

Abstract
Diverse cellular functions are controlled by RhoA-GTPases, which are activated by trimeric G proteins via RhoGEFs, among others. In this study, we focused on the signaling from GPCRs to RhoA via Gα13 and leukemia-associated RhoGEF (LARG). The activation of Gα13 was elucidated in living cells with high temporal and spatial resolution by means of FRET. The inactivation after agonist withdrawal occurred in the same range (t1/2 = 25.3 ± 2.2 s; mean ± sem; n = 22) as described for other Gα proteins. The interaction of Gα13 and LARG and the thereby-induced LARG translocation to the plasma membrane were at least 1 order of magnitude more stable after agonist withdrawal, exceeding Gα13 deactivation in the absence of LARG several fold. Consequently, we observed an almost 100-fold higher agonist sensitivity of the Gα13 LARG interaction compared to the Gα13 activation in the absence of LARG.—Bodmann, E.-L., Krett, A.-L., Bünemann, M. Potentiation of receptor responses induced by prolonged binding of Gα13 and leukemia-associated RhoGEF.
Keywords: ARHGEF12, G protein, thromboxane, FRET, Rho
Signals from the extracellular space can be transmitted into the cell by GPCRs. Inside the cells certain members of the family of heterotrimeric G proteins are recruited to activated GPCRs, dependent on their G-protein specificity. The Gα12/13 family of heterotrimeric G proteins is activated by several physiologically important GPCRs and mediates important effects, such as platelet degranulation (1), angiogenesis (2), and vascular smooth muscle contraction (3). However, to date, all GPCRs that have been reported to activate the Gα12/13 family also activate additional Gα families, such as Gq/11 (4). Thus, the correlation of intracellular effects with the specific activation of Gα12/13 family by GPCRs is challenging (reviewed in ref. 5). Real-time measurements of G-protein activity by means of Förster resonance energy transfer (FRET) have been successfully applied to Gαs, Gαi, and Gαq family members (6–8), but so far, not to Gα12/13 proteins.
The best understood downstream effect of active Gα12/13 family proteins is the activation of RhoGTPases via RhoGEFs (5). The Rho family (RhoGTPases) regulates actin dynamics and is involved in a variety of cellular functions, such as vesicular trafficking, cell proliferation, contractility, and gene expression (9). For example, the serum response element (SRE) is activated downstream of RhoA via actin and megakaryocytic acute leukemia (MAL)-serum response factor (SRF) and regulates targets such as immediate-early and muscle-specific genes (10).
Among other RhoGEFs, RH-RhoGEFs [namely, leukemia-associated RhoGEF (LARG), PDZ-RhoGEF, and p115-RhoGEF] are direct interaction partners of the Gα12/13 family and RhoA (reviewed in ref. 11). They share an RGS homology domain and a DH/PH domain common to many RhoGEFs. The RH-RhoGEF LARG, also known as ARHGEF12, was first described as part of a fusion protein in a patient with acute myeloid leukemia (12, 13). More recently, LARG has been found to be the most abundant RhoGEF in the heart and is claimed to be the central player in pressure-overload–induced hypertrophy (14). Although eGFP-labeled LARG was solely localized in the cytosol of MDCK cells, coexpression of constitutive active Gα12-induced LARG localization at the plasma membrane (15). It is still a matter of debate whether translocation induced by the Gα12/13 family alone activates RH-RhoGEFs, or whether interaction with the catalytic domain is needed in addition (16, 17).
Our purpose was to visualize receptor-mediated Gα13 activation and interaction with the effector LARG in intact cells to resolve the kinetics of crucial initial steps of Gα13-mediated signaling. Therefore, we established FRET-based assays and measured G-protein activation and interaction with its effector in living single cells upon agonist-mediated thromboxane A2 receptor (TPα-R) stimulation. Using these assays, we studied the dynamics and sensitivity of the signaling cascade and observed a strikingly long-lasting interaction between LARG and Gα13, accompanied by a dramatically enhanced agonist sensitivity of the LARG Gα13 interaction compared with the Gα13 activation observed in the absence of LARG, which is somewhat reminiscent of a much less pronounced high agonist sensitivity reported for the inhibition of adenylyl cyclase V by Gαi (18).
MATERIALS AND METHODS
Plasmids and agonist
Mouse Gα13-XFP [enhanced yellow fluorescent protein (eYFP) as well as monomer turquoise fluorescent protein (mTur2)] was cloned by insertion of an NheI restriction site between aa 127 and 128. Next, eYFP or mTur2 (19) was amplified with flanking NheI restriction sites and inserted into Gα13. All LARG constructs were cloned from human pHyg-LARG, provided by B. Moepps (20) (Institute of Pharmacology and Toxicology, University of Ulm, Ulm, Germany). YFP-LARG was cloned by replacing Gβ1 in Gβ1-N-YFP (21) with LARG, using the BamHI and XhoI restrictions sites. For LARG-insYFP, eYFP from YFP-LARG was inserted at an intrinsic AscI restriction site within wild-type LARG. This restriction site encodes a cut behind bp 2781 (aa 624). The monomer red fluorescent protein (mRFP)-LARG was cloned from YFP-LARG and pcDNA3-mRFP, which was a gift from D. Golenbock (Infectious Disease; University of Massachusetts Medical School, Worcester, MA, USA; plasmid 13032; Addgene, Cambridge, MA, USA) with KpnI and BamHI restriction enzymes. Further, we used human TPα-R [NM_001060.50, cDNA from T. Wieland (University of Heidelberg, Mannheim, Germany), cloned into pcDNA3 backbone vector for this study], human β2AR-mTur2 (C. Krasel, University of Marburg), mouse Gα13 (NM_010303.3, cDNA from N. Wettschureck, Max-Planck-Institute for Heart and Lung Research, Bad Nauheim, Germany), human constitutive active Gα13QL [a kind gift from T. Wieland (22)], human Gβ1 (8), human Gβ1-cerulean (Cer) (21), bovine Gγ2 (8), monomer cyan fluorescent protein (mCFP) (23), eYFP-β2-adrenoceptor-mTur2 cloned as described for similar constructs in Dorsch et al. (24), pRL-TK (Promega, GmbH, Mannheim, Germany), pSRE.L (luciferase) [provided by T. Wieland (25)], pcDNA3 (Thermo Fisher Scientific, Waltham, MA, USA) and human M3-R was obtained from the Missouri S&T cDNA Resource Center (http://www.cdna.org). All plasmids used were verified by sequencing.
The agonist U-46619 (16450; Cayman Chemical, Ann Arbor, MI, USA) was prepared as a stock solution of 2.5 mM solved in ethanol and stored at −20°C. On the day of the experiment, the solution was further diluted with Tyrode buffer [137 mM NaCl, 5.4 mM KCl, 2 mM CaCl2, 1 mM MgCl2, and 10 mM HEPES (pH 7.3)] containing 0.1% delipidized bovine serum albumin (BSA) (Sigma-Aldrich, Munich, Germany).
Cell culture and transfection
HEK (human embryonic kidney) 293T cells (kind gift from M. Lohse, University of Würzburg, Würzburg, Germany) were chosen as a suitable cellular system for expression of fluorescently labeled constructs and subsequent FRET measurements. They were maintained as described previously (6) for single-cell experiments and transfected with Effectene reagent (Qiagen, Hilden, Germany), according to the manufacturer’s protocol. MycoAlert LT07-118 (Lonza, Cologne, Germany) is used regularly to check for and avoid mycoplasma contamination.
Lipofectamine RNAiMax (Thermo Fisher Scientific) was used according to the manufacturer’s protocol to transfect small interfering RNA (siRNA) pools. At 6 to 8 h later, the cDNA transfection was performed as usual without removing ingredients for the siRNA transfection from the cells. The mock-siRNA pool and siRNA buffer were purchased from Dharmacon (SO-2480722G, D-001810-10-05; Lafayette, CO, USA), siRNAs against LARG, PDZ-RhoGEF, and p115-RhoGEF were purchased from Santa Cruz Biotechnology (sc 41800, sc 45823, sc-41734; Dallas, TX, USA). Forty-two picomoles for each siRNA was pooled and transfected to silence endogenous Rho-GEFs.
The activation of Gα13 was measured in HEK293T cells transfected with 0.5 µg TPα-R or 1 µg H1-R, 0.8 µg Gα13-YFP, 0.5 µg Gβ-Cer, or 0.2 µg Gγ (with TPα-R in Figs. 1, 5A, B, E, F, and 7A, C and H1-R in Fig. 1D). The influence of LARG on Gα13 activation was studied in cells additionally transfected with 1 µg mRFP-LARG (Figs. 6A, B, D, and 7D). The interaction of Gα13 and LARG was detected in cells transfected with 0.5 µg TPα-R, 1 µg Gα13-mTur2, 0.5 µg Gβ, 0.2 µg Gγ, or 1 µg YFP-LARG (Figs. 3A–C, 5A–C, and 7A, C) or LARG-insYFP (Figs. 3D and 5C). As the control, HEK293T cells were transfected with 0.5 µg TPα-R, 1 µg Gα13, 0.5 µg Gβ, 0.2 µg Gγ, 1 µg YFP-LARG, or 1 µg mCFP (Fig. 3C). For translocation experiments, similar conditions as described for the Gα13 LARG interaction were used [Gα13-mTur2 was replaced by Gα13 in some experiments (Figs. 4 and 5D)]. In cases in which muscarinic receptors were used as the control, we expressed human muscarinic M2 receptor (M2-R, C-terminally His-tagged, a kind gift from M. Marlene Hosey, Northwestern Medical School, Chicago, IL, USA) or human muscarinic M3 receptor [M3-R, obtained from the Missouri S&T cDNA Resource Center (http://www.cdna.org)]. For confocal images, cells were transfected as described in Confocal Microscopy.
Figure 1.

Activation and inactivation kinetics of Gα13 determined by means of FRET imaging. A) To measure FRET, eYFP was inserted between aa 127 and 128 of Gα13, as schematically displayed using the crystal structures of YFP- and Gα13-GDP and UCSF chimera (28, 43, 44) (A; gray, α-helical domain; cyan, Ras-like domain; green, linkers; yellow, YFP). Gα13-YFP localized at the plasma membrane in HEK293T cells transfected with TPα-R, Gα13-YFP, Gβ, and Gγ, as visualized by confocal microscopy (see Materials and Methods for DNA amounts; A, bottom). B, D) If Gβ-Cer was transfected, a rapid and reversible increase in YFP fluorescence and decrease in CFP fluorescence upon stimulation with the thromboxane analog U-46619 was detected [representative cell (B) and mean ± sem trace (D), black, n = 22]. The Gα13 activation did not occur upon stimulation of H1-R (D; red, n = 8). C) Direct illumination of Gα13-YFP and Gβ-Cer in a representative cell. E) As determined by donor recovery after acceptor photobleaching, FRET in unstimulated cells transfected as before was significantly less than in stimulated cells of the same kind. Compared to control transfected cells, the stimulated condition shows significantly higher FRET. Numbers of cells measured for each condition are shown in parentheses. ****P < 0.0001 (black asterisks, paired Student’s t test; gray asterisks, unpaired Student’s t test).
Figure 5.

LARG dissociated slower from Gα13 than Gα13 inactivated in the absence of cotransfected LARG. LARG Gα13 interaction (measured as described in Fig. 3) is prolonged compared to Gα13 inactivation (measured as described in Fig. 1) upon agonist withdrawal as shown in representative (A) and mean (B) recordings, which were normalized to their maximum response (black n = 30, red n = 10). C) To test for reversibility of the LARG Gα13 interaction, 10 minutes after washout, the agonist was applied a second time. For both, LARG constructs (black and red with N-terminally and inserted YFP label, respectively), some reversibility of the FRET signal between Gα13-mTur2 and LARG was detected. Depicted are average traces of n = 12 individual cells, measured at 0.5 Hz illumination frequency to reduce bleaching. D) Reversibility of U-46619-induced translocation of YFP-LARG was measured either with unlabeled (red trace) or mTur2-labeled Gα13 (black trace; n = 12 and 9, respectively). For better comparison the y axis was inverted. Data are shown as means ± sem (B–D).
Figure 7.

The EC50 of Gα13 LARG interaction is left shifted compared with Gα13 activation. A) Concentration–response curves for FRET between YFP-LARG and Gα13-mTur2 (black) and Gα13 activation (red) measured in the absence of exogenous LARG were determined by normalization to the indicated reference concentration. Every concentration was tested as often as indicated in brackets, and means ± sem are shown. B, C) Representative single-cell recordings for LARG Gα13 interaction (B) and Gα13 activation (C). D) To test for the effect of LARG on the Gα13 Gβγ assembly, cells either overexpressing mRFP-LARG (red) or siRNA-silenced for LARG (black, see Fig. 6E) were subjected to the Gα13-FRET assay (similar to that described in Fig. 1), and alterations in FRET were monitored upon exposure to the indicated low concentration of agonist. Traces show means ± sem for 12 (black trace) and 14 (red trace) individual cells. Bar graphs show the absolute value of the agonist-induced FRET change, determined by averaging the last 5 time points of the 1 nM U-46619 application. *P = 0.0129, unpaired Student’s t test (means ± sem).
Figure 6.

Impact of LARG expression on Gα13 activity, as indicated by FRET between Gα13-YFP and Gβ-Cer. A) Alteration of FRET between Gα13-YFP and Gβ-Cer induced by 25 nM U-46619 was compared in cells in absence (black) or presence of mRFP-LARG (red, both n = 16). The bar graphs depict the average decrease in F535/F480 of the last 5 time points before withdrawal of U-46619 (means ± sem). P = 0.0358, unpaired Student’s t test. B) In similar experiments as described in A, relaxation kinetics of FRET between Gα13-YFP and Gβ-Cer after withdrawal of agonist (either 10 nM or 1 µM, as indicated) were compared in the absence (black trace, n = 22) or presence of mRFP-LARG (red, n = 5). To reduce the impact of endogenous RH-RhoGEFs under control conditions (black data), endogenous LARG expression was attenuated by siRNA treatment. C) Analysis of relaxation halftimes of the experiments shown in B revealed much slower kinetics in the presence of mRFP LARG (means ± sem). P < 0.0001, unpaired Student’s t test. D) The agonist concentration-dependent alteration in FRET between Gα13-YFP and Gβ-Cer was compared in cells overexpressing mRFP-LARG (red trace), transfected with mock-siRNA (blue trace), or in cells with less endogenous LARG because of siRNA treatment (black trace). Traces were corrected for bleaching and blotted as means ± sem of 10 (blue trace) and 11 cells (black and red traces). E) The siRNA pool, consisting of siRNA against LARG, PDZ-RhoGEF, and p115-RhoGEF, reduced endogenous LARG expression significantly compared to mock-siRNA pool. P < 0.05 (paired Student’s t test), as shown for 7 individual Western blots (means ± sem) and a representative blot. Transfection of mRFP-LARG resulted in an ∼5-fold increase in LARG expression. Protein samples originate from the transfections measured in D and Fig. 7D. Traces are means ± sem (A, B, D).
Figure 3.

TPα-R-stimulated interactions between fluorophore-labeled Gα13 and LARG were resolved by single-cell FRET imaging. A, C) HEK293T cells transfected with 0.5 µg TPα-R, 1 µg Gα13-mTur2, 0.5 µg Gβ, 0.2 Gγ, and 1 µg N-terminally YFP-labeled LARG showed a rapid increase in FRET upon stimulation with 10 nM U-46619 [representative cell in A, and mean ± sem of 30 cells (black trace) in C]. B) YFP-LARG localized mainly to the cytosol, as visualized by confocal microscopy from HEK293T cells transfected with 0.5 µg TPα-R, 1 µg Gα13, 0.5 µg Gβ, 0.2 Gγ, and 1 µg YFP-LARG. C) FRET change upon stimulation with U-46619 was reduced if Gα13 and membrane-bound CFP were transfected instead of Gα13-mTur2 (red trace; mean ± sem of n = 13). D) Also LARG with a YFP inserted between the RH and the DH/PH domain (LARG-insYFP) showed an increase in FRET upon activation of TPα-R (means ± sem; n = 9).
Figure 4.

YFP-LARG translocated from the cytosol to the plasma membrane upon receptor-mediated activation of Gα13. A) Plasma membrane translocation of LARG, expressed as alterations of the ratio of cytosolic over whole-cell fluorescence, was monitored upon treatment with agonist, by confocal microscopy. Depicted is a representative recording from a single cell transfected with TPα-R, Gα13, Gβ, Gγ, and YFP-LARG. B) YFP images of the same cell at the time points indicated by 1 and 2 in A. C) Only minor translocation was observed without Gα13 cotransfection (red trace). Cotransfection of Gα13 led to an increase (black trace, mean ± sem of 8 individual cells each). The mean amplitudes originating from the last 5 time points of the U-46619 application of the cells measured were significantly different (means ± sem). Amplitudes were mirrored at the time axis. **P < 0.01, unpaired Student’s t test.
Luciferase reporter assay and subsequent Western blot analysis
The activation of Rho can be monitored with the reporter plasmid pSRE.L which is regulated by a modified transcriptional regulatory element (25, 26). The experiments were performed with a dual-luciferase reporter assay (Promega). Luciferase assays were performed in a 96-well format in accordance with the manufacturer’s protocol and as described elsewhere in more detail (6). For this study, the following plasmids were transfected: pSRE.L (21.6 ng/well), pRL-TK (3.4 ng/well), and TPα-R (0.25 ng/well), with or without Gα13, Gα13-YFP, or Gα13-mTur2 (0.8 ng/well) and were filled up to 125 ng/well with pcDNA3. Each condition was transfected in triplicate, and 24 h later, the indicated conditions were treated with 5 nM U-46619 dissolved in Tyrode buffer containing 0.00065% EtOH/ 0.1% BSA. An additional 24 h later, probes were measured with an EnVision Multilabel Reader (PerkinElmer, Boston, MA, USA) and by use of the Dual-Luciferase Reporter Assay (Promega). After cell lysis in 25 µl passive lysis buffer (Promega), a brief dose–response curve for the constitutive active Gα13 variant (Gα13Q226L) was measured, and the cell lysates were diluted to give a signal within the dynamic range of the plate reader. The shown experiment is representative for at least 3 independent experiments. The leftover lysates of each triplicate were pooled and used as a control for Gα13 expression by SDS/PAGE and immunoblotting. For detection of Gα13, a polyclonal anti-Gα13 antibody (sc-26788; Santa Cruz Biotechnology) was used; actin was detected by a monoclonal antibody (08691001; MP Biomedicals, Singapore), and the luminescence signals were visualized by ECL (AppliChem, Darmstadt, Germany) using the ChemiDoc XRS+ system (Bio-Rad Laboratories, Hercules, CA, USA).
Antibodies used for the Western blot depicted in Fig. 6E were sc-25638 to detect LARG and sc-562 to detect GRK2 (both from Santa Cruz Biotechnology). Transfected cells were split to be seeded on coverslips for FRET measurements (shown in Figs. 6D and 7C) and on 6-cm dishes for subsequent Western blot analysis.
Recording and analysis of microscopic FRET measurements
HEK293T cells, which were grown on coverslips, were maintained in Tyrode buffer 2 d after transfection at room temperature (20°C) and mounted on an inverted microscope (Axiovert 100; Zeiss, Jena, Germany) equipped with a Plan/Apo N ×60/1.45 oil objective (Nikon, Tokyo, Japan), a pressure-driven perfusion system (VC3-8xP Series; ALA Scientific Instruments, Farmingdale, NY, USA), LED light sources (precisExcite-100, 440 and 500 nm from CoolLED) and a high-performance CCD-camera (Spot Pursuit from Spot Imaging solutions; Sterling Heights, MI, USA) for all FRET measurements, except those shown in Supplemental Fig. S2, for which a previously described microscopic setup was used (18). During FRET measurement, the light path was restricted with a 436/20 filter (Chroma Technology, Bellows Falls, VT, USA) for excitation and for emission a dichroic (458LP; Semrock, Rochester, NY, USA), a beamsplitter (505LP, 416, 500, 582, and 657; Chroma), and emission filters (470/24, Chroma, and 525/39, Semrock) were used. Cells were constantly superfused with Tyrode buffer or U-46619, diluted as indicated in Tyrode buffer with 0.1% delipidized BSA (Sigma-Aldrich) or acetylcholine or histamine diluted in Tyrode buffer. Cells were illuminated with 2 Hz, except where otherwise indicated. Data were stored with the VisiView software (Visitron Systems, Puchheim, Germany) and further processed in Excel (Microsoft, Redmond, WA, USA).
All FRET data were corrected for background fluorescence, bleed-through, and false excitation, and the ones used in Figs. 1B, D, 3, 5A, B, F, and 6 were corrected for bleaching. With Origin Pro (Originlab, Northampton, MA, USA), on- and offset kinetics were evaluated by fitting a monoexponential trace to the individual FRET traces upon agonist stimulation or withdrawal, respectively.
The direct YFP- and CFP-illumination images in Fig. 1C were achieved as follows: stk files originating from direct YFP and CFP measurements with the VisiView software were opened with ImageJ (U.S. National Institutes of Health, Bethesda, MD, USA), and 5 images were background corrected and averaged.
Concentration–response relationships were evaluated for Gα13 activation and its interaction with LARG. Single cells were superfused with 1 or 2 test concentrations, followed by a reference concentration, and FRET amplitudes were evaluated relative to the FRET amplitude at the reference concentration. The individual FRET amplitudes were calculated as FYFP/FCFP during the agonist plateau subtracted by FYFP/FCFP before stimulation (see also red arrows in Fig. 7B, C). For each data point, at least 5 cells were measured, and a concentration–response curve was fitted with Prism (GraphPad, San Diego, CA, USA) with variable top, Hill slope, as well as EC50 and constrained bottom. FRET amplitudes as shown in Figs. 4C, 6A, and 7D were calculated as indicated, taking the average of the 5 last data points before agonist application and before agonist withdrawal (mean ± sem). For statistical comparison, we used an unpaired Student’s t test.
Quantification of the relative expression levels of fluorophore-labeled LARG and Gα13 was obtained by comparing the YFP and CFP emissions of each experiment with a reference construct that shows a 1-to-1 expression of eYFP or mTur2, similar to the experiments performed by Wolters et al. (27).
Measurement of donor recovery after acceptor bleaching
Steady-state FRET was measured as the increase in donor fluorescence upon bleaching of the FRET acceptor YFP (ΔFCFP). The YFP fluorescence was bleached with high LED intensity for 120 s, FCFP was monitored every 1.5 s and CFP fluorescence before (FCFP B) and after (FCFP A) bleaching was used to calculate the donor recovery (Eq. 1):
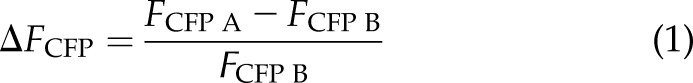 |
Microscopic translocation measurements
For translocation experiments, HEK293T cells were maintained in Tyrode buffer 2 d after transfection and mounted on an inverted fluorescence microscope (IX 7) equipped with a UPlanSApo ×100/1.40 (Olympus, Tokyo, Japan) oil objective, a high-performance EM-CCD digital camera (Hamamatsu, Hamamatsu City, Japan), and a confocal VT-HAWK FRAP imaging system (VisiTech International, Sunderland, United Kingdom). The excitation occurred through a laser (491 nm) and beam splitter (405/491/642 DC; both from VisiTech International) with 0.03 Hz for 200 ms. The emission light path was restricted and split with a beamsplitter (T495lpxr; Chroma) and emission filters (ET470/×40 and ET535/30 m; Chroma). The cells were superfused, as described for FRET measurements, and cytosolic (Fcytosol) and whole-cell YFP fluorescence (Fcell) was monitored over time. The data were corrected for background fluorescence, and YFP translocation to the plasma membrane was calculated using Eq. 2:
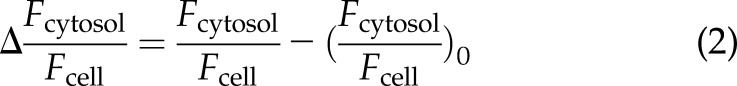 |
Confocal microscopy
Confocal images of Gα13-YFP or N-terminally YFP-labeled LARG were taken with an inverted fluorescence microscope (TCS SP5; Leica, Wetzlar, Germany) equipped with a Lambda Blue ×63/1.4 NA oil objective (Leica) 2 d after transfection (0.5 µg TPα-R, 0.8 µg Gα13-YFP, 0.5 µg Gβ, 0.2 µg Gγ, or 0.5 µg TPα-R, 1 µg Gα13, 0.5 µg Gβ, 0.2 µg Gγ, and 1 µg YFP-LARG). Excitation beam splitter FW DD 458/514 with a 514-nm diode laser was used for illumination, and the YFP emission was detected at a range from 525 to 600 nm. Images were taken with LAS AF software (Leica) in a 512 × 512 pixel format consisting of 4 averaged frame scans. The scan speed was set to 400 Hz, and the pinhole was set to 1 airy unit. Images were saved as .lif files and exported as JPEG with the LAS AF Lite software.
Isolation of cortical mouse neurons
Cortical mouse neurons were isolated from spare wild-type mice (C57BL/6), so that no ethics approval was needed. After anesthesia and cervical fracture, cortical neurons were isolated, resuspended in lysis buffer (0.25 M mannitol, 0.05 M Tris, 1 M EDTA, 1 M EGTA, 1 mM DTT, and 1% Triton-X) containing Complete Mini Protease Inhibitor Cocktail and PhosStop (11697498001 and 04906845001; Roche Diagnostics, Mannheim, Germany), and frozen in liquid nitrogen. After thawing, neurons were centrifuged for 15 min at 10,000 rpm and 4°C, and the supernatant was used to proceed with a Bradford assay.
Statistical analysis
All averaged data are shown as means ± sem of n independent experiments. (For FRET measurements, single cells from different coverslips were regarded as biologic replicates.) No statistical methods were used to predetermine the sample size. For statistical analysis, we used paired t tests for comparing values obtained with the same experiment (cell). The unpaired t test was used for all other statistical comparisons of 2 different conditions. An ANOVA test was used for comparison between multiple conditions.
RESULTS
Dynamics of receptor-induced Gα13 activity characterized by FRET
To study Gα13 activation in living cells, we inserted either eYFP or mTur2 into the α-helical domain of Gα13 between aa 127 and 128 in the αB–αC loop (Fig. 1A) (19). These constructs localized nicely at the plasma membrane, as was visible in confocal images of HEK293T cells transfected with the Gα13-coupled thromboxane A2 receptor Gα13-YFP and unlabeled Gβ and Gγ.
Because the stimulation of the TPα-R with the thromboxane analog U-46619 should lead to a conformational change in the trimeric Gα13 protein, we tested whether a change in FRET between Gα13-YFP and Gβ-Cer would be observed upon stimulation of single living cells with a saturating U-46619 concentration. In most of the cells, a quick, reversible increase in FRET was observed upon stimulation with 1 µM U-46619. The change in FRET reflected the increase in YFP mirrored by the decrease in CFP, demonstrated in a representative cell in Fig. 1B. Proper membrane localization of fluorescent constructs was confirmed by confocal microscopy, as shown in Fig. 1C. In Fig. 1D, a mean ± sem trace of 22 cells of the same condition as in Fig. 1B is shown in black. In contrast, upon stimulation of the Gαq-coupled histaminergic H1 receptor (H1-R) or M2 receptor (M2-R), no increase in FRET was observed between Gα13-YFP and Gβ-Cer (Fig. 1D; Supplemental Fig. S1A). Moreover, no change in FRET was observed upon stimulation of putative endogenous TPα-R with 1 µM U-46619 in HEK293T cells (Supplemental Fig. S1B). To exclude the possibility that bystander FRET (which reflects an unspecific change in distance between Gα13 and a random membrane-bound protein) caused the observed FRET increase, a membrane-bound mCFP was transfected as a putative FRET donor, together with the unlabeled Gβ. As shown in Supplemental Fig. S1C, no change in FRET was observed in this case. These observations show the specificity of the FRET assay between Gα13 and Gβ as a tool to study Gα13 activation in single living cells. The increase in FRET upon stimulation was also detected by donor recovery after acceptor photobleaching (Fig. 1E). As estimated with the Gα13 Gβγ FRET assay and increasing U-46619 concentrations, the Gα13 activation by TPα-R occurs with a maximum ± se kon of 8.01 ± 1.04 s−1, as assessed by fitting (Supplemental Fig. S2). The inactivation upon washout of 1 µM U-46619 took ∼25.3 ± 2.2 s (t1/2; n = 22), as evaluated by the monoexponential fit of the measurements summarized by the black trace in Fig. 1D.
The fluorophore-labeled Gα13 constructs activated downstream signaling in an agonist-dependent fashion, similar to the wild-type Gα13, as investigated by an SRE.L reporter gene assay (Fig. 2A) taking care of equal expression levels as determined by immunoblot analysis (Fig. 2B).
Figure 2.

SRE.L activation upon U-46619 stimulation by Gα13-YFP was comparable to wild-type Gα13. A) Basal (white) and U-46619 induced (red) SRE.L activation by unlabeled and fluorophore-labeled Gα13. Results are shown as relative (rel.) mean ± sem luciferase activity after normalization to nontransfected controls. Activation of the individual conditions by stimulation was tested with unpaired 2-tailed Student’s t tests (black lines and asterisks), and stimulation in the presence of the Gα13 constructs was compared to stimulation in the absence of Gα13 by ANOVA with Dunnet’s multiple comparison test (gray lines and asterisks). B) Expression of the different Gα13 constructs [wild-type (wt) Gα13≈44 kDa, GFP derivate≈28 kDa, top panel) during the SRE.L activation assay was confirmed by Western blot analysis. Actin (≈42 kDa, bottom panel) was used as the loading control. This experiment is representative of 3 individual experiments. ***P < 0.001, ****P < 0.0001.
Interactions between YFP-labeled LARG and Gα13-mTur2 were monitored by FRET
Gα13 activates and interacts with members of the Dbl-RhoGEF family that in turn activate RhoGTPases, the next step in the signaling cascade. Therefore, we sought to monitor the interaction of an effector with Gα13. The first Gα13 effector we chose to study in single living cells by FRET was the widely expressed LARG. Upon stimulation of TPα-R, an increase in FRET was observed between YFP-LARG and Gα13-mTur2 (Fig. 3A, C). The increase occurred with a t1/2 of 3.69 s upon stimulation with 1 µM U-46619 as measured in 9 cells (kon = 0.271 ± 0.023 s−1; mean ± sem). In contrast, if mCFP and unlabeled Gα13 were cotransfected with YFP-LARG, Gβ, Gγ, and TPα-R, almost no change in FRET was observed (Fig. 3C). The YFP-LARG construct is localized mainly to the cytosol (Fig. 3B). In addition, we generated a construct with YFP inserted between the RH and DH/PH domain of LARG (between aa 623 and 624; LARG-insYFP). In cells transfected with this construct, instead of YFP-LARG, an increase in FRET was observed upon stimulation with low agonist concentrations (Fig. 3D). When membrane-bound CFP was cotransfected with unlabeled trimeric G13 protein and LARG-insYFP a smaller increase attributable to translocation-induced bystander FRET was observed, which did not appear in the absence of the G protein (Supplemental Fig. S3). These data suggest that the FRET between Gα13 and LARG specifically reflects their interaction.
Receptor-induced Gα13 activation results in LARG translocation to the plasma membrane
An agonist-dependent translocation to the plasma membrane for p115-RhoGEF was described by Meyer and colleagues (15). In line with this, we found translocation of YFP-LARG upon agonist stimulation by confocal microscopy (Fig. 4A, B). This translocation was detected only in cells cotransfected with the trimeric Gα13 protein (Fig. 4C). The amplitudes of agonist-induced alterations of the signals were determined directly before agonist withdrawal (means ± sem, unpaired Student’s t test). In addition, LARG translocated only upon stimulation of the Gα12/13-coupled TPα-R, but did not travel to the plasma membrane upon stimulation of the Gαq-coupled muscarinic M3 receptor with saturating agonist concentrations (Supplemental Fig. S4).
Influence of LARG on dynamics of Gα13 Gβγ assembly
As shown for representative cells in Fig. 5A, the interaction of Gα13 and LARG occurred in the same time scale as the Gα13 protein activation, whereas the respective dissociation and deactivation differed (see also mean traces in Fig. 5B). When measuring deactivation of Gα13 upon agonist withdrawal in the absence of exogenous LARG (Fig. 5A, red trace), we measured kinetics that were ∼1 order of magnitude faster than those observed for the dissociation of LARG and Gα13 in cells expressing fluorescently labeled LARG and Gα13 (Fig. 5A, black trace). The LARG Gα13 interaction was slowly reversed over time, as detected by a decrease in FRET, and followed a similar time course as the LARG retranslocation to the cytosol (Fig. 5C, D). In these experiments, U-46619 was applied for a second time after 10 min of washout, and a second increase in FRET or plasma membrane localization was observed. The translocation of YFP-LARG in the presence of fluorophore-labeled Gα13 was indistinguishable from the one in the presence of wild-type Gα13 (Fig. 5D). Based on these findings, we reasoned that cells expressing high levels of LARG should also exhibit slowed inactivation kinetics in the Gα13 Gβγ FRET assay, because of the prolonged binding of Gα13 to LARG. We ensured high expression levels of LARG by cotransfection of N-terminally mRFP-labeled LARG, together with TPα-R, Gα13-YFP, Gβ-Cer, Gγ. and selection for bright red fluorescent cells. In these cells, a decrease in FRET was observed upon stimulation with 25 nM U-46619 (Fig. 6A) The bar graphs in Fig. 6A show the averaged amplitude of the agonist-induced alteration of the FRET signal. As expected, this response was slowly reversible only as shown in Fig. 6B, C. Compared to G-protein deactivation kinetics of 25.3 ± 2.2 s (t1/2, n = 22) calculated from the cells depicted in Fig. 1D (Fig. 6B, C), the deactivation time in the presence of mRFP-LARG was 527.0 ± 134.7 s (t1/2, n = 5) as evaluated by monoexponential fitting. To ensure that the decrease did not result from an artifact related to the mRFP, which can collect energy emitted from YFP, control measurements were performed. In these measurements, the YFP emission of the Gα13 Gβγ FRET assay was collected in the presence of either mRFP-LARG or unlabeled LARG (Supplemental Fig. S6A). No decrease in YFP emission associated with mRFP was detected. As HEK cells are known to express LARG endogenously, we used an siRNA approach to knockdown endogenous LARG expression to minimize an influence of endogenous LARG on the dissociation of Gα13 and Gβγ. Therefore, we tested for agonist-induced alterations in FRET between Gα13-YFP and Gβ-Cer in cells treated with a cocktail of specific siRNAs to knock down endogenous LARG expression (Fig. 6D). mock-siRNA-treated cells served as an appropriate control for endogenous LARG expression and mRFP-LARG–expressing cells as a positive control. Upon application of 1 nM U-46619, we detected only an agonist-induced FRET decline in cells transfected with mRFP-LARG. The subsequent application of 10 nM agonist decreased the FRET signal much more markedly in mRFP-LARG–expressing cells compared with mock-siRNA– or LARG-siRNA–treated cells. However, maximum Gα13 stimulation by 1 µM U-46619 resulted in an increase in FRET in all conditions, suggesting that G-protein subunit rearrangement occurs for the non–LARG-bound fraction. Western blot analysis confirmed the successful knockdown of LARG by siRNA treatment (Fig. 6E).
We summarized from the data that the presence of LARG leads to a full dissociation of the trimeric G protein, and because LARG stays with Gα13, the reassociation with Gβγ is delayed. Attempts to measure an agonist-induced FRET signal between Gβ-Cer and YFP-LARG revealed only a transient increase in FRET upon agonist stimulation that disappeared within 60 s of agonist exposure (Supplemental Fig. S6B), indicating the absence of a specific interaction of LARG and Gβγ.
Concentration–response curve of LARG Gα13 interaction is left shifted compared with G13 activation
With the described assays, we were able to monitor Gα13 activation and Gα13 LARG interaction in living cells in response to different U-46619 concentrations. Therefore, we measured and compared concentration–response curves for both Gα13 activation (in cells without exogenous LARG) and Gα13 interaction with its effector (Fig. 7A). The underlying data were obtained by comparing the response to 1 or 2 test concentrations with the response to a saturating agonist concentration in single living cells, as shown in Fig. 7B, C for representative cells. The EC50 of Gα13 activation was 29 nM, whereas the interaction of YFP-LARG and Gα13-mTur2 occurred with an EC50 of 0.36 nM (Fig. 7A). In the latter case, a lower apparent EC50 could be related to an extreme expression ratio of G protein vs. effector. We therefore determined the expression ratio of the fluorescent proteins in individual cells by comparing their yellow and cyan fluorescence (YFP/mTur2 fluorescence ratio: 0.069 ± 0.009; n = 30) to the fluorescence measured in control cells expressing a stoichiometer construct bearing both labels on the same protein (YFP/mTur2 fluorescence ratio: 0.089 ± 0.005; n = 10). This experiment indicated a 1.3-fold higher expression of Gα13-mTur2 compared to YFP-LARG. For interaction of Gα13 with LARG-insYFP, a similar sensitive concentration–response relationship was obtained (Supplemental Fig. S5). Overexpression of mRFP-LARG not only reversed the direction of the agonist-induced FRET response of the Gα13-YFP Gβ-Cer, but also transferred the drastically increased agonist sensitivity seen in the LARG G-protein interaction to the G-protein–derived signal (Figs. 6D, 7D). In the presence of heterologous LARG expression, the cells exhibited a small agonist-induced decrease in FRET between Gα13-YFP and Gβ-Cer compared with control measurements in which endogenous LARG expression was significantly reduced by an siRNA pool transfection (Figs. 6D, E, and 7D). The amplitude of the agonist-induced alteration of the FRET signal was determined at the end of agonist stimulation (Fig. 7D). Reduction of the endogenous LARG expression by siRNA resulted in a more stable FRET signal at low concentration of agonist and also after withdrawal of agonist (Fig. 6D). From this remarkable shift in agonist sensitivity observed in Fig. 7A, we reasoned that high agonist concentrations should lead to activation of all available G13 proteins, including the non-effector-bound population, which should outnumber the LARG-bound ones, whereas low agonist concentrations should preferentially lead to accumulation of active Gα13 bound to LARG. Moreover, the LARG level within the cell should influence the G-protein FRET response. Indeed, 1 µM U-46619 induced an increase in FRET, even in the presence of high mRFP-LARG expression, whereas silencing of endogenous LARG apparently reduced the decrease in FRET (Fig. 6D).
Because the relative expression levels of Gα13 and LARG will determine the sensitivity of this pathway, we estimated relative endogenous expression levels in HEK293T cells, in mouse cortical neurons, and in murine platelets by means of immunoblot analysis using calibrated antibodies (Supplemental Fig. S7). We found a moderately higher expression ratio of Gα13 over LARG (3-fold in HEK293T cells, 2–3-fold in murine thrombocytes, and 5–8-fold in cortical neurons from mice).
DISCUSSION
In this study, we temporally resolved the activation of Gα13 and its interaction with LARG in single living cells for the first time. The kinetics of LARG Gα13 dissociation was much slower than G-protein inactivation, if measured in the absence of exogenous LARG. These differences in kinetics were reflected also in the agonist sensitivity of G13 activation in the absence of LARG and Gα13 LARG interaction.
Dynamics of Gα13 activation
The FRET assay between Gα13-YFP and Gβ-Cer, monitors G-protein activity by means of alterations in the assembly of Gα13 and Gβ in response to receptor activation with high temporal and spatial specificity. As similarly described for Gαi1 proteins (8, 21), we found an increase in FRET upon receptor activation, suggesting that Gα13 and Gβγ undergo a subunit rearrangement instead of a full dissociation during activation. To verify the functionality of the fluorescent Gα13, we used several approaches: insertion of GFP derivatives into the α-helical domain of Gα13 between aa 127 and 128 did not change the G protein’s ability for stimulus-dependent SRE.L reporter gene regulation (Fig. 2). No Gα13 activation was observed with our FRET assay in the absence of TPα-R or upon stimulation of H1-R and M2-R receptors, which couple preferentially to Gq or Gi proteins (29–31) (Fig. 1D and Supplemental Fig. S1A). This observation was also true in the condition with TPα-R stimulation in the presence of unlabeled Gβ- and plasma-membrane–bound mCFP as a FRET donor for Gα13-YFP (Supplemental Fig. S1C). Thus, the increase in FRET between Gα13 and Gβ likely reflects a subunit rearrangement within the G protein and cannot be attributed to a bystander FRET because of the altered orientation of the YFP relative to the plane of the plasma membrane. In the case of Gαi1, the direction of the activation-induced alteration in FRET is dependent on whether the fluorophore is fused to the α-A loop (increase in FRET) or α-B loop (32). The newly generated fluorescent Gα13 carries the YFP at a site that corresponds to the αB loop, which for Gαi1 was found to be critical for the FRET increase (8, 21, 33, 34). Therefore, for G-protein FRET pairs that exhibit an increase in FRET upon activation, complete dissociation of Gα and Gβγ can be ruled out. The activation-induced decrease in FRET seen in other constructs including Gαq, Gαs, Gαo, and YFP (αB-αC loop)-Gαi1 (32) could reflect either an increase in the fraction of dissociated Gα and Gβγ or alternatively a subunit rearrangement leading to a fluorophore orientation that is less favorable for FRET. Classic biochemical studies and also elegant measurements of the lifetime of heterotrimeric state of G proteins in intact cells clearly demonstrated that the affinity between Gβγ and Gα is much lower in the active state compared with the inactive state (33). Therefore, a likely scenario could be that, even though activation dramatically reduces the affinity between Gα and Gβγ, it is still sufficient to keep most of the Gα and Gβγ together, however, with a much enhanced subunit exchange rate, allowing effectors for GTP-Gα or Gβγ to successfully compete with either Gα or Gβγ for binding of the other G-protein subunit. Our finding was that, in studies using overexpressed LARG, the FRET signal between Gα13-YFP and CFP-Gβγ decreased upon agonist-mediated activation in the case of low agonist concentrations (Figs. 6A, B, D, and 7D). This result is in line with an effector-induced dissociation of Gα13-GTP and Gβγ, which results from sequestering of active Gα13 by LARG. Accordingly, we failed to detect any prolonged FRET signal between CFP-Gβγ and LARG, arguing against a LARG Gα13βγ complex (Supplemental Fig. S6B). At very low concentrations of agonist, we observed varying effects on the G13 FRET amplitude; therefore, we tested whether the knockdown of endogenous RH-RhoGEF proteins may affect the G13 FRET response. We observed that the small FRET decline seen without RH-RhoGEF knockdown disappeared (Fig. 6D). Further support for the above-mentioned concept of G-protein assembly in intact cells comes from a previous study showing a dramatically enhanced decline in FRET between activated Gαq-YFP and CFP-Gβγ upon coexpression of GCPR kinase 2/3, which was prominent only if binding sites for Gαq and Gβγ were intact in the GRK2 molecule (27).
Furthermore, we observed a remarkable effect of LARG coexpression on the G-protein deactivation (reassembling) kinetics. Without heterologous expression of LARG, on- and offset kinetics of G13 activation were reminiscent of other members of the G-protein family (Fig. 1B, D) (6, 8); however, the relaxation of the G-protein FRET signal after withdrawal of agonist was much delayed when LARG was coexpressed (Fig. 6B, C).
Dynamics of Gα13 interaction with LARG
Studying interactions of G-protein subunits with their effectors by means of FRET enables the acquisition of detailed information about their dynamics. In the present study, we extend this method to monitor the interaction of a Gα12 family member with an effector in living cells. So far, only G-protein–mediated regulation of ion channels allowed for a time-resolved indirect measurement of G-protein effector interactions (35, 36). Results from these studies and studies based on FRET suggest that most G-protein effectors have no major impact on the dynamics of G-protein activation and deactivation, as shown for the interactions of GRK2 and p63RhoGEF with Gαq (6, 27). To date, only in the context of Gαi-mediated inhibition of the adenylate cyclase V was an ∼2-fold slower dissociation of the Gαi subunit adenylate cyclase V complex compared with G-protein deactivation observed (18). In this study, we demonstrate that the interaction between LARG and activated Gα13 outlasts the normal length of the G-protein cycle by at least 1 order of magnitude (Fig. 5A, B). This surprising finding is probably related to the fundamental properties of the Gα13 interaction with LARG. The slow dissociation of LARG and Gα13 in living cells measured by FRET (Fig. 5C) was mirrored by similar kinetics of YFP-LARG redistribution to the cytosol after agonist withdrawal both for wild-type and mTur2-labeled Gα13 which was measured by using an independent assay (Fig. 5D). Translocation of LARG to the plasma membrane seems to be key to its activation, because artificial membrane targeting of 2 other members of the RH-RhoGEF family was sufficient for proper Rho activation (16). Thus, we tested for Gα13-dependent YFP-LARG trafficking. In unstimulated HEK293T cells, we found YFP-LARG predominantly in the cytosol (Fig. 4B), in line with previous studies (15, 37–39). Upon stimulation of TPα-R with U-46619, YFP-LARG translocated to the plasma membrane only if Gα13 was cotransfected (Fig. 4C). This LARG translocation could not be induced by stimulation of Gαq via muscarinic M3 receptor (Supplemental Fig. S4). Furthermore, the translocation was not influenced by the fluorophore label on Gα13 itself (Fig. 5D).
In line with the requirement of Gα13 for LARG translocation, we found the robust agonist-induced FRET signal between Gα13-mTur2 and YFP-LARG to be specific and not attributable to translocation-induced alterations in bystander FRET (Fig. 3C). The kinetics of FRET signals between Gα13 and LARG were comparable for LARG constructs labeled at 2 different positions with YFP (Fig. 3C, D).
Agonist sensitivity of G13 activation is influenced by LARG
Even though GAP activity of LARG toward Gα13 was shown by in vitro measurements (40), our data clearly suggest a prolonged interaction of Gα13 and LARG upon agonist washout in intact cells. Currently, it is unclear whether LARG exhibits this prolonged interaction with the GTP-bound Gα13 or the GDP-bound Gα13 after GTP hydrolysis. Based on results of previous studies showing that membrane localization seems to be the major determinant of RH-RhoGEFs (16), it may be less important whether Gα13-GTP or Gα13-GDP induces membrane targeting of LARG. Either way, most likely as a consequence of the prolonged interaction, we found an almost 100-fold left shift in concentration–response relationship for the LARG Gα13 interaction compared with the Gα13 activation in the absence of LARG (Fig. 6 and Supplemental Fig. S5). This cannot be attributed to a vast overexpression of G proteins vs. LARG, as we controlled for relative expression levels in individual cells, which showed on average a 1.3-fold expression ratio for Gα13 vs. LARG. Comparison of relative expression levels of endogenous Gα13 vs. LARG in various cells revealed only a moderately higher expression ratio of Gα13 over LARG (2–3-fold in murine thrombocytes, 3-fold HEK293T cells, and 5–8-fold cortical mouseneurons) under physiologic conditions (Supplemental Fig. S7A, B).
Our finding that the Gα13-LARG interaction occurs at very low agonist concentrations corresponds well with the high agonist sensitivity of Gα13-mediated platelet activation and RhoA activation, which occur at low nanomolar concentrations of U-46619 in a murine model (1). Gq-mediated responses require much higher concentrations of U-46619 (1). Considering that U-46619-activated TPα-Rs couple equally well with Gq and G13 (41), together with the observation that LARG is involved in the Gα13-mediated platelet response (42), it seems likely that the high agonist sensitivity of the Gα13 response originates in its high-affinity interaction with LARG.
AUTHOR CONTRIBUTIONS
M. Bünemann designed and directed the project; A.-L. Krett and E.-L. Bodmann performed the experiments and analyzed the results, together with M. Bünemann; and E.-L. Bodmann and M. Bünemann wrote the manuscript.
Supplementary Material
ACKNOWLEDGMENTS
The pcDNA3-mRFP plasmid was kindly provided by Doug Golenbock (University of Massachusetts Medical School, Worcester, MA, USA), the pSRE.L was kindly provided by Barbara Moepps (Institute of Pharmacology and Toxicology, University of Ulm, Ulm, Germany), and the pHyg-LARG was a gift from T. Wieland (University of Heidelberg, Mannheim, Germany). The University of California, San Francisco (UCSF; San Francisco, CA, USA) chimera was developed by the Resource for Biocomputing, Visualization, and Informatics at UCSF (supported by U.S. National Institutes of Health, National Institute of General Medical Sciences Grant P41-GM103311). Mouse thrombocytes were kindly provided by Prof. Dr. Bernhard Nieswandt (Rudolf Virchow Research Center for Experimental Biomedicine, University of Würzburg, Würzburg, Germany). This work was funded by the Deutsche Forschungsgemeinschaft as part of the project A13 as part of the SFB 593. The authors declare no conflicts of interest.
Glossary
- BSA
bovine serum albumin
- CFP
cyan fluorescent protein
- Cer
cerulean
- eYFP
enhanced yellow fluorescent protein
- FRET
Förster resonance energy transfer
- LARG
leukemia-associated RhoGEF
- MAL
megakaryocytic acute leukemia
- mCFP
monomer cyan fluorescent protein
- mRFP
monomer red fluorescent protein
- mTur2
monomer turquoise fluorescent protein
- siRNA
small interfering RNA
- SRE
serum response element
- SRF
serum response factor
- TPα-R
thromboxane A2 receptor
- YFP
yellow fluorescent protein
Footnotes
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
REFERENCES
- 1.Moers A., Nieswandt B., Massberg S., Wettschureck N., Grüner S., Konrad I., Schulte V., Aktas B., Gratacap M.-P., Simon M. I., Gawaz M., Offermanns S. (2003) G13 is an essential mediator of platelet activation in hemostasis and thrombosis. Nat. Med. 9, 1418–1422 [DOI] [PubMed] [Google Scholar]
- 2.Sivaraj K. K., Takefuji M., Schmidt I., Adams R. H., Offermanns S., Wettschureck N. (2013) G13 controls angiogenesis through regulation of VEGFR-2 expression. Dev. Cell 25, 427–434 [DOI] [PubMed] [Google Scholar]
- 3.Gohla A., Schultz G., Offermanns S. (2000) Role for G(12)/G(13) in agonist-induced vascular smooth muscle cell contraction. Circ. Res. 87, 221–227 [DOI] [PubMed] [Google Scholar]
- 4.Riobo N. A., Manning D. R. (2005) Receptors coupled to heterotrimeric G proteins of the G12 family. Trends Pharmacol. Sci. 26, 146–154 [DOI] [PubMed] [Google Scholar]
- 5.Worzfeld T., Wettschureck N., Offermanns S. (2008) G(12)/G(13)-mediated signalling in mammalian physiology and disease. Trends Pharmacol. Sci. 29, 582–589 [DOI] [PubMed] [Google Scholar]
- 6.Bodmann E.-L., Rinne A., Brandt D., Lutz S., Wieland T., Grosse R., Bünemann M. (2014) Dynamics of Gαq-protein-p63RhoGEF interaction and its regulation by RGS2. Biochem. J. 458, 131–140 [DOI] [PubMed] [Google Scholar]
- 7.Hein P., Rochais F., Hoffmann C., Dorsch S., Nikolaev V. O., Engelhardt S., Berlot C. H., Lohse M. J., Bünemann M. (2006) Gs activation is time-limiting in initiating receptor-mediated signaling. J. Biol. Chem. 281, 33345–33351 [DOI] [PubMed] [Google Scholar]
- 8.Bünemann M., Frank M., Lohse M. J. (2003) Gi protein activation in intact cells involves subunit rearrangement rather than dissociation. Proc. Natl. Acad. Sci. USA 100, 16077–16082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Etienne-Manneville S., Hall A. (2002) Rho GTPases in cell biology. Nature 420, 629–635 [DOI] [PubMed] [Google Scholar]
- 10.Miralles F., Posern G., Zaromytidou A. I., Treisman R. (2003) Actin dynamics control SRF activity by regulation of its coactivator MAL. Cell 113, 329–342 [DOI] [PubMed] [Google Scholar]
- 11.Aittaleb M., Boguth C. A., Tesmer J. J. G. (2010) Structure and function of heterotrimeric G protein-regulated Rho guanine nucleotide exchange factors. Mol. Pharmacol. 77, 111–125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kourlas P. J., Strout M. P., Becknell B., Veronese M. L., Croce C. M., Theil K. S., Krahe R., Ruutu T., Knuutila S., Bloomfield C. D., Caligiuri M. A. (2000) Identification of a gene at 11q23 encoding a guanine nucleotide exchange factor: evidence for its fusion with MLL in acute myeloid leukemia. Proc. Natl. Acad. Sci. USA 97, 2145–2150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kuner R., Swiercz J. M., Zywietz A., Tappe A., Offermanns S. (2002) Characterization of the expression of PDZ-RhoGEF, LARG and G(alpha)12/G(alpha)13 proteins in the murine nervous system. Eur. J. Neurosci. 16, 2333–2341 [DOI] [PubMed] [Google Scholar]
- 14.Takefuji M., Krüger M., Sivaraj K. K., Kaibuchi K., Offermanns S., Wettschureck N. (2013) RhoGEF12 controls cardiac remodeling by integrating G protein- and integrin-dependent signaling cascades. J. Exp. Med. 210, 665–673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Meyer B. H., Freuler F., Guerini D., Siehler S. (2008) Reversible translocation of p115-RhoGEF by G(12/13)-coupled receptors. J. Cell. Biochem. 104, 1660–1670 [DOI] [PubMed] [Google Scholar]
- 16.Carter A. M., Gutowski S., Sternweis P. C. (2014) Regulated localization is sufficient for hormonal control of regulator of G protein signaling homology Rho guanine nucleotide exchange factors (RH-RhoGEFs). J. Biol. Chem. 289, 19737–19746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen Z., Guo L., Hadas J., Gutowski S., Sprang S. R., Sternweis P. C. (2012) Activation of p115-RhoGEF requires direct association of Gα13 and the Dbl homology domain. J. Biol. Chem. 287, 25490–25500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Milde M., Rinne A., Wunder F., Engelhardt S., Bünemann M. (2013) Dynamics of Gαi1 interaction with type 5 adenylate cyclase reveal the molecular basis for high sensitivity of Gi-mediated inhibition of cAMP production. Biochem. J. 454, 515–523 [DOI] [PubMed] [Google Scholar]
- 19.Goedhart J., von Stetten D., Noirclerc-Savoye M., Lelimousin M., Joosen L., Hink M. A., van Weeren L., Gadella T. W. J. Jr., Royant A. (2012) Structure-guided evolution of cyan fluorescent proteins towards a quantum yield of 93%. Nat. Commun. 3, 751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Reuther G. W., Lambert Q. T., Booden M. A., Wennerberg K., Becknell B., Marcucci G., Sondek J., Caligiuri M. A., Der C. J. (2001) Leukemia-associated Rho guanine nucleotide exchange factor, a Dbl family protein found mutated in leukemia, causes transformation by activation of RhoA. J. Biol. Chem. 276, 27145–27151 [DOI] [PubMed] [Google Scholar]
- 21.Frank M., Thümer L., Lohse M. J., Bünemann M. (2005) G protein activation without subunit dissociation depends on a Galpha(i)-specific region. J. Biol. Chem. 280, 24584–24590 [DOI] [PubMed] [Google Scholar]
- 22.Lutz S., Freichel-Blomquist A., Rümenapp U., Schmidt M., Jakobs K. H., Wieland T. (2004) p63RhoGEF and GEFT are Rho-specific guanine nucleotide exchange factors encoded by the same gene. Naunyn Schmiedebergs Arch. Pharmacol. 369, 540–546 [DOI] [PubMed] [Google Scholar]
- 23.Hein P., Frank M., Hoffmann C., Lohse M. J., Bünemann M. (2005) Dynamics of receptor/G protein coupling in living cells. EMBO J. 24, 4106–4114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dorsch S., Klotz K.-N., Engelhardt S., Lohse M. J., Bünemann M. (2009) Analysis of receptor oligomerization by FRAP microscopy. Nat. Methods 6, 225–230 [DOI] [PubMed] [Google Scholar]
- 25.Hill C. S., Wynne J., Treisman R. (1995) The Rho family GTPases RhoA, Rac1, and CDC42Hs regulate transcriptional activation by SRF. Cell 81, 1159–1170 [DOI] [PubMed] [Google Scholar]
- 26.Mao J., Yuan H., Xie W., Wu D., Simon M. I. (1998) Specific involvement of G proteins in regulation of serum response factor-mediated gene transcription by different receptors. J. Biol. Chem. 273, 27118–27123 [DOI] [PubMed] [Google Scholar]
- 27.Wolters V., Krasel C., Brockmann J., Bünemann M. (2015) Influence of gαq on the dynamics of m3-acetylcholine receptor-g-protein-coupled receptor kinase 2 interaction. Mol. Pharmacol. 87, 9–17 [DOI] [PubMed] [Google Scholar]
- 28.Kreutz B., Yau D. M., Nance M. R., Tanabe S., Tesmer J. J. G., Kozasa T. (2006) A new approach to producing functional G alpha subunits yields the activated and deactivated structures of G alpha(12/13) proteins. Biochemistry 45, 167–174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Marinissen M. J., Servitja J. M., Offermanns S., Simon M. I., Gutkind J. S. (2003) Thrombin protease-activated receptor-1 signals through Gq- and G13-initiated MAPK cascades regulating c-Jun expression to induce cell transformation. J. Biol. Chem. 278, 46814–46825 [DOI] [PubMed] [Google Scholar]
- 30.Chikumi H., Vázquez-Prado J., Servitja J.-M., Miyazaki H., Gutkind J. S. (2002) Potent activation of RhoA by Galpha q and Gq-coupled receptors. J. Biol. Chem. 277, 27130–27134 [DOI] [PubMed] [Google Scholar]
- 31.Pfreimer M., Vatter P., Langer T., Wieland T., Gierschik P., Moepps B. (2012) LARG links histamine-H1-receptor-activated Gq to Rho-GTPase-dependent signaling pathways. Cell. Signal. 24, 652–663 [DOI] [PubMed] [Google Scholar]
- 32.Gibson S. K., Gilman A. G. (2006) Gialpha and Gbeta subunits both define selectivity of G protein activation by alpha2-adrenergic receptors. Proc. Natl. Acad. Sci. USA 103, 212–217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Digby G. J., Lober R. M., Sethi P. R., Lambert N. A. (2006) Some G protein heterotrimers physically dissociate in living cells. Proc. Natl. Acad. Sci. USA 103, 17789–17794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Galés C., Van Durm J. J. J., Schaak S., Pontier S., Percherancier Y., Audet M., Paris H., Bouvier M. (2006) Probing the activation-promoted structural rearrangements in preassembled receptor-G protein complexes. Nat. Struct. Mol. Biol. 13, 778–786 [DOI] [PubMed] [Google Scholar]
- 35.Bünemann M., Bücheler M. M., Philipp M., Lohse M. J., Hein L. (2001) Activation and deactivation kinetics of alpha 2A- and alpha 2C-adrenergic receptor-activated G protein-activated inwardly rectifying K+ channel currents. J. Biol. Chem. 276, 47512–47517 [DOI] [PubMed] [Google Scholar]
- 36.Doupnik C. A., Davidson N., Lester H. A., Kofuji P. (1997) RGS proteins reconstitute the rapid gating kinetics of gbetagamma-activated inwardly rectifying K+ channels. Proc. Natl. Acad. Sci. USA 94, 10461–10466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Banerjee J., Wedegaertner P. B. (2004) Identification of a novel sequence in PDZ-RhoGEF that mediates interaction with the actin cytoskeleton. Mol. Biol. Cell 15, 1760–1775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Grabocka E., Wedegaertner P. B. (2007) Disruption of oligomerization induces nucleocytoplasmic shuttling of leukemia-associated rho guanine-nucleotide exchange factor. Mol. Pharmacol. 72, 993–1002 [DOI] [PubMed] [Google Scholar]
- 39.Siehler S. (2009) Regulation of RhoGEF proteins by G12/13-coupled receptors. Br. J. Pharmacol. 158, 41–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Suzuki N., Nakamura S., Mano H., Kozasa T. (2003) Galpha 12 activates Rho GTPase through tyrosine-phosphorylated leukemia-associated RhoGEF. Proc. Natl. Acad. Sci. USA 100, 733–738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang L., Brass L. F., Manning D. R. (2009) The Gq and G12 families of heterotrimeric G proteins report functional selectivity. Mol. Pharmacol. 75, 235–241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zou S., Teixeira A. M., Yin M., Xiang Y., Xavier-Ferrucio J., Zhang P. X., Hwa J., Min W., Krause D. S. (2016) Leukaemia-associated Rho guanine nucleotide exchange factor (LARG) plays an agonist specific role in platelet function through RhoA activation. Thromb. Haemost. 116, 506–516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wachter R. M., Elsliger M. A., Kallio K., Hanson G. T., Remington S. J. (1998) Structural basis of spectral shifts in the yellow-emission variants of green fluorescent protein. Structure 6, 1267–1277 [DOI] [PubMed] [Google Scholar]
- 44.Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., Ferrin T. E. (2004) UCSF chimera: a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.