

Abstract
Fast beta (20–28 Hz) electroencephalogram (EEG) oscillatory activity may be a useful endophenotype for studying the genetics of disorders characterized by neural hyperexcitability, including substance use disorders (SUDs). However, the genetic underpinnings of fast beta EEG have not previously been studied in a population of African-American ancestry (AA). In a sample of 2382 AA individuals from 482 families drawn from the Collaborative Study on the Genetics of Alcoholism (COGA), we performed a genome-wide association study (GWAS) on resting-state fast beta EEG power. To further characterize our genetic findings, we examined the functional and clinical/behavioral significance of GWAS variants. Ten correlated single-nucleotide polymorphisms (SNPs) (r2>0.9) located in an intergenic region on chromosome 3q26 were associated with fast beta EEG power at P<5 × 10−8. The most significantly associated SNP, rs11720469 (β: − 0.124; P<4.5 × 10−9), is also an expression quantitative trait locus for BCHE (butyrylcholinesterase), expressed in thalamus tissue. Four of the genome-wide SNPs were also associated with Diagnostic and Statistical Manual of Mental Disorders Alcohol Dependence in COGA AA families, and two (rs13093097, rs7428372) were replicated in an independent AA sample (Gelernter et al.). Analyses in the AA adolescent/young adult (offspring from COGA families) subsample indicated association of rs11720469 with heavy episodic drinking (frequency of consuming 5+ drinks within 24 h). Converging findings presented in this study provide support for the role of genetic variants within 3q26 in neural and behavioral disinhibition. These novel genetic findings highlight the importance of including AA populations in genetics research on SUDs and the utility of the endophenotype approach in enhancing our understanding of mechanisms underlying addiction susceptibility.
INTRODUCTION
Human electroencephalography (EEG) noninvasively measures ongoing resting-state brain electrical activity. These oscillations are divided into frequency bands (delta (1–3 Hz), theta (4–7 Hz), alpha (8–12 Hz), beta (13–28 Hz) and gamma (>29 Hz)), with each band reflecting a different global brain state (for example, alpha activity reflects a relaxed state while beta EEG reflects an alert awake state1–3). Although local excitatory–inhibitory interactions underlying sensory and motor functions involve gamma-band oscillations, cognitive functions mediated by long-range cortical interactions often involve EEG activity in the beta range.3 Beta EEG is also associated with several externalizing disorders,4–10 including alcohol and other substance use disorders (SUDs) and attention deficit hyperactivity disorder (ADHD). Given these associations, and the high degree of genetic influence observed in twin studies (49–85%11,12), beta EEG has been proposed as a useful endophenotype13 for identifying genetic factors underlying disorders characterized by disinhibitory traits.14
Previous studies report differences in the magnitude of fast (>19 Hz) beta EEG among individuals with alcohol use disorders (AUD)4–6,15,16,17 and related problems (that is, SUD, ADHD). Further, fast beta EEG was superior to the severity of illness, major depression and conduct problems in predicting relapse in abstinent individuals with a history of AUD.5,17,18 As elevated beta EEG is present in the offspring of alcoholics prior to the onset of risky drinking,4,15,19,20 researchers have hypothesized that excess beta power precedes the development of AUD and is likely related to an underlying genetic predisposition for developing AUD, rather than a consequence of heavy drinking. Begleiter and Porjesz4 have suggested that this may be an electrophysiological index of an imbalance in the excitation–inhibition homeostasis in the cortex, which underlies a predisposition to develop AUD and related disorders.4,14,20 Further supporting this hypothesis is the association of beta EEG and other disorders characterized by behavioral disinhibition: externalizing behavior in children19 and adolescents,7 ADHD,10,21,22 internet addiction,8,9 and binge drinking in emerging adults.23
Despite the high heritability estimates provided by twin and family studies (49–85%11,12), there have been relatively few genetic studies of beta EEG, and to date, only one finding has replicated. An early analysis found linkage between beta EEG and a region of chromosome 424 harboring variants in the gene that encodes GABA α2 (GABRA2).25 More recently, a study of 586 individuals of European ancestry with Diagnostic and Statistical Manual of Mental Disorders (DSM-IV) Alcohol Dependence (AD) and 603 controls26 replicated the association between beta activity and GABRA2 single-nucleotide polymorphisms (SNPs). To date, only two genome-wide association studies (GWASs) of beta EEG have been conducted.12,27 In a study of 322 Native-American individuals, there were no genome-wide significant associations reported for beta EEG.27 A recent GWAS of monopolar beta EEG in 4026 European ancestry adolescent twins and their parents12 did not report any genome-wide significant variants but replicated the previous associations observed with GABRA2.
Importantly, there have been no GWASs of EEG conducted in populations of AA, and thus the genetic architecture of EEG-related traits is not well described in AA populations. In addition to the public health importance of including all populations in research, conducting genetic studies in populations of AA is important because of the greater genetic diversity and the evolutionary differences in disease allele frequency and linkage disequilibrium (LD) patterns observed.28 Moreover, African-American drinkers consume less alcohol than Non-Hispanic whites but experience more alcohol-related problems, including social consequences, illness and death,29–32 indicating a need to identify factors that mitigate risk for problem drinking. Because research examining how basic brain functioning is related to human behavior and disorders has the ultimate goal of providing prevention and/or interventions for all individuals, this important gap in the literature needs to be addressed.
Given that beta EEG is highly heritable and is related to several externalizing behaviors and SUDs, genetic analysis of beta EEG may aid in our understanding of underlying brain function in individuals at risk for a range of externalizing disorders. The primary aim of this study was to conduct a GWAS of fast beta EEG power (bipolar occipital derivation, chosen due to high heritability observed in previous studies33) in families of AA from the Collaborative Study on the Genetics of Alcoholism (COGA), a recently ascertained family sample densely affected with AD and co-occurring externalizing disorders (for example, SUD, ADHD). The secondary aims of this study were to examine the functional and behavioral significance of GWAS findings.
MATERIALS AND METHODS
Collaborative Study on the Genetics of Alcoholism
COGA recruited AD probands from treatment facilities through seven participating sites, as described previously.34,35 Institutional review boards at all sites approved the study. Participants were administered the Semi-Structured Assessment for the Genetics of Alcoholism (SSAGA), a poly-diagnostic interview.36 Individuals aged <18 years were administered an adolescent SSAGA. In addition, DNA and EEG were collected. Principal components (PCs) derived from GWAS data were used to assign ancestry in the full genotyped sample and were the basis for the selection of AA families. A total of 99.5% of the AA (defined by PCs) individuals self-identify as ‘Black/African-American’ when given the following response options: ‘Native American/American Indian’, ‘Asian’, ‘Pacific Islander’, ‘African-American/Black’, ‘Caucasian/White’, and ‘Other’. Independent of their self-reported race/ethnicity, 11.1% of the sample endorsed being of ‘Hispanic or Latino descent’. The analytical sample consisted of all participants with EEG and GWAS data available, 2382 individuals from 482 families. The demographic characteristics of the full AA sample and the EEG subsample are comparable (Table 1). While 27.6% of the full AA sample met criteria for AD, rates of other co-occurring substance use and externalizing disorders (for example, cocaine dependence (CoD), ADHD) were also substantial.
Table 1.
Descriptive characteristics of analytical COGA samples
| AA fGWAS genotyped sample | AA fGWAS fast beta EEG subsample | AA fGWAS fast beta EEG subsample with DSM-IV data | AA prospective study subsample (at baseline) | |
|---|---|---|---|---|
| Genotyped (N) | 3414 | 2382 | 2242 | 892 |
| Families (N) | 598 | 482 | 480 | 212 |
| Female (%) | 52.7% | 50.3% | 53.4% | 52.3% |
| Self-identified as ‘Black/African-American’ (%) | 99.5% | 97.9% | 97.4% | 92.7% |
| Age (mean, s.d.), years | 32.4 (11.2) | 29.4 (12.3) | 29.9 (11.8) | 16.1 (3.3) |
| Ever drinkers | 92.2% | 92.3% | 89.5% | 33.8% |
| DSM-IV AD diagnosis (%) | 27.6% | 23.6% | 24.5% | 1.8% |
Abbreviations: AA, African-American ancestry; AD, alcohol dependence; COGA, Collaborative Study on the Genetics of Alcoholism; DSM-IV, Diagnostic and Statistical Manual of Mental Disorders; EEG, electroencephalography; fGWAS, function genome-wide association study.
EEG recording and processing
EEG recording and processing has been detailed previously.33 Briefly, resting (eyes closed) EEG was recorded for 4.25 min; a continuous interval of 256 s was analyzed. This study focused on log-transformed absolute fast beta power (20–28 Hz) at occipital bipolar leads (O1–O2; Supplementary Figure S1). EEG procedures were identical at all collection sites.
Genotyping, imputation and quality control
Genotyping, imputation and quality control has been previously reported.37 Genotyping of 3414 individuals from 598 families was performed at the Center for Inherited Disease Research using the Illumina 2.5M array (Illlumina, San Diego, CA, USA). SNPs with a genotyping rate <98% or that violated Hardy–Weinberg equilibrium (P<10−6) or with minor allele frequency (MAF) <3% were excluded from the analyses. Mendelian inconsistencies were removed,38 after which data were imputed to 1000 genomes (hg19) using SHAPEIT39 and IMPUTE2.40 Following imputation, genotype probabilities ⩾ 0.90 were changed to genotypes. Mendelian errors in the imputed SNPs were reviewed and resolved as described in Wetherill et al.37 All SNPs with an imputation information score <0.30 or MAF <0.03 were excluded from subsequent association analysis.40
GWAS
GWAS was conducted in GWAF (genome-wide association analyses with family) on 12 972 748 SNPs using a linear mixed model incorporating a genetic relationship matrix to control for the relatedness in the family sample.41 Sex and log-transformed age (at the time of EEG recording) were included as covariates in the model, as each of these variables were associated with beta EEG (P<0.001). The first 10 PCs (PC1–PC10) computed from SNPRelate42 were also included as covariates to reduce the risk of false-positives owing to population stratification. An additive genetic model was assumed. Established thresholds for genome-wide significance (P<5 × 10−8) were utilized. Genome-wide complex trait analysis (GCTA) was utilized to determine SNP heritability of fast beta EEG in the analytical sample. The genetic relatedness matrix was incorporated to adjust heritability estimates for familial clustering.
Functional analyses
We examined whether the most significantly associated variant for fast beta EEG was an expression quantitative trait locus (eQTL) in the UK Brain Expression Consortium (BRAINEAC; http://www.braineac.org/). Braineac draws on data from 134 neuropathologically normal individuals of European ancestry and assesses 10 different regions of the brain (Table 3).43 Only the single SNP most associated with fast beta EEG was examined in Braineac to minimize multiple testing. Further, a Bonferonni correction was applied to all P-values. Associations that withstood multiple testing were examined in the Genotype-Tissue Expression Project (GTEx) database (www.gtexportal.org) to confirm eQTL findings.
Table 3.
Summary (P-values) of mRNA expression results for rs11720469 in the Braineac database
| Gene | CRBL | FCTX | HIPP | MEDU | OCTX | PUTM | SNIG | TCTX | THAL | WHMT |
|---|---|---|---|---|---|---|---|---|---|---|
| BCHE | 0.29 | 0.98 | 0.17 | 0.53 | 0.015 | 0.27 | 0.87 | 0.72 | 0.001 | 0.34 |
| PDCD10 | 0.09 | 0.003 | 0.61 | 0.82 | 0.52 | 0.64 | 0.62 | 0.41 | 0.90 | 0.67 |
| SERPINI1 | 0.007 | 0.11 | 0.014 | 0.22 | 0.035 | 0.68 | 0.66 | 0.47 | 0.39 | 0.26 |
| WDR49 | 0.73 | 0.80 | 0.05 | 0.79 | 0.003 | 0.54 | 0.95 | 0.23 | 0.22 | 0.70 |
| ZBBX | 0.27 | 0.004 | 0.93 | 0.61 | 0.71 | 0.37 | 0.52 | 0.73 | 0.48 | 0.56 |
Note: A Bonferroni’s correction was used to adjust for multiple testing; bold font denotes P-values that withstood this correction. In the Braineac database (http://www.braineac.org/), rs11720469 was used for expression quantitative trait locus analysis (N = 134 individuals of European Ancestry) of 10 different regions of the brain, including: cerebellar cortex (CRBL), frontal cortex (FCTX), hippocampus (HIPP), medulla (specifically inferior olivary nucleus; MEDU), occipital cortex (specifically primary visual cortex; OCTX), putamen (PUTM), substantia nigra (SNIG), thalamus (THAL), and intralobular white matter (WHMT). A detailed description of the samples used in the study, tissue processing and dissection is provided in Trabzuni et al.43
Alcohol use behavior
We determined whether variants that were genome-wide associated with fast beta EEG were also associated with DSM-IV AD44 in the discovery sample (2242 individuals from 480 families (Table 1)). Non-drinkers, those aged <18 years and unaffected individuals with 2+ SUD criteria were excluded. Analyses were performed using SAS Version 9.4 (SAS Institute (http://search.ebscohost.com/login.aspx?direct=true&db=plh&AN=101476231&site=eds-live)). Logistic regression models were adjusted for age, sex, relatedness and PC1–PC10. Given that a single phenotype was tested, we examined association with all fast beta EEG genome-wide significant SNPs. Individual P-values were adjusted using the Pnorm procedure,45 which accounts for both the LD structure of the SNPs, and the sampling of relatives. Pnorm uses the multivariate normal distribution approximation to evaluate the significance of each test adjusting for simultaneous testing.
Next we examined whether GWAS variants were associated with DSM-IV AD in an independent sample of unrelated individuals recruited for studies of the genetics of opioid, cocaine or AD46 made publicly accessible on dbGaP (Accession no.: phs000425.v1.p1.c1). Only individuals of AA who had AD data available were included in the analyses (1346 AD cases and 461 unaffected controls).
Finally, because of prior evidence indicating a relationship between binge drinking in young adulthood and high fast beta EEG,23 we determined whether the top SNP meeting genome-wide criteria for fast beta EEG was associated with a measure similar to binge drinking, heavy-episodic drinking (frequency of consuming 5+ drinks within 24 h in the past year), in adolescent and young adult offspring from COGA families in COGA’s prospective study. This sample (ages 12–24 years at baseline) is longitudinally followed and has been described in detail previously.47 The present study utilizes data from the baseline assessment of each AA individual (Table 1). Participants were asked to “Think about the last 12 months. How often did you have 5 or more drinks in a 24-hour-period?” Thirteen response options, detailed in Supplementary Table S3, ranged from ‘Never’ to ‘Every Day’. Of the 892 individuals from 212 families, 33.8% had ever consumed a full drink of alcohol. The remaining 66.2% were coded as zero. Due to the relatively small sample size, only the single SNP most strongly associated with fast beta EEG was examined to minimize multiple testing. Association was tested with log-transformed heavy-episodic drinking, adjusted for relatedness, sex, age and PC1–PC10 in Mplus 7.4.48
Post hoc analyses
BCHE (and/or surrounding region, 3q26) has previously been associated with behavioral conditions relevant to fast beta EEG and AUD, including ADHD49–52 and cocaine use/problems.53 To determine whether the significant signal observed for fast beta EEG was accounted for by any of these disorders, we carried out three separate post hoc GWAS of fast beta EEG, each adjusted for one of the three disorders: DSM-IV AD, DSM-IV ADHD, and DSM-IV CoD by including it as a covariate in the model. All analyses conducted in the current study are summarized in Supplementary Table S1.
RESULTS
GWAS of beta EEG
Ten individual SNPs (pair-wise r2>0.9 for all 10 SNPs based on hg19 1000 Genomes from the sample of African ancestry), located in an intergenic region on 3q26 (Chr 3, 166 471 942–166 489 551) were associated with fast beta EEG at P<5 × 10−8 (Table 2, Supplementary Table S2, Figures 1 and 2, Supplementary Figure S4). The most significant SNP was rs11720469 (P<4.5 × 10−9); the minor allele (G) was negatively associated with fast beta EEG (β: − 0.124; Supplementary Figure S5). Figure 2 graphically illustrates this GWAS signal, as well as the known genes located upstream and downstream of this signal, including BCHE, PDCD10, WDR49, SERPINI1, SERPINI2 and ZBBX. GABRA2 was also associated with fast beta EEG but not at a genome-wide level (P<0.01). GCTA estimated that 33.8% (s.e.: 0.014, P<5.5 × 10−17) of the variance in fast beta EEG was due to genome-wide SNPs (narrow sense heritability).
Table 2.
Summary of association results with P<5 × 10−8 for fast beta EEG in COGA AA families
| SNP | Bp | Major allele | Minor allele | MAF | Effect size (β) | P-value |
|---|---|---|---|---|---|---|
| rs9859643 | 166 471 942 | G | A | 0.449 | −0.121 | 9.90E-09 |
| rs4680631 | 166 473 095 | G | A | 0.438 | −0.119 | 1.60E-08 |
| rs11720469 | 166 481 208 | G | A | 0.451 | −0.124 | 4.50E-09 |
| rs11705903 | 166 482 205 | G | A | 0.453 | −0.122 | 6.40E-09 |
| rs6806557 | 166 482 963 | C | T | 0.44 | −0.12 | 1.40E-08 |
| rs4680634 | 166 483 225 | T | G | 0.44 | −0.121 | 9.90E-09 |
| rs7428372 | 166 484 059 | A | C | 0.435 | −0.124 | 5.30E-09 |
| rs7430178 | 166 484 123 | G | C | 0.44 | −0.12 | 1.30E-08 |
| rs7430210 | 166 484 297 | G | A | 0.439 | −0.121 | 1.00E-08 |
| rs13093097 | 166 489 551 | T | C | 0.46 | −0.117 | 2.30E-08 |
Abbreviations: AA, African-American ancestry; Bp, base pair; COGA, Collaborative Study on the Genetics of Alcoholism; EEG, electroencephalography; MAF, minor allele frequency; SNP, single-nucleotide polymorphism.
Figure 1.
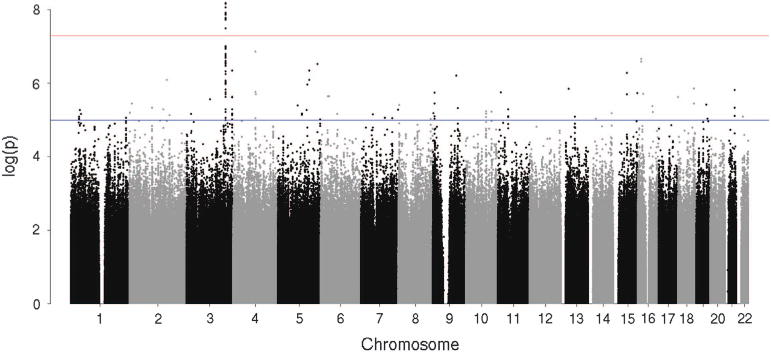
Genome-wide association results for fast beta electroencephalogram in the African-American ancestry function genome-wide association study. y Axis denotes the –log10(P-value) for association. x Axis is the physical position of the single-nucleotide polymorphisms across the genome. Note: Red line indicates the threshold of genome-wide significance (P<5 × 10−8), whereas the blue line indicates the threshold of P<5 × 10−5.
Figure 2.
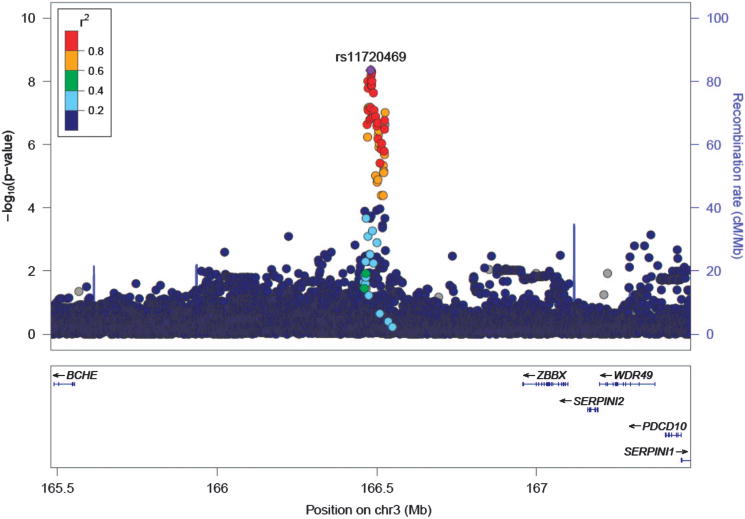
Association results for fast beta electroencephalogram on chromosome 3q26. y Axis denotes the –log10(P-value) for association. x Axis is the physical position on the chromosome (Mb). The most significantly associated single-nucleotide polymorphism (SNP; rs11720469) is shown in purple. The extent of linkage disequilibrium (LD; as measured by r2) between each SNP and the most significantly associated SNP is indicated by the color scale at the top left. Larger values of r2 indicate greater LD. Circles represent P-values from the African-American ancestry function genome-wide association study sample. LD is based on hg19 1000 Genomes from the African sample.
Functional analyses
In the Braineac database, rs11720469 is associated with the mRNA expression of BCHE, PDCD10, SERPINI1, WDR49 and ZBBX. Only one of these findings survived a Bonferroni multiple test correction: rs11720469 is an eQTL for BCHE expression in thalamus tissue (P = 4.20 × 10−4); the minor allele is associated with decreased mRNA expression (Table 3). In the GTEx database, rs11720469 is associated with the expression of BCHE in brain tissue: cortex (P<0.007) and caudate (P<0.005). HaploReg V4.154,55 indicated that rs11720469 alters regulatory motifs in some cell types in the ROADMAP Epigenomics data.56
Alcohol use behavior
Seven of the 10 variants associated with fast beta EEG were also associated with AD in a subset of the discovery GWAS sample (Table 4); the minor allele was associated with reduced AD risk, indicating a protective effect. Given the high LD observed among these 10 SNPs, these P-values were adjusted for the number of tests (1.2) as estimated using the Pnorm procedure.45 Four of these variants survived multiple test correction: rs7428372 (β: − 0.164, P-value<0.037), rs11705903 (β: − 0.161, P-value<0.039), rs6806557 (β: − 0.161, P-value<0.041), and rs13093097 (β: − 0.172, P-value<0.027).
Table 4.
Association of fast beta EEG variants and DSM-IV AD in subset of discovery sample (N = 2242 from 480 families) and replication sample (N = 1807)
| SNP |
COGA AA families
|
Gelernter et al.46
sample
|
||||
|---|---|---|---|---|---|---|
| MAF |
Effect size (β) |
P-value | MAF |
Effect size (β) |
P-value | |
| rs11720469 | 0.451 | −0.142 | 0.070 | 0.364 | −0.154 | 0.061 |
| rs7428372 | 0.435 | −0.164 | 0.037 | 0.364 | −0.167 | 0.042 |
| rs11705903 | 0.453 | −0.161 | 0.039 | 0.364 | −0.148 | 0.072 |
| rs9859643 | 0.449 | −0.146 | 0.060 | 0.364 | −0.149 | 0.069 |
| rs4680634 | 0.440 | −0.156 | 0.046 | 0.364 | −0.158 | 0.055 |
| rs7430210 | 0.439 | −0.157 | 0.046 | 0.364 | −0.153 | 0.063 |
| rs7430178 | 0.440 | −0.157 | 0.046 | 0.364 | −0.165 | 0.045 |
| rs6806557 | 0.440 | −0.161 | 0.041 | 0.364 | −0.158 | 0.057 |
| rs4680631 | 0.438 | −0.148 | 0.058 | 0.364 | −0.163 | 0.046 |
| rs13093097 | 0.460 | −0.172 | 0.027 | 0.364 | −0.179 | 0.029 |
Abbreviations: AA, African-American ancestry; AD, alcohol dependence; COGA, Collaborative Study on the Genetics of Alcoholism; DSM-IV, Diagnostic and Statistical Manual of Mental Disorders; EEG, electroencephalography; GWAS, genome-wide association study; MAF, minor allele frequency; SNP, single-nucleotide polymorphism. Note: In COGA AA families, analyses were performed using SAS Version 9.4 (SAS Institute). Models were adjusted for family relatedness, age, sex and PC1–PC10. Individual P-values were adjusted using the Pnorm procedure (Wang et al.45) that adjusts P-values for multiple testing in the context of both the linkage disequilibrium structure of the SNPs and the dependence structure owing to sampling of relatives. In the sample by Gelernter et al.46 (Accession no. phs000425.v1.p1.c1), P-values were extracted from a GWAS of DSM-IV AD (Gelernter et al.46). For both samples, individual P-values that withstood the multiple test correction using the Pnorm procedure (Wang et al.45) are represented in bold.
In an independent sample,46 4 of the 10 SNPs were associated with AD (Table 4). Two SNPs withstood the multiple test correction, rs7428372 (β: − 0.167, P-value<0.042) and rs13093097 (β: − 0.179, P-value<0.029); the minor allele was associated with reduced AD risk, indicating a protective effect.
Finally, having one or more copies of the minor allele was associated with reduction in heavy-episodic drinking (β: − 0.064, P<0.035) in the AA subsample of adolescents/young adult offspring from COGA families; however, the effect size was modest.
Post hoc analyses
In the primary GWAS sample, individuals with elevated fast beta EEG were more likely to meet criteria for DSM-IV AD, CoD and ADHD (all P-values <0.001, surviving Bonferroni’s correction). To determine whether the significant signal observed for fast beta EEG was accounted for by one of these disorders, we carried out three separate post hoc GWAS of fast beta EEG, each with one of these phenotypes included as a covariate in the model. Including AD as a covariate, the 3q26 association remains but is slightly diminished (rs11720469: β: − 0.120, P-value: 2.2 × 10−8; Supplementary Figure 3A). Including DSM-IV CoD as a covariate, the 3q26 association remains but is also slightly diminished (rs11720469: β: − 0.122, P-value: 1.3 × 10−8; Supplementary Figure 3B). Including DSM-IV ADHD as a covariate reduced the 3q26 association (rs11720469: β: − 0.088, P-value: 6 × 10−4; Supplementary Figure 3C).
DISCUSSION
Although previous studies have reported variation in beta EEG among individuals diagnosed with AD and related conditions, there have been relatively few studies examining genetic variants in relation to beta EEG and only one finding that has been replicated to date (GABRA212,26,57). Subsequently, associations between GABRA2, AD25,58–62 drug dependence58,63 and externalizing behavior64–67 have been observed, indicating the utility of beta EEG as an endophenotype for facilitating discovery of genes underlying disinhibitory behavior.
In what we believe is the first GWAS of fast beta EEG in families of AA, we report a genome-wide significant signal in an intergenic region on 3q26. The most significant SNP, rs11720469, was negatively associated with fast beta EEG (β: − 0.124). Interestingly, this same intergenic region has been previously implicated in a sub-threshold (that is, approaching genome-wide significant) association and gene-based association (C3orf57) in the report by Hodgkinson et al. of monopolar beta (13–30 Hz) EEG in 322 Native Americans.27 Note that, although the present study focused on fast beta (20–28 Hz), post hoc analyses indicated that the 3q26 signal is also observed at a genome-wide level for mid-beta (16–20 Hz) and at a sub-threshold level for low beta (12–16 Hz). We present Manhattan plots for the three beta frequency sub-bands in Supplementary Figure S2: (a) 12–16 Hz, (b) 16–20 Hz, and (c) 20–28, for comparison.
BCHE, thalamus and disinhibitory disorders
rs11720469 is an eQTL for BCHE expression in thalamus and related regions (that is, cortex, caudate). The thalamus plays a central role in relaying sensory and motor signals to the cerebral cortex,68 reflected in EEG dynamics. Dynamic coordination of lower frequencies (theta/alpha rhythms from subcortical regions) and higher frequencies (beta/gamma rhythms from cortical sites) through a mechanism of phase–amplitude coupling modulate thalamo-cortical and corticocortical communication in the brain.69,70 Steriade71 reports that neuronal oscillations result from synaptic interactions in corticothalamic neuronal loops and that intracellularly recorded thalamo-cortical neurons displayed fast oscillations involving beta rhythms. Thalamic volume and/or function contributes to higher-order cognitive functions, including inhibitory control, decision-making and disinhibitory disorders.72 Mackey et al.73 reported that greater temporal discounting was associated with greater volume in a subcortical region encompassing the ventral striatum, hypothalamus and anterior thalamus. Magnetic resonance imaging studies have found that structural variation in the thalamus is related to alcohol consumption74 and AUD,75 as well as relapse in treatment seeking individuals with AUD.76,77 Together, this literature suggests that the thalamus plays a key role in regulatory mechanisms underlying fast beta EEG and AUD.
In addition to AUD, BCHE (and/or surrounding region, 3q26) has previously been associated with behavioral conditions relevant to fast beta EEG, including ADHD and cocaine use/problems. In four independent studies, variations within or surrounding BCHE have also been associated with ADHD.49–52 Jacob et al.50 report that, when meta-analyzing the results of their study with three additional GWAS for copy number variations, they found that individuals with ADHD were more likely to have a deletion in the BCHE promoter region. Given several lines of evidence suggesting the involvement of BCHE in the etiology of ADHD,50 post hoc we re-examined the association between 3q26 variants and fast beta EEG, adjusting for ADHD. When the GWAS of fast beta EEG included ADHD as a covariate in the model, results (Supplementary Figure S3C) show that the 3q26 association is significantly reduced, with the top SNP no longer meeting genome-wide significance criteria. This suggests an important connection between ADHD, 3q26 and fast beta EEG that future studies with longitudinal designs should disentangle (for example, does fast beta EEG mediate the association between 3q26 and ADHD?).
In addition, BChE has a key role in the metabolism of various anesthetics, muscle relaxants and cocaine.78 Once absorbed, cocaine is rapidly transformed into two metabolites catalyzed by BChE, the enzyme produced by BCHE.79 BChE is synthesized primarily in the liver and is distributed throughout the intestinal mucosa, plasma and the brain.80 For these reasons, BChE has been conceptualized as a therapeutic agent for CoD.53 Researchers hypothesize that polymorphisms in BCHE lead to various enzyme profiles that allow different concentrations of cocaine to reach the reward system in the brain, thereby influencing susceptibility to developing addiction.81,82 As fast beta EEG has been associated with CoD in rodents83 and humans,84,85 the apparent links among BCHE, fast beta EEG and CoD should be explored further. Post hoc, we examined the association between 3q26 variants and fast beta EEG, adjusted for CoD. Results (Supplementary Figure S3B) show that the 3q26 association remains, although slightly diminished, suggesting that CoD is not driving the association among 3q26 and fast beta EEG in this sample. Also of potential relevance, variations within BCHE have also been implicated in association studies of learning and memory,86 cognitive functioning,87 schizophrenia88 and Alzheimer’s disease.89
3q26, fast beta EEG, substance use behaviors/disorders
In COGA AA families, individuals with elevated fast beta EEG were more likely to meet criteria for AD (P<0.01), and a small, but significant portion of the variance shared among fast beta EEG and AD is attributable to genome-wide variants (genetic correlation, as estimated by GCTA: 0.10, s.e.: 0.17). There is evidence of association between 4 of the 10 variants meeting genome-wide criteria for fast beta EEG and AD (rs7428372 and rs13093097 survived a multiple test correction), suggesting a potential protective role for fast beta EEG variants in AD in the primary GWAS sample. Further, 4 of the 10 SNPs meeting genome-wide criteria for fast beta EEG in COGA were nominally associated with AD in an independent sample,46 and 2 of these SNPs withstood multiple test correction. Interestingly, these two variants, rs7428372 and rs13093097 (r2>0.97), were two of the four variants that withstood a multiple test correction in the discovery sample.
Association analyses of the top genome-wide significant SNP associated with fast beta EEG in the prospective sample indicated that rs11720469 was associated with heavy-episodic drinking. As this sample has an average age of approximately 17 years, this suggests that individuals with a genetic predisposition toward neuronal hyperexcitability show differences in risky drinking in adolescence/young adulthood. Previous studies indicate that compared with non-binge drinkers or mild-binge drinkers, more severe-binge drinkers have increased fast beta EEG (20–35 Hz).23 As this fast beta power spectral pattern is also observed among those with AD,90 the authors suggested that fast beta EEG may be a biomarker for the development of future AUDs.
These findings provide additional support for the links between fast beta EEG and alcohol use problems. Taken further, this may suggest that 3q26 harbors variants that are related to both fast beta EEG and alcohol problems (or underlying externalizing behavior, for example, ADHD). In addition, this could suggest that fast beta EEG may mediate the associations between 3q26 and alcohol problems. To assess this, post hoc we examined the association between 3q26 variants and fast beta EEG, adjusted for AD. Results (Supplementary Figure S3A) show that the 3q26 association remains but is slightly diminished, suggesting that AD may have an indirect role in the association of 3q26 and fast beta EEG in this sample. Stated another way, fast beta EEG may be a risk factor for some, but not all individuals with AD. These findings, along with the ADHD findings, could also indicate that the association between 3q26 and fast beta EEG is more reflective of generalized neural disinhibition, best captured in this sample by ADHD as compared with CoD and AD. Future studies are needed to examine these hypotheses.
In light of the previous BCHE findings from the literature, and the evidence that rs11720469 is an eQTL for BCHE, this discussion has primarily focused on BCHE. However, there are several other potential candidate genes upstream and downstream of the GWAS signal that might harbor risk/protective variants influencing fast beta EEG and related disorders. These genes include, but are not limited to, PDCD10, WDR49, SERPINI1, SERPINI2 and ZBBX. Given that there are two recombination hotspots between rs11720469 and BCHE, it is possible that BCHE is not directly involved in the association of 3q26 and fast beta EEG observed in this study. WDR49 has previously been associated with quantity of visceral fat91 and SERPINI1 has previously been associated with variation in heart rate.92
Limitations
Most notable is the relatively small sample size and related lack of statistical power to detect subtle genotypic effects. However, GWAS results seem reliable based on corroborating information (that is, several genome-wide significant SNPs in high LD, biological plausibility, replication in an independent sample). Furthermore, given the nominal associations observed in eQTL analyses, these findings must be replicated in larger samples of individuals of AA. Finally, future studies should examine the effects of genetic variants on trajectories of beta EEG during development in order to delineate age-specific effects and the links between these effects and/or the onset of psychopathology (AUD, ADHD, CoD).
CONCLUSIONS
To date, there have been relatively few genetic studies examining beta EEG and only one finding that has been replicated. In addition, no previous gene identification study of beta EEG had been conducted in a population of AA. As the ultimate goal of this research is providing prevention and/or interventions for all individuals, it is crucial that AA populations are included in this work, especially because African-Americans are at greater risk for drinking-related consequences. This study found association between an intergenic signal on 3q26 and fast beta EEG in a sample of related individuals of AA. The most significant SNP is an eQTL for BCHE, a gene previously implicated in disinhibitory disorders and expressed in the thalamus, a brain region central to beta EEG and AUD. Further, fast beta EEG genome-wide associated variants (rs7428372 and rs13093097) were associated with AD both in the discovery sample and an independent sample. Converging data provide support for the role of genetic variants within 3q26 in neural hyperexcitability and disorders characterized by impulsivity. In addition, this study demonstrates the utility of the endophenotype approach13; genetic findings of fast beta EEG have provided an underlying biological hypothesis (that is, neural hyperexcitability) that can enhance our understanding of functional cerebral circuits and mechanisms underlying a predisposition to AUD and related behaviors.
Data availability
COGA data used in the current study are available from the website https://zork5.wustl.edu/coganew/contacts.html upon written request. Details regarding access to COGA data are available through the National Institute of Alcoholism and Abuse at http://www.niaaa.nih.gov.proxy.library.vcu.edu/research/major-initiatives/collaborative-studies-genetics-alcoholism-coga-study#Access. Following an embargo period, COGA data are also available from the publicly accessible dbGAP database at http://www-ncbi-nlm-nih-gov.proxy.library. vcu.edu/gap/?term = COGA (IDs: phs000763.v1.p1).
Supplementary Material
Acknowledgments
The Collaborative Study on the Genetics of Alcoholism (COGA), Principal Investigators B Porjesz, V Hesselbrock, H Edenberg, L Bierut, includes 11 different centers: University of Connecticut (V Hesselbrock); Indiana University (HJ Edenberg, J Nurnberger Jr, T Foroud); University of Iowa (S Kuperman, J Kramer); SUNY Downstate (B Porjesz); Washington University in St Louis (L Bierut, J Rice, K Bucholz, A Agrawal); University of California at San Diego (M Schuckit); Rutgers University (J Tischfield, A Brooks); University of Texas Rio Grand Valley (L Almasy), Virginia Commonwealth University (D Dick), Icahn School of Medicine at Mount Sinai (A Goate), and Howard University (R Taylor). Other COGA collaborators include: L Bauer (University of Connecticut); J McClintick, L Wetherill, X Xuei, Y Liu, D. Lai, S O’Connor, M Plawecki, S Lourens (Indiana University); G Chan (University of Iowa; University of Connecticut); J Meyers, D Chorlian, C Kamarajan, A Pandey, J Zhang (SUNY Downstate); J-C Wang, M Kapoor, S Bertelsen (Icahn School of Medicine at Mount Sinai); A Anokhin, V McCutcheon, S Saccone (Washington University); J Salvatore, F Aliev, B Cho (Virginia Commonwealth University); and Mark Kos (University of Texas Rio Grand Valley). A Parsian and M Reilly are the NIAAA Staff Collaborators. We continue to be inspired by our memories of Henri Begleiter and Theodore Reich, founding PI and Co-PI of COGA, and also owe a debt of gratitude to other past organizers of COGA, including Ting-Kai Li, P Michael Conneally, Raymond Crowe and Wendy Reich, for their critical contributions. This national collaborative study is supported by an NIH Grant U10AA008401 from the National Institute on Alcohol Abuse and Alcoholism (NIAAA) and the National Institute on Drug Abuse (NIDA). JLM is supported by K01DA037914 from the National Institute on Drug Abuse (NIDA), JES acknowledges support from K01AA024152 (NIAAA) and AA acknowledges support from K02DA032573 (NIDA). Funding support for GWAS genotyping performed at the Johns Hopkins University Center for Inherited Disease Research was provided by the National Institute on Alcohol Abuse and Alcoholism, the NIH GEI (U01HG004438), and the NIH contract ‘High throughput genotyping for studying the genetic contributions to human disease’ (HHSN268200782096C). GWAS genotyping was also performed at the Genome Technology Access Center in the Department of Genetics at Washington University School of Medicine, which is partially supported by NCI Cancer Center Support Grant no. P30 CA91842 to the Siteman Cancer Center and by ICTS/CTSA Grant no. UL1RR024992 from the National Center for Research Resources (NCRR), a component of the National Institutes of Health (NIH), and NIH Roadmap for Medical Research.
Footnotes
CONFLICT OF INTEREST
The authors declare no conflict of interest. However, unrelated to this work, AA (coauthor) received peer-reviewed funding, travel and an honorarium from ABMRF (end December 2012), which receives support from the brewing industry.
DISCLAIMER
This material is original research, has not been previously published and has not been submitted for publication elsewhere while under consideration.
Supplementary Information accompanies the paper on the Molecular Psychiatry website (http://www.nature.com/mp)
References
- 1.Niedermeyer E, Sherman DL, Geocadin RJ, Hansen HC, Hanley DF. The burst-suppression electroencephalogram. Clin Electroencephalogr. 1999;30:99–105. doi: 10.1177/155005949903000305. [DOI] [PubMed] [Google Scholar]
- 2.Pfurtscheller G, Lopes da Silva FH. Event-related EEG/MEG synchronization and desynchronization: basic principles. Clin Neurophysiol. 1999;110:1842–1857. doi: 10.1016/s1388-2457(99)00141-8. [DOI] [PubMed] [Google Scholar]
- 3.Donner TH, Siegel M. A framework for local cortical oscillation patterns. Trends Cogn Sci. 2011;15:191–199. doi: 10.1016/j.tics.2011.03.007. [DOI] [PubMed] [Google Scholar]
- 4.Begleiter H, Porjesz B. What is inherited in the predisposition toward alcoholism? A proposed model. Alcohol Clin Exp Res. 1999;23:1125–1135. doi: 10.1111/j.1530-0277.1999.tb04269.x. [DOI] [PubMed] [Google Scholar]
- 5.Bauer LO. Predicting relapse to alcohol and drug abuse via quantitative electroencephalography. Neuropsychopharmacology. 2001;25:332–340. doi: 10.1016/S0893-133X(01)00236-6. [DOI] [PubMed] [Google Scholar]
- 6.Rangaswamy M, Porjesz B, Chorlian DB, Wang K, Jones KA, Bauer LO, et al. Beta power in the EEG of alcoholics. Biol Psychiatry. 2002;52:831–842. doi: 10.1016/s0006-3223(02)01362-8. [DOI] [PubMed] [Google Scholar]
- 7.Gilmore CS, Malone SM, Iacono WG. Brain electrophysiological endophenotypes for externalizing psychopathology: a multivariate approach. Behav Genet. 2010;40:186–200. doi: 10.1007/s10519-010-9343-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Choi J-S, Park SM, Lee J, Hwang JY, Jung HY, Choi S-W, et al. Resting-state beta and gamma activity in Internet addiction. Int J Psychophysiol. 2013;89:328–333. doi: 10.1016/j.ijpsycho.2013.06.007. [DOI] [PubMed] [Google Scholar]
- 9.Lee J, Hwang JY, Park SM, Jung HY, Choi S-W, Kim DJ, et al. Differential resting-state EEG patterns associated with comorbid depression in Internet addiction. Prog Neuropsychopharmacol Biol Psychiatry. 2014;50:21–26. doi: 10.1016/j.pnpbp.2013.11.016. [DOI] [PubMed] [Google Scholar]
- 10.Barry RJ, Clarke AR, Johnstone SJ. A review of electrophysiology in attention-deficit/hyperactivity disorder: I Qualitative and quantitative electroencephalography. Clin Neurophysiol. 2003;114:171–183. doi: 10.1016/s1388-2457(02)00362-0. [DOI] [PubMed] [Google Scholar]
- 11.van Beijsterveldt CE, Molenaar PC, de Geus EJ, Boomsma DI. Heritability of human brain functioning as assessed by electroencephalography. Am J Hum Genet. 1996;58:562–573. [PMC free article] [PubMed] [Google Scholar]
- 12.Malone SM, Burwell SJ, Vaidyanathan U, Miller MB, McGue M, Iacono WG. Heritability and molecular-genetic basis of resting EEG activity: a genome-wide association study. Psychophysiology. 2014;51:1225–1245. doi: 10.1111/psyp.12344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gottesman II, Gould TD. The endophenotype concept in psychiatry: etymology and strategic intentions. Am J Psychiatry. 2003;160:636–645. doi: 10.1176/appi.ajp.160.4.636. [DOI] [PubMed] [Google Scholar]
- 14.Porjesz B, Rangaswamy M, Kamarajan C, Jones KA, Padmanabhapillai A, Begleiter H. The utility of neurophysiological markers in the study of alcoholism. Clin Neurophysiol. 2005;116:993–1018. doi: 10.1016/j.clinph.2004.12.016. [DOI] [PubMed] [Google Scholar]
- 15.Propping P, Krüger J, Mark N. Genetic disposition to alcoholism An EEG study in alcoholics and their relatives. Hum Genet. 1981;59:51–59. doi: 10.1007/BF00278854. [DOI] [PubMed] [Google Scholar]
- 16.Costa L, Bauer L. Quantitative electroencephalographic differences associated with alcohol, cocaine, heroin and dual-substance dependence. Drug Alcohol Depend. 1997;46:87–93. doi: 10.1016/s0376-8716(97)00058-6. [DOI] [PubMed] [Google Scholar]
- 17.Winterer G, Klöppel B, Heinz A, Ziller M, Dufeu P, Schmidt LG, et al. Quantitative EEG (QEEG) predicts relapse in patients with chronic alcoholism and points to a frontally pronounced cerebral disturbance. Psychiatry Res. 1998;78:101–113. doi: 10.1016/s0165-1781(97)00148-0. [DOI] [PubMed] [Google Scholar]
- 18.Saletu-Zyhlarz GM, Arnold O, Anderer P, Oberndorfer S, Walter H, Lesch OM, et al. Differences in brain function between relapsing and abstaining alcohol-dependent patients, evaluated by EEG mapping. Alcohol Alcohol. 39:233–240. doi: 10.1093/alcalc/agh041. [DOI] [PubMed] [Google Scholar]
- 19.Deckel AW, Hesselbrock V, Bauer L. Antisocial personality disorder, childhood delinquency, and frontal brain functioning: EEG and neuropsychological findings. J Clin Psychol. 1996;52:639–650. doi: 10.1002/(SICI)1097-4679(199611)52:6<639::AID-JCLP6>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 20.Rangaswamy M, Porjesz B, Chorlian DB, Wang K, Jones KA, Kuperman S, et al. Resting EEG in offspring of male alcoholics: beta frequencies. Int J Psychophysiol. 2004;51:239–251. doi: 10.1016/j.ijpsycho.2003.09.003. [DOI] [PubMed] [Google Scholar]
- 21.Bresnahan SM, Barry RJ. Specificity of quantitative EEG analysis in adults with attention deficit hyperactivity disorder. Psychiatry Res. 2002;112:133–144. doi: 10.1016/s0165-1781(02)00190-7. [DOI] [PubMed] [Google Scholar]
- 22.Markovska-Simoska S, Pop-Jordanova N. Quantitative EEG in children and adults with attention deficit hyperactivity disorder: comparison of absolute and relative power spectra and theta/beta ratio. Clin EEG Neurosci. 2016 May 11;:pii. doi: 10.1177/1550059416643824. advance online publication. 1550059416643824 [e-pub ahead of print] [DOI] [PubMed] [Google Scholar]
- 23.Courtney KE, Polich J. Binge drinking effects on EEG in young adult humans. Int J Environ Res Public Health. 2010;7:2325–2336. doi: 10.3390/ijerph7052325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Porjesz B, Almasy L, Edenberg HJ, Wang K, Chorlian DB, Foroud T, et al. Linkage disequilibrium between the beta frequency of the human EEG and a GABA A receptor gene locus. Proc Natl Acad Sci USA. 2002;99:3729–3733. doi: 10.1073/pnas.052716399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Edenberg HJ, Dick DM, Xuei X, Tian H, Almasy L, Bauer LO, et al. Variations in GABRA2, encoding the alpha 2 subunit of the GABA(A) receptor, are associated with alcohol dependence and with brain oscillations. Am J Hum Genet. 2004;74:705–714. doi: 10.1086/383283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lydall GJ, Saini J, Ruparelia K, Montagnese S, McQuillin A, Guerrini I, et al. Genetic association study of GABRA2 single nucleotide polymorphisms and electroencephalography in alcohol dependence. Neurosci Lett. 2011;500:162–166. doi: 10.1016/j.neulet.2011.05.240. [DOI] [PubMed] [Google Scholar]
- 27.Hodgkinson CA, Enoch M-A, Srivastava V, Cummins-Oman JS, Ferrier C, Iarikova P, et al. Genome-wide association identifies candidate genes that influence the human electroencephalogram. Proc Natl Acad Sci USA. 2010;107:8695–8700. doi: 10.1073/pnas.0908134107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rosenberg NA, Huang L, Jewett EM, Szpiech ZA, Jankovic I, Boehnke M. Genome-wide association studies in diverse populations. Nat Rev Genet. 2010;11:356–366. doi: 10.1038/nrg2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mukku VK, Benson TG, Alam F, Richie WD, Bailey RK. Overview of substance use disorders and incarceration of African American males. Front Psychiatry. 2012;3:98. doi: 10.3389/fpsyt.2012.00098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zapolski TCB, Pedersen SL, McCarthy DM, Smith GT. Less drinking, yet more problems: understanding African American drinking and related problems. Psychol Bull. 2014;140:188–223. doi: 10.1037/a0032113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mulia N, Ye Y, Greenfield TK, Zemore SE. Disparities in alcohol-related problems among white, black, and Hispanic Americans. Alcohol Clin Exp Res. 2009;33:654–662. doi: 10.1111/j.1530-0277.2008.00880.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zemore SE, Ye Y, Mulia N, Martinez P, Jones-Webb R, Karriker-Jaffe K. Poor, persecuted, young, and alone: toward explaining the elevated risk of alcohol problems among Black and Latino men who drink. Drug Alcohol Depend. 2016;163:31–39. doi: 10.1016/j.drugalcdep.2016.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tang Y, Chorlian DB, Rangaswamy M, Porjesz B, Bauer L, Kuperman S, et al. Genetic influences on bipolar EEG power spectra. Int J Psychophysiol. 2007;65:2–9. doi: 10.1016/j.ijpsycho.2007.02.004. [DOI] [PubMed] [Google Scholar]
- 34.Reich T, Edenberg HJ, Goate A, Williams JT, Rice JP, Van Eerdewegh P, et al. Genome-wide search for genes affecting the risk for alcohol dependence. Am J Med Genet. 1998;81:207–215. [PubMed] [Google Scholar]
- 35.Foroud T, Edenberg HJ, Goate A, Rice J, Flury L, Koller DL, et al. Alcoholism susceptibility loci: confirmation studies in a replicate sample and further mapping. Alcohol Clin Exp Res. 2000;24:933–945. [PubMed] [Google Scholar]
- 36.Bucholz KK, Cadoret R, Cloninger CR, Dinwiddie SH, Hesselbrock VM, Nurnberger JI, et al. A new, semi-structured psychiatric interview for use in genetic linkage studies: a report on the reliability of the SSAGA. J Stud Alcohol. 1994;55:149–158. doi: 10.15288/jsa.1994.55.149. [DOI] [PubMed] [Google Scholar]
- 37.Wetherill L, Agrawal A, Kapoor M, Bertelsen S, Bierut LJ, Brooks A, et al. Association of substance dependence phenotypes in the COGA sample. Addict Biol. 2015;20:617–627. doi: 10.1111/adb.12153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.O’Connell JR, Weeks DE. PedCheck: a program for identification of genotype incompatibilities in linkage analysis. Am J Hum Genet. 1998;63:259–266. doi: 10.1086/301904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Delaneau O, Marchini J. Integrating sequence and array data to create an improved 1000 Genomes Project haplotype reference panel. Nat Commun. 2014;5:3934. doi: 10.1038/ncomms4934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Howie BN, Donnelly P, Marchini J. A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet. 2009;5:e1000529. doi: 10.1371/journal.pgen.1000529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen M-H, Yang Q. GWAF: an R package for genome-wide association analyses with family data. Bioinformatics. 2010;26:580–581. doi: 10.1093/bioinformatics/btp710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zheng X, Levine D, Shen J, Gogarten SM, Laurie C, Weir BS. A high-performance computing toolset for relatedness and principal component analysis of SNP data. Bioinformatics. 2012;28:3326–3328. doi: 10.1093/bioinformatics/bts606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Trabzuni D, Ryten M, Walker R, Smith C, Imran S, Ramasamy A, et al. Quality control parameters on a large dataset of regionally dissected human control brains for whole genome expression studies. J Neurochem. 2011;119:275–282. doi: 10.1111/j.1471-4159.2011.07432.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 4th. Washington, DC: 2000. text rev. [Google Scholar]
- 45.Wang Z. Direct assessment of multiple testing correction in case-control association studies with related individuals. Genet Epidemiol. 2011;35:70–79. doi: 10.1002/gepi.20555. [DOI] [PubMed] [Google Scholar]
- 46.Gelernter J, Kranzler HR, Sherva R, Almasy L, Koesterer R, Smith AH, et al. Genome-wide association study of alcohol dependence:significant findings in African- and European-Americans including novel risk loci. Mol Psychiatry. 2014;19:41–49. doi: 10.1038/mp.2013.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dick DM, Cho S, Bin, Latendresse SJ, Aliev F, Nurnberger JI, Edenberg HJ, et al. Genetic influences on alcohol use across stages of development: GABRA2 and longitudinal trajectories of drunkenness from adolescence to young adulthood. Addict Biol. 2014;19:1055–1064. doi: 10.1111/adb.12066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Muthén LK, Muthén BO. Mplus User’s Guide. 7th. Muthén & Muthén; Los Angeles, CA: 1998–2015. [Google Scholar]
- 49.Elia J, Gai X, Xie HM, Perin JC, Geiger E, Glessner JT, et al. Rare structural variants found in attention-deficit hyperactivity disorder are preferentially associated with neurodevelopmental genes. Mol Psychiatry. 2010;15:637–646. doi: 10.1038/mp.2009.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jacob CP, Weber H, Retz W, Kittel-Schneider S, Heupel J, Renner T, et al. Acetylcholine-metabolizing butyrylcholinesterase (BCHE) copy number and single nucleotide polymorphisms and their role in attention-deficit/hyperactivity syndrome. J Psychiatr Res. 2013;47:1902–1908. doi: 10.1016/j.jpsychires.2013.08.006. [DOI] [PubMed] [Google Scholar]
- 51.Lesch K-P, Selch S, Renner TJ, Jacob C, Nguyen TT, Hahn T, et al. Genome-wide copy number variation analysis in attention-deficit/hyperactivity disorder: association with neuropeptide Y gene dosage in an extended pedigree. Mol Psychiatry. 2011;16:491–503. doi: 10.1038/mp.2010.29. [DOI] [PubMed] [Google Scholar]
- 52.Lionel AC, Crosbie J, Barbosa N, Goodale T, Thiruvahindrapuram B, Rickaby J, et al. Rare copy number variation discovery and cross-disorder comparisons identify risk genes for ADHD. Sci Transl Med. 2011;3:95ra–75. doi: 10.1126/scitranslmed.3002464. [DOI] [PubMed] [Google Scholar]
- 53.Lockridge O. Review of human butyrylcholinesterase structure, function, genetic variants, history of use in the clinic, and potential therapeutic uses. Pharmacol Ther. 2015;148:34–46. doi: 10.1016/j.pharmthera.2014.11.011. [DOI] [PubMed] [Google Scholar]
- 54.Ward LD, Kellis M. HaploReg v4: systematic mining of putative causal variants, cell types, regulators and target genes for human complex traits and disease. Nucleic Acids Res. 2016;44:D877–D881. doi: 10.1093/nar/gkv1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ward LD, Kellis M. HaploReg: a resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic Acids Res. 2012;40:D930–D934. doi: 10.1093/nar/gkr917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Consortium RE. Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, et al. Integrative analysis of 111 reference human epigenomes. Nature. 2015;518:317–330. doi: 10.1038/nature14248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Porjesz B, Begleiter H, Wang K, Almasy L, Chorlian DB, Stimus AT, et al. Linkage and linkage disequilibrium mapping of ERP and EEG phenotypes. Biol Psychol. 2002;61:229–248. doi: 10.1016/s0301-0511(02)00060-1. [DOI] [PubMed] [Google Scholar]
- 58.Enoch M-A, Hodgkinson CA, Yuan Q, Shen P-H, Goldman D, Roy A. The influence of GABRA2, childhood trauma, and their interaction on alcohol, heroin, and cocaine dependence. Biol Psychiatry. 2010;67:20–27. doi: 10.1016/j.biopsych.2009.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Roh S, Matsushita S, Hara S, Maesato H, Matsui T, Suzuki G, et al. Role of GABRA2 in moderating subjective responses to alcohol. Alcohol Clin Exp Res. 2011;35:400–407. doi: 10.1111/j.1530-0277.2010.01357.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ittiwut C, Yang B-Z, Kranzler HR, Anton RF, Hirunsatit R, Weiss RD, et al. GABRG1 and GABRA2 variation associated with alcohol dependence in African Americans. Alcohol Clin Exp Res. 2012;36:588–593. doi: 10.1111/j.1530-0277.2011.01637.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Li D, Sulovari A, Cheng C, Zhao H, Kranzler HR, Gelernter J. Association of gamma-aminobutyric acid A receptor α2 gene (GABRA2) with alcohol use disorder. Neuropsychopharmacology. 2014;39:907–918. doi: 10.1038/npp.2013.291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lappalainen J, Krupitsky E, Remizov M, Pchelina S, Taraskina A, Zvartau E, et al. Association between alcoholism and gamma-amino butyric acid alpha2 receptor subtype in a Russian population. Alcohol Clin Exp Res. 2005;29:493–498. doi: 10.1097/01.alc.0000158938.97464.90. [DOI] [PubMed] [Google Scholar]
- 63.Ehlers CL, Gizer IR. Evidence for a genetic component for substance dependence in Native Americans. Am J Psychiatry. 2013;170:154–164. doi: 10.1176/appi.ajp.2012.12010113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dick DM, Aliev F, Latendresse S, Porjesz B, Schuckit M, Rangaswamy M, et al. How phenotype and developmental stage affect the genes we find: GABRA2 and impulsivity. Twin Res Hum Genet. 2013;16:661–669. doi: 10.1017/thg.2013.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Salvatore JE, Meyers JL, Yan J, Aliev F, Lansford JE, Pettit GS, et al. Intergenerational continuity in parents’ and adolescents’ externalizing problems: The role of life events and their interaction with GABRA2. J Abnorm Psychol. 2015;124:709–729. doi: 10.1037/abn0000066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wang FL, Chassin L, Geiser C, Lemery-Chalfant K. Mechanisms in the relation between GABRA2 and adolescent externalizing problems. Eur Child Adolesc Psychiatry. 2016;25:67–80. doi: 10.1007/s00787-015-0703-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Trucco EM, Hicks BM, Villafuerte S, Nigg JT, Burmeister M, Zucker RA. Temperament and externalizing behavior as mediators of genetic risk on adolescent substance use. J Abnorm Psychol. 2016;125:565–575. doi: 10.1037/abn0000143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sherman SM, Guillery RW. Distinct functions for direct and transthalamic corticocortical connections. J Neurophysiol. 2011;106:1068–1077. doi: 10.1152/jn.00429.2011. [DOI] [PubMed] [Google Scholar]
- 69.Canolty RT, Knight RT. The functional role of cross-frequency coupling. Trends Cogn Sci. 2010;14:506–515. doi: 10.1016/j.tics.2010.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Malekmohammadi M, Elias WJ, Pouratian N. Human thalamus regulates cortical activity via spatially specific and structurally constrained phase-amplitude coupling. Cereb Cortex. 2015;25:1618–1628. doi: 10.1093/cercor/bht358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Steriade M. Grouping of brain rhythms in corticothalamic systems. Neuroscience. 2006;137:1087–1106. doi: 10.1016/j.neuroscience.2005.10.029. [DOI] [PubMed] [Google Scholar]
- 72.Mitchell AS, Sherman SM, Sommer MA, Mair RG, Vertes RP, Chudasama Y. Advances in understanding mechanisms of thalamic relays in cognition and behavior. J Neurosci. 2014;34:15340–15346. doi: 10.1523/JNEUROSCI.3289-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mackey S, Chaarani B, Kan K-J, Spechler PA, Orr C, Banaschewski T, et al. Brain regions related to impulsivity mediate the effects of early adversity on antisocial behavior. Biol Psychiatry. 2016 Jan 18;:pii. doi: 10.1016/j.biopsych.2015.12.027. advance online publication. S0006-3223(16)00043-3. [DOI] [PubMed] [Google Scholar]
- 74.Hu S, Zhang S, Chao HH, Krystal JH. Li C-SR association of drinking problems and duration of alcohol use to inhibitory control in nondependent young adult social drinkers. Alcohol Clin Exp Res. 2016;40:319–328. doi: 10.1111/acer.12964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Mon A, Durazzo TC, Abe C, Gazdzinski S, Pennington D, Schmidt T, et al. Structural brain differences in alcohol-dependent individuals with and without comorbid substance dependence. Drug Alcohol Depend. 2014;144:170–177. doi: 10.1016/j.drugalcdep.2014.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Segobin SH, Chételat G, Le Berre A-P, Lannuzel C, Boudehent C, Vabret F, et al. Relationship between brain volumetric changes and interim drinking at six months in alcohol-dependent patients. Alcohol Clin Exp Res. 2014;38:739–748. doi: 10.1111/acer.12300. [DOI] [PubMed] [Google Scholar]
- 77.Zahr NM, Carr RA, Rohlfing T, Mayer D, Sullivan EV, Colrain IM, et al. Brain metabolite levels in recently sober individuals with alcohol use disorder: Relation to drinking variables and relapse. Psychiatry Res. 2016;250:42–49. doi: 10.1016/j.pscychresns.2016.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kamendulis LM, Brzezinski MR, Pindel EV, Bosron WF, Dean RA. Metabolism of cocaine and heroin is catalyzed by the same human liver carboxylesterases. J Pharmacol Exp Ther. 1996;279:713–717. [PubMed] [Google Scholar]
- 79.Allderdice PW, Gardner HA, Galutira D, Lockridge O, LaDu BN, McAlpine PJ. The cloned butyrylcholinesterase (BCHE) gene maps to a single chromosome site, 3q26. Genomics. 1991;11:452–454. doi: 10.1016/0888-7543(91)90154-7. [DOI] [PubMed] [Google Scholar]
- 80.Goodall R. Cholinesterase: phenotyping and genotyping. Ann Clin Biochem. 2004;41:98–110. doi: 10.1258/000456304322879971. [DOI] [PubMed] [Google Scholar]
- 81.Brimijoin S, Gao Y, Anker JJ, Gliddon LA, Lafleur D, Shah R, et al. A cocaine hydrolase engineered from human butyrylcholinesterase selectively blocks cocaine toxicity and reinstatement of drug seeking in rats. Neuropsychopharmacology. 2008;33:2715–2725. doi: 10.1038/sj.npp.1301666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Negrão AB, Pereira AC, Guindalini C, Santos HC, Messas GP, Laranjeira R, et al. Butyrylcholinesterase genetic variants: association with cocaine dependence and related phenotypes. PLoS One. 2013;8:e80505. doi: 10.1371/journal.pone.0080505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kiyatkin EA, Smirnov MS. Rapid EEG desynchronization and EMG activation induced by intravenous cocaine in freely moving rats: a peripheral, non-dopamine neural triggering. Am J Physiol Regul Integr Comp Physiol. 2010;298:R285–R300. doi: 10.1152/ajpregu.00628.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Newton TF, Cook IA, Kalechstein AD, Duran S, Monroy F, Ling W, et al. Quantitative EEG abnormalities in recently abstinent methamphetamine dependent individuals. Clin Neurophysiol. 2003;114:410–415. doi: 10.1016/s1388-2457(02)00409-1. [DOI] [PubMed] [Google Scholar]
- 85.Binienda ZK, Beaudoin MA, Thorn BT, Ali SF. Analysis of electrical brain waves in neurotoxicology: γ-hydroxybutyrate. Curr Neuropharmacol. 2011;9:236–239. doi: 10.2174/157015911795017209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wan Q. Effect of Dipsacus total saponins on the ability of learning and memory and acetylcholine metabolism of hippocampus in AD rats. Zhongguo Ying Yong Sheng Li Xue Za Zhi. 2015;31:82–84. [PubMed] [Google Scholar]
- 87.Manoharan I, Kuznetsova A, Fisk JD, Boopathy R, Lockridge O, Darvesh S. Comparison of cognitive functions between people with silent and wild-type butyrylcholinesterase. J Neural Transm. 2007;114:939–945. doi: 10.1007/s00702-007-0631-x. [DOI] [PubMed] [Google Scholar]
- 88.Mabrouk H, Mechria H, Mechri A, Rahali H, Douki W, Gaha L, et al. Butyrylcholinesterase activity in schizophrenic patients. Ann Biol Clin (Paris) 69:647–652. doi: 10.1684/abc.2011.0634. [DOI] [PubMed] [Google Scholar]
- 89.Ramanan VK, Risacher SL, Nho K, Kim S, Swaminathan S, Shen L, et al. APOE and BCHE as modulators of cerebral amyloid deposition: a florbetapir PET genome-wide association study. Mol Psychiatry. 2014;19:351–357. doi: 10.1038/mp.2013.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Rangaswamy M, Porjesz B. Understanding alcohol use disorders with neuroelectrophysiology. Handb Clin Neurol. 2014;125:383–414. doi: 10.1016/B978-0-444-62619-6.00023-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Fox CS, Liu Y, White CC, Feitosa M, Smith AV, Heard-Costa N, et al. Genome-wide association for abdominal subcutaneous and visceral adipose reveals a novel locus for visceral fat in women. PLoS Genet. 2012;8:e1002695. doi: 10.1371/journal.pgen.1002695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Jeff JM, Ritchie MD, Denny JC, Kho AN, Ramirez AH, Crosslin D, et al. Generalization of variants identified by genome-wide association studies for electrocardiographic traits in African Americans. Ann Hum Genet. 2013;77:321–332. doi: 10.1111/ahg.12023. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
COGA data used in the current study are available from the website https://zork5.wustl.edu/coganew/contacts.html upon written request. Details regarding access to COGA data are available through the National Institute of Alcoholism and Abuse at http://www.niaaa.nih.gov.proxy.library.vcu.edu/research/major-initiatives/collaborative-studies-genetics-alcoholism-coga-study#Access. Following an embargo period, COGA data are also available from the publicly accessible dbGAP database at http://www-ncbi-nlm-nih-gov.proxy.library. vcu.edu/gap/?term = COGA (IDs: phs000763.v1.p1).