

Abstract
Transforming growth factor-β (TGF-β) is a cytokine produced by many cell types and implicated in cell growth, differentiation, apoptosis, and inflammation. It stimulates store-operated calcium entry (SOCE) through the calcium release-activated calcium (CRAC) channel Orai1/Stim1 in endometrial Ishikawa cells. Bone cells generate fibroblast growth factor (FGF) 23, which inhibits renal phosphate reabsorption and 1,25(OH)2D3 formation in concert with its co-receptor Klotho. Moreover, Klotho and FGF23 counteract aging and age-related clinical conditions. FGF23 production is dependent on Orai1-mediated SOCE and inflammation. Here, we explored a putative role of TGF-β2 in FGF23 synthesis. To this end, UMR106 osteoblast-like cells were cultured, Fgf23 transcript levels determined by qRT-PCR, FGF23 protein measured by ELISA, and SOCE analyzed by fluorescence optics. UMR106 cells expressed TGF-β receptors 1 and 2. TGF-β2 enhanced SOCE and potently stimulated the production of FGF23, an effect significantly attenuated by SB431542, an inhibitor of the transforming growth factor-β (TGF-β) type I receptor activin receptor-like kinases ALK5, ALK4, and ALK7. Furthermore, the TGF-β2 effect on FGF23 production was blunted by SOCE inhibitor 2-APB. We conclude that TGF-β2 induces FGF23 production, an effect involving up-regulation of SOCE.
Introduction
Transforming growth factor β (TGF-β) is a cytokine, which is produced by a broad range of cells1. It impacts on cell proliferation, growth, differentiation, migration, and apoptosis. TGF-β induces cell cycle arrest in normal cells. In cancer cells mutations of the gene encoding TGF-β may result in a loss of its anti-proliferative activity2. Moreover, TGF-β promotes fibrosis of many organs including the heart and kidney1. Its role is particularly well established in chronic kidney disease3. The cellular effects of TGF-β are mediated by membrane receptors including TGF-β receptors 1 and 24. When binding TGF-β, the two receptors form a heterodimer and phosphorylate cellular proteins4.
The hormone fibroblast growth factor 23 (FGF23) is a powerful regulator of several renal functions5–8: It is produced by osteocytes and osteoblasts9. FGF23 inhibits renal phosphate reabsorption by stimulating the internalization of NaPi-IIa, the main renal Na+-dependent phosphate transporter5. In addition, it inhibits CYP27B1 or 25-hydroxyvitamin D3 1-alpha-hydroxylase, the renal key enzyme for the synthesis of 1,25(OH)2D3 or calcitriol, the active form of vitamin D5. The consequence of these effects is a decrease of the plasma concentrations of both, phosphate and 1,25(OH)2D3. The major renal effects of FGF23 are mediated by a membrane receptor which requires the transmembrane protein Klotho as a co-receptor5. In addition, FGF23 has extrarenal effects, including the Klotho-independent induction of left ventricular hypertrophy10.
Analysis of transgenic mice has revealed a powerful influence of the two proteins on aging. Mice deficient in either FGF23 or Klotho age rapidly, have a life span of a few weeks only, and suffer from a plethora of aging-associated diseases including muscle, skin, neuronal, metabolic, and cardiovascular abnormalities11, 12. Notably, the aging of the mice can be almost normal if the mice are fed a low vitamin D or low phosphate diet pointing to a hitherto underestimated impact of phosphate metabolism on aging and age-associated diseases13.
Regulation of FGF23 formation is still incompletely understood. Recent research suggests that the plasma FGF23 concentration is a valuable and sensitive biomarker in several acute and chronic disorders14. In line with this the plasma FGF23 level correlates with disease activity in renal, cardiovascular, metabolic, and inflammatory diseases14.
Known regulators of FGF23 production include dietary phosphorus intake, parathyroid hormone (PTH)15, 1,25(OH)2D3 16, the iron status17, and inflammation17. In detail, activation of the key inflammatory transcription factor NFκB up-regulates the calcium release-activated calcium (CRAC) channel Orai1/STIM1 which mediates store-operated calcium entry (SOCE)18. Orai1/STIM1-mediated SOCE then triggers FGF23 production. Of note, TGF-β has also been shown to up-regulate Orai1/STIM1 in endometrial tumor cells19. Moreover, the production of TGF-β is also enhanced in some of the aforementioned disorders associated with high serum FGF23 levels (e.g. chronic kidney disease3).
The present study addressed the question whether the TGF-β-isoform TGF-β2 is a regulator of FGF23 formation.
Results
TGF-β2 induced Fgf23 transcription in UMR106 cells
Our first series of experiments aimed to explore whether TGF-β2 is capable of influencing the production of FGF23. To this end, we treated UMR106 osteoblast-like cells with and without TGF-β2 for 24 h and determined Fgf23 transcripts by qRT-PCR to estimate FGF23 synthesis. Figure 1A illustrates that the presence of TGF-β2 significantly up-regulated Fgf23 transcript levels, pointing to enhanced FGF23 production. Next, we sought to investigate whether the increase in Fgf23 transcript levels upon incubation with TGF-β2 indeed translated into enhanced FGF23 protein formation. Thus, we determined C-terminal FGF23 protein in the supernatant retrieved from cells incubated with and without TGF-β2 for 24 h using ELISA. As shown in Fig. 1B, TGF-β2 significantly stimulated FGF23 production. Figure 1C depicts the dose response effect of TGF-β2 on Fgf23 transcripts in UMR106 cells.
Figure 1.
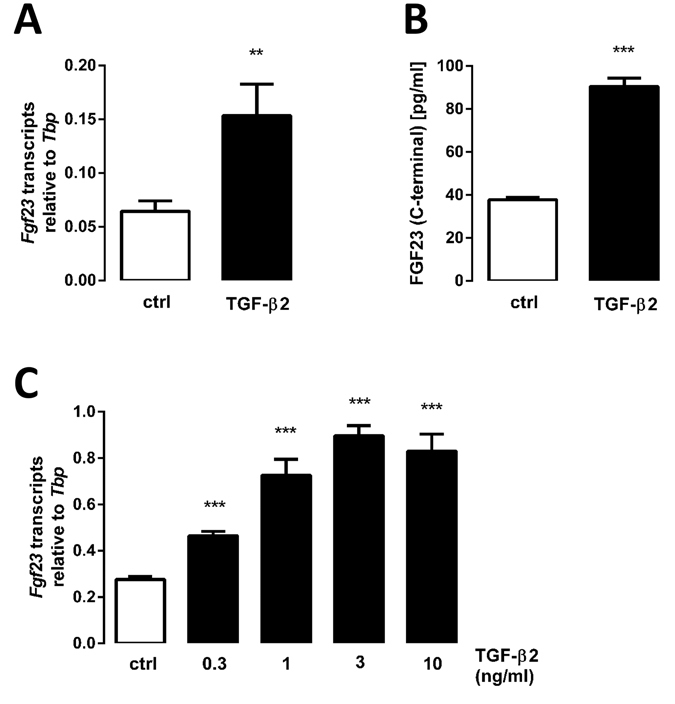
TGF-β2 stimulates Fgf23 gene expression and FGF23 protein synthesis in UMR106 cells. Arithmetic means ± SEM (n = 7) of (A) Fgf23 mRNA abundance (relative to Tbp mRNA) and (B) FGF23 (C-terminal) protein concentration (pg/ml) in the cell culture medium of UMR106 cells incubated without (ctrl, white bars) or with TGF-β2 (black bars, 10 ng/ml, 24 h). **p < 0.01, ***p < 0.001 indicate significant difference compared to UMR106 cells treated with vehicle only (paired t-test). (C) Arithmetic mean ± SEM (n = 5) of Fgf23 mRNA levels (relative to Tbp mRNA) in UMR106 cells after 24-hour treatment without (ctrl, white bar) or with TGF-β2 (0.3–10 ng/ml, black bars). ***p < 0.001 indicates significant difference compared to UMR106 cells treated with vehicle only (ANOVA).
The TGF-β2 effect on Fgf23 expression was mediated by downstream TGF-β receptor signaling
The cellular effects of TGF-β2 are typically mediated by the joint action of TGF-β receptors 1 and 2. Employing RT-PCR, we therefore studied the expression of these receptors in UMR106 cells. It is demonstrated in Fig. 2 that UMR106 cells expressed TGF-β receptors 1 and 2. Moreover, cells retrieved from mouse bone also expressed both receptors (Fig. 2). As a control, expression of TGF-β receptors 1 and 2 in rat NRK-52E cells is provided (Fig. 2).
Figure 2.
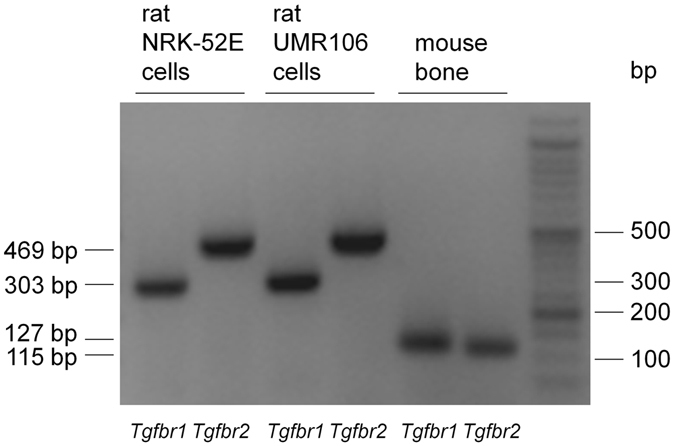
Expression of TGF-β receptor 1 and 2 in UMR106 cells and murine bone tissue. Original agarose gel photo demonstrating amplified Tgfbr1 and Tgfbr2-specific cDNA in UMR106 cells and murine bone tissue. NRK-52E cells were used as a positive control.
As a next step, we explored whether the stimulating effect of TGF-β2 on the formation of FGF23 was dependent on downstream TGF-β receptor signaling which involves TGF-β receptor-related kinases. Hence, we determined the TGF-β2 effect on Fgf23 transcripts in the presence and absence of SB431542, an antagonist of TGF-β receptor-related kinases. As shown in Fig. 3, the effect of TGF-β2 on Fgf23 mRNA was significantly attenuated in the presence of SB431542. This result demonstrates that TGF-β receptor-related kinases were required for TGF-β2 to enhance FGF23 generation.
Figure 3.
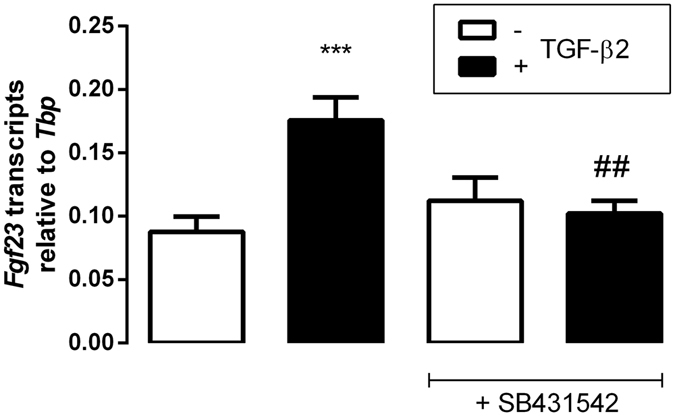
Effect of TGF-β2 on Fgf23 transcript levels in UMR106 cells is blunted by TGF-β receptor-related kinase inhibitor SB431542. Arithmetic means ± SEM (n = 9) of Fgf23 mRNA abundance (relative to Tbp mRNA) in UMR106 cells incubated without (white bars) or with (black bars) TGF-β2 (10 ng/ml) in the absence or presence of downstream TGF-β receptor-related kinase inhibitor SB431542 (10 μM) for 24 h. ***p < 0.001 indicates significant difference between cells treated with or without TGF-β2. ## p < 0.01 indicates significant difference between cells treated with TGF-β2 alone (2nd bar) and cells treated with TGF-β2 and SB431542 (4th bar) (ANOVA).
Store-operated calcium entry (SOCE) was needed for the TGF-β2 effect on Fgf23 expression
Store-operated calcium entry through Orai1/STIM1 has been shown to induce FGF23 formation in UMR106 cells18. Moreover, TGF-β stimulates Orai1/STIM1 expression and thus SOCE in other cell types19. We therefore sought to clarify whether SOCE participates in TGF-β2-induced FGF23 release. To this end, we utilized Fura-2-dependent Ca2+ imaging in UMR106 cells after depletion of sarcoplasmic Ca2+ stores with thapsigargin to assess SOCE. As illustrated in Fig. 4, treatment with TGF-β2 enhanced the thapsigargin-induced increase of cellular Ca2+ and SOCE in UMR106 cells.
Figure 4.
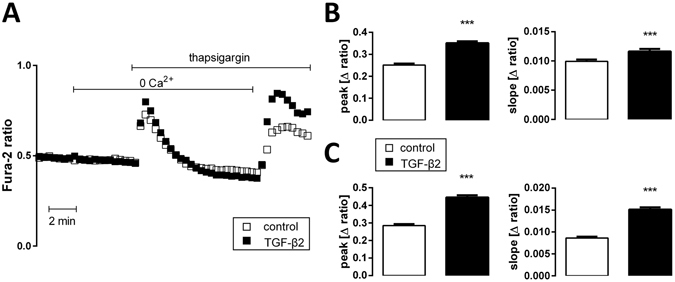
TGF-β2 induces store-operated calcium entry (SOCE) in UMR106 cells. (A) Representative original tracings (left panel) showing intracellular Ca2+ concentrations ([Ca2+]i) in Fura-2/AM-loaded UMR106 cells before and after removal of extracellular Ca2+, addition of the sarco-endoplasmic Ca2+-ATPase (SERCA) inhibitor thapsigargin (1 µM), and readdition of extracellular Ca2+ in the absence (open squares) and presence (closed squares) of TGF-β2 (10 ng/ml, 1 h). (B) Arithmetic means ± SEM (n = 124–173 cells measured on three different days) of the peak (left) and slope (right) values of [Ca2+]i increase after addition of thapsigargin reflecting Ca2+ release from intracellular stores. (C) Arithmetic means ± SEM (n = 124–173 cells measured on three different days) of the peak (left) and slope (right) values of [Ca2+]i increase following readdition of extracellular Ca2+ reflecting store-operated Ca2+ entry. White bars: without, black bars: with TGF-β2 (10 ng/ml, 1 h). ***p < 0.001 indicates significant difference (unpaired t-test).
Given the stimulating effect of TGF-β2 on SOCE in UMR106 cells, we addressed the question, whether SOCE is required for the induction of Fgf23 transcription by TGF-β2. In order to test this, we carried out experiments with SOCE inhibitor 2-APB. According to Fig. 5, the induction of FGF23 synthesis by TGF-β2 was significantly blunted by 2-APB. This result suggests that SOCE contributes to the TGF-β2 effect on FGF23 production.
Figure 5.
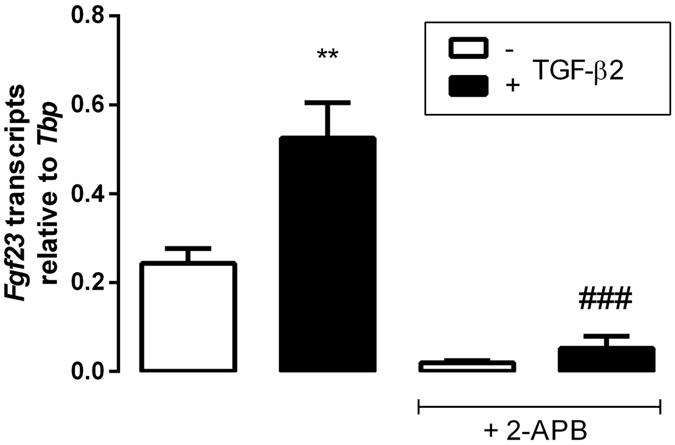
Store-operated calcium entry (SOCE) inhibitor 2-APB attenuates the TGF-β2 effect on Fgf23 transcript levels in UMR106 cells. Arithmetic means ± SEM (n = 4) of Fgf23 mRNA abundance (relative to Tbp mRNA) in UMR106 cells incubated without (white bars) or with TGF-β2 (black bars, 10 ng/ml) in the absence or presence of SOCE inhibitor 2-APB (25 μM) for 24 h. **p < 0.01 indicates significant difference from the absence of TGF-β2. ### p < 0.001 indicates significant difference between cells treated with TGF-β2 alone (2nd bar) and cells treated with TGF-β2 and 2-APB (4th bar) (ANOVA).
Finally we sought to explore whether the extracellular Ca2+ concentration influenced Fgf23 transcription in UMR106 cells. To this end, we incubated the cells at different Ca2+ concentrations (adjusted by the addition of Ca2+ chelator EGTA or CaCl2, resp.) and determined Fgf23 transcripts after 24 h. As a result, in the absence of TGF-β2 relative Fgf23 transcripts were (n = 3 each) 0.27 ± 0.01 arbitrary units (a.u.) at 1.8 mM Ca2+ (control), a value not significantly different from 0 mM Ca2+ (0.29 ± 0.04 a.u.), 0.6 mM Ca2+ (0.27 ± 0.02 a.u.), 1.2 mM Ca2+ (0.26 ± 0.02 a.u.), 2.4 mM Ca2+ (0.24 ± 0.02 a.u.), and 3.0 mM Ca2+ (0.25 ± 0.01 a.u.).
Discussion
According to our observations, transforming growth factor-β2 (TGF-β2) up-regulated the expression of fibroblast growth factor 23 (FGF23) in UMR106 osteoblast-like cells.
As a versatile cytokine, TGF-β is involved in a myriad of cellular effects in most tissues and organs including bone, the main site of FGF23 formation. TGF-β is required for both, the formation of bone during development as well as the maintenance and continuous remodeling throughout life20. Bone structures such as perichondrium, periosteum, and the epiphyseal growth plate express all TGF-β isoforms20. The analysis of transgenic mouse models has, however, revealed that only Tgfb2-deficient mice have an abnormal skeleton whereas Tgfb1- or Tgfb3-deficient mice do not exhibit a bone-related phenotype20. Moreover, it is TGF-β2 that is essential for the development of the skeleton during embryonic development20. Therefore, we used TGF-β2 in this study. A putative role of the other TGF-β isoforms in the regulation of FGF23 synthesis must be addressed in a future study. In later life, TGF-β is involved in bone resorption and formation and has several effects on osteoblast differentiation20.
Fibroblast growth factors (FGF) have already been demonstrated to be part of downstream TGF-β signaling21, and FGF2, FGF4, and FGF6 share the ability with TGF-ß to induce osteoblast proliferation22. Our study adds to these results demonstrating that bone-derived FGF23 is directly controlled by TGF-β2.
An important regulator of FGF23 formation is PTH which increases FGF23 expression15. Notably, TGF-β receptor 2 phosphorylates the PTH type I receptor thereby attenuating PTH signaling resulting in more trabecular and less cortical bone23. Thus, TGF-β is an important modulator of PTH action in bone24. Our present results demonstrate that the expression of FGF23, an important target of PTH, is also modulated by TGF-β.
Another major inducer of FGF23 expression is 1,25(OH)2D3, the active vitamin D metabolite. 1,25(OH)2D3 inhibits downstream TGF-β signaling25. Vitamin D supplementation lowers the availability of TGF-β26. Downstream TGF-β and 1,25(OH)2D3 signaling are characterized by intense crosstalk27. Our study demonstrates that TGF-β2 shares with 1,25(OH)2D3 the ability to induce FGF23 expression. It is intriguing to speculate that the regulation of FGF23 expression may also be subject to the intense 1,25(OH)2D3 – TGF-β signaling crosstalk.
Similar to what has been demonstrated in other cells types19, TGF-β2 induced SOCE, but also the thapsigargin-induced elevation in cytosolic Ca2+ in UMR106 cells. In addition, our study shows that TGF-β2-induced gene expression of Fgf23 was sensitive to 2-APB, an inhibitor of SOCE. These results suggest that the full effect of TGF-β2 required SOCE. In line with this, UMR106 cells have been demonstrated to express Orai1/STIM18, and Fgf23 gene expression is enhanced upon Orai1-mediated calcium entry18. Moreover, TGF-β up-regulates Orai119 and induces cellular calcium influx28.
Our results suggest that in the absence of TGF-β2 the extracellular Ca2+ concentration does not per se influence Fgf23 transcription in UMR106 cells. In contrast, Orai1-mediated Ca2+ influx from the extracellular space is required for the full TGF-β2 effect on FGF23.
Notably, the stimulating effect of advanced glycation end products (AGEs)29 and aldosterone30 on Fgf23 transcription was also significantly blunted by SOCE inhibitor 2-APB, and NFκB has similarly been shown to induce Fgf23 gene expression by enhancing SOCE18. Therefore, it appears to be possible that other regulators of SOCE similarly influence Fgf23 transcription.
An elevated FGF23 plasma level is observed in many acute and chronic disorders14. Among them, the role of FGF23 has most extensively been studied in chronic kidney disease (CKD) where the FGF23 plasma concentration rises even before the plasma phosphate concentration increases31. Notably, CKD and CKD-associated renal fibrosis are associated with enhanced TGF-β formation32. Given our present results, it appears likely that enhanced TGF-β production in CKD and other disorders contributes to the high FGF23 plasma concentration common in those diseases. According to a recent study, FGF23 also influences TGF-β expression33. Together with our findings, this paper suggests that FGF23 and TGF-β signaling are closely intertwined.
Taken together, TGF-β2 enhanced gene expression and protein synthesis of FGF23 in osteoblast-like cells. The effect was paralleled by enhanced SOCE which was required for the full effect on Fgf23 gene expression. TGF-β2-induced FGF23 formation presumably contributes to high plasma FGF23 levels in acute and chronic diseases.
Methods
Cell culture
Cell culture experiments were conducted as previously described34. Briefly, UMR106 rat osteoblast-like cells and NRK-52E rat tubular epithelial kidney cells were cultured in DMEM high glucose medium (Gibco, Life Technologies, Darmstadt, Germany) containing 1.8 mM Ca2+ supplemented with 10% FBS (Gibco, Life Technologies) (or 5% BCS (Sigma, Schnelldorf, Germany) for NRK-52E cells, resp.) and 100 U/mL penicillin/100 μg/ml streptomycin (Gibco, Life Technologies) under standard culture conditions. UMR106 cells do not have appreciable amounts of Fgf23 mRNA per se. Fgf23 expression was therefore induced by pre-treatment with 100 nM 1,25(OH)2D3 (Tocris, Bristol, UK)35. After 24 h, cells were additionally treated with TGF-β2 with or without 25 μM 2-APB or 10 µM SB431542 (all from Sigma) for another 24 h or treated with vehicle only. For the calcium measurements, cells were treated with or without 10 ng/ml TGF-β2 for 1 h.
Calcium concentration in the medium of pretreated UMR106 cells was modified by adding EGTA (0.6–1.8 mM) or CaCl2 (0.6 and 1.2 mM) for another 24 h.
ELISA
To assess FGF23 release into the cell culture medium, UMR106 cells were treated for 24 h without or with TGF-β2 (10 ng/ml). Cell culture media were collected for subsequent FGF23 (C-terminal) measurements using an ELISA kit (Immutopics, San Clemente, CA, USA) according to the manufacturer’s protocol.
Quantitative real-time PCR
Total RNA was extracted from UMR106 cells in Tri-Fast (Peqlab, Erlangen, Germany). Messenger RNA was transcribed with GoScript™ Reverse Transcription System (Promega, Mannheim, Germany) using 1.2 μg of total RNA and random primers. For qRT-PCR analysis, the final volume of the RT-PCR reaction mixture was 20 µl and contained: 2 µl cDNA, 1 µM of each primer, 10 µl GoTaq® qPCR Master Mix (Promega), and sterile water up to 20 µl. PCR conditions were 95 °C for 3 min, followed by 40 cycles of 95 °C for 10 s, 58 °C for 30 s and 72 °C for 45 s. Quantitative RT-PCR was performed on a Rotor-Gene Q (QIAGEN, Hilden, Germany).
The following primers were used:
Rat Tbp (TATA box-binding protein)
forward (5′-3′): ACTCCTGCCACACCAGCC
reverse (5′-3′): GGTCAAGTTTACAGCCAAGATTCA
Rat Fgf23
forward (5′-3′): TGGCCATGTAGACGGAACAC
reverse (5′-3′): GGCCCCTATTATCACTACGGAG
Calculated mRNA expression levels were normalized to the expression levels of Tbp of the same cDNA sample as internal reference. Relative quantification of gene expression was performed using the ΔCt method.
Qualitative expression analysis
Total RNA was extracted from UMR106 cells treated for 48 h with 100 nM 1,25(OH)2D3 only or from NRK-52E cells. For analysis of TGF-β receptor type 1 and 2 expression in murine bone tissue, femur and tibia were scrubbed removing residual soft tissue. Both epiphyses from femur and tibia were removed and the bone marrow was flushed out of the bone with 0.9% NaCl. Bone tissue was homogenized in liquid nitrogen using a mortar and pestle. Total RNA from bone was extracted with Tri-Fast (Peqlab). Next, the RNeasy Mini Kit (QIAGEN) was employed according to the manufacturer’s protocol.
PCR was carried out using 2 μl cDNA, 1 μM of each primer, 10 μl GoTaq® Green Master Mix (Promega) and sterile water up to 20 μl. The PCR conditions were: 95 °C for 3 min, followed by 35 cycles of 95 °C for 30 s, annealing at primer-specific temperature (56–60 °C) for 30 s, and 72 °C for 45 s (PCR Thermocycler BiometraTone, Analytik Jena, Jena, Germany). PCR products were analyzed by electrophoresis on a 1.5% agarose gel and visualized by Midori Green (Biozym, Hessisch Oldendorf, Germany) staining.
The following primers were used:
Rat Tgfbr1 (TGF-β receptor 1)
forward (5′-3′): CTGCCTGCTTCTCATCGTGT
reverse (5′-3′): TGCTTTTCTGTAGTTGGGAGTTCT
Rat Tgfbr2 (TGF-β receptor 2)
forward (5′-3′): CCCAAGTCGGTTAACAGCGAT
reverse (5′-3′): TGGCAATGACAGCTATGGCA
Mouse Tgfbr1
forward (5′-3′): CCTGAAGTTCTAGATGATTCC
reverse (5′-3′): CTTCATGGATTCCACCAATAG
Mouse Tgfbr2
forward (5′-3′): CCAGGATGAATCTGGAAAAC
reverse (5′-3′): TAATCCTTCACTTCTCCCAC
Calcium measurements
The measurements were performed as described previously18. Fura-2 fluorescence was utilized to estimate the cytosolic Ca2+ concentration ([Ca2+]i). Cells were loaded with Fura-2/AM (1 μM, Biomol, Hamburg, Germany) for 15 min at 37 °C. Fluorescence of the cells was excited alternatively at 340 nm and 380 nm (emission 510/87 nm, Fura-2 filter set) through an objective (CFI S-Fluor 40 x/1.30 oil) in an inverted microscope (Eclipse TS 100, all of Nikon, Düsseldorf, Germany) by a Lumen 220 Pro lamp (Prior Scientific Instruments, Cambridge, UK). Emitted fluorescence intensity was recorded by an Orca Flash 4.0 camera (Hamamatsu Photonics, Herrsching, Germany). System control and data acquisition were accomplished by µManager software. Cytosolic Ca2+ activity was estimated from the 340 nm/380 nm ratio. SOCE was determined after extracellular Ca2+ removal and subsequent Ca2+ readdition in the presence of thapsigargin (1 µM, Sigma). For quantification of Ca2+ entry, the slope (delta ratio/s) and peak (delta ratio) following readdition of Ca2+ were calculated. Experiments were performed in Ringer solution containing (in mM): 125 NaCl, 5 KCl, 1.2 MgSO4, 2 CaCl2, 2 Na2HPO4, 32 HEPES, 5 glucose, pH 7.4. To reach nominally Ca2+-free conditions, Ca2+-free Ringer solution was used (in mM: 125 NaCl, 5 KCl, 1.2 MgSO4, 2 Na2HPO4, 32 HEPES, 0.5 EGTA, 5 glucose, pH 7.4).
Statistics
Data are provided as means ± SEM, n represents the number of independent experiments. All data were tested for significance using paired or unpaired Student’s t-test or one-way ANOVA. Only results with p < 0.05 were considered statistically significant.
Acknowledgements
The authors acknowledge the technical assistance of S. Ross. We thank Carsten Wagner, University of Zürich, for helping with the isolation of Tgfbr1 and Tgfbr2 mRNA from mouse bone. The study was supported by the Deutsche Forschungsgemeinschaft (Fo 695/2-1, La 315/15-1).
Author Contributions
Ma.F., P.H., B.Z., F.H. and P.G. conducted experiments, F.L. and M.F. designed the study, Ma.F., M.F., and P.H. wrote the paper. All authors reviewed the manuscript.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Martina Feger and Philipp Hase contributed equally to this work.
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Meng X-M, Nikolic-Paterson DJ, Lan HY. TGF-beta: the master regulator of fibrosis. Nature reviews. Nephrology. 2016;12:325–338. doi: 10.1038/nrneph.2016.48. [DOI] [PubMed] [Google Scholar]
- 2.Drabsch Y, Dijke Pten. TGF-beta signalling and its role in cancer progression and metastasis. Cancer metastasis reviews. 2012;31:553–568. doi: 10.1007/s10555-012-9375-7. [DOI] [PubMed] [Google Scholar]
- 3.Munoz-Felix JM, Gonzalez-Nunez M, Martinez-Salgado C, Lopez-Novoa JM. TGF-beta/BMP proteins as therapeutic targets in renal fibrosis. Where have we arrived after 25 years of trials and tribulations? Pharmacology & therapeutics. 2015;156:44–58. doi: 10.1016/j.pharmthera.2015.10.003. [DOI] [PubMed] [Google Scholar]
- 4.Xu P, Liu J, Derynck R. Post-translational regulation of TGF-beta receptor and Smad signaling. FEBS letters. 2012;586:1871–1884. doi: 10.1016/j.febslet.2012.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kuro-O, M. & Moe, O. W. FGF23-alphaKlotho as a paradigm for a kidney-bone network. Bone (2016). [DOI] [PubMed]
- 6.Raya AI, et al. Energy-dense diets increase FGF23, lead to phosphorus retention and promote vascular calcifications in rats. Scientific reports. 2016;6:36881. doi: 10.1038/srep36881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hu X, et al. Elevation in fibroblast growth factor 23 and its value for identifying subclinical atherosclerosis in first-degree relatives of patients with diabetes. Scientific reports. 2016;6:34696. doi: 10.1038/srep34696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sun N, et al. FGF23 neutralization improves bone quality and osseointegration of titanium implants in chronic kidney disease mice. Scientific reports. 2015;5:8304. doi: 10.1038/srep08304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Martin A, David V, Quarles LD. Regulation and function of the FGF23/klotho endocrine pathways. Physiological reviews. 2012;92:131–155. doi: 10.1152/physrev.00002.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Faul, C. Cardiac actions of fibroblast growth factor 23. Bone (2016). [DOI] [PMC free article] [PubMed]
- 11.Kuro-o M, et al. Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature. 1997;390:45–51. doi: 10.1038/36285. [DOI] [PubMed] [Google Scholar]
- 12.Shimada T, et al. Targeted ablation of Fgf23 demonstrates an essential physiological role of FGF23 in phosphate and vitamin D metabolism. The Journal of clinical investigation. 2004;113:561–568. doi: 10.1172/JCI200419081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kuro-O M. Klotho, phosphate and FGF-23 in ageing and disturbed mineral metabolism. Nature reviews. Nephrology. 2013;9:650–660. doi: 10.1038/nrneph.2013.111. [DOI] [PubMed] [Google Scholar]
- 14.Schnedl C, Fahrleitner-Pammer A, Pietschmann P, Amrein K. FGF23 in Acute and Chronic Illness. Disease markers. 2015;2015:358086. doi: 10.1155/2015/358086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lavi-Moshayoff V, Wasserman G, Meir T, Silver J, Naveh-Many T. PTH increases FGF23 gene expression and mediates the high-FGF23 levels of experimental kidney failure: a bone parathyroid feedback loop. American journal of physiology. Renal physiology. 2010;299:F882–9. doi: 10.1152/ajprenal.00360.2010. [DOI] [PubMed] [Google Scholar]
- 16.Masuyama R, et al. Vitamin D receptor in chondrocytes promotes osteoclastogenesis and regulates FGF23 production in osteoblasts. The Journal of clinical investigation. 2006;116:3150–3159. doi: 10.1172/JCI29463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.David V, et al. Inflammation and functional iron deficiency regulate fibroblast growth factor 23 production. Kidney international. 2016;89:135–146. doi: 10.1038/ki.2015.290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang B, et al. NFkappaB-sensitive Orai1 expression in the regulation of FGF23 release. Journal of molecular medicine (Berlin, Germany) 2016;94:557–566. doi: 10.1007/s00109-015-1370-3. [DOI] [PubMed] [Google Scholar]
- 19.Schmidt S, et al. TGFbeta1 and SGK1-sensitive store-operated Ca2+ entry and Orai1 expression in endometrial Ishikawa cells. Molecular human reproduction. 2014;20:139–147. doi: 10.1093/molehr/gat066. [DOI] [PubMed] [Google Scholar]
- 20.Wu M, Chen G, Li Y-P. TGF-beta and BMP signaling in osteoblast, skeletal development, and bone formation, homeostasis and disease. Bone research. 2016;4:16009. doi: 10.1038/boneres.2016.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sasaki T, et al. TGFbeta-mediated FGF signaling is crucial for regulating cranial neural crest cell proliferation during frontal bone development. Development (Cambridge, England) 2006;133:371–381. doi: 10.1242/dev.02200. [DOI] [PubMed] [Google Scholar]
- 22.Bosetti M, Boccafoschi F, Leigheb M, Cannas MF. Effect of different growth factors on human osteoblasts activities: a possible application in bone regeneration for tissue engineering. Biomolecular engineering. 2007;24:613–618. doi: 10.1016/j.bioeng.2007.08.019. [DOI] [PubMed] [Google Scholar]
- 23.Qiu T, et al. TGF-beta type II receptor phosphorylates PTH receptor to integrate bone remodelling signalling. Nature cell biology. 2010;12:224–234. doi: 10.1038/ncb2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen G, Deng C, Li Y-P. TGF-beta and BMP signaling in osteoblast differentiation and bone formation. International journal of biological sciences. 2012;8:272–288. doi: 10.7150/ijbs.2929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zerr P, et al. Vitamin D receptor regulates TGF-beta signalling in systemic sclerosis. Annals of the rheumatic diseases. 2015;74:e20. doi: 10.1136/annrheumdis-2013-204378. [DOI] [PubMed] [Google Scholar]
- 26.Irani M, et al. Vitamin D supplementation decreases TGF beta-1 bioavailability correlating with clinical improvement in Vitamin D deficient women with PCOS. A randomized placebo-controlled trial. Fertility and Sterility. 2015;104:e105. doi: 10.1016/j.fertnstert.2015.07.324. [DOI] [Google Scholar]
- 27.Subramaniam N, et al. Cross-talk between 1,25-dihydroxyvitamin D3 and transforming growth factor-beta signaling requires binding of VDR and Smad3 proteins to their cognate DNA recognition elements. The Journal of biological chemistry. 2001;276:15741–15746. doi: 10.1074/jbc.M011033200. [DOI] [PubMed] [Google Scholar]
- 28.McGowan TA, et al. TGF-beta-induced Ca(2+) influx involves the type III IP(3) receptor and regulates actin cytoskeleton. American journal of physiology. Renal physiology. 2002;282:F910–20. doi: 10.1152/ajprenal.00252.2001. [DOI] [PubMed] [Google Scholar]
- 29.Bar, L. et al. Advanced glycation end products stimulate gene expression of fibroblast growth factor 23. Molecular nutrition & food research (2017). [DOI] [PubMed]
- 30.Zhang B, et al. Up-regulation of FGF23 release by aldosterone. Biochemical and biophysical research communications. 2016;470:384–390. doi: 10.1016/j.bbrc.2016.01.034. [DOI] [PubMed] [Google Scholar]
- 31.Isakova T, et al. Fibroblast growth factor 23 is elevated before parathyroid hormone and phosphate in chronic kidney disease. Kidney international. 2011;79:1370–1378. doi: 10.1038/ki.2011.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vega G, Alarcon S, San Martin R. The cellular and signalling alterations conducted by TGF-beta contributing to renal fibrosis. Cytokine. 2016;88:115–125. doi: 10.1016/j.cyto.2016.08.019. [DOI] [PubMed] [Google Scholar]
- 33.Dai B, et al. A comparative transcriptome analysis identifying FGF23 regulated genes in the kidney of a mouse CKD model. PloS one. 2012;7:e44161. doi: 10.1371/journal.pone.0044161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fajol A, et al. Enhanced FGF23 production in mice expressing PI3K-insensitive GSK3 is normalized by beta-blocker treatment. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2016;30:994–1001. doi: 10.1096/fj.15-279943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Saini RK, et al. 1,25-dihydroxyvitamin D(3) regulation of fibroblast growth factor-23 expression in bone cells: evidence for primary and secondary mechanisms modulated by leptin and interleukin-6. Calcified tissue international. 2013;92:339–353. doi: 10.1007/s00223-012-9683-5. [DOI] [PMC free article] [PubMed] [Google Scholar]