

Abstract
Objective
To describe a novel macular phenotype that is associated with normal visual function.
Design
Retrospective observational case series.
Participants
36 affected individuals from 23 unrelated families.
Methods
This was a retrospective study of patients who had a characteristic macular phenotype. Subjects underwent a full ocular examination, electrophysiological studies, spectral domain optical coherence tomography (OCT) and fundus autofluorescence imaging. Genomic analyses were performed using haplotype sharing analysis and whole-exome sequencing.
Main Outcome Measures
Visual acuity; Retinal features; Electroretinography; Whole-exome sequencing; Haplotype sharing analysis.
Results
Twenty-six of 36 subjects were female. The median age at presentation was 15 years, range 5-59 years. The majority of subjects were asymptomatic and either presented following a routine eye examination (22/36 subjects), following screening due to a positive family history (13/36 subjects) or from another ophthalmologist (1/36 subjects). Of the three symptomatic subjects, 2 had reduced visual acuity. Reduced vision was attributed to diagnoses of non-organic visual loss and bilateral ametropic amblyopia with strabismus. Visual acuity was 0.18 LogMAR or better in 30/33 subjects. Color vision was normal in all subjects tested, except for the subject with non-organic visual loss.
All subjects had bilateral symmetric multiple yellow dots at the macula. In the majority these were evenly distributed throughout the fovea, but in nine subjects they were concentrated in the nasal parafoveal area. The dots were hyperautofluorescent on fundus autofluorescence imaging. OCT imaging was generally normal, but in 6 subjects subtle irregularities at the inner segment ellipsoid band were seen. Electrophysiological studies identified normal macular function in 17/19 subjects and normal full-field retinal function in all subjects. Whole-exome analysis across 3 unrelated families found no pathogenic variants in known macular dystrophy genes. Haplotype sharing analysis in one family excluded linkage with the North Carolina macular dystrophy (MCDR1) locus.
Conclusions
A new retinal phenotype is described, which is characterised by bilateral multiple early onset yellow dots at the macula. Visual function is normal and the condition is non-progressive. In familial cases, the phenotype appears to be inherited in an autosomal dominant manner, but a causative gene is yet to be ascertained.
Introduction
The inherited macular dystrophies are a clinically and genetically heterogeneous group of disorders in which there are structural and functional abnormalities of the central retina [1], [2]. These disorders usually occur in isolation, but they may be associated with a variety of systemic abnormalities. All of the Mendelian and mitochondrial inheritance patterns have been described [3]. Most forms of macular dystrophy present in later childhood or in adult life after a period of normal visual development, and are usually progressive. The exception is a rare group of disorders that present with visual impairment in infancy and where there is abnormal foveal or macular development [4]. Such disorders do not commonly progress.
Although most macular dystrophies present with central visual loss, some patients with normal visual acuity are referred to ophthalmologists when a macular abnormality is noted on routine optometric examination. Whatever the mode of presentation, the specific diagnosis is made on the basis of the macular appearance, along with retinal imaging, electrophysiological studies, inheritance patterns and, increasingly, the results of molecular genetic testing [5]. Some clinical phenotypes do not easily fit into well-characterised disorders.
The present report describes a novel macular phenotype that may occur in isolation, or as a familial trait, which is non-progressive, and which is associated with normal visual function.
Methods
Subjects
Subjects were ascertained based upon the presence of a specific macular phenotype, and were recruited from the pediatric and adult medical retina clinics of three ophthalmologists (one UK, two USA). Informed consent was obtained from all subjects and family members involved in this study. The study had IRB approval from Cincinnati Children's Hospital, Bascom Palmer Eye Hospital and the Moorfields Eye Hospital Local Research Ethics Committee, and all investigations were conducted in accordance with the principles of the Declaration of Helsinki.
Clinical Examination
Best-corrected monocular visual acuity (VA) was measured using a logMAR scale and color vision was assessed using Ishihara pseudoisochromatic plates, Hardy Rand Rittler (HRR) color plates and the Farnsworth-Munsell 100 Hue Test. Funduscopy and slit-lamp biomicroscopy were performed. Color fundus photography was undertaken in all subjects; in the majority this was carried out using a Topcon TRC 501A retinal camera (Topcon Corporation, Tokyo, Japan), but in some individuals, seen early in the study period, a Zeiss retinal film camera was used. Spectral domain optical coherence tomography (sd-OCT) using a Heidelberg SPECTRALIS® Spectral domain OCT scanner (Heidelberg Engineering, Dossenheim, Germany) and fundus autofluorescence (FAF) imaging (Heidelberg Engineering, Dossenheim, Germany) were also performed. Electrophysiological assessment including full-field electroretinography (ERG), pattern electroretinography (PERG) and Electro-Oculograms (EOG) were performed in the subjects from the UK according to the recommendations of the International Society for Clinical Electrophysiology of Vision (ISCEV) [6]-[7, 8]. Fundus fluorescein angiography was also undertaken in selected subjects.
Genomic analyses
DNA was extracted from whole blood by standard methods. Whole-exome sequencing (WES) was performed for Family 4 (Subjects 8, 9, 11), Moorfields Eye Hospital Genetic Clinic (GC) number GC14302, Family 8 (Subjects 19, 20, 21) and Family 22 (Subject 35), as previously described [9]. Briefly, dsDNA was sheared by sonication to an average size of 200 bp. After nine cycles of PCR amplification using the Clontech Advantage II kit, 1 μg of genomic library was recovered for exome enrichment using the NimbleGen EZ Exome V2 kit. Libraries were sequenced on an Illumina HiSeq2500. Data analysis used the Broad Institute's Genome Analysis Toolkit (GATK) [10]. Reads were aligned with the Illumina Chastity Filter with the Burrows Wheeler Aligner [11]. Variant sites were called using the GATK UnifiedGenotyper module [10]. Variant filtering and group analysis was performed using Qiagen Ingenuity Variant Analysis.
Haplotype sharing analysis was performed on SNP data from 5 affected members of Family 4 genotyped using the Illumina HumanOmniExpress-24 v1.0 beadchip (Illumina, Inc., San Diego, CA, USA) that includes over 715,000 SNPs. Genotypes were determined using the Genotyping Module in the Illumina GenomeStudio v2011.1 software. Build hg19/GRCh37 was used to annotate chromosomal coordinates. The haplotype sharing analysis was carried out using the non-parametric Homozygosity Haplotype (HH) method that searches for chromosomal segments sharing the same haplotype across affected individuals (as an indication of genetic linkage with the disease) [12]. The HH is a type of haplotype described by the homozygous SNPs only (all heterozygous SNPs are removed). Since affected family members who inherited the same mutation from a common ancestor share a chromosomal segment identical-by-descent (IBD) around the disease gene, they should not have discordant homozygous calls in the IBD region and thus they should share the same HH. The HH approach predicts IBD regions through the identification of regions with a conserved HH (RCHHs) defined as those regions with a shared HH among patients and a genetic length longer than a certain cut-off value (recommended cut-off for Illumina array is 2.5/3.0 cM).
Results
Thirty-six affected individuals were identified from 23 unrelated families. Subjects were either referred from community optometrists (22/36), from another ophthalmologist (1/36) or following screening due to a positive family history (13/36). Of the 36 subjects, 15 were sporadic. Twelve of the 15 sporadic cases were Caucasian, 1 was of West African origin, 1 of South Asian descent, and 1 of African-Caribbean descent. Eight families (all Caucasian) demonstrated an autosomal dominant inheritance pattern (Figure 1, pedigree of Family 4). The median age at presentation was 15 years, range 5-59 years. Twenty-six of 36 subjects were female (Table 1).
Figure 1.
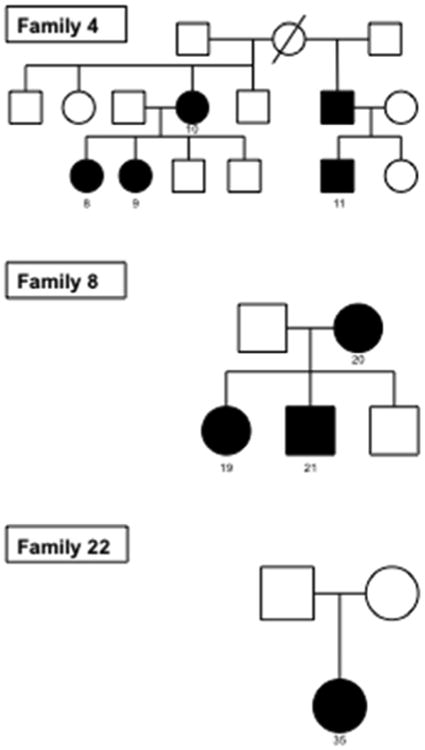
Pedigrees of Familes 4, 8 and 22 that underwent molecular analysis. Family 4 had 5 affected individuals in two successive generations; Family 8 had 2 affected individuals in two successive generations; Family 22 had only one affected individual. Black circles / squares denote affected individuals
Table 1. Clinical features of Benign Yellow Dot Maculopathy Subjects.
| Subject no, Family no (GC number) | Proband? | Presenting age (years) | Gender | Mode of Presentation | Family history | BCVA LogMAR RE, LE (Age, years) | Past Ocular History |
|---|---|---|---|---|---|---|---|
|
| |||||||
| 1, 1 | Proband | 5 | F | Community | Dominant | 0, 0 (5) | Nil |
| 2, 1 | Mother | 28 | F | FH | Dominant | 0, -0.08 (28) | Nil |
| 3, 1 | Maternal G.mother | 50 | F | FH | Dominant | -0.08, -0.08 (50) | Nil |
| 4, 2 | Proband | 14 | M | Community | Dominant | -0.08, 0 (14) | Hypermet, ET, strab surgery |
| 5, 2 | Mother | 43 | F | FH | Dominant | 0.18, 0.18 (43) | Refractive error |
| 6, 3 | Proband | 45 | F | Community | Dominant | -0.08, -0.08 (45) | Nil |
| 7, 3 | Son | 6 | M | FH | Dominant | -0.08, -0.08 (6) | Nil |
| 8, 4 (GC14302) | Proband | 15 | F | Community | Dominant | 0.78, 0.78 (15) | Non organic visual loss |
| 9, 4 (GC14302) | Sister | 18 | F | Community | Dominant | 0, 0, (18) | Nil |
| 10, 4 (GC14302) | Mother | 45 | F | FH | Dominant | 0.18, 0.18 (45) | Refractive error |
| 11, 4 (GC14302) | Maternal 1st cousin | 10 | M | FH | Dominant | -0.08, -0.08 (10) | Nil |
| 12, 5 (GC16206) | Proband | 22 | F | Community | Dominant | -0.08, -0.08 (22) | Refractive error |
| 13, 5 (GC16206) | Mother | Unr | F | FH | Dominant | 0, 0, (Unr) | Nil |
| 14, 6 | Proband | 7 | F | Community | Dominant | 0, 0 (7) | Nil |
| 15, 6 | Father | Unr | M | FH | Dominant | Unr, Unr (Unr) | Nil |
| 16, 6 | Paternal G.mother | 59 | F | FH | Dominant | Unr, Unr (Unr) | Nil |
| 17, 7 | Proband | 11 | F | Community | Dominant | 0, 0 (11) | Refractive error |
| 18, 7 | Father | Unr | M | FH | Dominant | Unr, Unr (Unr) | Nil |
| 19, 8 | Proband | 7 | F | Community | Dominant | 0, 0 (7) | Refractive error |
| 20, 8 | Mother | 45 | F | FH | Dominant | 0, 0 (45) | Nil |
| 21, 8 | Brother | 13 | M | FH | Dominant | 0, 0 (13) | Nil |
| 22, 9 | Proband | 8 | F | Community | Sporadic | 0, 0 (8) | Nil |
| 23, 10 (GC17650) | Proband | 6 | M | Another Ophthalmologist | Sporadic | 0.7, 0.8 (6) | Hypermet, ET, bilat Ametropic amblyopia |
| 24, 11 (GC19048) | Proband | 10 | F | Community | Sporadic | 0, 0 (10) | Refractive error |
| 25, 12 (GC19084) | Proband | 5 | F | Community | Sporadic | 0, 0 (10) | Bilateral optic neuropathy |
| 26, 13 | Proband | 10 | F | Community | Sporadic | 0, 0 (10) | Nil |
| 27, 14 | Proband | 12 | F | Community | Sporadic | 0.4, 0 (14) | Hypermet, Anisometropic amblyopia |
| 28, 15 | Proband | 14 | M | Community | Sporadic | 0.08, 0.14 (14) | Hypermet, Ametropic amblyopia |
| 29, 16 (GC17091) | Proband | 15 | F | Community | Sporadic | 0, 0 (15) | Refractive error |
| 30, 17 | Proband | 15 | M | Community | Sporadic | 0, 0 (15) | Nil |
| 31, 18 (GC18107) | Proband | 28 | M | Community | Sporadic | 0, 0 (29) | Nil |
| 32, 19 | Proband | 28 | F | Community | Sporadic | -0.08, -0.08 (29) | Refractive error |
| 33, 20 | Proband | 29 | F | Community | Sporadic | 0, 0 (29) | Refractive error |
| 34, 21 | Proband | 8 | F | Community | Sporadic | 0, 0 (8) | Refractive error |
| 35, 22 | Proband | 16 | F | Community | Sporadic | 0, 0 (16) | Nil |
| 36, 23 | Proband | 5.5 | F | Community | Sporadic | 0, 0 (5.5) | Refractive error |
Key: GC number – Genetics Clinic number (Subjects recruited from Moorfields Eye Hospital, UK); BCVA – Best Corrected Visual Acuity; RE – Right Eye; LE – Left Eye; F – Female; FH – Family History; G.mother – Grandmother; M – Male; Hypermet – Hypermetropia; ET – Esotropia; Strab – Strabismus; Unr – Unrecorded; Bilat – Bilateral
Thirty-three subjects (91.7%) were asymptomatic. One subject complained of floaters (Subject 35) but had normal visual acuities and a normal peripheral retinal examination. Reduced visual acuity was the presenting complaint in the other two symptomatic subjects (Subjects 8 and 23). No cause was found for the reduced vision in Subject 8, who presented at age 16 years, and in whom multiple electrophysiological studies over a number of years were normal. A diagnosis of ‘non-organic’ visual loss was made. Subject 23 presented to another ophthalmologist at age 4 years with reduced vision, attributed to bilateral ametropic amblyopia due to hypermetropic astigmatism; macular yellow dots were not identified until age 6 years. His vision eventually improved with refractive correction and occlusion therapy to 0.18 OD and 0.26 OS.
Refractive error (identified in 15 subjects) was the predominant finding in the 17 subjects who had any past ocular history (Table 1). Two of 17 had been treated for strabismus and 3/17 had been treated for amblyopia. One subject developed bilateral optic neuropathy of unknown etiology, during the follow-up period, four and a half years after presentation with the macular phenotype, which resolved spontaneously; and one had non-organic visual loss. General health was good in all, except for Subject 33, who was taking antidepressants.
Visual acuity (VA) at presentation was 0.18 LogMAR or better in both eyes in 30/33 subjects (Table 1). In 3 subjects the VA was unrecorded (these were all affected family members of probands). Subject 8, with non-organic visual loss, had a presenting VA of 0.78 LogMAR in either eye. Amblyopia affected Subjects 23 (bilateral), 27 (unilateral right eye) and 28 (unilateral left eye) (Table 1). Successful amblyopia therapy improved the acuity in Subject 28 to better than 0.18 LogMAR OU. The finding of amblyopia and refractive error in a subset of patients likely reflects the fact that the majority of subjects were ascertained in ophthalmology or optometry clinics. Color vision was normal in 23 of 24 subjects examined, using a variety of color vision tests. Only one subject had mild colour vision abnormalities (Subject 8). She failed two of the HRR screening plates at age 16 years. Anterior segments and ocular mediae were normal in all subjects.
Funduscopy in all subjects revealed characteristic bilateral macular changes consisting of yellow dots at the level of the retinal pigment epithelium (RPE), concentrated around the fovea. These were symmetrical between each eye in all subjects (Figure 2). In the majority of subjects the dots were fine and discrete but in 9 subjects some of the dots were confluent (Table 2). The yellow dots were distributed evenly around the fovea in 13 subjects; in 10 they were concentrated in the nasal parafoveal region. In Subjects 22 and 24 a few additional dots were visible outside the temporal vascular arcades in the right eye. In all but 1 subject (Subject 23) where detailed images were available (26/36), a yellow crescent was visible to varying degrees around the optic disc, which was otherwise normal in all (Table 2, Figure 2). The retinal periphery and vasculature were otherwise normal in all subjects. Funduscopy was normal in all parents of the sporadic cases that were examined.
Figure 2.
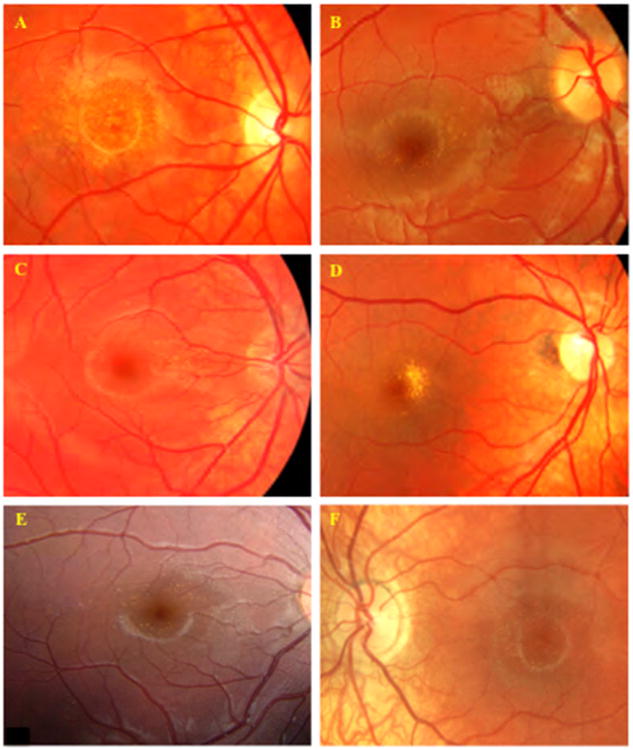
Fundus images of Benign Yellow Dot Maculopathy Subjects. A – Familial benign yellow dot maculopathy, Subject 11; B – Sporadic benign yellow dot maculopathy, fine, discrete, dots, Subject 24; C – Sporadic benign yellow dot maculopathy, concentrated in the nasal parafoveal region, Subject 26; D – Sporadic benign yellow dot maculopathy, confluent, Subject 28; E - Sporadic benign yellow dot maculopathy, Subject 34; F – Familial benign yellow dot maculopathy, Subject 19
Table 2. Fundus features in Benign Yellow Dot Maculopathy.
| Subject no, Family no | Macula | Optic disc | |
|---|---|---|---|
| 1, 1 | Discrete, Fine, Yellow dots | Concentrated in temporal parafovea | Fine yellow crescent around temporal border of disc |
| 2, 1 | Discrete, Fine, Yellow dots | ||
| 3, 1 | Discrete, Fine, Yellow dots | ||
| 4, 2 | Discrete, Fine, Yellow dots | Concentrated in nasal parafovea | |
| 5, 2 | Discrete, Fine, Yellow dots | ||
| 6, 3 | Discrete, Fine, Yellow dots | ||
| 7, 3 | Discrete, Fine, Yellow dots | ||
| 8, 4 | Discrete, Yellow dots, Variable sizes, Some confluent | Even distribution | Yellow crescent around nasal border of disc between 12-6 o'clock |
| 9, 4 | Discrete, Fine, Yellow dots | Even distribution | Yellow crescent around disc 360 degrees |
| 10, 4 | Discrete, Yellow dots, Larger | Even distribution | Yellow crescent around temporal border of disc between 12-6 o'clock |
| 11, 4 | Discrete, Yellow dots, Variable sizes, Some confluent | Even distribution | Yellow crescent around disc 360 degrees |
| 12, 5 | Discrete, Fine, Yellow dots | Concentrated in nasal parafovea | Yellow crescent around disc temporally |
| 13, 5 | Discrete, Yellow dots, Variable sizes, Some confluent | Concentrated in nasal parafovea | Yellow crescent around disc 360 degrees |
| 14, 6 | Discrete, Yellow dots, Variable sizes, Some confluent | Concentrated in superior parafovea | Yellow crescent around disc temporally - left eye |
| 15, 6 | Discrete, Fine, Yellow dots | Concentrated in superior parafovea | Yellow crescent around nasal border of disc between 12-6 o'clock |
| 16, 6 | Discrete, Fine, Yellow dots, Some confluent | Concentrated in superior parafovea | Peri-papillary atrophy |
| 17, 7 | Discrete, Fine, Yellow dots, Some confluent | Even distribution | Halo around disc resembling peri-papillary atrophy |
| 18, 7 | Discrete, Fine, Yellow dots | Concentrated in nasal parafovea | Yellow crescent around disc 360 degrees |
| 19, 8 | Discrete, Fine, Yellow dots | Concentrated in nasal parafovea | Yellow crescent around disc 360 degrees |
| 20, 8 | Discrete, Fine, Yellow dots | Concentrated in nasal parafovea | Not visible |
| 21, 8 | Discrete, Fine, Yellow dots | Concentrated in nasal parafovea | Not visible |
| 22, 9 | Discrete, Yellow dots, Variable sizes, Some confluent | Even distribution, some extra-macula in RE | Yellow crescent around disc 360 degrees |
| 23, 10 | Discrete, Fine, Yellow dots | Even distribution | No crescent |
| 24, 11 | Discrete, Fine, Yellow dots | Even distribution, some extra-macula in RE | Yellow crescent around disc 360 degrees |
| 25, 12 | Discrete, Fine, Yellow dots | Even distribution | Yellow crescent around disc 360 degrees |
| 26, 13 | Discrete, Fine, Yellow dots | Concentrated in nasal parafovea | Yellow crescent around disc temporally; full discs - no swelling |
| 27, 14 | Discrete, Fine, Yellow dots | Concentrated in nasal parafovea | Yellow crescent around disc 360 degrees |
| 28, 15 | Discrete, Yellow dots, Many confluent | Concentrated in nasal parafovea | Halo around disc resembling peri-papillary atrophy |
| 29, 16 | Discrete, Fine, Yellow dots | Even distribution | Halo around disc, temporally |
| 30, 17 | Discrete, Fine, Yellow dots | Concentrated in nasal parafovea | Peri-papillary atrophy temporal |
| 31, 18 | Discrete, Yellow dots, Variable sizes, Some confluent | Concentrated in nasal parafovea | Yellow crescent around disc, temporally |
| 32, 19 | Discrete, Fine, Yellow dots | Even distribution | Yellow crescent around disc 360 degrees |
| 33, 20 | Discrete, Fine, Yellow dots | Even distribution | Peri-papillary atrophy |
| 34, 21 | Discrete, Fine, Yellow dots | Even distribution | Yellow crescent around disc 360 degrees |
| 35, 22 | Discrete, Fine, Yellow dots | Concentrated in nasal parafovea | Not visible |
| 36, 23 | Discrete, Fine, Yellow dots | Concentrated in temporal parafovea RE | Not visible |
Key: RE – Right eye
Fundus autofluorescence imaging, available for 22/36 subjects, revealed foci of hyperautofluorescence corresponding to the yellow dots on otherwise normal background autofluorescence (Figure 3). The autofluorescence imaging did not reveal any lesions other than those visible on funduscopy.
Figure 3.
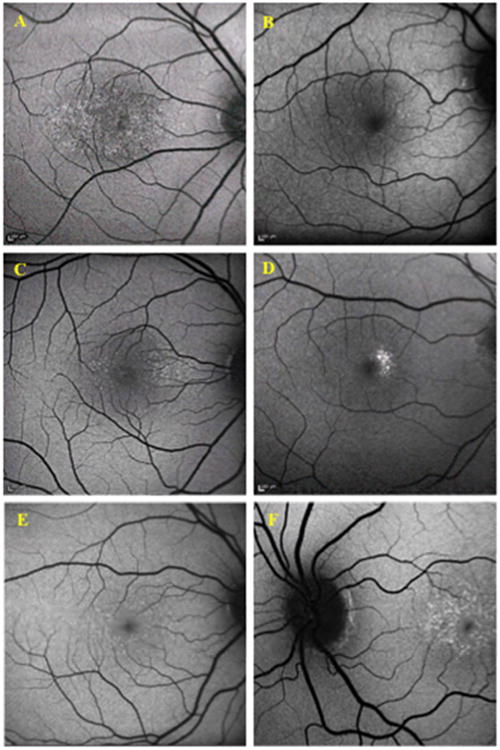
Fundus autofluorescence of Benign Yellow Dot Maculopathy Subjects. The yellow dots show hyperautofluorescence on fundus autofluorescence imaging. A –Subject 11; B –Subject 24; C –Subject 26; D – Subject 28; E – Subject 34; F – Subject 19
Sd-OCT imaging was performed in 18 subjects and was normal in 11. There was minimal irregularity of the inner segment ellipsoid (ISe) band in 6 subjects (Subjects 11, 14, 16, 17, 23, 26), corresponding to the locations of the yellow dots, as observed on the infrared reflectance image (Figure 4). In one additional subject the only change seen on OCT imaging was a mild irregularity of the RPE layer (Subject 28).
Figure 4.
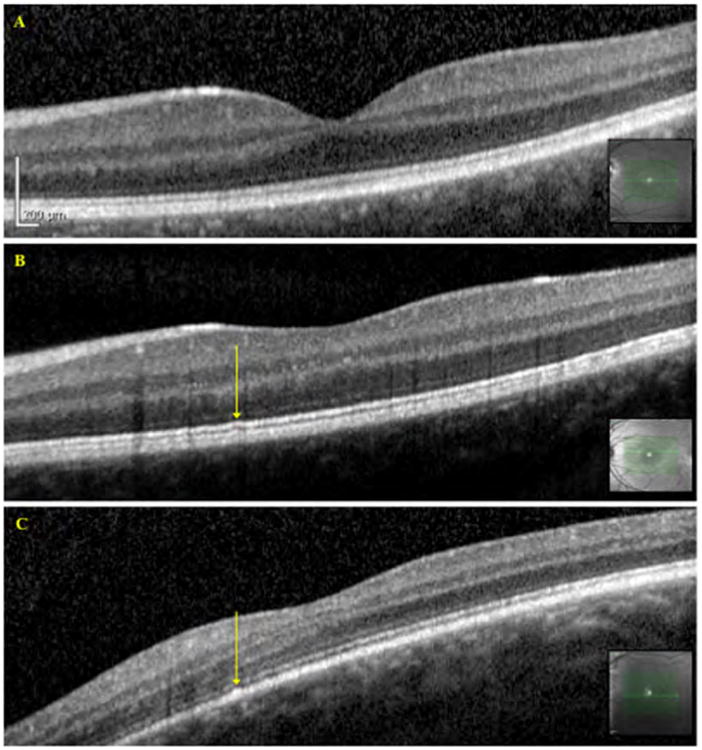
OCT images of Benign Yellow Dot Maculopathy Subjects. A – Normal OCT, Subject 24; B – Slight irregularity of the inner segment ellipsoid band, indicated by the arrow, Subject 26; C – Slight irregularity of the RPE layer, indicated by the arrow, Subject 11. Arrows correspond to the location of the dots as identified from the infrared reflectance images obtained during fundus autofluorescence image acquisition
Electroretinography was performed in 19 subjects. The ERG was normal in all 19 subjects, and the PERG was abnormal in 2. There was a mildly subnormal PERG P50 component amplitude in subject 4. In Subject 28 the PERG was moderately subnormal in each eye, indicative of moderate macular dysfunction. EOGs in 9 of 10 subjects demonstrated a normal light rise. In Subject 28 the EOG was of poor technical quality, which precluded accurate quantification of the light rise.
Fundus fluorescein angiography performed in 3 subjects (Subjects 2, 14 and 17) demonstrated early hyperfluorescence of the dots with no change in size or intensity over time.
WES was performed in affected members of Family 4, Family 8 and Family 22. After filtering for retinal dystrophy-associated genes and genes within the MCDR1 locus on chromosome 6 and MCDR3 locus on chromosome 5, no rare variants were found to consistently segregate with the macular phenotype in any of the known retinal disease genes. Additionally, gene-level analysis did not detect exonic or splice site variants in the same gene for two or more families. Therefore, non-coding causes were predicted as a common mechanism, as has been reported for MCDR1 and MCDR3 [13, 14]. Genome-wide analysis was performed for family 4 using SNP chips. A search for a shared IBD chromosomal segment among the five affected individuals with SNP chip data in family 4 (GC14302) was performed using the HH method. The haplotype sharing analysis revealed no evidence of linkage at the MCDR1 locus (Supplementary Table 1 and Supplementary Figure 1). There were 10 regions (on chromosome 2, 3, 5, 6, 8, 11, 17, 18) with a conserved HH, including a shared segment from marker rs7703994 to marker rs879143 (GRCh37/hg19 chr5:3339142-10620274) that overlaps partially with the MCDR3 locus. No rare exonic variants were detected within this interval.
Discussion
This report describes a novel phenotype consisting of characteristic yellow dots at the macula, which are first evident in childhood and are usually associated with good visual function. The condition is commonly sporadic, although it may also segregate in an autosomal dominant manner.
Most of the affected individuals were female. The macular abnormalities were identified as an incidental finding on routine funduscopy in the majority of subjects, or were discovered during examination of the family members of affected individuals. The condition appears to be non-progressive and, in familial cases, the macular appearance was similar in children and older adults, suggesting that the disorder is stationary. The phenotype is associated with normal visual acuity and normal color vision in the majority of affected individuals. The full-field ERG showed no evidence of generalised retinal dysfunction in any subject, although in one subject the PERG indicated moderate macular dysfunction and in another, very mild macular dysfunction. Overall the normal visual acuity, color vision and retinal electrophysiology are consistent with normal macular function, although it is possible that more detailed psychophysical testing, for example microperimetry, may have revealed subtle loss of retinal sensitivity. OCT of the central retina showed normal retinal thickness and, in the majority, a normal ISe band, in keeping with the good visual acuity. In a small minority there were subtle irregularities at the ISe band outside the fovea.
Many different inherited disorders are associated with ‘deposits’ at the macula. However, they may be distinguished from the phenotype described here by the age of onset, associated visual loss, disease progression, retinal electrophysiology and results of OCT imaging. A similar non-progressive retinal phenotype with drusenoid deposits that are present from childhood can be seen in North Carolina Macular Dystrophy (NCMD), a dominantly inherited macular dystrophy which has been mapped to chromosome 6q16 (MCDR1 locus) [15]. The ERG and EOG are also normal in NCMD, with dysfunction being confined to the macula. Recently, Small et al. identified rare variants upstream of the retinal transcription factor gene PRDM13 in families with NCMD that link to the MCDR1 locus [14]. An identical phenotype has been mapped to chromosome 5p15 (MCDR3 locus) [16], [17]. Although the disorder reported here is also of early onset, is non-progressive and is associated with a normal ERG and EOG, there are a number of differences between the two. There may be considerable phenotypic heterogeneity in NCMD, with some family members having normal visual acuity with drusen-like deposits, whilst others have large ectatic macular lesions causing central visual loss. Furthermore, there is generally a family history consistent with an autosomal dominant inheritance pattern. Most of the present cases were sporadic or from small families. Those family members of sporadic cases who were examined had normal fundi. WES in 7 subjects failed to reveal any mutations in known retinal dystrophy genes, and SNP-based haplotype sharing analysis excluded linkage to the MCDR1 locus and a portion of the MCDR3 locus, adding support to the hypothesis that this is a new macular condition. Further molecular analysis is required to determine the etiology.
Drusen-like deposits at the macula may be seen in children and young adults in a variety of ocular and syndromic disorders (for review see Khan et al 2016) [18]. Small yellow deposits may be present in the early stages of the macular dystrophy associated with mutations in PRPH2, but the macular abnormalities, which are rarely seen before teenage years, are progressive and are often associated with full-field ERG abnormalities, particularly in late disease [19, 20]. Macular drusen are seen in autosomal dominant drusen but again, those have a later age of onset, a different retinal distribution and a different appearance on retinal imaging [21-23]. Drusen-like deposits at the macula have also been reported in systemic disorders such as mesangioproliferative glomerulonephritis type 2 and lipodystrophy, but in those disorders the drusen are larger, more extensive and increase in number with age [24]-[25, 26]. Similar macular deposits have been reported in association with other systemic findings in individuals with ring chromosome 17 and trisomy 10q [27-30]. However, none of the present subjects had any relevant systemic abnormalities.
The affected individuals in the present study were identified at routine optometric visits or by examination of other affected family members. Visual acuity was normal in the majority, and in all but one subject this was accompanied by normal electroretinographic findings that investigated both the detailed macular function (PERG) and global retinal function (ERG). Although no significant longitudinal data are available, none of the subjects showed progression over time, and the similarity of fundus appearance across generations in familial cases suggests the phenotype to be non-progressive.
The novel macular phenotype described here has a characteristic appearance on fundus examination and retinal imaging. However in order to exclude other disorders with a similar appearance early in the disease process, it is important to demonstrate normal retinal function. Investigations should include a full-field ERG and pattern ERG or multifocal ERG. OCT is also useful to exclude other phenotypes with drusen-like deposits [18]. The major differential diagnosis is from NCMD, where there is also a normal ERG, but examination of other family members should allow this disorder to be excluded.
To conclude, a novel childhood onset macular phenotype is described in which there are multiple yellow dots at the macula associated with normal macular function. The condition is of early onset and may be developmental in origin; it appears to be distinct from other developmental macular dystrophies. The yellow dots are hyperautofluorescent on FAF imaging and may show subtle irregularities at the inner segment ellipsoid band on OCT imaging. Such a phenotype has not, to our knowledge, been previously reported. Affected individuals can be reassured that the condition is benign and unlikely to be associated with any significant visual loss.
Supplementary Material
Supplementary Table 1. Homozygosity Haplotype analysis of Family 4
Supplementary Figure 1. Identification of the candidate regions for Family 4 (GC14302) using the Homozygosity Haplotype approach. Five affected family members with SNP chip data were included in the analysis. A densitogram of the genomic Regions with a Conserved Homozygosity Haplotype (RCHHs) is depicted. The darker the color, the more individuals share a HH in the region. Black regions indicate RCHHs that are shared by all 5 affected family members included in the analysis
{kind=link}
Acknowledgments
We thank Moshin Shazad for help with linkage data. The authors are grateful for support from the National Institute for Health Research Biomedical Research Centre at Moorfields Eye Hospital NHS Foundation Trust and UCL Institute of Ophthalmology (UK), Fight for Sight, Foundation Fighting Blindness (USA) and Research to Prevent Blindness (USA). The views expressed in this publication are those of the authors and not necessarily those of the Department of Health.
Footnotes
Financial Disclosures: None
Conflict of Interest: No conflicting relationship exists for any author
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Moore AT. Childhood macular dystrophies. Current opinion in ophthalmology. 2009;20(5):363–8. doi: 10.1097/ICU.0b013e32832f8002. [DOI] [PubMed] [Google Scholar]
- 2.North V, Gelman R, Tsang SH. Juvenile-onset macular degeneration and allied disorders. Dev Ophthalmol. 2014;53:44–52. doi: 10.1159/000357293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Michaelides M, Hunt DM, Moore AT. The genetics of inherited macular dystrophies. Journal of medical genetics. 2003;40(9):641–50. doi: 10.1136/jmg.40.9.641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Michaelides M, Jeffery G, Moore AT. Developmental macular disorders: phenotypes and underlying molecular genetic basis. Br J Ophthalmol. 2012;96(7):917–24. doi: 10.1136/bjophthalmol-2011-300994. [DOI] [PubMed] [Google Scholar]
- 5.O'Sullivan J, et al. A paradigm shift in the delivery of services for diagnosis of inherited retinal disease. Journal of medical genetics. 2012;49(5):322–6. doi: 10.1136/jmedgenet-2012-100847. [DOI] [PubMed] [Google Scholar]
- 6.Marmor MF, et al. ISCEV Standard for full-field clinical electroretinography (2008 update) Documenta ophthalmologica Advances in ophthalmology. 2009;118(1):69–77. doi: 10.1007/s10633-008-9155-4. [DOI] [PubMed] [Google Scholar]
- 7.Holder GE, et al. ISCEV standard for clinical pattern electroretinography--2007 update. Documenta ophthalmologica Advances in ophthalmology. 2007;114(3):111–6. doi: 10.1007/s10633-007-9053-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brown M, et al. ISCEV Standard for Clinical Electro-oculography (EOG) 2006. Documenta ophthalmologica Advances in ophthalmology. 2006;113(3):205–12. doi: 10.1007/s10633-006-9030-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hufnagel RB, et al. Neuropathy target esterase impairments cause Oliver-McFarlane and Laurence-Moon syndromes. J Med Genet. 2015;52(2):85–94. doi: 10.1136/jmedgenet-2014-102856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.DePristo MA, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43(5):491–8. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25(14):1754–60. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Miyazawa H, et al. Homozygosity haplotype allows a genomewide search for the autosomal segments shared among patients. Am J Hum Genet. 2007;80(6):1090–102. doi: 10.1086/518176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bowne SJ, et al. North Carolina macular dystrophy (MCDR1) caused by a novel tandem duplication of the PRDM13 gene. Mol Vis. 2016;22:1239–1247. [PMC free article] [PubMed] [Google Scholar]
- 14.Small KW, et al. North Carolina Macular Dystrophy Is Caused by Dysregulation of the Retinal Transcription Factor PRDM13. Ophthalmology. 2016;123(1):9–18. doi: 10.1016/j.ophtha.2015.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Small KW. North Carolina macular dystrophy: clinical features, genealogy, and genetic linkage analysis. Trans Am Ophthalmol Soc. 1998;96:925–61. [PMC free article] [PubMed] [Google Scholar]
- 16.Rosenberg T, et al. Clinical and genetic characterization of a Danish family with North Carolina macular dystrophy. Mol Vis. 2010;16:2659–68. [PMC free article] [PubMed] [Google Scholar]
- 17.Michaelides M, et al. An early-onset autosomal dominant macular dystrophy (MCDR3) resembling North Carolina macular dystrophy maps to chromosome 5. Investigative ophthalmology & visual science. 2003;44(5):2178–83. doi: 10.1167/iovs.02-1094. [DOI] [PubMed] [Google Scholar]
- 18.Khan KN, et al. Differentiating drusen: Drusen and drusen-like appearances associated with ageing, age-related macular degeneration, inherited eye disease and other pathological processes. Prog Retin Eye Res. 2016;53:70–106. doi: 10.1016/j.preteyeres.2016.04.008. [DOI] [PubMed] [Google Scholar]
- 19.Piguet B, et al. Full characterization of the maculopathy associated with an Arg-172-Trp mutation in the RDS/peripherin gene. Ophthalmic Genet. 1996;17(4):175–86. doi: 10.3109/13816819609057891. [DOI] [PubMed] [Google Scholar]
- 20.Boon CJ, et al. The spectrum of retinal dystrophies caused by mutations in the peripherin/RDS gene. Prog Retin Eye Res. 2008;27(2):213–35. doi: 10.1016/j.preteyeres.2008.01.002. [DOI] [PubMed] [Google Scholar]
- 21.Holz FG, et al. Ultrastructural findings in autosomal dominant drusen. Arch Ophthalmol. 1997;115(6):788–92. doi: 10.1001/archopht.1997.01100150790017. [DOI] [PubMed] [Google Scholar]
- 22.Edwards AO, et al. Malattia leventinese: refinement of the genetic locus and phenotypic variability in autosomal dominant macular drusen. Am J Ophthalmol. 1998;126(3):417–24. doi: 10.1016/s0002-9394(98)00097-x. [DOI] [PubMed] [Google Scholar]
- 23.Zweifel SA, et al. Multimodal imaging of autosomal dominant drusen. Klin Monbl Augenheilkd. 2012;229(4):399–402. doi: 10.1055/s-0031-1299404. [DOI] [PubMed] [Google Scholar]
- 24.D'Souza Y, et al. Long-term follow-up of drusen-like lesions in patients with type II mesangiocapillary glomerulonephritis. Br J Ophthalmol. 2008;92(7):950–3. doi: 10.1136/bjo.2007.130138. [DOI] [PubMed] [Google Scholar]
- 25.Duvall-Young J, MacDonald MK, McKechnie NM. Fundus changes in (type II) mesangiocapillary glomerulonephritis simulating drusen: a histopathological report. Br J Ophthalmol. 1989;73(4):297–302. doi: 10.1136/bjo.73.4.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Davis TM, et al. Retinal pigment epithelial change and partial lipodystrophy. Postgrad Med J. 1988;64(757):871–4. doi: 10.1136/pgmj.64.757.871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Charles SJ, et al. Flecked retina associated with ring 17 chromosome. Br J Ophthalmol. 1991;75(2):125–7. doi: 10.1136/bjo.75.2.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gass JD, Taney BS. Flecked retina associated with cafe au lait spots, microcephaly, epilepsy, short stature, and ring 17 chromosome. Arch Ophthalmol. 1994;112(6):738–9. doi: 10.1001/archopht.1994.01090180036013. [DOI] [PubMed] [Google Scholar]
- 29.Neely K, et al. Ocular findings in partial trisomy 10q syndrome. Am J Ophthalmol. 1988;106(1):82–7. doi: 10.1016/s0002-9394(14)76393-7. [DOI] [PubMed] [Google Scholar]
- 30.Tabandeh H, et al. Drusen-like macular deposits in partial trisomy 10q. Retina. 2000;20(6):678–9. doi: 10.1097/00006982-200006000-00020. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 1. Homozygosity Haplotype analysis of Family 4
Supplementary Figure 1. Identification of the candidate regions for Family 4 (GC14302) using the Homozygosity Haplotype approach. Five affected family members with SNP chip data were included in the analysis. A densitogram of the genomic Regions with a Conserved Homozygosity Haplotype (RCHHs) is depicted. The darker the color, the more individuals share a HH in the region. Black regions indicate RCHHs that are shared by all 5 affected family members included in the analysis