

Abstract
Low nephron endowment at birth has been associated with an increased risk for developing hypertension and chronic kidney disease. We demonstrated in an earlier study that conditional deletion of the microRNA (miRNA)-processing enzyme Dicer from nephron progenitors results in premature depletion of the progenitors and increased expression of the proapoptotic protein Bim (also known as Bcl-2L11). In this study, we generated a compound mouse model with conditional deletion of both Dicer and Bim, to determine the biologic significance of increased Bim expression in Dicer-deficient nephron progenitors. The loss of Bim partially restored the number of nephron progenitors and improved nephron formation. The number of progenitors undergoing apoptosis was significantly reduced in kidneys with loss of a single allele, or both alleles, of Bim compared to mutant kidneys. Furthermore, 2 miRNAs expressed in nephron progenitors (miR-17 and miR-106b) regulated Bim levels in vitro and in vivo. Together, these data suggest that miRNA-mediated regulation of Bim controls nephron progenitor survival during nephrogenesis, as one potential means of regulating nephron endowment.—Cerqueira, D. M., Bodnar, A. J., Phua, Y. L., Freer, R., Hemker, S. L., Walensky, L. D., Hukriede, N. A., Ho, J. Bim gene dosage is critical in modulating nephron progenitor survival in the absence of microRNAs during kidney development.
Keywords: nephrogenesis, metanephros, Bcl-2L11, Dicer, apoptosis
The kidney consists of functional units, called nephrons, that are essential for the regulation of extracellular fluid volume and electrolyte balance (1). The development of this highly complex organ commences with reciprocal inductive interactions between the metanephric mesenchyme and ureteric bud. This process results in the metanephric mesenchyme condensing, which forms a cap of nephron progenitors around the ureteric bud tip, and ureteric bud branching, which generates the collecting ducts. Nephrogenesis proceeds with a subpopulation of the nephron progenitors undergoing a mesenchymal–epithelial transition (MET) to form renal vesicles and then generate comma- and S-shaped body structures. Finally, the S-shaped body fuses with the collecting duct to form a functional nephron (2–4). Another subpopulation of nephron progenitors does not undergo MET, but continues to proliferate. Thus, a fine balance between nephron progenitor self-renewal and differentiation is critical in generating an appropriate number of nephrons during kidney development.
Nephrogenesis is completed at approximately the 36th week of gestation in humans (5) and at approximately postnatal d 4 in mice (6–8), and, henceforth, no new nephrons are formed. The number of nephron progenitors appears to be a limiting factor during nephrogenesis, as depletion of a fraction of embryonic nephron progenitors results in reduced nephron endowment (9), a condition that has been associated with adult-onset hypertension and chronic kidney disease (10–12). Numerous genetic factors can impact nephron endowment; for example, mutations in sine oculis homeobox homolog 2 (Six2), Pax2, and Ret, lead to renal hypodysplasia in mice and humans (13–17). Moreover, adverse intrauterine conditions, such as intrauterine hypoxia, also result in a reduced number of nephrons (18).
Recent studies have identified microRNAs (miRNAs) as an additional regulatory layer for kidney development (19–22). By binding to complementary sequences largely in the 3′-UTR of a target mRNA, miRNAs negatively regulate gene expression (23, 24). Conditional knockout of the miRNA-processing enzyme, Dicer, in nephron progenitors results in increased apoptosis and premature depletion of this cell population (19, 22). The increase in apoptosis was accompanied by a higher level of expression of the proapoptotic protein Bim (also known as Bcl-2L11) (19). In this study, the loss of Bim activity impaired apoptosis and partially restored the number of nephron progenitors and developing nephrons in Dicer-deficient kidneys. By combining in vitro assays and in vivo experiments in Xenopus laevis embryos, we found that Bim was under tight post-transcriptional regulation by miR-17 and miR-106b. Together, these data provide evidence for a model in which miRNA-mediated regulation of Bim controls the balance between apoptosis and survival in nephron progenitors, as a means for regulating the number of nephrons.
MATERIALS AND METHODS
Mouse strains
Transgenic (Tg) mice with the Six2-TGC transgene (Six2-TGCTg/+), strain Tg(Six2-EGFP/cre)1Amc/J, from The Jackson Laboratory (Bar Harbor, ME, USA) (25) were maintained on a 129/SvJae background. These mice were crossed with a conditionally floxed (flx) Dicer allele that is necessary for the production of mature miRNAs, from The Jackson Laboratory (strain B6;129S7-Dicer1tm1Smr/J) (26), and with a conditionally floxed (flx) Bim allele, obtained from L.D.W. (27, 28). The mice were crossed to generate the following genotypes: control (Six2-TGCTg/+, Bim+/+, Dicer+/+), mutant (Six2-TGCTg/+, Bim+/+, Dicerflx/flx), heterozygous rescue (Six2-TGCTg/+, Bimflx/+, Dicerflx/flx), and homozygous rescue (Six2-TGCTg/+, Bimflx/flx, Dicerflx/flx) kidneys. In addition, Six2-TGCTg/+, Dicerflx/+ mice were crossed with Bim-null mice from The Jackson Laboratory (strain B6;129S1-Bc2L11 < tm.1AST > /J) to generate the following genotypes: Six2-TGCTg/+, Dicerflx/+, Bim+/−; Six2-TGCTg/+, Dicerflx/flx, Bim+/+ and Six2-TGCTg/+, Dicerflx/flx, Bim+/−. Kidneys were collected from animals (male and female) at postnatal day 0 (P0) and from weaned animals at 1 mo of age. Animals were genotyped with genomic DNA isolated from tail clippings by PCR with the following primers: Six2-TGC forward (F): 5′-ATGCTCATCCGGAGTTCCGTATG-3′, (R): 5′-CACCTTGTCGCCTTGCGTATAA-3′; Bimflx (F): 5′-GAATTCATGGCAAAGCAACCTCTGA-3′, (R): 5′-GTCGACTCAATGCATTCTCCACACC-3′; Dicerflx (F): 5′-CCTGACAGTGACGGTCCAAAG-3′, (R): 5′-CATGACTCTTCAACTCAAACT-3′; and Bim-null (F): 5′- GTGAAACAGCATTGCTGTCACTT-3′, (R): 5′-CTAGGCCACAGAATTGAAAGATCT-3′. All animals were housed in the vivarium at the Rangos Research Center at the Children’s Hospital of Pittsburgh of (University of Pittsburgh Medical Center, Pittsburgh, PA, USA), and all animal experiments were performed in accordance with the policies of the Institutional Animal Care and Use Committee at the University of Pittsburgh.
Real-time quantitative PCR
Total RNA was isolated using the RNeasy Mini Kit (Qiagen, Valencia, CA, USA). First-strand cDNA was synthesized from total RNA with the SuperScript III First-Strand Synthesis System (Thermo Fisher Scientific, Waltham, MA, USA), according to the manufacturer’s protocol. Real-time quantitative PCR (qPCR) was performed in a 96-well C100 Thermal Cycler (Bio-Rad, Hercules, CA, USA), with SYBR Green Master Mix (Thermo Fisher Scientific). The expression of Dicer exon 24 and Dicer exon 21 (internal control) was assessed with the following primers: Dicer exon 21 (F): 5′-GAACATGCTGCACATCAAGG-3′, (R): 5′-GCAACCTTTTGCAGTTCACA-3′; and Dicer exon 24 (F): 5′-TCCAGGGGTCTTGACTGACT-3′ (R): 5′-CCAATGATGCAAAGATGGTG-3′. Expression levels were normalized to that of endogenous control, GAPDH (F): 5′-AGGTCGGTGTGAACGGATTTG-3′, (R): 5′-TGTAGACCATGTAGTTGAGGTCA-3′, using the cycle threshold value (Ct). Data were analyzed by the 2 −ΔΔCt method (29).
Histopathology and immunohistochemical staining
For morphologic analysis, kidneys were fixed in 4% paraformaldehyde overnight, embedded in paraffin, sectioned at 4–6 μm, and stained with hematoxylin and eosin (H&E), periodic acid-Schiff (PAS), and Masson’s trichrome stain.
For immunohistochemical staining, after deparaffinization, rehydration, and permeabilization in PBS-Tween (PBS-T), antigen retrieval was performed by boiling in 10 mM sodium citrate pH 6.0 buffer for 30 min. Sections were then blocked in 5% bovine serum albumin (BSA) before incubating overnight with the primary antibody. On the next day, sections were washed with PBS-T, incubated with secondary antibody and washed again with PBS-T before visualizing with a Leica DM2500 microscope (Leica, Buffalo Grove, IL, USA) and photographed with a QImaging Qiacam Fast 1394 camera, using QCapture software (QImaging, Surrey, BC, Canada). The following primary antibodies were used at the dilution recommended by the manufacturers: anti-Six2 (30) (11562-1-AP; Proteintech, Rosemont, IL, USA), anti-Bim (19) (2819; Cell Signaling Technology, Danvers, MA, USA), and anti-Pax2 (21) (71-6000; Thermo Fisher Scientific). Anti-neural cell adhesion molecule (NCAM) (31) (C9672) and anti-phosphorylated histone H3 (pHH3) (32) (H9161) were purchased from Sigma-Aldrich (St. Louis, MO, USA). Anti-Jagged1 (33) (sc-8303) and anti-Wilm’s tumor 1 (WT1) (34) (sc-192) were purchased from Santa Cruz Biotechnology (Dallas, TX, USA). All secondary antibodies were used at the dilution of 1:200. Anti-rabbit 594 (35) (111-515-144) and anti-mouse 488 (36) (715-545-151) were purchased from Jackson ImmunoResearch Laboratories (West Grove, PA, USA). Anti-rabbit horseradish peroxidase (HRP) conjugated (37) (SA1-9510) was purchased from Thermo Fisher Scientific.
To determine the number of early epithelial structures, kidney sections were immunostained with an anti-Jagged1 antibody as previously described. Ten digital images per kidney were captured, and the number of Jagged1+ structures was counted per image.
Western blot analysis
Protein extracts from P0 kidneys or human embryonic kidney (HEK) 293 cells were homogenized in RIPA buffer [p20 mM Tris-HCl (pH 7.5); 150 mM NaCl, 1 mM Na2 EDTA, 1 mM EGTA, 1% NP-40, 1% sodium deoxycholate, 2.5 mM sodium pyrophosphate, 1 mM β-glycerophosphate, 1 mM Na3VO4 and 1 μg/ml leupeptin) and the protein concentration was determined using the Pierce BCA Protein Assay Kit (Thermo Fisher Scientific). Ten micrograms of protein was run on a reducing 12% SDS-polyacrylamide gel and blotted to an ImmunoBlot PVDF membrane (Bio-Rad, Hercules, CA, USA). All primary and secondary antibodies were used at the concentration of 1:1000 and 1:4000, respectively. Anti-Bcl2 (38) (sc-7382) was purchased from Santa Cruz Biotechnology, anti-GAPDH (39) (2275-PC-100) from Trevigen (Gaithersburg, MD, USA), and anti-goat-HRP conjugate (35) (A5420) and anti-mouse-HRP conjugate (40) (A4416) from Sigma-Aldrich.
The signals were developed with the Amersham ECL Advance Western blot detection kit (GE Healthcare, Pittsburgh, PA, USA). Densitometric analyses of bands on Western blot gels were performed with ImageJ software (National Institutes of Health, Bethesda, MD, USA; https://imagej.nih.gov/ij/).
Quantification of glomerular number
Kidneys were collected at P0, fixed for 1 h in 4% paraformaldehyde and processed according to the methods outlined by Cullen-McEwen and colleagues (41). In brief, entire kidneys were serially sectioned at 4 μm. The sections were immunostained with a rabbit anti-WT1 antibody, as noted above, and visualized using the Vectastain ABC kit (Vector Laboratories, Burlingame, CA, USA), as per the manufacturer’s directions. For quantification of glomerular number, a pair of consecutive sections out of every 20 sections was chosen, and all of the WT1-stained structures were identified with a Wacom drawing tablet (Wacom, Portland, OR, USA) and Stereoinvestigator v.9.04 software (MBF Bioscience, Williston, VT, USA). The total glomerular number (nglom) was calculated using Eq. 1, described by Cullen-McEwen and colleagues (41):
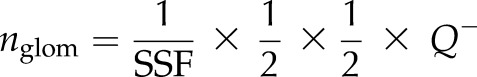 |
where nglom is the total of WT1+ structures in the entire kidney, 1/SSF is the reciprocal of the section-sampling fraction (the number of sections advanced between section pairs) and 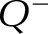 is the total number of WT1+ structures appearing and disappearing between 2 consecutive sections.
is the total number of WT1+ structures appearing and disappearing between 2 consecutive sections.
Size measurements
Pups at P0 and their kidneys were photographed with a scale bar using a QImaging QICAM Fast 1394 camera coupled to the Leica M165FC Stereo Microscope, with QCapture software (QImaging). ImageJ software was used to determine kidney length, surface area, and crown–rump length in each digital image.
Flow cytometric analysis and cell sorting
Kidneys were dissected at P0, treated with 3 mg/ml of Collagenase A (Roche Diagnostics, Indianapolis, IN, USA) for 30 min at 37°C and dissociated by passing through a 25-gauge needle. Single cell suspensions were analyzed by fluorescence-activated cell sorting (BD FACSAria II; BD Biosciences, Franklin Lakes, NJ, USA) to isolate green fluorescent protein (GFP)+ nephron progenitors, as this cell population expresses a GFP-Cre fusion protein driven by Six2 promoter.
Apoptosis assays
TUNEL assay was used for detecting apoptotic programmed cell death. In brief, kidneys were embedded in Tissue-Tek Optimal Cutting Temperature Compound (OCT; Sakura, Torrance, CA, USA) and sectioned at 10 μm. TUNEL staining was performed with the ApopTag Plus Fluorescein In Situ Apoptosis Detection kit (EMD Millipore Corp., Billerica, MA, USA), according to the manufacturer’s instructions. Costaining with a rabbit anti-Six2 antibody was performed sequentially after the TUNEL staining, as previously described. Eight digital images per kidney were captured with a Leica DM2500 microscope coupled to a QImaging Qicam Fast 1394 camera. The numbers of Six2+, TUNEL+ and Six2+/TUNEL+ double-positive cells were counted per image. The number of cells across all 8 images was summed, and the percentage of Six2+/TUNEL+ double-positive cells was calculated.
For kidney explants, kidneys were dissected from timed-mated embryonic day (E)12.5 CD-1 wild-type mice and grown on 0.4 μm polyethylene terephthalate membrane insert (Corning, Inc., Corning, NY, USA) in a 12-well plate in a 37°C incubator. Kidney explants were cultured in IMEM medium (Thermo Fisher Scientific) containing 50 μg/ml of transferrin (Sigma-Aldrich, St. Louis, MO, USA) in the presence of 4.8 μM of either a murine BIM-stabilized α-helix of BCL-2 domains (SAHB)A1 peptide (IRIAQELRXIGDXFNETYTRR) or its R153D mutant control (IRIAQELDXIGDXFNETYTRR) (obtained from author L.D.W) (42). 0.48% DMSO was used as a vehicle. After 48 h of treatment, explants were fixed in 4% paraformaldehyde overnight, processed for the TUNEL assay, and costained with an anti-Six2 and a mouse anti-Calbindin (43) (C9848; Sigma-Aldrich) antibodies overnight at 4°C. On the next day, explants were washed with PBS-Tween, incubated with secondary anti-rabbit 594 and anti-mouse 647 (44) (A-31571; Thermo Fisher Scientific) antibodies and washed before they were visualized with an Olympus FluoView FV1000 confocal laser scanning microscope and Fluoview v.2.1c software (Olympus, Center Valley, PA, USA).
miRNA expression in nephron progenitors
CD-1 wild-type females were time mated with Six2-TGCTg/+ males to generate Six2-TGCTg/+ E14.5 kidneys. The kidneys were dissected from the pregnant dams, dissociated into a single-cell suspension and subjected to fluorescence-activated cell sorting (BD Biosciences) to isolate GFP+ nephron progenitors, as published (19). In brief, the kidneys were tritiated through a 23-gauge needle, dissociated in 1 mg/ml Dispase (Thermo Fisher Scientific) for 10 min at 37°C, washed in PBS, and sorted. Nephron progenitors were pooled from 6–7 embryos across 3 litters.
Total RNA was extracted from GFP+ cells using the Qiagen miRNeasy Mini kit (Qiagen). The expression of miRNAs in Six2+ GFP+ embryonic kidney cells was profiled with Exiqon’s miRCURY LNA Array miRNA profiling services (Exiqon, Vedbaek, Denmark) (45, 46). RNA samples were labeled with Hy3 and hybridized in triplicate to miRcury LNA Arrays ver. 11. The quantified signals were normalized with the global Lowess regression algorithm. Spike-in controls confirmed that labeling and hybridization to the arrays were successful. The miRNAs expressed in nephron progenitors were listed in descending order based on the average signal intensities for each miRNA from the 3 replicates (Supplemental Table S1). The microarray results are publicly available through Gene Expression Omnibus (GEO Accession No.: GSE86217).
The presence of potential miRNA binding sites on the mouse Bim 3′-UTR was predicted by in silico analyses using 2 algorithms: microRNA.org (http://www.microrna.org/microrna) and TargetScan (http://www.targetscan.org), with default parameters.
Cell culture experiments
For dual luciferase assays, the intact 3′-UTR of Bim was amplified from genomic DNA [(F) primer: 5′-GCTAGCAGCGCTCTGCACTGTGTCGATGTGAACGG-3′; (R) primer: 5′-CTCGAGTTTTGAAAGCTAGTCGCAAGTTT-3] and cloned into the pmiRGLO Dual-Luciferase miRNA Target Expression Vector (Promega, Madison, WI, USA). HEK 293 cells were transfected with the jetPRIME Transfection Reagent (Polyplus, New York, NY, USA), with 1 μg of either empty pmiRGLO or pmiRGLO-Bim 3′UTR in the presence of 1 μg of short hairpin RNA (shRNA) control or Dicer shRNA (Sigma-Aldrich). Two hours later, cells were transfected with 17 nM of either miR-10a, miR-17, miR-24-1, or miR-106b MISSION microRNA mimics (Sigma-Aldrich) and incubated at 37°C in 5% CO2. The luciferase activity was measured 48 h after transfection with the Dual Luciferase Assay Kit (Promega).
For determination of endogenous Bim expression levels, HEK 293 cells were transfected with 2 μg of either shRNA control or Dicer shRNA in the presence of 17 nM microRNA mimics, according to the protocol described above. Six hours later, the cells were washed with PBS and lysed for Western blot analysis, as noted below. HEK293 cells were authenticated by the Applied Biosystems AmpflSTR Identifiler PCR Amplification kit (Thermo Fisher Scientific) with an evaluation value of 0.82, and confirmed to have no Mycoplasma contamination by the University of Pittsburgh Cell Culture and Cytogenetics Facility in August 2016.
Validation of miRNA targets in Xenopus embryos
All frogs were housed in the aquarium at the University of Pittsburgh School of Medicine, and all experiments were performed in accordance with the Institutional Animal Care and Use Committee at the University of Pittsburgh. Xenopus laevis embryos were obtained by in vitro fertilization (47) and staged according to Nieuwkoop and Faber (48).
For in vivo validation of miRNA activity, embryos were injected animally at the 2-cell stage with 1 ng of enhanced green fluorescent protein (EGFP)-mouse Bim 3′-UTR RNA in the presence of 2 ng of mmu-pri-miR-10a, mmu-pri-miR-17, mmu-pri-miR-24-1, or mmu-pri-miR-106b and cultured in 0.1× Barth (47). At stage 10, embryos were fixed with MEMFA fixative (47), and EGFP expression was visualized with a Leica M165FC Stereo Microscope and photographed with a QImaging Qicam Fast 1394 camera.
To generate the EGFP-mouse Bim 3′-UTR RNA, the EGFP sequence was amplified by PCR from the CMV-d2eGFP vector (Addgene, Cambridge, MA, USA) using a pair of primers [(F): 5′-ATCGATTTAAGCTTGGTACCGAGCTCG-3′; (R): 5′-AGGCCTATGGCTAAGCTTCTTGTACAGCTC-3′] that introduce a 5′ ClaI site and a 3′ StuI site, and are subcloned in the pGEM-T easy vector. The EGFP gene was then excised with ClaI and StuI (New England BioLabs, Inc., Ipswich, MA, USA) and inserted into the pCS2+ vector. Next, the Bim 3′-UTR was amplified from mouse genomic DNA by PCR [(F) primer: 5′-CTCGAGAGCGCTCTGCACTGTGTC-3′, (R) primer: 5′-TACGTATTTTGAAAGCTAGTCGCAAGTTT-3′], and cloned into the pCS2+-EGFP vector (between the XhoI and XbaI sites) downstream of the EGFP gene. Primary miRNA (pri-miR) transcripts flanked by a sequence of 150 nucleotides, which is essential for efficient processing (49), were amplified by PCR from mouse genomic DNA, using the following primers: pri-miR-10a (F): 5′-CTCGAGCTTGTAATCCCAAGAACGGAC-3′, (R): 5′-TCTAGAAAGCTTCCAGGGGTGTGC-3′; pri-miR-17 (F): 5′-CTCGAGCCCCTTGGGTATAAGCTGTAATT-3′, (R): 5′-TCTAGAACCAACGAAAGCAATAGAAATCA-3′; pri-miR-24-1 (F): 5′-CTCGAGACTCTACAAATCCCCACCTCG-3′, (R): 5′-TCTAGAATGCCACACGTGATGGGT-3′; and pri-miR-106b (F): 5′-CTCGAGATGCCACCTATACTTCTGCCC-3′, (R): 5′-TCTAGATAAGGTCCAAGAGGGGAGGA-3′. Next, pri-miRs were cloned into the pCS2+ vector. Finally, the pCS2+-EGFP-mouse Bim 3′-UTR, pCS2+-mmu-pri-miR-10a, pCS2+-mmu-pri-miR-17, pCS2+-mmu-pri-miR-24-1, and pCS2+-mmu-pri-miR-106b vectors were linearized with NotI (New England BioLabs, Inc.) and in vitro transcribed with the mMessage mMachine SP6 Transcription Kit (Thermo Fisher Scientific).
To test the efficiency of the miRNA processing, Xenopus embryos were microinjected at the 2-cell stage with 2 ng of each pri-miR transcript and cultured until sibling control embryos reached stage 10. Next, embryos were harvested with Trizol Reagent (Thermo Fisher Scientific) for RNA extraction and first cDNA strands were generated with the TaqMan MicroRNA Reverse Transcription Kit (Thermo Fisher Scientific), according to the manufacturer’s protocol. Finally, mature miRNAs were detected by qPCR using the following TaqMan-based assays (Thermo Fisher Scientific): RT000387 (hsa-miR-10a), RT002308 (hsa-miR-17), RT000402 (hsa-miR-24-1-3p), and RT 000442 (hsa-miR-106b). U6 small nuclear RNA was used as the housekeeping gene (RT001973).
Statistical analysis
All experiments were performed independently at least 3 times. A Student’s t test or 1-way ANOVA was used to determine statistical significance. When applicable, Tukey’s post hoc test was used for multiple comparison analysis.
RESULTS
Conditional deletion of Dicer and Bim from nephron progenitors
Conditional deletion of Dicer in nephron progenitors results in a premature loss of nephron progenitors associated with increased expression of Bim (19, 22). To investigate the biologic significance of elevated Bim activity, we conditionally deleted Bim in Six2-TGCTg/+;Dicerflx/+ mice to generate compound mice with the following genotypes: Six2-TGCTg/+;Bim+/+;Dicer+/+ (control), Six2-TGCTg/+;Dicerflx/flx;Bim+/+ (mutant), Six2-TGCTg/+;Dicerflx/flx;Bimflx/+ (heterozygous rescue), and Six2-TGCTg/+;Dicerflx/flx;Bimflx/flx (homozygous rescue) (Fig. 1A). Cre-mediated excision of Dicer exon 24 from the Dicerflx allele was confirmed by qPCR, with mutant, heterozygous rescue and homozygous rescue kidneys exhibiting a significant decrease in Dicer exon 24 expression compared with controls (Fig. 1B). Conversely, expression of Dicer exon 21, which is not flanked by LoxP sites, was unchanged.
Figure 1.

Loss of Bim in nephron progenitors results in decreased Bim expression in Six2-TGCTg/+, Dicerflx/flx nephron progenitors. A) PCR genotyping showing Dicer wild type (Wt) (350 bp), Dicerflx (400 bp), Bim Wt (250 bp), and Bimflx (300 bp) alleles. B) qPCR analysis demonstrating reduced expression of Dicer exon 24 in mutant (Six2-TGCTg/+, Bim+/+, Dicerflx/flx), heterozygous rescue (Six2-TGCTg/+, Bimflx/+, Dicerflx/flx) and homozygous rescue (Six2-TGCTg/+, Bimflx/flx, Dicerflx/flx), when compared to control (Six2-TGCTg/+, Bim+/+, Dicer+/+) kidneys. There was no statistically significant difference between these 4 genotypes for Dicer exon 21, an internal control (n = 3 embryos per genotype derived from 3 litters). Error bars ± sem. **P ≤ 0.01 (1-way ANOVA with Tukey’s post hoc test). C–F) Immunostaining on P0 kidney sections demonstrate that Bim expression is increased in nephron progenitors of mutant kidneys (D) and decreased in heterozygous rescue (E) and homozygous rescue (F) nephron progenitors, compared to control (C). Images shown are representative of 3 independent experiments (n ≥ 3 embryos per genotype derived from at least 3 litters). Scale bar, 50 μm. G) Western blot analysis comparing the levels of the Bim isoforms (BimEL, BimL, and BimS) and Bcl-2 in P0 kidneys from control, mutant, heterozygous rescue, and homozygous rescue mice. The expression of BimEL was decreased in homozygous rescue kidneys, confirming the efficiency of Cre-mediated Bim deletion. GAPDH was used as a loading control.
Furthermore, Bim immunostaining in P0 kidney sections confirmed increased Bim expression in nephron progenitors that lack Dicer activity (19), whereas the deletion of 1 or both alleles of Bim decreased Bim expression, as was predicted (Fig. 1C–F). To verify these data, Western blot analysis detected expression of all 3 Bim isoforms [Bim short (BimS), Bim long (BimL) and Bim extra-long (BimEL)] (50) in P0 kidneys from control, mutant, homozygous, and heterozygous rescue mice (Fig. 1G). As expected, mutant kidneys displayed increased BimEL compared to control kidneys, and in homozygous rescue kidneys, BimEL levels were reduced in a dose-dependent manner (Fig. 1G; Supplemental Fig. S1).
Loss of Bim expression in nephron progenitors results in a partial rescue of nephron number in Six2-TGCTg/+;Dicerflx/flx kidneys
If miRNA-mediated regulation of Bim expression is important in determining nephron progenitor survival (and hence, nephron number), altering Bim gene dosage in the absence of Dicer function in nephron progenitors would result in changes in nephron number. H&E staining of P0 kidney sections from control, mutant, heterozygous rescue, and homozygous rescue mice indeed demonstrated various numbers of glomeruli (Fig. 2A–D). Stereological analyses of sections immunostained for WT1 revealed a reduction of ∼80% of the total number of glomeruli in mutant kidneys compared to the control (Fig. 2E–I). Furthermore, the loss of Bim partially restored the quantity of those structures. Heterozygous and homozygous rescue kidneys unexpectedly exhibited a similar number of glomeruli. In addition, mutant kidney lengths were significantly smaller than control littermates, with homozygous rescue and heterozygous rescue kidneys exhibiting an intermediate length (Fig. 2J). No significant differences in pup weight (data not shown) or crown–rump length (Fig. 2K) were observed between these genotypes. Notably, this phenotype was replicated when Six2-TGCTg/+;Dicerflx/flx mice were crossed with the Bim-null allele, suggesting that this observation does not simply reflect less efficient Cre-mediated excision of the Dicerflx allele when the Bimflx allele is present (Supplemental Fig. S2). Moreover, although Six2-TGCTg/+;Dicerflx/flx mutant mice die perinatally universally (22), we observed incompletely penetrant survival of homozygous rescue and heterozygous rescue mice at 1 mo of age (Supplemental Fig. S3). The removal of Bim was not capable of preventing cyst formation in Dicer-deficient mice (Fig. 2B–D and Supplemental Fig. S2), suggesting that other molecules contribute to cystogenesis in these mice. Taken together, these data demonstrate that Dicer deficiency decreases the number of nephrons formed in mutant kidneys, and that this is dependent, at least in part, on Bim expression.
Figure 2.

Loss of Bim function results in a partial rescue of the defects in nephron formation in mice that lack Dicer in nephron progenitors. A–D) H&E staining of P0 kidney sections from control (A), mutant (B), homozygous rescue (C), and heterozygous rescue (D) kidneys demonstrate fewer developing nephron structures and glomeruli in mutant kidneys (B) compared to controls (A). In contrast, heterozygous (C) or homozygous (D) loss of Bim function results in a partial rescue of the phenotype, with an increased number of developing nephron structures and glomeruli. Black arrows: glomeruli. E–H) Immunostaining of P0 kidneys from control (E), mutant (F), homozygous rescue (G), and heterozygous rescue (H) mice for WT1 expression. Red arrows: glomeruli. Images shown are representative of 3 independent experiments (n ≥ 3 embryos per genotype derived from at least 3 litters). Scale bars, 100 μm. I) Stereological quantification of glomerular number demonstrates that mutant kidneys exhibit a decreased number of glomeruli when compared to control, and the loss of Bim partially restores the number of these structures in Dicer-deficient kidneys (n = 4 embryos per genotype derived from at least 3 litters). J) Kidney length is significantly smaller in mutant, heterozygous and homozygous rescue kidneys at P0 when compared to control (Six2-TGCTg/+, Bim+/+, Dicerflx/+) kidneys. However, both heterozygous and homozygous rescue kidneys are intermediate in size between control and mutant kidneys, and rescue kidneys are significantly longer than mutant kidneys (n ≥ 14 embryos per genotype derived from at least 10 litters). K) There was no statistically significant difference between any of the genotypes in crown–rump length (n ≥ 7 embryos per genotype derived from at least 4 litters). Error bars ± sem. N.s., nonsignificant. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001 (1-way ANOVA with Tukey’s post hoc test).
Conditional deletion of Bim partially restores the number of nephron progenitors and developing nephrons in Six2-TGCTg/+;Dicerflx/flx kidneys
To assess the effects of Dicer and Bim deletion on the kidney progenitor population, sections of P0 kidneys from control, mutant, heterozygous rescue, and homozygous rescue mice were immunostained for the nephron progenitor markers Six2 and Pax2. Control kidneys display Six2+ progenitors clustering as a robust cap of cells around the ureteric tips, whereas the Six2+ cell population was dramatically reduced in mutant kidneys (Fig. 3A-H). Six2 staining of homozygous and heterozygous rescue kidneys revealed improved progenitor numbers around the ureteric tips. The Pax2 staining is also consistent with these findings. To further confirm these results, GFP+ nephron progenitors were isolated and counted by fluorescence-activated cell sorting. The percentage of nephron progenitors in mutant kidneys (0.46%) was dramatically reduced when compared to control kidneys (5.79%). The percentage of nephron progenitors in heterozygous rescue and homozygous rescue kidneys was consistent with a partial rescue, at ∼0.75 and 1.18%, respectively (Fig. 3I).
Figure 3.

Decreased Bim expression in nephron progenitors of Six2-TGC Tg/+; Dicerflx/flx kidneys partially rescues depletion of nephron progenitors in mutant kidneys. A–H) Immunofluorescence was performed on control (A, B), mutant (C, D), heterozygous rescue (E, F), and homozygous rescue (G, H) P0 kidneys. Staining with Six2 (red; A, C, E, G) and Pax2 (green; B, D, F, H) revealed improved progenitor capping around the ureteric tips in heterozygous and homozygous rescue kidneys when compared to mutant kidneys. Notably, the few forming nephrons in mutant kidneys appear dysplastic. Pax2 also labels ureteric bud and collecting duct. Images shown are representative of 3 independent experiments (n = 3 embryos per genotype derived from 3 litters). Scale bar, 50 μm. I) Quantification of GFP+ cells sorted by fluorescence-activated cell sorting demonstrated a dramatic reduction in the percentage of nephron progenitors in mutant kidneys (∼0.46%) when compared to control kidneys (∼5.79%). The percentage of nephron progenitors in heterozygous rescue and homozygous rescue kidneys was ∼0.75% and 1.18%, respectively (n = 5 embryos per genotype derived from 3 litters, except for heterozygous rescue, from 1 litter). Error bars ± sem. N.s., nonsignificant. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001 (1-way ANOVA with Tukey’s post hoc test).
Having observed variations in the nephron progenitors between the 4 genotypes, we next interrogated whether the number of early epithelial derivatives of nephron progenitors (renal vesicle, comma-shaped body, and S-shaped body) is also perturbed. We immunostained sections from P0 kidneys for Jagged1 and NCAM, both of which mark these early epithelial derivatives. Early developing nephrons were present throughout the cortical area in control kidneys, whereas only a few renal vesicles were present in mutant kidneys (Fig. 4A–H). As expected, the loss of Bim partially restored the number of developing nephrons (Fig. 4I) and the expression of Jagged1 (Fig. 4J–K) in homozygous and heterozygous rescue kidneys. Together, these data demonstrate that the loss of Bim partially restores the number of nephron progenitors and improves nephron formation in kidneys with Dicer-deficient nephron progenitors.
Figure 4.

Decreased Bim activity partially restores the number of early epithelial structures in Six2-TGCTg/+;Dicerflx/flx kidneys. A–H) Early epithelial derivatives of nephron progenitors were detected in kidney sections from P0 control (A, B), mutant (C, D), homozygous rescue (E, F) and heterozygous rescue (G, H) mice using immunostaining for Jagged1 (red; A, C, E, G) and NCAM (green; B, D, F, H). NCAM expression also marks nephron progenitors and the renal interstitium. White arrows: early developing nephrons. Images are representative of 3 independent experiments (n = 3 embryos per genotype derived from 3 litters). Scale bar, 50 μm. I) Semiquantitative analysis of the number of Jagged1+ structures demonstrated that mutant kidneys exhibited only a few developing nephrons, compared to control kidneys. An intermediate number of early epithelial structures was observed in heterozygous and homozygous rescue kidneys. The number of Jagged1+ structures was calculated by using 10 digital images per kidney (n = 3 embryos per genotype derived from 3 litters). Error bars ± sem. N.s., nonsignificant. *P ≤ 0.05, **P ≤ 0.01 (1-way ANOVA with Tukey’s post hoc test). J) Western blot analysis comparing the levels of the Jagged1 in P0 kidneys from control, mutant, heterozygous rescue and homozygous rescue mice. GAPDH was used as a loading control. K) Densitometric analyses of the bands on the Western blot shown in J. The relative levels of Jagged1 were calculated by normalizing with GAPDH.
Inhibition of Bim expression maintains nephron progenitors
We next evaluated whether Bim is sufficient to promote apoptosis in nephron progenitors. To address this question, kidney explants from wild-type CD-1 E12.5 embryos were treated with a murine BIM SAHBA1, a SAHB domains peptide modeled after the BIM BH3 domain, which effectively replaces the proapoptotic functionality of the BIM protein (42). Apoptosis was measured using a TUNEL assay. The number of Six2+/TUNEL+ double-positive cells was markedly increased in kidney explants treated with BIM SAHBA1, compared to explants treated with vehicle (0.48% DMSO) or the control peptide, BIM SAHBA1 R153D, which impairs BH3-binding and functional activity (42) (Fig. 5A–D).
Figure 5.

Increased Bim activity is sufficient to induce apoptosis in nephron progenitors, whereas decreased Bim expression reduces apoptosis in nephron progenitors. A) Sequences of the murine BIM SAHBA1 peptide and its R153D mutant control. B–D) TUNEL assay (green) followed by immunostaining for the nephron progenitor marker, Six2 (red) and the ureteric bud marker calbindin (blue) in E12.5 wild-type kidney explants. Increased apoptotic Six2+/TUNEL+ double-positive cells were observed in kidneys explants treated with 4.8 μM of BIM SAHBA1 peptide (C), when compared to those treated with the vehicle (0.48% DMSO) (B) or 4.8 μM of the mutant control peptide, BIM SAHBA1 R153D (D). E–J) Immunofluorescence of kidney sections from P0 control, mutant, and homozygous rescue mice for Six2 (red). Apoptotic (E–G) and proliferating (H–J) cells were detected with TUNEL assay (green) and anti-pHH3 antibody (green), respectively. Images are representative of 3 independent experiments (n = 3 embryos per genotype derived from at least 3 litters). Scale bars, 25 μm. K) Quantification of the Six2+/TUNEL+ double-positive cells demonstrated an increased number of nephron progenitors undergoing apoptosis in mutant kidneys compared to control kidneys. The loss of Bim significantly reduces apoptosis in homozygous rescue kidneys. L) Quantification of the Six2+/pHH3+ double-positive cells demonstrated no significant difference in the number of proliferating nephron progenitors between the 3 genotypes. Numbers across 8 images per kidney (n = 3 embryos per genotype derived from at least 3 litters) were summed, and the percentage of Six2+/TUNEL+ and Six2+/pHH3+ cells was calculated. Error bars ± sem. N.s., nonsignificant. *P ≤ 0.05 (1-way ANOVA with Tukey’s post hoc test).
Next, we examined whether the conditional deletion of Bim affects apoptosis in Dicer-deficient nephron progenitors. In P0 control kidneys, only a few apoptotic cells were detected, predominantly in the developing renal stroma, whereas the number of Six2+/TUNEL+ double-positive cells was significantly increased in mutant kidneys, corroborating our previous results at E14.5 (19) (Fig. 5E, F). Analysis of homozygous rescue kidneys demonstrated reduced apoptosis in the nephrogenic zone, with the number of Six2+/TUNEL+ double-positive cells similar to control kidneys (Fig. 5E–G, K). No significant differences in the number of apoptotic cells were observed in the ureteric bud structures, confirming the specificity of Six2-TGC-mediated conditional deletion (data not shown). The rate of cell proliferation in nephron progenitors remained constant in the 4 genotypes, as measured by Six2+/phosphorylated histone H3+ double-positive staining (Fig. 5H–J,L). Together these results suggest that Bim plays an important role in the maintenance of nephron progenitors during kidney development.
Post-transcriptional regulation of Bim by miRNAs
Since Bim is a potent inducer of apoptosis, its expression is tightly regulated by a variety of transcriptional, post-transcriptional, and post-translational mechanisms, including miRNAs (for a review, see ref. 51). To define the miRNAs that regulate Bim activity in nephron progenitors, we profiled miRNA expression in progenitors by using a miRNA microarray. Supplemental Table S1 shows in green the miRNAs predicted to target the 3′-UTR of mouse Bim mRNA. From these miRNAs, we selected those, which are known to be expressed in nephron progenitors (19), and to repress Bim (52–57). Based on these criteria, mmu-miR-24-1, mmu-miR-17, mmu-miR-106b, and mmu-miR-10a were selected for further analysis.
In silico analysis with microRNA.org and TargetScan revealed that mmu-miR-24-1, mmu-miR-17, mmu-miR-106b, and mmu-miR-10a contain conserved binding sites within the 3′-UTR of Bim mRNA (Supplemental Fig. S4). To validate these findings, luciferase assays were performed in HEK 293 cells with the pmiRGLO luciferase reporter vector containing the full-length Bim 3′-UTR. To block the biogenesis of endogenous miRNAs and sensitize the assay, cells were cotransfected with Dicer shRNA (58). Luciferase reporter activity was decreased approximately 10, 11, and 18%, in the presence of the miR-24-1, miR-17, and miR-106b mimics, respectively (Fig. 6A). In contrast, the miR-10a mimic was not sufficient to repress luciferase activity. In addition, we evaluated endogenous Bim expression in HEK 293 cells transfected with either shRNA control or Dicer shRNA in the presence of miRNA mimics. The expression of BimEL was reduced upon transfection of cells with the miR-24-1, miR-17, miR-106b, and miR-10a mimics (Fig. 6B,C). We did not detect the presence of the other isoforms of Bim, BimL, and BimS, after a short exposure of blot to film.
Figure 6.

miR-24-1, miR-17, miR-106b, and miR-10a repress the expression of Bim in vitro and in vivo. A) Schematic of the pmiRGLO-Bim 3′-UTR luciferase reporter vector containing the mouse Bim 3′-UTR. Potential miRNA binding sites predicted by in silico analyses are represented by vertical bars. Blue: predicted binding sites for miR-24-1, miR-17, miR-106b, and miR-10a. Luciferase reporter assays performed in HEK 293 cells transfected with pmiRGLO-Bim 3′-UTR in the presence of Dicer shRNA and miRNA mimics (miR-24-1, miR-17, miR-106b, and miR-10a). There was a reduction of ∼10, 11, and 18% in luciferase activity in the presence of miR-24-1, miR-17, and miR-106b, respectively. No significant change in luciferase activity was observed in the presence of miR-10a mimic (n = 3 independent experiments). Error bars ± sem. N.s., nonsignificant. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001 (1-way ANOVA with Turkey’s post hoc test). B) Western blot analysis comparing Bim levels in protein extracts of HEK 293 cells transfected with either shRNA control or Dicer shRNA, in the presence of miRNA mimics. The miR-24-1, miR-17, miR-106b, and miR-10a mimics decreased the expression of BimEL. GAPDH was used as a loading control. C) Densitometric analyses of the bands on the Western blot shown in (B). The relative levels of BimEL were calculated by normalizing with GAPDH. D–K) Xenopus laevis embryos were microinjected with a synthetic full-length Bim 3′-UTR fused to the EGFP reporter gene, in the presence or absence of pri-mmu-miR-24-1, pri-mmu-miR-17, pri-mmu-miR-106b, or pri-mmu-miR-10a. D) EGFP expression was assessed at stage 10. E–K) Representative images (E–J) and the quantification (K) of 3 independent experiments are shown. The total number of embryos analyzed is indicated above the individual bars. L) qPCR analysis demonstrating the levels of mature miRNAs (mmu-miR-24-1, mmu-miR-17, mmu-miR-106b, and mmu-miR-10a) in embryos injected with the primary transcripts, pri-mmu-miR-24-1, pri-mmu-miR-17, pri-mmu-miR-106b, and pri-mmu-miR-10a (n = 3 biologic replicates per condition from 3 independent experiments. Each replicate represented 4 embryos that were pooled). Error bars ± sem. N.s., nonsignificant. *P ≤ 0.05, **P ≤ 0.01 (Student’s unpaired Student’s t test).
To test the ability of these miRNAs to regulate Bim in vivo, Xenopus laevis embryos were injected with an RNA containing the EGFP sequence fused to the mouse Bim 3′-UTR, in the presence or absence of the mouse pri-miR transcripts pri-mmu-miR-24-1, pri-mmu-miR-17, pri-mmu-miR-106b, or pri-mmu-miR-10a (Fig. 6D). EGFP expression was repressed by each one of the 4 pri-miRs tested (Fig. 6E–J), although the efficiency of repression varied (Fig. 6K). One potential explanation for this variability is that there is differential processing of primary transcripts into mature miRNAs. To test this, we injected Xenopus embryos with 2 ng of each pri-miR transcript and analyzed the levels of mature miRNAs by qPCR. Injected embryos expressed elevated levels of miR-17, miR-106b, and miR-10a compared to uninjected control embryos. However, no significant changes in the levels of mature miR-24-1 were detected between uninjected and injected embryos (Fig. 6L). These data are consistent with the idea that miR-17 and miR-106b act in vitro and in vivo as rheostats to fine tune Bim expression in nephron progenitors.
DISCUSSION
We have reported that conditional knockdown of Dicer in murine nephron progenitors results in increased apoptosis and increased Bim expression (19, 22). In the current study, we examined the biological significance of augmented Bim expression in Dicer-deficient nephron progenitors during kidney development. The loss of Bim was sufficient to decrease apoptosis of Dicer-deficient nephron progenitors and partially rescue the number of developing nephrons in mutant kidneys. Moreover, we provide evidence that Bim expression is regulated by miR-17 and miR-106b. These data support a model in which miRNA-mediated regulation of Bim expression regulates nephron progenitor survival, and hence nephron number, during kidney development (Fig. 7).
Figure 7.

Proposed model for Bim function in nephron progenitors during kidney development. A–C) In control kidneys, nephron progenitors (red) form a cap of cells surrounding the ureteric bud tips (blue) (A); whereas in mutant kidneys, the deletion of Dicer from nephron progenitors results in premature ablation of this cell population (B). The loss of Bim partially restores the number of progenitors in Dicer-deficient kidneys (C). A′–C′) Pre-miRNAs are cleaved by Dicer to produce mature miRNAs. Mature miRNAs recognize specific binding sites on the 3′-UTR of the Bim mRNA, recruit the RNA-induced silencing complex (RISC), causing either Bim mRNA degradation or its translational repression. During kidney development, low Bim levels in nephron progenitors allow this cell population to be maintained (A′). In mutant kidneys, the absence of Dicer impairs the formation of mature miRNAs, resulting in increased Bim expression, and nephron progenitors preferentially undergo apoptosis (B′). In heterozygous rescue and homozygous rescue kidneys, nephron progenitors lacking mature miRNAs also display reduced or absent expression of Bim mRNA, which in turn favors cell survival (C′).
Reduced nephron endowment has been strongly correlated with adult-onset hypertension and chronic kidney disease (10–12). Several other mouse models have illustrated the importance of tight control of apoptosis to complete nephrogenesis during development. For example, the fibroblast growth factor (FGF) signaling pathway has been shown to be necessary for maintenance of the self-renewing nephron progenitor population. Loss of Fgf8 or Fgf9/Fgf20 in mice results in massive apoptosis of the metanephric mesenchyme cells and deficient nephron formation (59–61). Moreover, deletion of the FGF receptors, Fgfr1 and Fgfr2, in the metanephric mesenchyme leads to reduced survival of this progenitor cell population and consequently, renal dysgenesis (62, 63). Likewise, mice lacking Six2 (14), Pax2 (64), WT1 (65), or Sall1 (66) exhibit severe renal dysgenesis, as these transcription factors are necessary for survival, proliferation, and subsequent differentiation of nephron progenitors. These findings demonstrate that the tight regulation of apoptosis is necessary for nephron progenitor survival and to complete nephrogenesis.
Although in this study we tested the functional significance of Bim expression in the context of Dicer-deficient nephron progenitors, findings in previous studies support the concept that Bim gene dosage is critical in normal kidney development as well. Embryonic kidneys from Bcl-2−/− mice are hypodysplastic because of increased apoptosis in nephron progenitors, and after birth, these mice develop renal cysts and kidney disease (67–70). Removal of a single allele of Bim in Bcl-2−/− mice restores nephron formation and prevents cystogenesis (71). Bim−/− mice progress to renal failure with age, although the failure is caused by a systemic autoimmune disease rather than renal developmental defects (68, 72–74). Although Bim is not necessary for normal kidney development, it may play a crucial role in establishing a threshold for activation of apoptosis in nephron progenitors, in combination with the activity of Bcl-2. Indeed, our results corroborate this conclusion, as the treatment of kidney explants from E12.5 wild-type mice with a stapled BIM BH3 peptide (BIM SAHBA1) was sufficient to induce apoptosis in nephron progenitors. Moreover, the conditional deletion of Bim diminished apoptosis of nephron progenitors and partially restored the number of nephrons, in the kidneys of Dicer-deficient mice. Of note, there remains the possibility that Bim also mediates downstream events of nephrogenesis, such as MET; this possibility is the subject of future studies.
No significant differences in the number of nephrons were observed between heterozygous and homozygous rescue kidneys. These findings suggest that compensatory mechanisms are needed to ensure a basal activity of Bim in the nephron progenitor population, probably to counteract the activity of prosurvival proteins (e.g., Bcl-2). Thus, at least 2 potential scenarios are possible. First, it has been shown that phosphorylation by MAPKs influences Bim protein stability, its interaction with other Bcl2 family proteins, and its apoptotic activity (75, 76). Second, expression or activation of proapoptotic proteins functionally redundant with Bim (e.g., Bid and Puma) may act as a compensatory mechanism (77). Indeed, the involvement of these other coregulators of apoptosis may be the reason for only a partial, rather than a complete rescue phenotype in this model.
Bim is an important initiator of apoptosis in both physiologic and pathologic conditions. Dysregulation of Bim has been implicated in type I diabetes (79–81), neurodegenerative disorders (82–85), cancer, and resistance to chemotherapy (for a review see refs. 51, 86). Given this, it is not surprising that Bim activity must be strictly modulated to ensure the appropriate balance between apoptosis and cell survival. In fact, several studies have demonstrated that Bim expression is regulated at transcriptional (87–93), post-transcriptional (94, 95), and post-translational (96–98) levels. In this study, Bim expression was post-transcriptionally modulated by miR-17 and miR-106b. Of the miRNAs we tested, miR-10a, miR-17, and miR-24-1 have been shown to regulate Bim expression in other organ systems: cardiac myocytes (53–55), pancreatic carcinoma (99), ovarian cells (52), lymphocytes (100, 101), or acute lymphocytic leukemia (56). Contrary to our findings, Kan and colleagues (57) did not detect changes in Bim expression in the presence of either the miR-106b mimic or the miR-106b inhibitor, probably because of differences in endogenous miRNA expression in different tissues. Indeed, the predicted miR-106b binding site on the 3′-UTR of the Bim mRNA is shared by other miRNAs (e.g., miR-17, miR-20, and miR-93) and is located in proximity with other predicted target sites (miR-291-3p/294-3p/295-3p/302-3p). We hypothesize that miR-106b functions in concert with another nephron progenitor miRNA, miR-17, to set a threshold of Bim expression in this cell population. However, we have not excluded the possibility that transcriptional mechanisms account, in part, for upregulation of Bim expression. It has been shown that several transcription factors, including forkhead box-containing protein, class O (FoxO) (88, 102–105), nuclear factor erythroid derived 2, like 2 (Nrf2) (106), C/ERB homologous protein (CHOP)-C/EBPα (107) and E2F1 (108), bind directly to the Bim promoter, inducing Bim expression and apoptosis in different cell types. Further experiments are needed to determine whether these transcription factors are activated in Dicer-deficient nephron progenitors.
A reduced number of nephrons at birth has been associated with chronic kidney disease (10–12), and the number of nephrons is related to the number of nephron progenitors generated during kidney development (9). The cessation of nephrogenesis is thought to begin with decreased proliferation and accelerated differentiation of nephron progenitors, which is accompanied by age-dependent changes in gene expression of progenitors (8, 109). We conclude that miRNA-mediated modulation of the proapoptotic protein Bim is a potential means of regulating nephron number and suggest that it may represent a critical mechanism in disease settings that would otherwise result in nephron progenitor apoptosis.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank Dr. Oliver Wessely (Cleveland Clinic Lerner Research Institute, Cleveland, OH, USA) for intellectual input; the Kidney Imaging Core of the Pittsburgh Center for Kidney Research for providing expert pathological and 3-dimensional stereological support [funded by U. S. National Institutes of Health (NIH), National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) Grant P30DK079307]; the Children’s Hospital of Pittsburgh of UPMC’s Histology Core Laboratory, University of Pittsburgh Flow Cytometry Core Laboratory; the Kansas State University Veterinary Diagnostic Laboratories for technical assistance; and the University of Pittsburgh Cell Culture and Cytogenetics Facility for cell line authentication. This work was supported by NIH NIDDK Grants DK087922 and DK103776 (to J.H.); NIH, NIDDK Grants DK069403, DK079307, and Eunice Kennedy Shriver National Institute of Child Health and Human Development HD053287 (to N.A.H.); NIH, NIDDK Grant T32 DK061296 (to S.L.H.); a Children’s Hospital of Pittsburgh Research Advisory Council Postdoctoral Fellowship (to D.M.C.); and a George B. Rathmann Research Fellowship supported by the American Society of Nephrology Ben J. Lipps Research Fellowship Program (to Y.L.P.). L.D.W is a scientific advisory member and consultant for Aileron Therapeutics. The remaining authors declare no conflicts of interest.
Glossary
- EGFP
enhanced green fluorescent protein
- flx
floxed
- Fgf
fibroblast growth factor
- Fgfr
fibroblast growth factor receptor
- GFP
green fluorescent protein
- HEK
human embryonic kidney
- H&E
hematoxylin and eosin
- HRP
horseradish peroxidase
- MET
mesenchymal–epithelial transition
- miRNA
microRNA
- NCAM
neural cell adhesion molecule
- P0
postnatal day 0
- PAS
periodic acid-Schiff
- pHH3
phosphorylated histone H3
- pri-miR
primary miRNA
- qPCR
quantitative PCR
- RISC
RNA-induced silencing complex
- SAHB
stabilized α-helix of BCL-2 domains
- shRNA
short hairpin RNA
- Six2
sine oculis homeobox homolog 2
- Tg
transgenic
- WT1
Wilm’s tumor 1
Footnotes
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
AUTHOR CONTRIBUTIONS
D. M. Cerqueira, A. J. Bodnar, and J. Ho, designed the experiments; D. M. Cerqueira, A. J. Bodnar, Y. L. Phua, S. L. Hemker, and R. Freer performed the experiments; D. M. Cerqueira, A. J. Bodnar, Y. L. Phua, S. L. Hemker, R. Freer, and J. Ho analyzed and interpreted data; L. D. Walensky assisted in the design and analysis of experiments using the BIM SAHBA1 peptides and provided these reagents; N. A. Hukriede assisted in the design and analysis of the Xenopus experiments; and D. M. Cerqueira and J. Ho wrote the paper.
REFERENCES
- 1.Vize P. D., Woolf A. S., Bard J. (2003) The Kidney: From Normal Development to Congenital Disease, Academic, Amsterdam/London [Google Scholar]
- 2.Lechner M. S., Dressler G. R. (1997) The molecular basis of embryonic kidney development. Mech. Dev. 62, 105–120 [DOI] [PubMed] [Google Scholar]
- 3.Saxén L., Sariola H. (1987) Early organogenesis of the kidney. Pediatr. Nephrol. 1, 385–392 [DOI] [PubMed] [Google Scholar]
- 4.Dressler G. R. (2009) Advances in early kidney specification, development and patterning. Development 136, 3863–3874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hinchliffe S. A., Sargent P. H., Howard C. V., Chan Y. F., van Velzen D. (1991) Human intrauterine renal growth expressed in absolute number of glomeruli assessed by the disector method and Cavalieri principle. Lab. Invest. 64, 777–784 [PubMed] [Google Scholar]
- 6.Hartman H. A., Lai H. L., Patterson L. T. (2007) Cessation of renal morphogenesis in mice. Dev. Biol. 310, 379–387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brunskill E. W., Lai H. L., Jamison D. C., Potter S. S., Patterson L. T. (2011) Microarrays and RNA-Seq identify molecular mechanisms driving the end of nephron production. BMC Dev. Biol. 11, 15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rumballe B. A., Georgas K. M., Combes A. N., Ju A. L., Gilbert T., Little M. H. (2011) Nephron formation adopts a novel spatial topology at cessation of nephrogenesis. Dev. Biol. 360, 110–122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cebrian C., Asai N., D’Agati V., Costantini F. (2014) The number of fetal nephron progenitor cells limits ureteric branching and adult nephron endowment. Cell Rep. 7, 127–137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brenner B. M., Garcia D. L., Anderson S. (1988) Glomeruli and blood pressure. Less of one, more the other? Am. J. Hypertens. 1, 335–347 [DOI] [PubMed] [Google Scholar]
- 11.Luyckx V. A., Brenner B. M. (2010) The clinical importance of nephron mass. J. Am. Soc. Nephrol. 21, 898–910 [DOI] [PubMed] [Google Scholar]
- 12.Bertram J. F., Cullen-McEwen L. A., Egan G. F., Gretz N., Baldelomar E., Beeman S. C., Bennett K. M. (2014) Why and how we determine nephron number. Pediatr. Nephrol. 29, 575–580 [DOI] [PubMed] [Google Scholar]
- 13.Weber S., Taylor J. C., Winyard P., Baker K. F., Sullivan-Brown J., Schild R., Knüppel T., Zurowska A. M., Caldas-Alfonso A., Litwin M., Emre S., Ghiggeri G. M., Bakkaloglu A., Mehls O., Antignac C., Network E., Schaefer F., Burdine R. D. (2008) SIX2 and BMP4 mutations associate with anomalous kidney development. J. Am. Soc. Nephrol. 19, 891–903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Self M., Lagutin O. V., Bowling B., Hendrix J., Cai Y., Dressler G. R., Oliver G. (2006) Six2 is required for suppression of nephrogenesis and progenitor renewal in the developing kidney. EMBO J. 25, 5214–5228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schuchardt A., D’Agati V., Pachnis V., Costantini F. (1996) Renal agenesis and hypodysplasia in ret-k- mutant mice result from defects in ureteric bud development. Development 122, 1919–1929 [DOI] [PubMed] [Google Scholar]
- 16.Zhang Z., Quinlan J., Hoy W., Hughson M. D., Lemire M., Hudson T., Hueber P. A., Benjamin A., Roy A., Pascuet E., Goodyer M., Raju C., Houghton F., Bertram J., Goodyer P. (2008) A common RET variant is associated with reduced newborn kidney size and function. J. Am. Soc. Nephrol. 19, 2027–2034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Quinlan J., Lemire M., Hudson T., Qu H., Benjamin A., Roy A., Pascuet E., Goodyer M., Raju C., Zhang Z., Houghton F., Goodyer P. (2007) A common variant of the PAX2 gene is associated with reduced newborn kidney size. J. Am. Soc. Nephrol. 18, 1915–1921 [DOI] [PubMed] [Google Scholar]
- 18.Ingelfinger J. R. (2008) Disparities in renal endowment: causes and consequences. Adv. Chronic Kidney Dis. 15, 107–114 [DOI] [PubMed] [Google Scholar]
- 19.Ho J., Pandey P., Schatton T., Sims-Lucas S., Khalid M., Frank M. H., Hartwig S., Kreidberg J. A. (2011) The pro-apoptotic protein Bim is a microRNA target in kidney progenitors. J. Am. Soc. Nephrol. 22, 1053–1063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chu J. Y. S., Sims-Lucas S., Bushnell D. S., Bodnar A. J., Kreidberg J. A., Ho J. (2014) Dicer function is required in the metanephric mesenchyme for early kidney development. Am. J. Physiol. Renal Physiol. 306, F764–F772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Marrone A. K., Stolz D. B., Bastacky S. I., Kostka D., Bodnar A. J., Ho J. (2014) MicroRNA-17~92 is required for nephrogenesis and renal function. J. Am. Soc. Nephrol. 25, 1440–1452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nagalakshmi V. K., Ren Q., Pugh M. M., Valerius M. T., McMahon A. P., Yu J. (2011) Dicer regulates the development of nephrogenic and ureteric compartments in the mammalian kidney. Kidney Int. 79, 317–330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bartel D. P. (2004) MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116, 281–297 [DOI] [PubMed] [Google Scholar]
- 24.Wilczynska A., Bushell M. (2015) The complexity of miRNA-mediated repression. Cell Death Differ. 22, 22–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kobayashi A., Valerius M. T., Mugford J. W., Carroll T. J., Self M., Oliver G., McMahon A. P. (2008) Six2 defines and regulates a multipotent self-renewing nephron progenitor population throughout mammalian kidney development. Cell Stem Cell 3, 169–181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Harfe B. D., McManus M. T., Mansfield J. H., Hornstein E., Tabin C. J. (2005) The RNaseIII enzyme Dicer is required for morphogenesis but not patterning of the vertebrate limb. Proc. Natl. Acad. Sci. USA 102, 10898–10903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Katz S. G., Labelle J. L., Meng H., Valeriano R. P., Fisher J. K., Sun H., Rodig S. J., Kleinstein S. H., Walensky L. D. (2014) Mantle cell lymphoma in cyclin D1 transgenic mice with Bim-deficient B cells. Blood 123, 884–893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Takeuchi O., Fisher J., Suh H., Harada H., Malynn B. A., Korsmeyer S. J. (2005) Essential role of BAX,BAK in B cell homeostasis and prevention of autoimmune disease. Proc. Natl. Acad. Sci. USA 102, 11272–11277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Livak K. J., Schmittgen T. D. (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 25, 402–408 [DOI] [PubMed] [Google Scholar]
- 30.Takasato M., Er P. X., Becroft M., Vanslambrouck J. M., Stanley E. G., Elefanty A. G., Little M. H. (2014) Directing human embryonic stem cell differentiation towards a renal lineage generates a self-organizing kidney. Nat. Cell Biol. 16, 118–126 [DOI] [PubMed] [Google Scholar]
- 31.Chi L., Galtseva A., Chen L., Mo R., Hui C. C., Rosenblum N. D. (2013) Kif3a controls murine nephron number via GLI3 repressor, cell survival, and gene expression in a lineage-specific manner. PLoS One 8, e65448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Montagne C., Gonzalez-Gaitan M. (2014) Sara endosomes and the asymmetric division of intestinal stem cells. Development 141, 2014–2023 [DOI] [PubMed] [Google Scholar]
- 33.Neves J., Parada C., Chamizo M., Giráldez F. (2011) Jagged 1 regulates the restriction of Sox2 expression in the developing chicken inner ear: a mechanism for sensory organ specification. Development 138, 735–744 [DOI] [PubMed] [Google Scholar]
- 34.Gebeshuber C. A., Kornauth C., Dong L., Sierig R., Seibler J., Reiss M., Tauber S., Bilban M., Wang S., Kain R., Böhmig G. A., Moeller M. J., Gröne H. J., Englert C., Martinez J., Kerjaschki D. (2013) Focal segmental glomerulosclerosis is induced by microRNA-193a and its downregulation of WT1. Nat. Med. 19, 481–487 [DOI] [PubMed] [Google Scholar]
- 35.Henderson-Smith A., Chow D., Meechoovet B., Aziz M., Jacobson S. A., Shill H. A., Sabbagh M. N., Caviness J. N., Adler C. H., Driver-Dunckley E. D., Beach T. G., Yin H., Dunckley T. (2013) SMG1 identified as a regulator of Parkinson’s disease-associated alpha-synuclein through siRNA screening. PLoS One 8, e77711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brunjes P. C., Osterberg S. K. (2015) Developmental markers expressed in neocortical layers are differentially exhibited in olfactory cortex. PLoS One 10, e0138541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhong B., Liu X., Wang X., Chang S. H., Liu X., Wang A., Reynolds J. M., Dong C. (2012) Negative regulation of IL-17-mediated signaling and inflammation by the ubiquitin-specific protease USP25. Nat. Immunol. 13, 1110–1117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Aerbajinai W., Giattina M., Lee Y. T., Raffeld M., Miller J. L. (2003) The proapoptotic factor Nix is coexpressed with Bcl-xL during terminal erythroid differentiation. Blood 102, 712–717 [DOI] [PubMed] [Google Scholar]
- 39.Fuchs C., Rosner M., Dolznig H., Mikula M., Kramer N., Hengstschläger M. (2012) Tuberin and PRAS40 are anti-apoptotic gatekeepers during early human amniotic fluid stem-cell differentiation. Hum. Mol. Genet. 21, 1049–1061 [DOI] [PubMed] [Google Scholar]
- 40.Mao Z., Sun J., Feng B., Ma J., Zang L., Dong F., Zhang D., Zheng M. (2013) The metastasis suppressor, N-myc downregulated gene 1 (NDRG1), is a prognostic biomarker for human colorectal cancer. PLoS One 8, e68206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cullen-McEwen L. A., Armitage J. A., Nyengaard J. R., Bertram J. F. (2012) Estimating nephron number in the developing kidney using the physical disector/fractionator combination. Methods Mol. Biol. 886, 333–350 [DOI] [PubMed] [Google Scholar]
- 42.LaBelle J. L., Katz S. G., Bird G. H., Gavathiotis E., Stewart M. L., Lawrence C., Fisher J. K., Godes M., Pitter K., Kung A. L., Walensky L. D. (2012) A stapled BIM peptide overcomes apoptotic resistance in hematologic cancers. J. Clin. Invest. 122, 2018–2031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Georgas K., Rumballe B., Valerius M. T., Chiu H. S., Thiagarajan R. D., Lesieur E., Aronow B. J., Brunskill E. W., Combes A. N., Tang D., Taylor D., Grimmond S. M., Potter S. S., McMahon A. P., Little M. H. (2009) Analysis of early nephron patterning reveals a role for distal RV proliferation in fusion to the ureteric tip via a cap mesenchyme-derived connecting segment. Dev. Biol. 332, 273–286 [DOI] [PubMed] [Google Scholar]
- 44.Bernardo-Garcia F. J., Fritsch C., Sprecher S. G. (2016) The transcription factor Glass links eye field specification with photoreceptor differentiation in Drosophila. Development 143, 1413–1423 [DOI] [PubMed] [Google Scholar]
- 45.Castoldi M., Benes V., Hentze M. W., Muckenthaler M. U. (2007) miChip: a microarray platform for expression profiling of microRNAs based on locked nucleic acid (LNA) oligonucleotide capture probes. Methods 43, 146–152 [DOI] [PubMed] [Google Scholar]
- 46.Castoldi M., Schmidt S., Benes V., Noerholm M., Kulozik A. E., Hentze M. W., Muckenthaler M. U. (2006) A sensitive array for microRNA expression profiling (miChip) based on locked nucleic acids (LNA). RNA 12, 913–920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sive H. L., Grainger R. M., Harland R. M. (1998) Early Development of Xenopus laevis: A Laboratory Manual, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, USA [Google Scholar]
- 48. Nieuwkoop, P. D. E., and Faber, J. E. (1956) Normal Table of Xenopus laevis (Daudin): A Systematical and Chronological Survey of the Development From the Fertilized Egg Till the End of Metamorphosis, North-Holland Publishing Co., Amsterdam. [Google Scholar]
- 49.Bonev B., Papalopulu N. (2012) Methods to analyze microRNA expression and function during Xenopus development. Methods Mol. Biol. 917, 445–459 [DOI] [PubMed] [Google Scholar]
- 50.Bouillet P., Zhang L. C., Huang D. C., Webb G. C., Bottema C. D., Shore P., Eyre H. J., Sutherland G. R., Adams J. M. (2001) Gene structure alternative splicing, and chromosomal localization of pro-apoptotic Bcl-2 relative Bim. Mamm. Genome 12, 163–168 [DOI] [PubMed] [Google Scholar]
- 51.Sionov R. V., Vlahopoulos S. A., Granot Z. (2015) Regulation of Bim in health and disease. Oncotarget 6, 23058–23134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Xiao G. Y., Cheng C. C., Chiang Y. S., Cheng W. T., Liu I. H., Wu S. C. (2016) Exosomal miR-10a derived from amniotic fluid stem cells preserves ovarian follicles after chemotherapy. Sci. Rep. 6, 23120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Guo C., Deng Y., Liu J., Qian L. (2015) Cardiomyocyte-specific role of miR-24 in promoting cell survival. J. Cell. Mol. Med. 19, 103–112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Qian L., Van Laake L. W., Huang Y., Liu S., Wendland M. F., Srivastava D. (2011) miR-24 inhibits apoptosis and represses Bim in mouse cardiomyocytes. J. Exp. Med. 208, 549–560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang L., Qian L. (2014) miR-24 regulates intrinsic apoptosis pathway in mouse cardiomyocytes. PLoS One 9, e85389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Harada M., Pokrovskaja-Tamm K., Söderhäll S., Heyman M., Grander D., Corcoran M. (2012) Involvement of miR17 pathway in glucocorticoid-induced cell death in pediatric acute lymphoblastic leukemia. Leuk. Lymphoma 53, 2041–2050 [DOI] [PubMed] [Google Scholar]
- 57.Kan T., Sato F., Ito T., Matsumura N., David S., Cheng Y., Agarwal R., Paun B. C., Jin Z., Olaru A. V., Selaru F. M., Hamilton J. P., Yang J., Abraham J. M., Mori Y., Meltzer S. J. (2009) The miR-106b-25 polycistron, activated by genomic amplification, functions as an oncogene by suppressing p21 and Bim. Gastroenterology 136, 1689–1700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Romaker D., Kumar V., Cerqueira D. M., Cox R. M., Wessely O. (2014) MicroRNAs are critical regulators of tuberous sclerosis complex and mTORC1 activity in the size control of the Xenopus kidney. Proc. Natl. Acad. Sci. USA 111, 6335–6340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Barak H., Huh S. H., Chen S., Jeanpierre C., Martinovic J., Parisot M., Bole-Feysot C., Nitschké P., Salomon R., Antignac C., Ornitz D. M., Kopan R. (2012) FGF9 and FGF20 maintain the stemness of nephron progenitors in mice and man. Dev. Cell 22, 1191–1207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Grieshammer U., Cebrián C., Ilagan R., Meyers E., Herzlinger D., Martin G. R. (2005) FGF8 is required for cell survival at distinct stages of nephrogenesis and for regulation of gene expression in nascent nephrons. Development 132, 3847–3857 [DOI] [PubMed] [Google Scholar]
- 61.Perantoni A. O., Timofeeva O., Naillat F., Richman C., Pajni-Underwood S., Wilson C., Vainio S., Dove L. F., Lewandoski M. (2005) Inactivation of FGF8 in early mesoderm reveals an essential role in kidney development. Development 132, 3859–3871 [DOI] [PubMed] [Google Scholar]
- 62.Poladia D. P., Kish K., Kutay B., Hains D., Kegg H., Zhao H., Bates C. M. (2006) Role of fibroblast growth factor receptors 1 and 2 in the metanephric mesenchyme. Dev. Biol. 291, 325–339 [DOI] [PubMed] [Google Scholar]
- 63.Sims-Lucas S., Cusack B., Baust J., Eswarakumar V. P., Masatoshi H., Takeuchi A., Bates C. M. (2011) Fgfr1 and the IIIc isoform of Fgfr2 play critical roles in the metanephric mesenchyme mediating early inductive events in kidney development. Dev. Dyn. 240, 240–249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rothenpieler U. W., Dressler G. R. (1993) Pax-2 is required for mesenchyme-to-epithelium conversion during kidney development. Development 119, 711–720 [DOI] [PubMed] [Google Scholar]
- 65.Kreidberg J. A., Sariola H., Loring J. M., Maeda M., Pelletier J., Housman D., Jaenisch R. (1993) WT-1 is required for early kidney development. Cell 74, 679–691 [DOI] [PubMed] [Google Scholar]
- 66.Nishinakamura R., Matsumoto Y., Nakao K., Nakamura K., Sato A., Copeland N. G., Gilbert D. J., Jenkins N. A., Scully S., Lacey D. L., Katsuki M., Asashima M., Yokota T. (2001) Murine homolog of SALL1 is essential for ureteric bud invasion in kidney development. Development 128, 3105–3115 [DOI] [PubMed] [Google Scholar]
- 67.Kamada S., Shimono A., Shinto Y., Tsujimura T., Takahashi T., Noda T., Kitamura Y., Kondoh H., Tsujimoto Y. (1995) bcl-2 deficiency in mice leads to pleiotropic abnormalities: accelerated lymphoid cell death in thymus and spleen, polycystic kidney, hair hypopigmentation, and distorted small intestine. Cancer Res. 55, 354–359 [PubMed] [Google Scholar]
- 68.Hutcheson J., Scatizzi J. C., Siddiqui A. M., Haines G. K. III, Wu T., Li Q. Z., Davis L. S., Mohan C., Perlman H. (2008) Combined deficiency of proapoptotic regulators Bim and Fas results in the early onset of systemic autoimmunity. Immunity 28, 206–217 [DOI] [PubMed] [Google Scholar]
- 69.Nakayama K., Nakayama K., Negishi I., Kuida K., Sawa H., Loh D. Y. (1994) Targeted disruption of Bcl-2 alpha beta in mice: occurrence of gray hair, polycystic kidney disease, and lymphocytopenia. Proc. Natl. Acad. Sci. USA 91, 3700–3704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sorenson C. M., Rogers S. A., Korsmeyer S. J., Hammerman M. R. (1995) Fulminant metanephric apoptosis and abnormal kidney development in bcl-2-deficient mice. Am. J. Physiol. 268, F73–F81 [DOI] [PubMed] [Google Scholar]
- 71.Bouillet P., Cory S., Zhang L. C., Strasser A., Adams J. M. (2001) Degenerative disorders caused by Bcl-2 deficiency prevented by loss of its BH3-only antagonist Bim. Dev. Cell 1, 645–653 [DOI] [PubMed] [Google Scholar]
- 72.Strasser A. (2005) The role of BH3-only proteins in the immune system. Nat. Rev. Immunol. 5, 189–200 [DOI] [PubMed] [Google Scholar]
- 73.Chen M., Huang L., Wang J. (2007) Deficiency of Bim in dendritic cells contributes to overactivation of lymphocytes and autoimmunity. Blood 109, 4360–4367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bouillet P., Metcalf D., Huang D. C., Tarlinton D. M., Kay T. W., Köntgen F., Adams J. M., Strasser A. (1999) Proapoptotic Bcl-2 relative Bim required for certain apoptotic responses, leukocyte homeostasis, and to preclude autoimmunity. Science 286, 1735–1738 [DOI] [PubMed] [Google Scholar]
- 75.Hübner A., Barrett T., Flavell R. A., Davis R. J. (2008) Multisite phosphorylation regulates Bim stability and apoptotic activity. Mol. Cell 30, 415–425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ley R., Ewings K. E., Hadfield K., Cook S. J. (2005) Regulatory phosphorylation of Bim: sorting out the ERK from the JNK. Cell Death Differ. 12, 1008–1014 [DOI] [PubMed] [Google Scholar]
- 77.Giam M., Huang D. C. S., Bouillet P. (2008) BH3-only proteins and their roles in programmed cell death. Oncogene 27(Suppl 1), S128–S136 [DOI] [PubMed] [Google Scholar]
- 78.Eichhorn J. M., Alford S. E., Sakurikar N., Chambers T. C. (2014) Molecular analysis of functional redundancy among anti-apoptotic Bcl-2 proteins and its role in cancer cell survival. Exp. Cell Res. 322, 415–424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Barthson J., Germano C. M., Moore F., Maida A., Drucker D. J., Marchetti P., Gysemans C., Mathieu C., Nuñez G., Jurisicova A., Eizirik D. L., Gurzov E. N. (2011) Cytokines tumor necrosis factor-α and interferon-γ induce pancreatic β-cell apoptosis through STAT1-mediated Bim protein activation. J. Biol. Chem. 286, 39632–39643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ren D., Sun J., Mao L., Ye H., Polonsky K. S. (2014) BH3-only molecule Bim mediates β-cell death in IRS2 deficiency. Diabetes 63, 3378–3387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wali J. A., Rondas D., McKenzie M. D., Zhao Y., Elkerbout L., Fynch S., Gurzov E. N., Akira S., Mathieu C., Kay T. W., Overbergh L., Strasser A., Thomas H. E. (2014) The proapoptotic BH3-only proteins Bim and Puma are downstream of endoplasmic reticulum and mitochondrial oxidative stress in pancreatic islets in response to glucotoxicity. Cell Death Dis. 5, e1124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Biswas S. C., Shi Y., Vonsattel J. P., Leung C. L., Troy C. M., Greene L. A. (2007) Bim is elevated in Alzheimer’s disease neurons and is required for beta-amyloid-induced neuronal apoptosis. J. Neurosci. 27, 893–900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Sanphui P., Biswas S. C. (2013) FoxO3a is activated and executes neuron death via Bim in response to β-amyloid. Cell Death Dis. 4, e625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Leon R., Bhagavatula N., Ulukpo O., McCollum M., Wei J. (2010) BimEL as a possible molecular link between proteasome dysfunction and cell death induced by mutant huntingtin. Eur. J. Neurosci. 31, 1915–1925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Perier C., Vila M. (2012) Mitochondrial biology and Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2, a009332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Akiyama T., Dass C. R., Choong P. F. M. (2009) Bim-targeted cancer therapy: a link between drug action and underlying molecular changes. Mol. Cancer Ther. 8, 3173–3180 [DOI] [PubMed] [Google Scholar]
- 87.Bouillet P., Zhang L. C., Huang D. C. S., Webb G. C., Bottema C. D. K., Shore P., Eyre H. J., Sutherland G. R., Adams J. M. (2001) Gene structure alternative splicing, and chromosomal localization of pro-apoptotic Bcl-2 relative Bim. Mamm. Genome 12, 163–168 [DOI] [PubMed] [Google Scholar]
- 88.Gilley J., Coffer P. J., Ham J. (2003) FOXO transcription factors directly activate bim gene expression and promote apoptosis in sympathetic neurons. J. Cell Biol. 162, 613–622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Yang Y., Zhao Y., Liao W., Yang J., Wu L., Zheng Z., Yu Y., Zhou W., Li L., Feng J., Wang H., Zhu W. G. (2009) Acetylation of FoxO1 activates Bim expression to induce apoptosis in response to histone deacetylase inhibitor depsipeptide treatment. Neoplasia 11, 313–324 IN1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Gogada R., Yadav N., Liu J., Tang S., Zhang D., Schneider A., Seshadri A., Sun L., Aldaz C. M., Tang D. G., Chandra D. (2013) Bim, a proapoptotic protein, up-regulated via transcription factor E2F1-dependent mechanism, functions as a prosurvival molecule in cancer. J. Biol. Chem. 288, 368–381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Biswas S. C., Shi Y., Sproul A., Greene L. A. (2007) Pro-apoptotic Bim induction in response to nerve growth factor deprivation requires simultaneous activation of three different death signaling pathways. J. Biol. Chem. 282, 29368–29374 [DOI] [PubMed] [Google Scholar]
- 92.Campone M., Noël B., Couriaud C., Grau M., Guillemin Y., Gautier F., Gouraud W., Charbonnel C., Campion L., Jézéquel P., Braun F., Barré B., Coqueret O., Barillé-Nion S., Juin P. (2011) c-Myc dependent expression of pro-apoptotic Bim renders HER2-overexpressing breast cancer cells dependent on anti-apoptotic Mcl-1. Mol. Cancer 10, 110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ridinger-Saison M., Evanno E., Gallais I., Rimmelé P., Selimoglu-Buet D., Sapharikas E., Moreau-Gachelin F., Guillouf C. (2013) Epigenetic silencing of Bim transcription by Spi-1/PU.1 promotes apoptosis resistance in leukaemia. Cell Death Differ. 20, 1268–1278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Matsui H., Asou H., Inaba T. (2007) Cytokines direct the regulation of Bim mRNA stability by heat-shock cognate protein 70. Mol. Cell 25, 99–112 [DOI] [PubMed] [Google Scholar]
- 95.Dávila D., Jiménez-Mateos E. M., Mooney C. M., Velasco G., Henshall D. C., Prehn J. H. M. (2014) Hsp27 binding to the 3'UTR of bim mRNA prevents neuronal death during oxidative stress-induced injury: a novel cytoprotective mechanism. Mol. Biol. Cell 25, 3413–3423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Lei K., Davis R. J. (2003) JNK phosphorylation of Bim-related members of the Bcl2 family induces Bax-dependent apoptosis. Proc. Natl. Acad. Sci. USA 100, 2432–2437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Lu J., Quearry B., Harada H. (2006) p38-MAP kinase activation followed by BIM induction is essential for glucocorticoid-induced apoptosis in lymphoblastic leukemia cells. FEBS Lett. 580, 3539–3544 [DOI] [PubMed] [Google Scholar]
- 98.Zhang L., Insel P. A. (2004) The pro-apoptotic protein Bim is a convergence point for cAMP/protein kinase A- and glucocorticoid-promoted apoptosis of lymphoid cells. J. Biol. Chem. 279, 20858–20865 [DOI] [PubMed] [Google Scholar]
- 99.Liu R., Zhang H., Wang X., Zhou L., Li H., Deng T., Qu Y., Duan J., Bai M., Ge S., Ning T., Zhang L., Huang D., Ba Y. (2015) The miR-24-Bim pathway promotes tumor growth and angiogenesis in pancreatic carcinoma. Oncotarget 6, 43831–43842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Xiao C., Srinivasan L., Calado D. P., Patterson H. C., Zhang B., Wang J., Henderson J. M., Kutok J. L., Rajewsky K. (2008) Lymphoproliferative disease and autoimmunity in mice with increased miR-17-92 expression in lymphocytes. Nat. Immunol. 9, 405–414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Ventura A., Young A. G., Winslow M. M., Lintault L., Meissner A., Erkeland S. J., Newman J., Bronson R. T., Crowley D., Stone J. R., Jaenisch R., Sharp P. A., Jacks T. (2008) Targeted deletion reveals essential and overlapping functions of the miR-17 through 92 family of miRNA clusters. Cell 132, 875–886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Barreyro F. J., Kobayashi S., Bronk S. F., Werneburg N. W., Malhi H., Gores G. J. (2007) Transcriptional regulation of Bim by FoxO3A mediates hepatocyte lipoapoptosis. J. Biol. Chem. 282, 27141–27154 [DOI] [PubMed] [Google Scholar]
- 103.Dijkers P. F., Medema R. H., Lammers J. W. J., Koenderman L., Coffer P. J. (2000) Expression of the pro-apoptotic Bcl-2 family member Bim is regulated by the forkhead transcription factor FKHR-L1. Curr. Biol. 10, 1201–1204 [DOI] [PubMed] [Google Scholar]
- 104.Essafi A., Fernández de Mattos S., Hassen Y. A. M., Soeiro I., Mufti G. J., Thomas N. S. B., Medema R. H., Lam E. W. F. (2005) Direct transcriptional regulation of Bim by FoxO3a mediates STI571-induced apoptosis in Bcr-Abl-expressing cells. Oncogene 24, 2317–2329 [DOI] [PubMed] [Google Scholar]
- 105.Sunters A., Fernández de Mattos S., Stahl M., Brosens J. J., Zoumpoulidou G., Saunders C. A., Coffer P. J., Medema R. H., Coombes R. C., Lam E. W. F. (2003) FoxO3a transcriptional regulation of Bim controls apoptosis in paclitaxel-treated breast cancer cell lines. J. Biol. Chem. 278, 49795–49805 [DOI] [PubMed] [Google Scholar]
- 106.Köhler U. A., Kurinna S., Schwitter D., Marti A., Schäfer M., Hellerbrand C., Speicher T., Werner S. (2014) Activated Nrf2 impairs liver regeneration in mice by activation of genes involved in cell-cycle control and apoptosis. Hepatology 60, 670–678 [DOI] [PubMed] [Google Scholar]
- 107.Puthalakath H., O’Reilly L. A., Gunn P., Lee L., Kelly P. N., Huntington N. D., Hughes P. D., Michalak E. M., McKimm-Breschkin J., Motoyama N., Gotoh T., Akira S., Bouillet P., Strasser A. (2007) ER stress triggers apoptosis by activating BH3-only protein Bim. Cell 129, 1337–1349 [DOI] [PubMed] [Google Scholar]
- 108.Hershko T., Ginsberg D. (2004) Up-regulation of Bcl-2 homology 3 (BH3)-only proteins by E2F1 mediates apoptosis. J. Biol. Chem. 279, 8627–8634 [DOI] [PubMed] [Google Scholar]
- 109.Chen S., Brunskill E. W., Potter S. S., Dexheimer P. J., Salomonis N., Aronow B. J., Hong C. I., Zhang T., Kopan R. (2015) Intrinsic age-dependent changes and cell-cell contacts regulate nephron progenitor lifespan. Dev. Cell 35, 49–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.