genome scaffolding proteins
| OMIM *gene |
Function | Syndrome | OMIM #disease |
|---|---|---|---|
|
NIPPED-B-LIKE (NIPBL) *608668 |
Cohesin loading onto chromatin | heterozygous deletion and mutations account for ~50% of cases diagnosed with Cornelia de Lange Syndrome (CdLS) including intellectual disability (reference 4,28 in text) | #122470 |
|
STRUCTURAL MAINTENANCE OF CHROMOS. 1A (SMC1A) *300040 |
Cohesin core unit, component of tripartite ring to entrap chromatin fibers | <5% of CdLS, gene duplication syndrome with behavioral alterations, intellectual disability (PMID 19052029) (references 29 in text) | |
|
STRUCTURAL MAINTENANCE OF CHROMOS. 3 (SMC3) *606062 |
Cohesin core unit, component of tripartite ring to entrap chromatin fibers | <5% of CdLS, gene duplication syndrome with behavioral alterations, intellectual disability (PMID 19052029)(reference 29 in text) | |
|
CCCTC-binding factor (CTCF) *604167 |
Chromosomal loop organizer and scaffold, insulator protein separating chromosomal domains, transcriptional regulator | heterozygous deletion and mutations microcephaly, mild mental retardation (PMID 23746550) (references 31,32 in text) | |
|
SPECIAL AT-RICH SEQUENCE BINDING PROTEIN 2 (SATB2) *608148 |
chromosomal domain organizer and scaffold | heterozygous mutations and deletions: Glass Syndrome (GLASS) including intellectual disability, seizure disorder, speech delay (references 36, 38 in text) | #612313 |
|
LAMIN B1 (LMNB1) *150340 |
Nuclear lamina protein tethering heterochromatin chromosomal domains to nuclear periphery | LMNB1 gene duplication: adult-onset autosomal dominant leukodystrophy (ADLD), progressive CNS demyelination. (reference 27 in text) | #169500 |
Genetic mutations in CTCF, as a key organizer for chromosomal loopings, have been linked to monogenic causes of microcephaly and cognitive disorder38,39. Consistent with these findings from clinical genetics, selective ablation of Ctcf in postnatal mouse brain causes behavioral alterations and dysregulated transcription of hundreds of neuronal transcripts. These include a deficit in Protocadherin cell adhesion molecule expression, resulting in altered connectivity40. It remains to be shown, however, whether the neurological manifestations of cohesin or CTCF gene mutations are associated with widespread 3D genome alterations in brain.
In addition to the aforementioned CTCF and cohesin complex, other examples of neurodevelopmental disease resulting from mutations in genes encoding 3D genome organizer proteins have been identified (Table 1). These include Special AT-rich Sequence Binding Proteins 1 and 2 (SATB1, SATB2) that govern chromosomal territories extending across hundreds of kilobases41 and anchor chromatin fibers into the nuclear matrix42. Of note, SATB2 is essential for craniofacial development and proper differentiation of transcallosal cortical projection neurons43,44. The gene has also been linked to some cases of Glass Syndrome (OMIM 612313) and mental retardation43,45. The related protein SATB1 is essential for connectivity and maturation of GABAergic interneurons in the cerebral cortex46,47. Furthermore, proteins encoded by the X-linked ATRX, and the Rett syndrome gene MECP2 (ATRX, OMIM #301040; MECP2, OMIM #312750), regulate looping structures at a subset of imprinted (parent-of-origin-specific) loci including H19/Igf2 and Dlk1/Gtl248. In addition, the ATRX and MECP2 proteins regulate higher order chromatin at the Dlx5/Dlx6 homeobox transcription factor locus 49 (but see also50). Moreover, ATRX is recruited by MECP2 to heterochromatic sequences51. However, because these proteins regulate a multitude of transcriptional and chromatin-associated functions inside the cell nucleus52, the precise role of chromosomal contacts and loopings in neurodevelopmental disorders with ATRX or MECP2 mutations remains to be elucidated.
Non-coding sequences implicated in autism
In the preceding chapter, we discussed examples of neurological disease caused by mutations or duplications of genes encoding protein scaffolds for the 3D genome. Here, we discuss a second type of brain disorder that is associated with locus-specific alterations of the 3D genome. A recent whole genome sequencing (WGS) study in families on the autism spectrum identified individuals with autism who did not carry disruptive mutations and copy number variations in known neurodevelopmental risk genes53. Instead, these cases carried deletions or duplications in non-coding DNA regions that in control samples were sensitive to digestion by DNAse I53 (note that DNAse I is an indicator for nucleosome-free (‘naked’) DNA and a widely used method for transcription factor footprinting). This would imply that chromatin around these sequences is involved in transcriptional regulation elsewhere in the genome53. These cases showed, as a group, significant enrichment for microdeletions in blocks of sequence 25–100kilobase (kb) upstream (i.e. in the regulatory non-coding region) of genes assigned with critical roles for normal human brain development. These genes included DSCAM, encoding Down Syndrome Cell Adhesion Molecule, and SCN2A encoding a sodium voltage-gated channel subunit implicated in autism and seizure disorder 53. Indeed, these non-coding sequences with apparently high disease penetrance are primarily located within 25–100kb from the 5′ end of genes associated with autism and related disease53. On a genome-wide scale, at least one half of all chromosomal loop formations targeting gene transcription start sites extend across 25–150kb (albeit some promoter-enhancer loopings can extend far beyond that range) 20,21,54 and therefore, it is possible that some of the autism-associated non-coding sequence mutations and deletions53 result in altered 3D regulation of neurodevelopmental risk genes. Similar principles may apply to other types of neurological disease. For example, aganglionic megacolon (hypertrophy and dilation of the colon) in TashT mutant mice, a model for Hirschsprung’s disease, is caused by a 700kb transgenic insertion into a gene desert extending across 3.3 Mb. The insertion disrupts silencer elements governing the expression of multiple target genes via long-range intrachromosomal loopings55.
The above mentioned deletions and mutations in intergenic non-coding sequence of specific individuals diagnosed with autism53 are in line with the ‘major gene’ model, which assumes either a single highly or a limited number moderately penetrant mutations in the etiology of autism56. Other disease cases may be better defined by the polygenic risk model, implying many inherited common variants in genetic susceptibility, each with small effect56. The majority of this type of risk-associated DNA single nucleotide polymorphisms and haplotypes do not directly affect coding sequence and protein structures56. There is indirect evidence that some of these risk-associated non-coding polymorphisms involve chromosomal loop formations. For example, brain-specific enhancer RNAs (eRNAs), which are short non-coding RNAs transcribed in cis at the site of active enhancer elements, are significantly enriched for genetic variants associated with autism spectrum disorder57. Because active enhancers are likely to positively regulate transcription of distant target genes via loop-bound chromosomal conformations, it is plausible to speculate that at least a portion of autism-associated DNA polymorphisms57 affect regulation of neuronal or glial 3D genomes.
Adult-onset neuropsychiatric disease
In addition to the an involvement of juvenile-onset neurological disorders, there is also evidence suggesting a role for the 3D Genome in adult-onset human cognitive disorders. For example, significant over-representation of enhancer sequences has been observed within the pool of polymorphisms and haplotypes associated with schizophrenia, an adult onset cognitive disorder often accompanied by delusions and hallucinations3,58. Among such type of risk-associated sequences are enhancer elements in an intragenic and intronic portion of the CACNA1C calcium channel gene. This gene also ranks prominently in the polygenic risk maps of other common psychiatric disease including depression59. Risk alleles within the CACNA1C enhancer sequences have also been associated with decreased reporter gene activity3. Remarkably, these CACNA1C intragenic enhancer sequences were found to be physically bound to the gene transcription start site via a 185kb spanning chromosomal loop formation in human prefrontal cortex (Table 2) 3.
Table 2.
Examples of chromosomal conformations implicated in the regulation of human cognition
| Locus | Gene Function | Chromosomal conformation in Brain |
|---|---|---|
| INSULIN GROWTH FACTOR 2 (IGF2/H19) | imprinted genes, controlling metabolism, growth (including postnatal brain) | Loss of ATRX and MECP2 proteins decreases local occupancy of CTCF and disrupts long- range chromosomal contacts across several Mb at Igf2/H19 locus, and additional loci, in mouse forebrain. PMID 24990380. References 41, 42 in text. |
| DIPEPITIDYL-AMINOPEPTIDASE-LIKE PROTEIN 10 (DPP10) | Kv4 channel auxiliary subunit regulating neuronal excitability | DPP10 transcription start site in prefrontal cortex neurons connected via 0.5Mb long-range loop with non-coding sequences showing histone modification profile unique to human (not shared with non-human primates incl. chimpanzee). PMID: 23185133. Reference 65 in text. |
| CALCIUM VOLTAGE- GATED CHANNEL SUBUNIT ALPHA1 C (CACNA1C) | Ion channel, possible role in synaptic plasticity (PMID 20573883) | 0.2Mb promoter- bound loop formation includes enhancer sequences with single nucleotide polymorphisms linked to heritable risk for schizophrenia. PMID: 25453756. Reference 51 in text. |
| GLUTAMATE RECEPTOR, IONOTROPIC, N-METHYL-D- ASPARTATE, SUBUNIT 2B (GRIN2B) | Ion channel broadly relevant for in developing and adult brain function | GRIN2B promoter targeted by multiple loop formations bypassing up to 0.5Mb linear genome in the context of transcriptional regulation; loop-bound DNA confers liability for working memory and cognitive disease. PMID: 25467983. Reference 17 in text. |
| GLUTAMIC ACID DECARBOXYLASE 1 (GAD1) | GABA synthesis gene | Human/mouse conserved 50 kb promoter-enhancer loop, sensitive to changes in neuronal activity and altered in prefrontal cortex of some cases with schizophrenia. PMID: 23864674. Reference 18 in text. |
Other studies have suggested that alterations in DNA methylation and other types of epigenetic dysregulation of enhancer sequences could contribute to the neuropathology underlying mood and psychosis spectrum disorders. For example, astrocyte dysfunction in depression60 and altered neuronal gene expression in schizophrenia61,62 have been linked to dysregulated enhancers.
Likewise, common sequence variants conferring genetic risk for Alzheimer’s neurodegeneration show strong overlap with mammalian enhancers and other cis-regulatory elements previously implicated in immune functions63. In an animal model of Alzheimer’s disease which involves hippocampal injury triggered by the Cdk5 kinase activator protein p25, m, any enhancer sequences harboring Alzheimer’s risk variants became activated 63. Furthermore, it was recently reported that heterochromatic compartments are disorganized in neuronal nuclei of Alzheimer’s cerebral cortex collected postmortem, accompanied by with improper re-expression of the normally silenced genes64,65. It is reasonable to speculate that the underlying molecular mechanisms involve reorganization and reactivation of enhancer-bound loop formations.
These findings, taken together, point to a strong representation of enhancer elements among the genetic and epigenetic risk architectures of some of the common neuropsychiatric disorders, including autism, schizophrenia, depression, and Alzheimer’s disease. The field now eagerly awaits studies undertaking comprehensive, genome-scale (Hi-C) maps from brain tissue of diseased subjects66. 3D genome and higher order chromatin mapping is technically feasible in postmortem brain tissue67 and this should clarify whether chromosomal loop formations that ‘recruit’ enhancer sequences in the context of transcriptional regulation are dysregulated on a genome-wide scale. Chromosome conformation capture studies performed at specific candidate gene loci already have indicated that the conformation of the 3D genome might be altered in cognitive disorders (Table 2). For example, a 50kb loop formation connecting enhancer elements enriched with IEG transcription factor motifs into physical proximity of the GAD1/GAD67 GABA synthesis gene promoter, is disrupted in prefrontal cortex of subjects with schizophrenia24. These alterations in the GAD1 chromatin loop occurred in conjunction with decreased gene expression and deficits in open chromatin-associated histone H3-lysine 4 methylation24,68. Given that dysregulated GAD1 expression contributes to disorganized inhibitory circuitry and altered synchronization of cortical networks in psychosis69,70, the reported alterations in GAD1 higher order chromatin24 illustrate the importance of proper 3D genome regulation for human cognition.
Additional examples of chromosomal conformations of potential importance for human cognition exist. For example, long range (0.5–1Mb) contacts within chromosomes 2q14.1 and 16p11.271. Interestingly, these loop formations connect regulatory sequences defined by human-specific histone methylation ‘peaks’ that are absent from non-human primate brain (Table 2) 71. This finding hints at the possibility that some of the cognitive abilities (and vulnerabilities) unique to human are associated with specific configurations in the 3D genome of cortical neurons71.
Dynamic NMDA receptor gene loopings affect cognition
In this section, we focus on a single gene locus, or ~1Mb of human (mouse) chromosome 12p31.1 (6, 66.36cM) encompassing the N-methyl-D-aspartate (NMDA) receptor subunit GRIN2B. This locus has been particularly well studied in the context of chromosomal contacts and conformations, and it provides an interesting example for the evolving view on the highly dynamic and multilayered regulation of the 3D genome as pertains to cognition and complex behavior. Furthermore, deleterious GRIN2B mutations rank prominently in exome sequencing studies on monogenic forms of intellectual disability, epilepsy, autism and psychosis72–75, which underscores the importance of this gene for human cognition. Until recently, however, little was known about the role of non-coding DNA at the GRIN2B locus in health or disease. Here, we review studies on GRIN2B higher order chromatin and chromosomal contacts in human postmortem brain and animal models, including the complex roles of loop-bound non-coding DNA in the regulation of NMDA receptor gene expression and cognition.
Chromosome conformation studies in human and mouse prefrontal cortex identified multiple loop-bound intronic and intergenic DNA sequences, up to 450kb downstream from the GRIN2B/Grin2b transcription start site. These sequences were loaded with multiple transcription factors and interacted via loop formations with the GRIN2B/Grin2b promoter and transcription start site23. In addition to such types of long-range promoter-enhancer loopings, the GRIN2B/Grin2b promoter physically interacts with intragenic repressive chromatin embedded in intron sequence23. These repressive elements were defined by high levels of SETDB1 histone H3K9 methyltransferase and heterochromatin-associated protein 1 (HP1) 23. Therefore, it was proposed that transcriptional regulation at the GRIN2B/Grin2b locus involves a dynamic and competitive interplay of multiple loop formations, each of which could engage with the GRIN2B promoter23. These include facilitative loopings with CTCF loop organizer protein bound at the loop contacts. The role of these loopings is to deliver IEGs and additional types of transcription factors to the GRIN2B promoter 23 (Figure 2C, Figure 3). However, this process is counterbalanced by promoter-bound higher order chromatin involving repressive intronic sequences 30kb downstream from the transcription start site23,76 (Figure 3). A subset of these chromosomal conformations were at least partially conserved in human and mouse brain and were sensitive to changes in neuronal activity23.
Figure 3. Dynamic model of chromomosomal conformation at GRIN2B NMDA receptor gene locus.
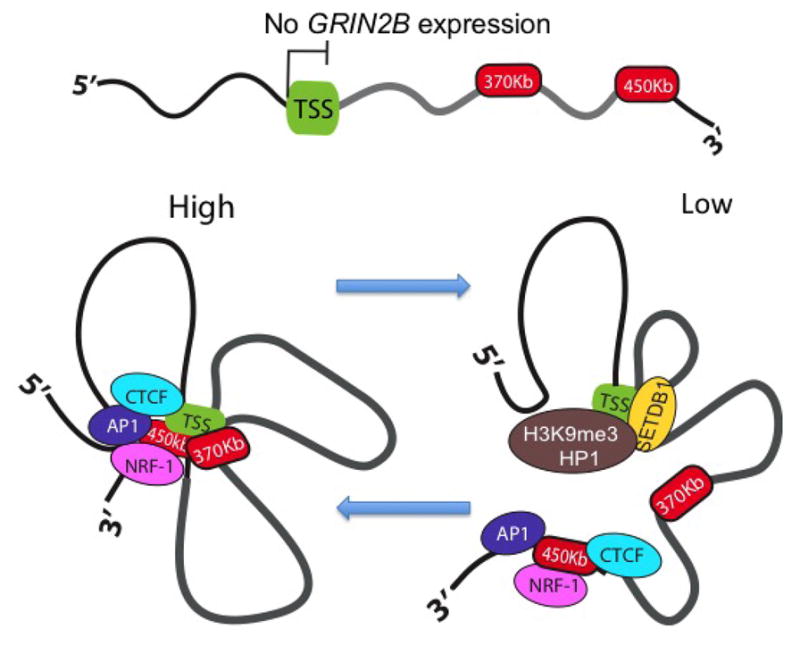
(top) Long-range chromosomal loop formations at the GRIN2B locus are not detectable cells and tissues that do not express the GRIN2B protein. However, in cells that express GRIN2B, the expression can be finely tuned by dynamic competition between multiple chromosomal conformations competing for access to the GRIN2B promoter and transcription start site. Transcription is increased by a loop-bound enhancer formation of DNA, to which IEG transcription factors, NRF-1 and additional activator proteins are bound as a complex. This is counterbalanced by additional chromosomal conformations that carry repressive chromatin into physical proximity to the GRIN2B promoter, This repressive chromatin is characterized by localized enrichments of HP1 (heterochromatin-associated protein 1) and repressive SETDB1 histone methyltransferase, (see text and reference23 for details).
Interestingly, multiple loop-bound sequences interacting with the GRIN2B promoter harbor single nucleotide polymorphisms (SNP) previously implicated in determining working memory efficiency23. These included SNP rs117578877, positioned in a 450kb loop connecting the GRIN2B 5′end to intergenic DNA downstream from the gene’s 3′end. Indeed, this polymorphism could contribute to risk for schizophrenia and personality traits associated with schizophrenia23. Notably, the risk-associated SNP allele was associated with decreased nucleoprotein binding and motif loss for the CCAAT/Enhancer Binding Protein CEBPB (C/EBPβ)23. CEBPB is a transcription factor implicated in consolidation of cortical and hippocampal learning and memory77–79. Because many enhancer elements are defined by sequential linear alignment of multiple transcription factors within short distance80,81, additional activator proteins may synergistically cooperate with loop-bound CEBPB to regulate GRIN2B expression23. Taken together, these findings point to a complex and multilayered regulation of chromosomal conformations within 1Mb surrounding the GRIN2B gene. Therefore, loop-bound DNA targeting the GRIN2B promoter could either facilitate or repress expression, depending on the protein ‘cargo’ (Figure 3). Multiple distal loop formations compete in a highly dynamic and activity-dependent manner for access to the GRIN2B promoter sequences23. It remains to be explored whether these findings from the GRIN2B locus also apply to other neuronal genes. It is possible, therefore, that neuronal (and glial) 3D genomes are defined by thousands of dynamically regulated chromosomal conformations that are poised to quickly re-configure in response to synaptic signaling and other stimuli.
Chromosomal Conformations – A Novel Therapeutic Target?
A number of chromatin modifying drugs have shown promising therapeutic effects in preclinical models of cognitive disease. These include histone deacetylase inhibitors and other drugs altering the balance between acetylation and deacetylation82–85, histone methyltransferase inhibitors86, topoisomerase inhibitors87, and compounds targeting the DNA methylome88,89. However, such types of drugs alter chromatin structure and function across widespread areas of the genome. Unsurprisingly then, both global and locus specific alterations in chromosomal conformations and spatial genome organization have been reported for small molecule drugs interfering with histone acetylation90,91 or methylation92. For example, exposure to histone deacetylase inhibitor drug, trichostatin A, is associated with repositioning of expressed genes towards the nuclear center and away from repressive environment such as the nuclear lamina90,91. Furthermore, drug-induced inhibition of the repressive H3K9 methyltransferase, G9a, induced a spatial reconfiguration of the β-globin gene cluster in hematopoetic cells, thereby shifting expression pattern towards the fetal γ–globin genes92. Much of this work, however, was limited to cell lines. It remains to be explored whether drug-induced changes in 3D genome organization could be harnessed for therapeutic interventions in brain disorders. Moreover, the safety profile for broadly acting chromatin-modifying drugs remains unknown in the context of neuropsychiatric disease93.
Recently, a novel perspective has been provided by an expanding repertoire of epigenomic editing tools that include the possibility for targeted, locus-specific interventions. For example, engineered zinc finger proteins (Zfp) were fused to the p65-NFκB domain to recruit p300/CBP histone acetyltransferase and targeted to highly specific 18–20bp sequence motifs. This approach was used successfully to specifically activate the promoters of glial-derived neurotrophic factor (Gdnf) and ΔFosB/fosb transcription factor in striatal neurons in vivo94,95. These interventions provided robust neuroprotective effects (Gdnf) 94 and produced changes in reward- and depression-related behaviors (ΔFosB) 95, thus confirming the feasibility of in vivo epigenomic editing in a preclinical model. However, in vivo epigenomic editing of loop-bound sequences has not yet been reported in brain. To this end, there is promising evidence from in vitro studies. For example, in primary neuronal culture, loading the VP64 transcriptional activator onto loop-bound distal regulatory elements bypassing 475kb of linear genome resulted in increased transcription of the Grin2b NMDA receptor subunit23. Although the resulting changes in Grin2b expression were very modest23, further technical improvements may deliver more robust effects. For example, a p300 histone acetyltansferase catalytic core fused to various designer DNA binding proteins, including Zfp, Transcription Activator-like Effector (TALE) or RNA-guided CRISPR/Cas could be used to regulate expression via loop-bound long range enhancer and repressor sequences96. These epigenomic editing approaches will complement genetic studies that introduce small 97,98 or large 99 genetic lesions of loop-bound DNA. The technique allows for correction of genetic mutations 98,100 and testing the resulting effects on target gene transcription101–104. Epigenomic editing approaches, in conjunction with ex vivo cell culture work, will be required to test causality and to develop therapeutic interventions based on loop-bound enhancer and other regulatory DNA elements that contribute to the genetic risk architecture of psychiatric and neurological disease. By combining the ability of human induced pluripotent stem cells (hIPSCs) to generate sufficient quantities of nearly any cell type with these novel methodologies to precisely manipulate human cells, it should be possible to test comprehensively the effect of disease-related genetic and epigenetic manipulations on the 3D genome105–107. The engineering of isogenic cell lines in this way have already been used across a variety of brain disorders to precisely indicate the causal effect of a disease associated mutation 71,108–112, including enhancer sequences associated with neurological disease113. We believe these approaches are poised to be used to further elucidate the functional consequences of manipulating the epigenome, including chromosomal loop formations, in human neurons and glia.
Summary and Future Directions
Regulation of 3-dimensional chromosomal conformations and spatial genome architectures (3D genome) is critical across the entire lifespan of the human brain. Mutations affecting the function of protein scaffolds and global organizers for the 3D genome frequently are associated with neurodevelopmental defects. Furthermore, many clinically relevant structural DNA variants positioned in intergenic or intragenic non-coding sequences could impact neuronal gene expression by bypassing the linear genome in order to directly interact with the target gene. The field now eagerly awaits comprehensive mapping of the 3D genome in psychiatric disorders. Multiple mechanistic types of loop-based promoter-enhancer interactions already have been identified in neurons, hinting at an unexpected functional diversity of transcriptional regulation in the context of synaptic plasticity. Furthermore, in vivo applications of (epi)genomic editing techniques will explore how specific manipulations of the brain’s 3D genome will affect cognition and behavior. Finally, it will be exciting to apply emerging technologies for 3D genome live-imaging, such as ‘Real-time Observation of Localization and Expression’ (ROLEX) 114, to the brain. Such an approach could provide deep and unprecedented insights into the dynamic reconfigurations of neuronal and glial genomes in response to internal or external stimuli.
Acknowledgments
This work was supported by NIMHs grant P50MH096890 and R01MH106056. The Authors report no conflict.
Glossary
- Common sequence variants
DNA polymorphisms with minor allele frequencies exceeding 0.05 in the general population
- Microdeletion
a loss of a fragment of a chromosome
- Microduplication
a duplication of a fragment of a chromosome
- Copy number variation (CNV)
genomic sequence, typically in kilo- or megabase range, that either is duplicated or deleted.
- Immediate
early gene transcription factors- Activator proteins such as Fos, Jun, Egr are expressed in response to stimulus-based triggering of cAMP and other intracellular signaling cascades and are rapidly expressed within 15 min of a stimulus in sensitive neurons.
- Intergenic
genome sequence between two annotated genes.
- Methylome
the genome-wide distribution of DNA cytosine methylation in specific cells or tissues.
- CRISPR/Cas
Clustered regularly-interspaced short palindromic repeats/CRISPR-associated protein 9, an RNA-guided DNA endonuclease enzyme in bacteria that is increasingly used a tool for targeted genomic and epigenomic editing in multicellular organisms.
- Nucleolus
a specialized nuclear organelle for the production of ribosome subunits
- Eukaryote
a life form with a well delineated nucleus separated from the cytoplasm by a nuclear membrane
- Transcriptional unit
a stretch of DNA being transcribed into RNA molecules
- Chromosomal scaffold
a three-dimensional chromatin structure whose integrity is maintained by non-histone scaffolding proteins (e.g., cohesin, CTCF).
- Nucleosomal arrays
chromatin packaging in the form of repeating units of DNA-bound core histone octamers connected by linker DNA and linker histone proteins
- Preinitiation complex
a large collection of proteins that are essential to begin DNA transcription, acting by recruiting RNA polymerase II, denaturing DNA, and proper positioning of DNA in the active site of polymerase.
- Cis-regulatory sequence
non-coding portions of the genome that regulate transcription of nearby or distal genes (e.g., promoters, enhancers).
Biographies
Prashanth Rajaranan trained as an undergraduate in psychology at Michigan State University in East Lansing, Michigan. Presently, he is an MD, PhD student jointly mentored by Kristen Brennand and Schahram Akbarian. He is interested in stem cell biology and the regulation of the three-dimensional genome in normal and diseased human brain.
Sergio Espeso Gil trained as an undergraduate in neuroscience at the School of Medicine-Universidad Autónoma de Madrid and the Université Pierre and Marie Curie. Presently, he is a PhD student in Dr. Stephan Ossowski’s Group at the Centre for Genomic Regulation in Barcelona. He is broadly interested in neuroepigenetics and bioinformatics.
Kristen Brennand, PhD, is an Assistant Professor of Psychiatry at the Icahn School of Medicine at Mount Sinai, in New York, New York. She trained in developmental and stem cell biology at Harvard University and in neurobiology at the Salk Institute for Biological Studies. By combining expertise in stem cell biology and neurobiology, she has pioneered a new approach by which to study psychiatric disease.
Schahram Akbarian, MD, PhD, is Professor and Chief of the Division of Psychiatric Epigenomics in the Friedman Brain Institute at the Icahn School of Medicine at Mount Sinai in New York. He studies epigenomic determinants of cognition and behavior in the mouse model and his laboratory is generating cell-type specific epigenome maps from human brain for the NIH-sponsored PsychENCODE consortium.
Footnotes
Competing Interests Statement: There is NO Competing Interest.
References
- 1.Stamatoyannopoulos JA. What does our genome encode? Genome Res. 2012;22:1602–1611. doi: 10.1101/gr.146506.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Loviglio MN, et al. Chromosomal contacts connect loci associated with autism, BMI and head circumference phenotypes. Mol Psychiatry. 2016 doi: 10.1038/mp.2016.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Roussos P, et al. A role for noncoding variation in schizophrenia. Cell Rep. 2014;9:1417–1429. doi: 10.1016/j.celrep.2014.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lieberman-Aiden E, et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science (New York, NY) 2009;326:289–293. doi: 10.1126/science.1181369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rao SS, et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell. 2014;159:1665–1680. doi: 10.1016/j.cell.2014.11.021. This paper provides chromosomal contact maps at ultrahigh resolution (1kb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Padeken J, Heun P. Nucleolus and nuclear periphery: velcro for heterochromatin. Curr Opin Cell Biol. 2014;28:54–60. doi: 10.1016/j.ceb.2014.03.001. [DOI] [PubMed] [Google Scholar]
- 7.Nasmyth K, Haering CH. Cohesin: its roles and mechanisms. Annu Rev Genet. 2009;43:525–558. doi: 10.1146/annurev-genet-102108-134233. [DOI] [PubMed] [Google Scholar]
- 8.Wendt KS, et al. Cohesin mediates transcriptional insulation by CCCTC-binding factor. Nature. 2008;451:796–801. doi: 10.1038/nature06634. [DOI] [PubMed] [Google Scholar]
- 9.Dowen JM, et al. Control of cell identity genes occurs in insulated neighborhoods in mammalian chromosomes. Cell. 2014;159:374–387. doi: 10.1016/j.cell.2014.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vietri Rudan M, et al. Comparative Hi-C reveals that CTCF underlies evolution of chromosomal domain architecture. Cell Rep. 2015;10:1297–1309. doi: 10.1016/j.celrep.2015.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Guo Y, et al. CRISPR Inversion of CTCF Sites Alters Genome Topology and Enhancer/Promoter Function. Cell. 2015;162:900–910. doi: 10.1016/j.cell.2015.07.038. This paper illustrates how the 3D genome orchestrates transcription at the Protocadherin cell adhesion molecule gene cluster. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vernimmen D, Bickmore WA. The Hierarchy of Transcriptional Activation: From Enhancer to Promoter. Trends Genet. 2015;31:696–708. doi: 10.1016/j.tig.2015.10.004. [DOI] [PubMed] [Google Scholar]
- 13.Zhou VW, Goren A, Bernstein BE. Charting histone modifications and the functional organization of mammalian genomes. Nat Rev Genet. 2011;12:7–18. doi: 10.1038/nrg2905. [DOI] [PubMed] [Google Scholar]
- 14.Ruthenburg AJ, Allis CD, Wysocka J. Methylation of lysine 4 on histone H3: intricacy of writing and reading a single epigenetic mark. Mol Cell. 2007;25:15–30. doi: 10.1016/j.molcel.2006.12.014. [DOI] [PubMed] [Google Scholar]
- 15.Hatzis P, Talianidis I. Dynamics of enhancer-promoter communication during differentiation-induced gene activation. Mol Cell. 2002;10:1467–1477. doi: 10.1016/s1097-2765(02)00786-4. [DOI] [PubMed] [Google Scholar]
- 16.Mueller-Storm HP, Sogo JM, Schaffner W. An enhancer stimulates transcription in trans when attached to the promoter via a protein bridge. Cell. 1989;58:767–777. doi: 10.1016/0092-8674(89)90110-4. [DOI] [PubMed] [Google Scholar]
- 17.Gorkin DU, Leung D, Ren B. The 3D genome in transcriptional regulation and pluripotency. Cell Stem Cell. 2014;14:762–775. doi: 10.1016/j.stem.2014.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kagey MH, et al. Mediator and cohesin connect gene expression and chromatin architecture. Nature. 2010;467:430–435. doi: 10.1038/nature09380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Frank CL, et al. Regulation of chromatin accessibility and Zic binding at enhancers in the developing cerebellum. Nat Neurosci. 2015;18:647–656. doi: 10.1038/nn.3995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sanyal A, Lajoie BR, Jain G, Dekker J. The long-range interaction landscape of gene promoters. Nature. 2012;489:109–113. doi: 10.1038/nature11279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jin F, et al. A high-resolution map of the three-dimensional chromatin interactome in human cells. Nature. 2013;503:290–294. doi: 10.1038/nature12644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Andersson R, et al. An atlas of active enhancers across human cell types and tissues. Nature. 2014;507:455–461. doi: 10.1038/nature12787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bharadwaj R, et al. Conserved higher-order chromatin regulates NMDA receptor gene expression and cognition. Neuron. 2014;84:997–1008. doi: 10.1016/j.neuron.2014.10.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bharadwaj R, et al. Conserved chromosome 2q31 conformations are associated with transcriptional regulation of GAD1 GABA synthesis enzyme and altered in prefrontal cortex of subjects with schizophrenia. J Neurosci. 2013;33:11839–11851. doi: 10.1523/JNEUROSCI.1252-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Joo JY, Schaukowitch K, Farbiak L, Kilaru G, Kim TK. Stimulus-specific combinatorial functionality of neuronal c-fos enhancers. Nat Neurosci. 2016;19:75–83. doi: 10.1038/nn.4170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shepherd JD, Bear MF. New views of Arc, a master regulator of synaptic plasticity. Nat Neurosci. 2011;14:279–284. doi: 10.1038/nn.2708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schaukowitch K, et al. Enhancer RNA facilitates NELF release from immediate early genes. Mol Cell. 2014;56:29–42. doi: 10.1016/j.molcel.2014.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Madabhushi R, et al. Activity-Induced DNA Breaks Govern the Expression of Neuronal Early-Response Genes. Cell. 2015;161:1592–1605. doi: 10.1016/j.cell.2015.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim TK, et al. Widespread transcription at neuronal activity-regulated enhancers. Nature. 2010;465:182–187. doi: 10.1038/nature09033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Camps J, Erdos MR, Ried T. The role of lamin B1 for the maintenance of nuclear structure and function. Nucleus. 2015;6:8–14. doi: 10.1080/19491034.2014.1003510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Peric-Hupkes D, et al. Molecular maps of the reorganization of genome-nuclear lamina interactions during differentiation. Mol Cell. 2010;38:603–613. doi: 10.1016/j.molcel.2010.03.016. This elegant paper highlights the dynamics of the chromosomal contacts with the nuclear lamina during the process of astrocyte differentiation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yoon KH, et al. Olfactory receptor genes expressed in distinct lineages are sequestered in different nuclear compartments. Proc Natl Acad Sci U S A. 2015;112:E2403–2409. doi: 10.1073/pnas.1506058112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Monahan K, Lomvardas S. Monoallelic expression of olfactory receptors. Annu Rev Cell Dev Biol. 2015;31:721–740. doi: 10.1146/annurev-cellbio-100814-125308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Boyle MI, Jespersgaard C, Brondum-Nielsen K, Bisgaard AM, Tumer Z. Cornelia de Lange syndrome. Clin Genet. 2015;88:1–12. doi: 10.1111/cge.12499. [DOI] [PubMed] [Google Scholar]
- 35.Yan J, et al. Genomic duplication resulting in increased copy number of genes encoding the sister chromatid cohesion complex conveys clinical consequences distinct from Cornelia de Lange. J Med Genet. 2009;46:626–634. doi: 10.1136/jmg.2008.062471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Finnsson J, Sundblom J, Dahl N, Melberg A, Raininko R. LMNB1-related autosomal-dominant leukodystrophy: Clinical and radiological course. Ann Neurol. 2015;78:412–425. doi: 10.1002/ana.24452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nolen LD, Boyle S, Ansari M, Pritchard E, Bickmore WA. Regional chromatin decompaction in Cornelia de Lange syndrome associated with NIPBL disruption can be uncoupled from cohesin and CTCF. Hum Mol Genet. 2013;22:4180–4193. doi: 10.1093/hmg/ddt265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Watson LA, et al. Dual effect of CTCF loss on neuroprogenitor differentiation and survival. J Neurosci. 2014;34:2860–2870. doi: 10.1523/JNEUROSCI.3769-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gregor A, et al. De novo mutations in the genome organizer CTCF cause intellectual disability. Am J Hum Genet. 2013;93:124–131. doi: 10.1016/j.ajhg.2013.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hirayama T, Tarusawa E, Yoshimura Y, Galjart N, Yagi T. CTCF is required for neural development and stochastic expression of clustered Pcdh genes in neurons. Cell Rep. 2012;2:345–357. doi: 10.1016/j.celrep.2012.06.014. [DOI] [PubMed] [Google Scholar]
- 41.Cai S, Lee CC, Kohwi-Shigematsu T. SATB1 packages densely looped, transcriptionally active chromatin for coordinated expression of cytokine genes. Nat Genet. 2006;38:1278–1288. doi: 10.1038/ng1913. [DOI] [PubMed] [Google Scholar]
- 42.Britanova O, Akopov S, Lukyanov S, Gruss P, Tarabykin V. Novel transcription factor Satb2 interacts with matrix attachment region DNA elements in a tissue-specific manner and demonstrates cell-type-dependent expression in the developing mouse CNS. Eur J Neurosci. 2005;21:658–668. doi: 10.1111/j.1460-9568.2005.03897.x. [DOI] [PubMed] [Google Scholar]
- 43.Docker D, et al. Further delineation of the SATB2 phenotype. Eur J Hum Genet. 2014;22:1034–1039. doi: 10.1038/ejhg.2013.280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Alcamo EA, et al. Satb2 regulates callosal projection neuron identity in the developing cerebral cortex. Neuron. 2008;57:364–377. doi: 10.1016/j.neuron.2007.12.012. [DOI] [PubMed] [Google Scholar]
- 45.Leoyklang P, et al. Heterozygous nonsense mutation SATB2 associated with cleft palate, osteoporosis, and cognitive defects. Hum Mutat. 2007;28:732–738. doi: 10.1002/humu.20515. [DOI] [PubMed] [Google Scholar]
- 46.Close J, et al. Satb1 is an activity-modulated transcription factor required for the terminal differentiation and connectivity of medial ganglionic eminence-derived cortical interneurons. J Neurosci. 2012;32:17690–17705. doi: 10.1523/JNEUROSCI.3583-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Denaxa M, et al. Maturation-promoting activity of SATB1 in MGE-derived cortical interneurons. Cell Rep. 2012;2:1351–1362. doi: 10.1016/j.celrep.2012.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kernohan KD, Vernimmen D, Gloor GB, Berube NG. Analysis of neonatal brain lacking ATRX or MeCP2 reveals changes in nucleosome density, CTCF binding and chromatin looping. Nucleic Acids Res. 2014;42:8356–8368. doi: 10.1093/nar/gku564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Horike S, Cai S, Miyano M, Cheng JF, Kohwi-Shigematsu T. Loss of silent-chromatin looping and impaired imprinting of DLX5 in Rett syndrome. Nat Genet. 2005;37:31–40. doi: 10.1038/ng1491. [DOI] [PubMed] [Google Scholar]
- 50.Schule B, Li HH, Fisch-Kohl C, Purmann C, Francke U. DLX5 and DLX6 expression is biallelic and not modulated by MeCP2 deficiency. Am J Hum Genet. 2007;81:492–506. doi: 10.1086/520063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nan X, et al. Interaction between chromatin proteins MECP2 and ATRX is disrupted by mutations that cause inherited mental retardation. Proc Natl Acad Sci U S A. 2007;104:2709–2714. doi: 10.1073/pnas.0608056104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jakovcevski M, Akbarian S. Epigenetic mechanisms in neurological disease. Nat Med. 2012;18:1194–1204. doi: 10.1038/nm.2828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Turner TN, et al. Genome Sequencing of Autism-Affected Families Reveals Disruption of Putative Noncoding Regulatory DNA. Am J Hum Genet. 2016;98:58–74. doi: 10.1016/j.ajhg.2015.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dekker J, Marti-Renom MA, Mirny LA. Exploring the three-dimensional organization of genomes: interpreting chromatin interaction data. Nat Rev Genet. 2013;14:390–403. doi: 10.1038/nrg3454. An authorative overview on genome-scale chromosome interactions and interpretation of Hi-C data. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bergeron KF, et al. Male-biased aganglionic megacolon in the TashT mouse line due to perturbation of silencer elements in a large gene desert of chromosome 10. PLoS Genet. 2015;11:e1005093. doi: 10.1371/journal.pgen.1005093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.de la Torre-Ubieta L, Won H, Stein JL, Geschwind DH. Advancing the understanding of autism disease mechanisms through genetics. Nat Med. 2016;22:345–361. doi: 10.1038/nm.4071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yao P, et al. Coexpression networks identify brain region-specific enhancer RNAs in the human brain. Nat Neurosci. 2015;18:1168–1174. doi: 10.1038/nn.4063. [DOI] [PubMed] [Google Scholar]
- 58.Gusev A, et al. Partitioning heritability of regulatory and cell-type-specific variants across 11 common diseases. Am J Hum Genet. 2014;95:535–552. doi: 10.1016/j.ajhg.2014.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cross-Disorder Group of the Psychiatric Genomics, C. Identification of risk loci with shared effects on five major psychiatric disorders: a genome-wide analysis. Lancet. 2013;381:1371–1379. doi: 10.1016/S0140-6736(12)62129-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nagy C, et al. Astrocytic abnormalities and global DNA methylation patterns in depression and suicide. Mol Psychiatry. 2015;20:320–328. doi: 10.1038/mp.2014.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jaffe AE, et al. Mapping DNA methylation across development, genotype and schizophrenia in the human frontal cortex. Nat Neurosci. 2016;19:40–47. doi: 10.1038/nn.4181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Roussos P, et al. The Relationship of Common Risk Variants and Polygenic Risk for Schizophrenia to Sensorimotor Gating. Biol Psychiatry. 2015 doi: 10.1016/j.biopsych.2015.06.019. [DOI] [PubMed] [Google Scholar]
- 63.Gjoneska E, et al. Conserved epigenomic signals in mice and humans reveal immune basis of Alzheimer’s disease. Nature. 2015;518:365–369. doi: 10.1038/nature14252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Frost B, Bardai FH, Feany MB. Lamin Dysfunction Mediates Neurodegeneration in Tauopathies. Curr Biol. 2016;26:129–136. doi: 10.1016/j.cub.2015.11.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Frost B, Hemberg M, Lewis J, Feany MB. Tau promotes neurodegeneration through global chromatin relaxation. Nat Neurosci. 2014;17:357–366. doi: 10.1038/nn.3639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Psych EC, et al. The PsychENCODE project. Nat Neurosci. 2015;18:1707–1712. doi: 10.1038/nn.4156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mitchell AC, et al. The genome in three dimensions: a new frontier in human brain research. Biol Psychiatry. 2014;75:961–969. doi: 10.1016/j.biopsych.2013.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Huang HS, et al. Prefrontal dysfunction in schizophrenia involves mixed-lineage leukemia 1-regulated histone methylation at GABAergic gene promoters. J Neurosci. 2007;27:11254–11262. doi: 10.1523/JNEUROSCI.3272-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Rocco BR, Lewis DA, Fish KN. Markedly Lower Glutamic Acid Decarboxylase 67 Protein Levels in a Subset of Boutons in Schizophrenia. Biol Psychiatry. 2015 doi: 10.1016/j.biopsych.2015.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lewis DA. Inhibitory neurons in human cortical circuits: substrate for cognitive dysfunction in schizophrenia. Curr Opin Neurobiol. 2014;26:22–26. doi: 10.1016/j.conb.2013.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Shulha HP, et al. Human-specific histone methylation signatures at transcription start sites in prefrontal neurons. PLoS Biol. 2012;10:e1001427. doi: 10.1371/journal.pbio.1001427. An early paper combining chromosome conformation capture assays with cell-type specific epigenomic profiling in human and non-human primate brain. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Talkowski ME, et al. Sequencing chromosomal abnormalities reveals neurodevelopmental loci that confer risk across diagnostic boundaries. Cell. 2012;149:525–537. doi: 10.1016/j.cell.2012.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Epi KC, et al. De novo mutations in epileptic encephalopathies. Nature. 2013;501:217–221. doi: 10.1038/nature12439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hamdan FF, et al. De novo mutations in moderate or severe intellectual disability. PLoS Genet. 2014;10:e1004772. doi: 10.1371/journal.pgen.1004772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.O’Roak BJ, et al. Exome sequencing in sporadic autism spectrum disorders identifies severe de novo mutations. Nat Genet. 2011;43:585–589. doi: 10.1038/ng.835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Jiang Y, et al. Setdb1 histone methyltransferase regulates mood-related behaviors and expression of the NMDA receptor subunit NR2B. J Neurosci. 2010;30:7152–7167. doi: 10.1523/JNEUROSCI.1314-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Taubenfeld SM, Milekic MH, Monti B, Alberini CM. The consolidation of new but not reactivated memory requires hippocampal C/EBPbeta. Nat Neurosci. 2001;4:813–818. doi: 10.1038/90520. [DOI] [PubMed] [Google Scholar]
- 78.Taubenfeld SM, et al. Fornix-dependent induction of hippocampal CCAAT enhancer-binding protein [beta] and [delta] Co-localizes with phosphorylated cAMP response element-binding protein and accompanies long-term memory consolidation. J Neurosci. 2001;21:84–91. doi: 10.1523/JNEUROSCI.21-01-00084.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Merhav M, et al. Behavioral interference and C/EBPbeta expression in the insular-cortex reveal a prolonged time period for taste memory consolidation. Learn Mem. 2006;13:571–574. doi: 10.1101/lm.282406. [DOI] [PubMed] [Google Scholar]
- 80.Dickel DE, Visel A, Pennacchio LA. Functional anatomy of distant-acting mammalian enhancers. Philos Trans R Soc Lond B Biol Sci. 2013;368:20120359. doi: 10.1098/rstb.2012.0359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Stampfel G, et al. Transcriptional regulators form diverse groups with context-dependent regulatory functions. Nature. 2015;528:147–151. doi: 10.1038/nature15545. [DOI] [PubMed] [Google Scholar]
- 82.Valor LM, Viosca J, Lopez-Atalaya JP, Barco A. Lysine acetyltransferases CBP and p300 as therapeutic targets in cognitive and neurodegenerative disorders. Curr Pharm Des. 2013;19:5051–5064. doi: 10.2174/13816128113199990382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Tsai LH, Graff J. On the resilience of remote traumatic memories against exposure therapy-mediated attenuation. EMBO Rep. 2014;15:853–861. doi: 10.15252/embr.201438913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Schroeder FA, Lin CL, Crusio WE, Akbarian S. Antidepressant-like effects of the histone deacetylase inhibitor, sodium butyrate, in the mouse. Biol Psychiatry. 2007;62:55–64. doi: 10.1016/j.biopsych.2006.06.036. [DOI] [PubMed] [Google Scholar]
- 85.Covington HE, 3rd, Maze I, Vialou V, Nestler EJ. Antidepressant action of HDAC inhibition in the prefrontal cortex. Neuroscience. 2015;298:329–335. doi: 10.1016/j.neuroscience.2015.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Maze I, et al. Essential role of the histone methyltransferase G9a in cocaine-induced plasticity. Science. 2010;327:213–216. doi: 10.1126/science.1179438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Huang HS, et al. Topoisomerase inhibitors unsilence the dormant allele of Ube3a in neurons. Nature. 2012;481:185–189. doi: 10.1038/nature10726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Day JJ, Kennedy AJ, Sweatt JD. DNA methylation and its implications and accessibility for neuropsychiatric therapeutics. Annu Rev Pharmacol Toxicol. 2015;55:591–611. doi: 10.1146/annurev-pharmtox-010814-124527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Halder R, et al. DNA methylation changes in plasticity genes accompany the formation and maintenance of memory. Nat Neurosci. 2016;19:102–110. doi: 10.1038/nn.4194. [DOI] [PubMed] [Google Scholar]
- 90.Zink D, et al. Transcription-dependent spatial arrangements of CFTR and adjacent genes in human cell nuclei. J Cell Biol. 2004;166:815–825. doi: 10.1083/jcb.200404107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Pickersgill H, et al. Characterization of the Drosophila melanogaster genome at the nuclear lamina. Nat Genet. 2006;38:1005–1014. doi: 10.1038/ng1852. [DOI] [PubMed] [Google Scholar]
- 92.Krivega I, et al. Inhibition of G9a methyltransferase stimulates fetal hemoglobin production by facilitating LCR/gamma-globin looping. Blood. 2015;126:665–672. doi: 10.1182/blood-2015-02-629972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Hasan A, Mitchell A, Schneider A, Halene T, Akbarian S. Epigenetic dysregulation in schizophrenia: molecular and clinical aspects of histone deacetylase inhibitors. Eur Arch Psychiatry Clin Neurosci. 2013;263:273–284. doi: 10.1007/s00406-013-0395-2. [DOI] [PubMed] [Google Scholar]
- 94.Laganiere J, et al. An engineered zinc finger protein activator of the endogenous glial cell line-derived neurotrophic factor gene provides functional neuroprotection in a rat model of Parkinson’s disease. J Neurosci. 2010;30:16469–16474. doi: 10.1523/JNEUROSCI.2440-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Heller EA, et al. Locus-specific epigenetic remodeling controls addiction- and depression-related behaviors. Nat Neurosci. 2014;17:1720–1727. doi: 10.1038/nn.3871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Hilton IB, et al. Epigenome editing by a CRISPR-Cas9-based acetyltransferase activates genes from promoters and enhancers. Nat Biotechnol. 2015;33:510–517. doi: 10.1038/nbt.3199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Cho SW, Kim S, Kim JM, Kim JS. Targeted genome engineering in human cells with the Cas9 RNA-guided endonuclease. Nat Biotechnol. 2013;31:230–232. doi: 10.1038/nbt.2507. [DOI] [PubMed] [Google Scholar]
- 98.Cong L, et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Tai DJ, et al. Engineering microdeletions and microduplications by targeting segmental duplications with CRISPR. Nat Neurosci. 2016 doi: 10.1038/nn.4235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Jiang W, Bikard D, Cox D, Zhang F, Marraffini LA. RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nat Biotechnol. 2013;31:233–239. doi: 10.1038/nbt.2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Maeder ML, et al. CRISPR RNA-guided activation of endogenous human genes. Nat Methods. 2013;10:977–979. doi: 10.1038/nmeth.2598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Perez-Pinera P, et al. RNA-guided gene activation by CRISPR-Cas9-based transcription factors. Nat Methods. 2013;10:973–976. doi: 10.1038/nmeth.2600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Cheng AW, et al. Multiplexed activation of endogenous genes by CRISPR-on, an RNA-guided transcriptional activator system. Cell research. 2013;23:1163–1171. doi: 10.1038/cr.2013.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Larson MH, et al. CRISPR interference (CRISPRi) for sequence-specific control of gene expression. Nat Protoc. 2013;8:2180–2196. doi: 10.1038/nprot.2013.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Hsu PD, Lander ES, Zhang F. Development and applications of CRISPR-Cas9 for genome engineering. Cell. 2014;157:1262–1278. doi: 10.1016/j.cell.2014.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Li M, Suzuki K, Kim NY, Liu GH, Izpisua Belmonte JC. A cut above the rest: targeted genome editing technologies in human pluripotent stem cells. J Biol Chem. 2014;289:4594–4599. doi: 10.1074/jbc.R113.488247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Hotta A, Yamanaka S. From Genomics to Gene Therapy: Induced Pluripotent Stem Cells Meet Genome Editing. Annu Rev Genet. 2015;49:47–70. doi: 10.1146/annurev-genet-112414-054926. [DOI] [PubMed] [Google Scholar]
- 108.Ding Q, et al. A TALEN genome-editing system for generating human stem cell-based disease models. Cell Stem Cell. 2013;12:238–251. doi: 10.1016/j.stem.2012.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Martinez RA, et al. Genome engineering of isogenic human ES cells to model autism disorders. Nucleic Acids Res. 2015;43:e65. doi: 10.1093/nar/gkv164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Wen Z, et al. Synaptic dysregulation in a human iPS cell model of mental disorders. Nature. 2014 doi: 10.1038/nature13716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Chen H, et al. Modeling ALS with iPSCs reveals that mutant SOD1 misregulates neurofilament balance in motor neurons. Cell Stem Cell. 2014;14:796–809. doi: 10.1016/j.stem.2014.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Liu GH, et al. Progressive degeneration of human neural stem cells caused by pathogenic LRRK2. Nature. 2012;491:603–607. doi: 10.1038/nature11557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Soldner F, et al. Parkinson-associated risk variant in distal enhancer of alpha-synuclein modulates target gene expression. Nature. 2016 doi: 10.1038/nature17939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Ochiai H, Sugawara T, Yamamoto T. Simultaneous live imaging of the transcription and nuclear position of specific genes. Nucleic Acids Res. 2015;43:e127. doi: 10.1093/nar/gkv624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Fudenberg G, Mirny LA. Higher-order chromatin structure: bridging physics and biology. Curr Opin Genet Dev. 2012;22:115–124. doi: 10.1016/j.gde.2012.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Rippe K. Making contacts on a nucleic acid polymer. Trends Biochem Sci. 2001;26:733–740. doi: 10.1016/s0968-0004(01)01978-8. [DOI] [PubMed] [Google Scholar]
- 117.Williamson I, et al. Spatial genome organization: contrasting views from chromosome conformation capture and fluorescence in situ hybridization. Genes Dev. 2014;28:2778–2791. doi: 10.1101/gad.251694.114. This paper provides a scholarly and competent discussion on the advantages and limitations of commonly used techniques to map chromosomal contacts. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Yang J, Corces VG. Chromatin Insulators: A Role in Nuclear Organization and Gene Expression. Advances in cancer research. 2011;110:43–76. doi: 10.1016/b978-0-12-386469-7.00003-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Phillips-Cremins JE, et al. Architectural protein subclasses shape 3D organization of genomes during lineage commitment. Cell. 2013;153:1281–1295. doi: 10.1016/j.cell.2013.04.053. [DOI] [PMC free article] [PubMed] [Google Scholar]