

Abstract
Aims
Our previous studies have established a role for 12/15-lipoxygenase (LO) in mediating the inflammatory response in diabetic retinopathy (DR). However, the extent at which the local or systemic induction of 12/15-LO activity involved is unclear. Thus, the current study aimed to characterize the relative contribution of retinal endothelial versus monocytic/macrophagic 12/15-LO to inflammatory responses in DR.
Materials & Methods
We first generated a clustered heat map for circulating bioactive lipid metabolites in the plasma of streptozotocin (STZ)-induced diabetic mice using liquid chromatography coupled with mass-spectrometry (LC-MS) to evaluate changes in circulating 12/15-LO activity. This was followed by comparing the in vitro mouse endothelium-leukocytes interaction between leukocytes isolated from 12/15-LO knockout (KO) versus those isolated from wild type (WT) mice using the myeloperoxidase (MPO) assay. Finally, we examined the effects of knocking down or inhibiting endothelial 12/15-LO on diabetes-induced endothelial cell activation and ICAM-1 expression.
Results
Analysis of plasma bioactive lipids’ heat map revealed that the activity of circulating 12/15-LO was not altered by diabetes as evident by no significant changes in the plasma levels of major metabolites derived from 12/15-lipoxygenation of different PUFAs, including linoleic acid (13-HODE), arachidonic acid (12- and 15- HETEs), eicosapentaenoic acid (12- and 15- HEPEs), or docosahexaenoic acid (17-HDoHE). Moreover, leukocytes from 12/15-LO KO mice displayed a similar increase in adhesion to high glucose (HG)-activated endothelial cells as do leukocytes from WT mice. Furthermore, abundant proteins of 12-LO and 15-LO were detected in human retinal endothelial cells (HRECs), while it was undetected (15-LO) or hardly detectable (12-LO) in human monocyte-like U937 cells. Inhibition or knock down of endothelial 12/15-LO in HRECs blocked HG-induced expression of ICAM-1, a well-known identified important molecule for leukocyte adhesion in DR.
Conclusion
Our data support that endothelial, rather than monocytic/macrophagic, 12/15-LO has a critical role in hyperglycemia-induced ICAM-1 expression, leukocyte adhesion, and subsequent local retinal barrier dysfunction. This may facilitate the development of more precisely targeted treatment strategies for DR.
Keywords: Diabetic Retinopathy, 12/15-Lipoxygenase, 12–HETE, 15-HETE, leukostasis, ICAM-1, eicosanoids, bioactive lipids, blood retinal barrier, permeability
INTRODUCTION
Diabetic retinopathy (DR) remains the leading cause of blindness among working-age populations worldwide, even though with the advent of many effective treatments [1]. It is characterized by breakdown of the blood retinal barrier (BRB) in the early stage of the disease, followed by capillary degeneration, and subsequent neovascularization (NV) at the late stages [2, 3]. The current therapies are heavily relying on controlling systemic hyperglycemia. However, due to the metabolic memory effect, many diabetic patients develop retinopathy despite their tight glycemic control [4]. Furthermore, the existing therapeutic strategies; corticosteroids, anti-vascular endothelial growth factor (VEGF) agents, ranibizumab and aflibercept, as well as laser photocoagulation, are limited by their side effects [5]. Therefore, it is worthwhile to explore new therapeutic avenues to prevent DR via deeper understanding of its pathophysiology.
Compelling evidence now indicates that DR is a multifactorial disease that involves chronic inflammation at every stage, from initiation to progression and eventually to ischemia and NV [6, 7]. Data from animal studies suggest that leukocyte-endothelial cell adhesion and entrapment (leukostasis) are rate-limiting steps for initiation of retinal inflammatory response prior to any clinical sign of DR [8, 9]. Further studies using human tissues demonstrated an increase in leukocyte density in human eyes with DR and strong relationship between leukocyte-endothelial cell adhesion and retinal capillary damage in diabetes [10]. Once leukocytes have attached to the endothelial cells, pro-inflammatory cytokines and chemokines are released. These mediators are known to alter endothelial cell junctional proteins, allowing leukocytic infiltration into the retina, with concomitant compromise of the BRB [11]. Attachment and extravasation of these leukocytes are mediated by adhesive interactions between molecules present on leukocytes and their counter receptors expressed on activated endothelial cells such as the E- and P-selectins and intercellular cell adhesion molecule (ICAM)-1 [12, 13]. ICAM-1 is directly up-regulated by diabetes [10] in the retinal vasculature where its blockade has highlighted it as a potential target for DR therapies, both clinically [14] and experimentally [15]. Yet, the exact underlying mechanisms of how hyperglycemia mediates ICAM-1 upregulation and hence leukocyte-endothelial cell adhesion need to be further elucidated.
Several mechanisms have been suggested to account for the causal association of ICAM-1 induction and leukocyte adhesion with DR, including oxidative stress [16], NF-κB [17], PKC [18], and bioactive lipids [19]. Our previous studies highlighted the role of bioactive lipid metabolites derived from 12/15-lipoxygenase (LO) in pathogenesis of the inflammatory response in early DR. We have shown an increase in the retinal 12/15-LO expression and activity, evident by elevated levels of 12/15-LO-derived hydroxyeicosatetraenoic acid (HETE) locally; in the vitreous of patients with DR [20], diabetic mouse retinas [21], and human retinal endothelial cells (HRECs) incubated with high glucose [22]. Additionally, we have reported a reduction in diabetes-induced retinal ICAM-1 expression by the pharmacological inhibition of 12/15-LO [21]. Furthermore, our very recent studies have demonstrated the ability of intravitreally injected 12-HETE to compromise endothelial barrier function in the retina associated with the induction of a pro-inflammatory phenotype (increased in retinal ICAM-1 expression and leukocyte adhesion) [22, 23]. As a further support for the role of 12/15-LO in early DR, we have shown by using fluorescein angiography (FA) and retinal albumin leakage assay a significant reduction in retinal barrier dysfunction in diabetic mice lacking global expression of 12/15-LO compared to diabetic wild type (WT) mice [22].
Two distinct mammalian 12/15-Lipoxygenases have been characterized on the basis of their products from arachidonic acid (AA); 15-lipoxygenase (15-LO) in humans [24] and rabbits [25], and its orthologue the “leukocyte-type” known as 12-lipoxygenase (12-LO) in pig, rat, and mouse [26]. Leukocyte 12-LO and 15-LO are highly related in their enzymological characteristics as well as primary structures, and both are collectively called 12/15-LO [27]. A variety of cells, including vascular and myeloid lineage cells such as endothelial cells, smooth muscle cells, platelets, and monocytes/macrophages, express 12/15-LO [28]. By looking at the 12/15-LO activity in different cell types, it has been shown that 12/15-LO may have opposing effects. For instance, overexpression of 12/15-LO in endothelial cells enhances atherogenic responses [29], while overexpression of 12/15-LO in monocytes/macrophages protects against atherogenesis [30]. These observations raised the possibility that different cellular expression of 12/15-LO may have different effects on pathogenesis of DR and this has not been previously addressed. Therefore, the current study aimed to characterize the relative contribution of retinal endothelial versus monocytic/macrophagic 12/15-LO to inflammatory responses in DR. To achieve this goal, we first evaluated the changes in circulating 12/15-LO activity by measuring its derived metabolites in the plasma of diabetic WT mice compared to non-diabetic controls. This was followed by comparing the in vitro endothelium-leukocytes interaction between leukocytes isolated from 12/15-LO−/− versus those isolated from WT mice. Finally, we examined the effects of knocking down or inhibiting endothelial 12/15-LO on diabetes-induced HREC activation and ICAM-1 expression.
MATERIALS AND METHODS
Animal Preparation and Experimental Design
All procedures with animals were conducted in accordance with the principles of Public Health Service Guide for the Care and Use of Laboratory Animals (Department of Health, Education, and Welfare publication, NIH 80-23) and Augusta University guidelines. Eight-week-old male 12/15-lipoxygenase knockout (B6.129S2-Alox15tm1Fun/J, 15-LO−/−) and corresponding littermate control wild-type (WT) mice in C57BL/6J background (Jackson Laboratory, Bar Harbor, ME), matched according to age and weight, were used in this study. For diabetic experiments, WT mice were kept fasting at least four hours before intraperitoneal (IP) injections of freshly prepared Streptozotocin (STZ, 50 mg/kg, Sigma, St. Louis, MO) for 5 consecutive days. Mice with blood glucose levels >300 mg ⁄dL were diagnosed as diabetic. Six months after establishment of diabetes, plasma samples were collected and subjected to lipidomic analysis using liquid chromatography–mass spectrometry (LC/MS).
Lipidomic analysis for bioactive lipid metabolites
This was done by LC/MS in Lipidomics Core Facility (Wayne State University, Detroit, MI) as described before [31]. Briefly, plasma samples were spiked with 10 ng of five internal standards covering all classes of oxygenated polyunsaturated fatty acid (PUFA) metabolites. These internal standards are [15(S)-HETE-d8, Leukotriene B4-d4, Prostaglandin E1-d4, RvD2-d5, and 14(15)-EpETrE-d11], and they used for recovery and quantitation. The spiked samples were mixed thoroughly and then loaded on C18 columns for extracting PUFA metabolites. LC-MS analysis was performed on a Prominence XR system (Shimadzu) using Luna C18 (3μ, 2.1×150 mm) column. The mobile phase consisted of a gradient between A: methanol-water-acetonitrile (10:85:5 v/v) and B: methanol-water-acetonitrile (90:5:5 v/v), both containing 0.1% ammonium acetate. The gradient program with respect to the composition of B was as follows: 0–1 min, 50%; 1–8 min, 50–80%; 8–15 min, 80–95%; and 15–17 min, 95%. The flow rate was 0.2 ml/min. The eluate was directly introduced to ESI source of QTRAP5500 mass analyzer (ABSCIEX) in the negative ion mode with following conditions: Curtain gas: 35 psi, GS1: 35 psi, GS2: 65 psi, Temperature: 600 °C, Ion Spray Voltage: −1500 V, Collision gas: low, Declustering Potential: −60 V, and Entrance Potential: −7 V. The eluate was monitored by Multiple Reaction Monitoring (MRM) method to detect unique molecular ion –daughter ion combinations. Optimized Collisional Energies (18–35 eV) and Collision Cell Exit Potentials (7–10 V) were used for each MRM transition. The data was collected using Analyst 1.5.2 software and the MRM transition chromatograms were quantitated by MultiQuant software (both from ABSCIEX). The internal standard signals in each chromatogram were used for normalization, recovery and relative quantitation of each analyte. The average recovery using this procedure was > 90%. Metabolite concentrations were normalized to the total plasma protein content. Matrix2png Viewer (http://www.chibi.ubc.ca/matrix2png/) was used to analyze and represent the lipid metabolomic data as a heat map.
Isolation of leukocytes from whole mouse blood
Whole blood was collected using heparinated microtubes. Erythrocytes lysed using ACK lysing buffer (BioWhittaker, Walkersville, MD) prior to sorting and leukocytes isolation. For direct isolation, the cell suspension was incubated with a cocktail of the following antibodies CD45/CD11b/CD2/CD4/CD8/CD19 (eBioScience) for 20 min (4°C). Preparative cell sorting was performed using a Mo-Flo flow cytometer to collect leukocytes using DakoCytomation Summit™ software (DakoCytomation, Fort Collins, CO). Leukocytes were selected at high purity (90–95%), which was achieved by setting sorting gates to collect cells unambiguously stained by antibodies. Isotype-matched controls were analyzed to set the appropriate gates.
Mouse retinal endothelial cell (mREC)-leukocyte adhesion assay
The mRECs, generated from immorto-mice as previously described [32], were grown in 24 well gelatin coated plates in DMEM containing 10% fetal bovine serum (FBS; Atlanta Biologicals, Atlanta, GA), 2 mM L-glutamine, 20 mM HEPES, 100 μg/ml streptomycin, 1% non-essential amino acids, 2mM sodium pyruvate, 100 U/ml penicillin, 55 U/ml of freshly added heparin at (Sigma), 100 μg/ml of endothelial growth supplement (Sigma), and 44 units/ml of murine recombinant interferon-γ (R & D, Minneapolis, MN) at 33°C with 5% CO2. When mRECs were 80% confluent, they were treated with high glucose (HG; D-Glucose, 30 mM) or normo-osmotic control (LG, 5mM D-glucose + 25 mM L-glucose) for 1–3 days. As a positive control, mRECs were exposed to 1 μg/ml of the endotoxin Escherichia coli lipopolysaccharide (LPS; Sigma) for 24 hours. Thereafter, leukocytes (purified from the blood as described above) from WT or 12/15-LO−/− mice were added to the mREC confluent monolayer and incubated for 90 minutes. After incubation, non-adherent leukocytes were carefully removed by gentle washing before measuring the activity of Myeloperoxidase (MPO), a specific marker for leukocytes, using Fluro MPO Myeloperoxidase Detection Kit (Catalog# MPO100-3, Cell Technology, Mountain View, CA,) according to the manufacturer’s instructions.
Microvascular human retinal endothelial cells (HRECs)
HRECs (Cell Systems Cooperation, Kirkland, WA) were grown on gelatin-coated plates in EBM2 Medium (Catalog #190860, Lonza, walkersville, MD) supplemented with 5% FBS, 1% Penicillin Streptomycin (PS, Catalog # 30-004-CI, Corning Inc, NY). After the cells were 80–90% confluent, they were serum starved overnight, then treated with HG for 5 days in the presence or absence of vehicle (DMSO) or 10 μM of baicalein. The osmolarity of control group was adjusted using L-Glucose. Cell lysates were collected and analyzed for ICAM-1 or 12/15-LO protein expression using Western blot analysis using antibodies for 12- or 15-LO (Novus biologicals, Littleton, CO), β-actin (Millipore, Billerica, MA), and ICAM-1(Santa Cruz, CA) according to a previous procedure [33].
HREC-leukocyte Adhesion Assay
HRECs were grown in 24 well plates until being 70% confluent then treated with HG or LG for 5 days in presence or absence of baicalein or Vehicle (DMSO). In another set of experiment, HRECs were treated with amadori glycated albumin (AGA, 500 μg/mL) or albumin (500 μg/mL) for 24 hours. Following the treatment, human monocytes (CD14+ monocytes purchased from lifeline cell technology (Frederick, MD) or human monocyte-like cells (U937; ATCC® CRL-1593.2)) maintained in ATCC formulated RPMI-1640 medium (Catalog #30-2001) containing 10% FBS and 1% PS, were added at (300,000–400,000 cells/well) to the confluent monolayer of HRECs and incubated at 37°C for 90 minutes. As a positive control, HRECs were exposed to 1 μg/ml of LPS for 24 hours. Then non-adherent cells were removed before measuring MPO activity as described earlier.
Transcellular Electrical Resistance (TER) using Electric Cell-substrate Impedance Sensing (ECIS)
Normalized TER was measured using ECIS® Zθ (theta) instrument (Applied Biophysics Inc, Troy, NY, USA). Briefly, a 96-wells array (catalog # 96W20idf PET, Applied Biophysics Inc) was used and coated with cysteine for 30 minutes then with gelatin for another 30 minutes. Thereafter, HRECs were seeded at a density of 7500 cells/well in EBM2 media with 5% FBS and 1% PS. After HRECs reached the confluency, indicated by a capacitance reading below 10F, they were serum starved then treated with HG or LG for 3 days then CD14+ monocytes were added. TER was recorded at 4000 Hz current frequency. Resistance value for each well was normalized as the ratio of measured resistance at each time point to baseline resistance and plotted as a function of time.
Quantitative Real-Time -PCR
Total RNAs were isolated from mouse leukocytes purified from the pooled blood of either five WT or 12/15-LO KO mice using QIAGEN spin-column kits. Subsequently, they subjected to a 50 cycle PCR amplification using TaqMan one-step RT-PCR kit (#4392938) obtained from (Applied Biosystems (ABI), Foster City, CA) with pre-designed TaqMan primers (Catalog#: 12-LO: Mm00545833_m1; 15-LO: Mm00507789_m1; 18S: Mm03928990_g1). 18S was used as the endogenous reference gene.
Transfection with short interfering RNA (siRNA)
HRECs were transfected with 12/15LO or control Dicer-substrate RNA (DsiRNA) duplexes (Catalog # HSC.RNAI.N001140. Integrated DNA Technologies) using lipofectamine 2000 (Invitrogen) per the manufacturer’s instructions.
Data Analysis
The results are expressed as mean ± SD. Differences among experimental groups were evaluated by using the two-tailed t test or one way analysis of variance (ANOVA). When statistical differences were observed using ANOVA, a post hoc Tukey’s test was performed to determine which groups differed.
RESULTS
Lipidomic profile of circulating bioactive lipid metabolites in STZ-induced diabetic mice
Our previous studies have established a role for 12/15-LO in mediating the inflammatory responses in DR. However, the extent at which the local or systemic induction of 12/15-LO activity is involved in inflammatory responses is unclear. Thus, in the current study we first sought to determine the broad profiling activities of various PUFA metabolizing enzymes, including 12/15-LO, cyclooxygenase, cytochrome P 450(CYP), and soluble epoxide hydrolase (sEH; EPHX) in the plasma of STZ-induced diabetic mice using LC-MS. Figure 1 shows a clustered heat map of bioactive lipid metabolites in the plasma of diabetic mice that is presented as fold-change relative to normal non diabetic mice. These metabolites were initially clustered into four major groups according to their PUFA origin; linoleic acid (LA), arachidonic acid (AA), eicosapentaenoic acid (EPA), or docosahexaenoic acid (DHA). This primary clustering was followed by a secondary subclustering of bioactive lipid metabolites within each PUFA group according to their putative enzymatic biosynthesis pathways, and hence chemical structure similarity. Intriguingly, the statistical analysis of the heat map (Table 1) revealed that out of 106 bioactive lipids screened, only 6 metabolites (9,10- dihydroxyoctadecamonoenoic (DiHOME), 5,6- Dihydroxyeicosatrienoic acids (DiHETrE), 11,12-DiHETrE, 14,15-DiHETrE, 15d-D12,14- prostaglandin (PG) J3, and Resolvin (Rv) D2) were significantly increased during diabetes. On the other hand and in contrary to what has been reported of the local increase in retinal levels of 12/15-LO-derived metabolites, 12- and 15-HETE [20, 21], the activity of circulating 12/15-LO was not altered by diabetes (Figure 2A). This was evident by no significant changes in the plasma levels of major metabolites derived from 12/15-lipoxygenation of different PUFAs, including LA (13-HODE), AA (12- and 15- HETEs), EPA (12- and 15- HEPEs), or DHA (17-HDoHE). In contrast to 12/15-LO metabolites, levels of CYP/sEH metabolites derived from different PUFAs showed an increasing trend in diabetes. This trend was significantly obvious among 9,10-DiHOME, 5,6-DiHETrE, 11,12-DiHETrE, 14,15-DiHETrE (Figure 2B). These data are showing that the circulating lipidomic profile of diabetic mice is dominated by CYP/sEH products rather than 12/15-LO products.
Figure 1. Clustered heat map of circulating bioactive lipid metabolites in the plasma of diabetic mice.
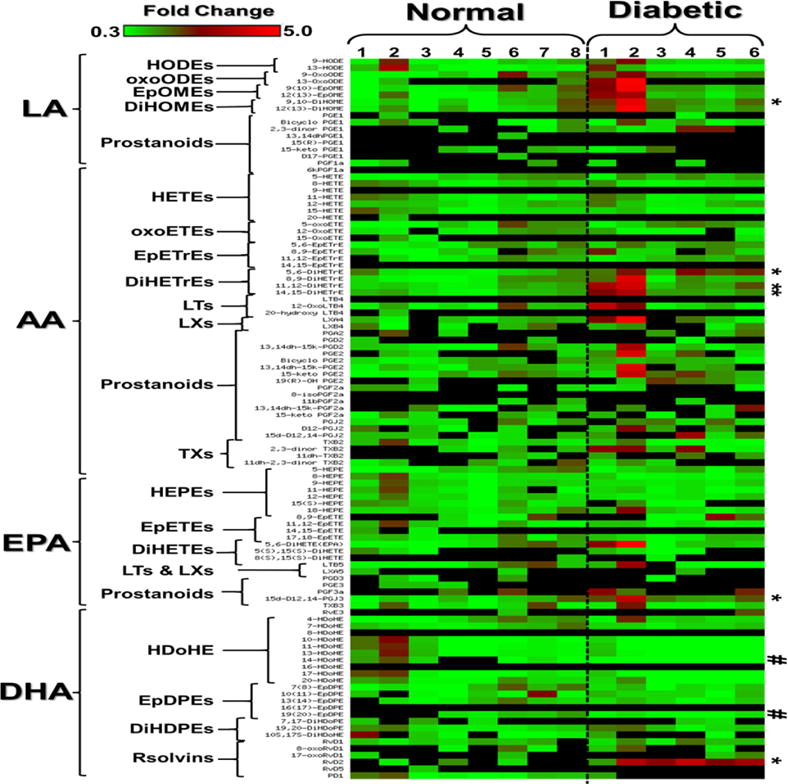
The metabolites were initially clustered into four major groups according to their polyunsaturated fatty acid (PUFA) origin; linoleic acid (LA), arachidonic acid (AA), eicosapentaenoic acid (EPA), or docosahexaenoic acid (DHA). This primary clustering is followed by secondary subclustering bioactive lipid metabolites within each PUFA group according to their putative enzymatic biosynthesis pathways and hence chemical structure similarity. The LA group includes the following subclusters: hydroxyoctadecadienoic acids (HODEs), oxooctadecadienoic acids (oxoODEs), epoxyoctadecamonoenoic acids (EpOME), dihydroxyoctadecamonoenoic acids (DiHOME), and LA-derived 1-series prostanoids. The AA group includes: hydroxyeicosatetraenoic acids (HETEs), oxoeicosatetraenoic acids (oxoETEs), epoxyeicosatrienoic acids (EpETrEs), Dihydroxyeicosatrienoic acids (DiHETrEs), AA-derived 4-series leukotrienes (LTs), AA-derived 4-series lipoxins (LXs), and AA-derived 2-series prostanoids and thromboxane (TXs). The EPA group includes: hydroxyeicosapentaenoic acids (HEPEs), epoxyeicosatetraenoic acids (EpETEs), dihydroxyeicosatetraenoic acids (DiHETEs), EPA-derived 5-series LTs, EPA-derived 5-series LXs, EPA-derived 3-series prostanoids, and E-series Resolvins (Rvs). The DHA group includes: hydroxydecosahexaenoic acids (HDoHEs), epoxydecosapentaenoic acids (EpDOPEs), Dihydoxydecosapentaenoic acids (DIHDOPEs), and D-series Rvs. The significant (P<0.05) increased metabolites (9,10-DiHOME, 5,6-DiHETrE, 11,12-DiHETrE, 14,15-DiHETrE, 15d-D12,14-PGJ3, and RvD2) were indicated by * while decreased metabolites were indicated by #. Data shown for the comparison are the fold change of 6 diabetic and 8 nondiabetic mice relative to the average of nondiabetic mice ± SD. The highest fold change is indicated by the red color, the lowest fold change is indicated by the green color, and the undetected metabolite is represented by the black color.
Table 1.
P values of statistical analysis of plasma bioactive lipid metabolites’ heat map between diabetic and normal non-diabetic mice.
| Bioactive Lipid | P value | Bioactive Lipid | P value | Bioactive Lipid | P value |
|---|---|---|---|---|---|
| 9-HODE | 0.334 | 12-OxoLTB4 | 0.268 | 5,6-DiHETE(EPA) | 0.212 |
| 13-HODE | 0.851 | 20-hydroxy LTB4 | NA | 5(S),15(S)-DiHETE | 0.585 |
| 9-OxoODE | 0.086 | LXA4 | 0.118 | 8(S),15(S)-DiHETE | NA |
| 13-OxoODE | 0.079 | LXB4 | 0.564 | LTB5 | 0.771 |
| 9(10)-EpOME | 0.091 | PGA2 | 0.557 | LXA5 | NA |
| 12(13)-EpOME | 0.128 | PGD2 | NA | PGD3 | NA |
| 9,10-DiHOME | 0.030 | 13,14dh-15k-PGD2 | 0.546 | PGE3 | NA |
| 12(13)-DiHOME | 0.067 | PGE2 | 0.286 | PGF3a | 0.133 |
| PGE1 | NA | Bicyclo PGE2 | 0.567 | 15d-D12,14-PGJ3 | 0.017 |
| Bicyclo PGE1 | 0.890 | 13,14dh-15k-PGE2 | 0.103 | TXB3 | 0.984 |
| 2,3-dinor PGE1 | 0.371 | 15-keto PGE2 | 0.070 | RvE3 | NA |
| 13,14dhPGE1 | NA | 19(R)-OH PGE2 | NA | 4-HDoHE | 0.746 |
| 15(R)-PGE1 | NA | PGF2a | 0.177 | 7-HDoHE | 0.485 |
| 15-keto PGE1 | 0.951 | 8-isoPGF2a | NA | 8-HDoHE | NA |
| D17-PGE1 | NA | 11bPGF2a | NA | 10-HDoHE | 0.121 |
| PGF1a | 0.630 | 13,14dh-15k-PGF2a | 0.223 | 11-HDoHE | 0.118 |
| 6kPGF1a | NA | 15-keto PGF2a | 0.638 | 13-HDoHE | 0.152 |
| 5-HETE | 0.506 | PGJ2 | 0.226 | 14-HDoHE | 0.037 |
| 8-HETE | 0.453 | D12-PGJ2 | 0.236 | 16-HDoHE | NA |
| 9-HETE | NA | 15d-D12,14-PGJ2 | 0.084 | 17-HDoHE | 0.146 |
| 11-HETE | 0.832 | TXB2 | 0.430 | 20-HDoHE | 0.253 |
| 12-HETE | 0.555 | 2,3-dinor TXB2 | 0.190 | 7(8)-EpDPE | 0.608 |
| 15-HETE | 0.205 | 11dh-TXB2 | 0.149 | 10(11)-EpDPE | 0.767 |
| 20-HETE | NA | 11dh-2,3-dinor TXB2 | 0.859 | 13(14)-EpDPE | 0.473 |
| 5-oxoETE | 0.539 | 5-HEPE | 0.874 | 16(17)-EpDPE | NA |
| 12-OxoETE | 0.085 | 8-HEPE | 0.204 | 19(20)-EpDPE | 0.018 |
| 15-OxoETE | NA | 9-HEPE | 0.306 | 7,17-DiHDoPE | 0.654 |
| 5,6-EpETrE | 0.160 | 11-HEPE | 0.313 | 19,20-DiHDoPE | 0.336 |
| 8,9-EpETrE | 0.333 | 12-HEPE | 0.294 | 10S,17S-DiHDoHE | 0.793 |
| 11,12-EpETrE | 0.577 | 15(S)-HEPE | 0.565 | RvD1 | 0.332 |
| 14,15-EpETrE | NA | 18-HEPE | 0.492 | 8-oxoRvD1 | 0.706 |
| 5,6-DiHETrE | 0.001 | 8,9-EpETE | 0.561 | 17-oxoRvD1 | 0.380 |
| 8,9-DiHETrE | 0.153 | 11,12-EpETE | 0.381 | RvD2 | 0.010 |
| 11,12-DiHETrE | 0.019 | 14,15-EpETE | NA | RvD5 | NA |
| 14,15-DiHETrE | 0.023 | 17,18-EpETE | 0.520 | PD1 | NA |
| LTB4 | NA |
Figure 2. Circulating lipidomic profile in diabetes is dominated by cytochrome P 450 (CYP) /sEH pathway derived metabolites rather than 12/15-LO metabolites.
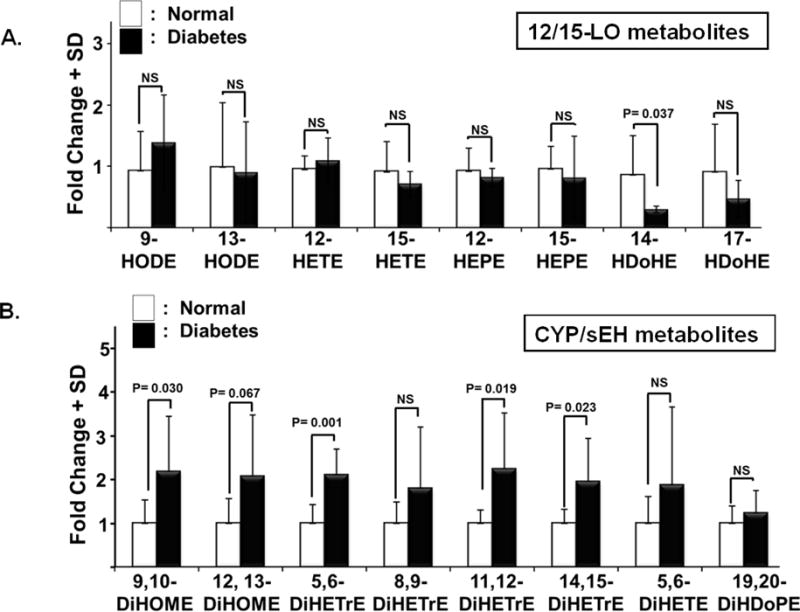
A) The activity of circulating 12/15-LO was not altered during diabetes as indicated by no significant changes in the plasma levels of major metabolites derived from 12/15-lipoxygenation of different PUFAs, including LA (9- and 13-HODEs), AA (12- and 15- HETEs), EPA (12- and 15- HEPEs), or DHA (14- and 17-HDoHE). B) In contrast to 12/15-LO metabolites, levels of CYP/sEH metabolites derived from different PUFAs showed an increasing trend in diabetes. This trend was significantly evident among 9,10-DiHOME, 5,6-DiHETrE, 11,12-DiHETrE, 14,15-DiHETrE. Data shown for the comparison are from the same experimental groups used in figure 1 and represented here by the mean fold change of 6 diabetic mice relative to the average of 8 nondiabetic mice ± SD.
Role of endothelial versus monocytic/macrophagic 12/15-LO in mediating high glucose-induced retinal leukostasis
Initial experiments were conducted to check whether mouse peripheral blood mononuclear cells (PBMCs) express 12/15-LO at basal condition. Toward this goal, 12- and 15-LO mRNAs were evaluated by RT-PCR in leukocytes isolated from WT mice in comparison with those isolated from 12/15-LO KO mice. As shown in Figure 3A, basal 12-LO and 15-LO mRNAs were detectable with RT-PCR at 20 to 30 cycles in which 12-LO is more abundant than 15-LO (as indicated by lower Ct value, i.e., higher level of mRNA transcription). Next, whether or not this basal expression of 12 or 15-LO mRNA plays a role in leukocyte adhesion was examined. For this purpose, we carried out leukocyte adhesion assay on HG- or LPS-activated mRECs using leukocytes isolated from 12/15-LO KO versus those isolated from WT mice. HG or LPS-activated mRECs significantly augmented the number of adherent leukocytes prepared from 12/15-LO KO relatively to the same extent as those derived from WT mice (Figures 3B & C, respectively).
Figure 3.
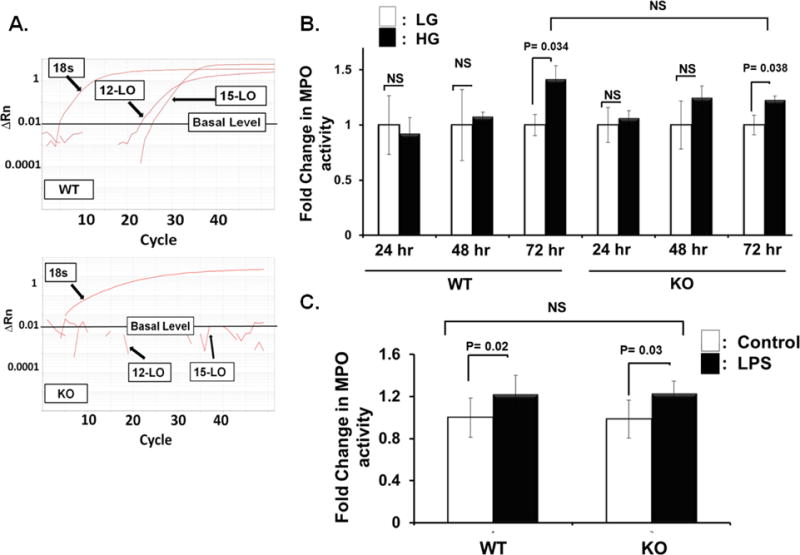
Monocytic/macrophagic 12/15-LO does not play a role in high glucose-induced leukocyte adhesion to retinal endothelial cells. A) Real-time (RT)-PCR expression of 12-LO and 15-LO mRNAs in mouse leukocytes. Delta Rn versus cycle number plots showing amplification curves from wild-type (WT) mouse peripheral blood mononuclear cells (PBMCs) versus knockout (KO) PBMCs. Rn represents the normalized fluorescent signal of the reporter divided by the passive reference dye, ROX. B) and C) Leukocyte adhesion assay was performed on high glucose, HG- (B), or LPS- (C) activated mouse retinal endothelial cells (mRECs) using leukocytes isolated from either 12/15-LO knockout (KO) or wild type (WT) mice. mRECs activated either by 72-hours HG treatment (B) or LPS (C) significantly increased the number of adherent leukocytes isolated from KO mice relatively to the same extent as those derived from WT mice. The data are presented as the fold change in Myeloperoxidase (MPO) activity (an indicator for leukocyte adhesion) ± SD relative to the corresponding control from non-activated mRECs, n =4–6 for each group.
This proof-of-concept was extended to include human retinal endothelial cells (HRECs). Initial studies were performed to optimize our experimental procedures in which HRECs were treated with different hyperglycemic insults, HG or glycated albumin (AGA), then subjected to leukocyte adhesion assay using either U937 monocyte-like cells or purified CD14+ monocytes prepared from PBMCs. As shown in Figure 4A, the number of adherent leukocytes to HRECs, indicated by elevated MPO activity, was increased under HG compared to normo-osmotic control (LG) and this effect of HG was more robustly compared with that obtained with AGA. Furthermore, CD14+ monocytes showed a better response to the activated HRECs than U937 cells. Given the fact that leukocyte adhesion is closely linked to endothelial barrier integrity, we proceeded to investigate the propensity of these adherent leukocytes to disrupt barrier function in our model of HREC monolayer using real-time analysis of TER, an indicator of monolayer integrity. To this end, HRECs were treated with HG or LG for 3–5-days. Thereafter, CD14+ monocytes were added and changes in TER were monitored over time. As shown in Figure 4B, there was no drop in TER with direct HG-treatment compared to LG-treatment. However, after adding CD14+ monocytes, a significant decrease in TER was observed in HRECs incubated with HG compared to LG (Figure 4C), demonstrating a key role for leukostasis in mediating diabetes- induced alterations of BRB homeostasis.
Figure 4. Activation of human retinal endothelial cells (HRECs) for leukocyte adhesion followed by hyperpermeability under different hyperglycemic conditions.
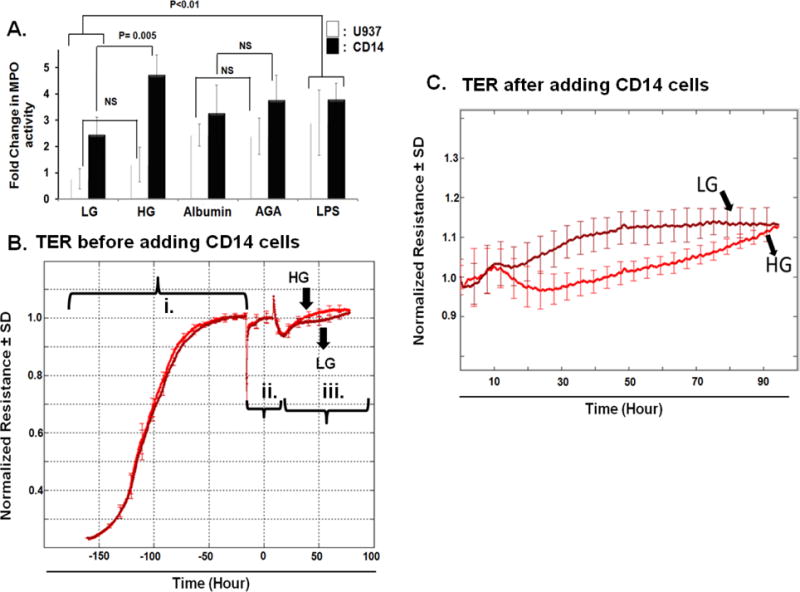
A) HRECs were treated, as described in material and method, with HG, glycated albumin (AGA), or LPS as a positive control, then subjected to leukocyte adhesion using either U937 monocyte-like cells or purified CD14+ monocytes. The number of adherent leukocytes to HRECs was significantly increased by HG-treatment compared to normo-osmotic control and the effect of HG was more robust compared with that obtained with the AGA. Additionally, CD14+ monocytes showed a better response towards activated HRECs than U937. The data presented are the fold changes in Myeloperoxidase (MPO) activity (an indicator for leukocyte adhesion) ± SD relative to the corresponding control from non-activated HRECs, n =4–6 in each group. B & C) Normalized TER resistance ± SD over time for HRECs incubated with HG or LG before and after CD14+ monocytes addition, respectively. The experiment was carried out after HRECs formed confluent mature monolayers as indicated by plateau of electronic resistance under (i) full media, (ii) serum free media (SFM). Thereafter, HG or LG in SFM was added (iii) to confluent mature monolayers before adding CD14+ monocytes in C), n=4–6 in each experimental group.
To discern the relative importance of human endothelial versus monocyte/macrophage-derived 12/15-LO in diabetes induced leukostasis, we compared protein level of 12/15-LO between HRECs and human U937 monocyte like cell line. Although abundant 12-LO and 15-LO were detected in HRECs, it was undetected (15-LO) or hardly detectable (12-LO) in human monocyte cell line, indicating a minimal contribution of leukocytic 12/15-LO to the observed inflammatory response (Figure 5A). Next, we examined the contribution of endothelial 12/15-LO to the observed inflammatory response at the molecular level by studying the effect of HG on ICAM-1, a well-established important molecule for leukocyte adhesion. As shown in Figure 5B, HG significantly enhanced ICAM-1 expression in HRECs, as assessed by Western blot analysis. However, inhibition of endothelial 12/15-LO by pre-treating HRECs with baicalein (10 μM) significantly reduced the elevation of ICAM-1 expression induced by HG treatment. Concordantly, knocking down of 12/15-LO from HRECs using siRNA (Figure 5C) significantly inhibited HG-induced upregulation of ICAM-1 expression compared to scrambled siRNA transfection (Figure 5D). To provide functional evidence for 12/15-LO involvement in hyperglycemia- induced enhancement of leukocyte adhesion to HRECs, the effect of inhibiting endothelial 12/15-LO by baicalein on leukocyte/endothelial cell adhesion under HG-treatment was determined. A shown in Figure 5E, baicalein significantly attenuated the number of adherent CD14+ monocytes to HG-activated HRECs compared to the vehicle (DMSO) treatment. Collectively, these data suggest that endothelium-derived 12/15-LO activity plays an essential role in mediating leukocyte adhesion and endothelial barrier dysfunction during DR.
Figure 5. Contribution of endothelial 12/15-LO to high glucose (HG)-induced ICAM-1 expression in HRECs.
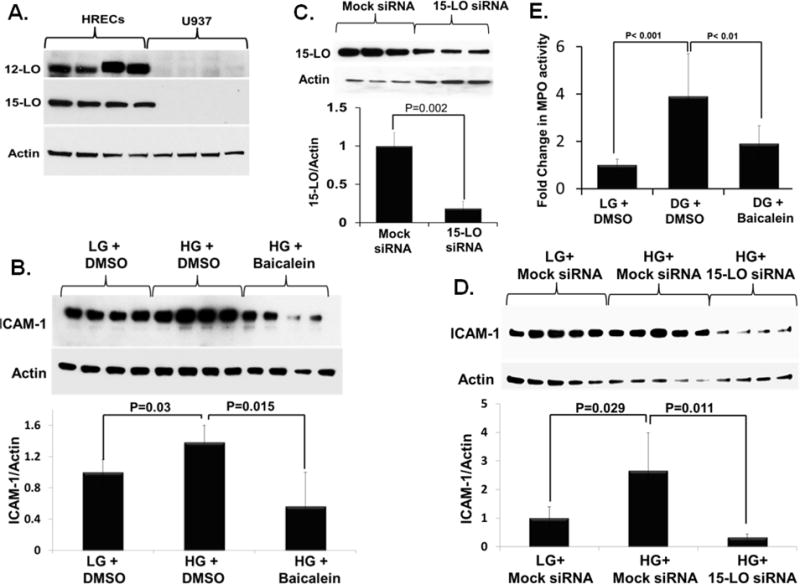
A) Expression of 12-LO and 15-LO in HRECs and human monocyte-like U937 cells. B) Inhibition of HG-induced ICAM-1 expression, as assessed by Western blot analysis, in HRECs pre-treated with the broad 12/15-LO pharmacological inhibitor, baicalein (10 μM). Cells were pretreated with baicalein for 30 min, and then incubated with (HG; D-Glucose, 30 mM) or normo-osmotic control (LG, 5mM D-glucose+25 mM L-glucose) for 5-days. C&D) Inhibition of HG-induced ICAM-1 expression, as assessed by Western blot analysis, in HRECs transfected with 15-LO siRNA. Cells were transfected with siRNA or mock siRNA for 24 hours and then treated with (HG; D-Glucose, 30 mM) or normo-osmotic control (LG, 5mM D-glucose+25 mM L-glucose) for 5-days. C) Measurement of 15-LO expression relative to actin by Western blot after transfection with 15-LO siRNA. D) Relative ICAM-1 expression in HRECs incubated with HG or LG in the presence or absence of 15-LO siRNA normalized to that of LG and mocked siRNA-treated cells, which was arbitrarily set at 1, n=4–5. E) HRECs were incubated in (HG; D-Glucose, 30 mM) or normo-osmotic control (LG, 5mM D-glucose+25 mM L-glucose) for 5-days in presence of baicalein (10 μM) or its vehicle (DMSO), then subjected to leukocyte adhesion using CD14+ monocytes. The data presented are the fold changes in Myeloperoxidase (MPO) activity (an indicator for leukocyte adhesion) ± SD relative to the corresponding control LG-treated HRECs, n =4 in each group.
DISCUSSION
The novel and central finding of the current study is that endothelial 12/15-LO rather than the monocytic/macrophagic 12/15-LO has a critical role in hyperglycemia-induced leukocyte adhesion and retinal endothelial barrier dysfunction. The following evidence, in addition to what we have published for the elevated levels of 12/15-LO metabolites locally under diabetic conditions, support this conclusion: (a) No significant change in the systemic plasma levels of primary metabolites derived from the activity of 12/15-LO, including 12- and 15-HETE, in STZ-diabetic mice. (b) Leukocytes from 12/15-LO−/− mice displayed a similar increase in adhesion to activated endothelial cells as did leukocytes from WT mice. (c) Abundant proteins of 12-LO and 15-LO were detected in HRECs, while it was undetected (15-LO) or hardly detectable (12-LO) in human monocyte-like U937 cells. (d) Inhibition of endothelial 12/15-LO in HRECs attenuated hyperglycemia-induced leukocyte adhesion and ICAM-1 expression, a well-known identified important molecule for leukocyte adhesion in DR.
Several in vivo and in vitro studies have demonstrated changes in bioactive lipid profiles under hyperglycemic conditions and have linked these changes with increased leukocyte adhesion and vascular dysfunction during diabetes. This input has originated partly from lipidomic studies that showed elevated levels of LO products including 5-, 12-, and 15-HETE released from aortic endothelial cells (AECs) and smooth muscle cells cultured under hyperglycemic conditions [34, 35]. Enhanced expression of 12/15-LO and production of HETEs have also been reported in patients with diabetic vascular complications [20, 36], in vessels from infants of diabetic mothers [37], and in experimental animal models of diabetes [38, 39]. These findings have been extended by Patriacia et al study, where 12- and 15-HETEs per se induced a similar increase in monocyte adhesion to human AECs as seen with glucose alone [34]. Furthermore, the specific role of 12-LO in glucose-induced monocyte-endothelial interactions was reinforced by additional studies showing a marked inhibition of high glucose-stimulated monocyte adhesion in AECs treated with a catalytically active ribozyme directed against 12-LO [40]. In support of the relevance of this pathway in vivo, inhibition of 12/15-LO in isolated primary AECs from db/db mice reduced the monocyte adhesion to db/db endothelium [38]. All of these reports strengthen the concept that modulation of the 12/15-LO pathway in endothelial cells may provide a therapeutic benefit for early vascular inflammation in diabetes. Consistent with these previous studies, we and others [41] have observed a similar pro-inflammatory phenotype, characterized by increased ICAM-1 expression and monocyte adhesion, in human retinal endothelial cells (HRECs) under hyperglycemic conditions. However, this study is the first to show that glucose regulates the acquisition of this phenotype in retinal endothelial cells through 12/15-LO. Furthermore, our study suggests that endothelial 12/15-LO rather than the monocytic/macrophagic 12/15-LO plays an important role in this process. Evidence for such differential activity of systemic and retinal 12/15-LO during diabetes was inferred from our in vivo and in vitro studies. In our in vivo study, there were no significant changes in the plasma levels of major metabolites derived from 12/15-lipoxygenation of different PUFAs, including LA (13-HODE), AA (12- and 15- HETEs), EPA (12- and 15- HEPEs), or DHA (17-HDoHE). In addition, there were no significant changes in the plasma levels of other metabolites derived from 12/15-lipoxygenase, including LXA4, LXB4, 5,15-DiHETE, 8,15-DiHETE (Table 1). Of note, the only increased metabolite in the plasma of diabetic mice among those derived from 15-LO pathway was RvD2 (7S,16R,17S-trihydroxy-DHA) and this point needs further discussion because RvD2 is formed by multiple enzymes including 15-LO. It is formed by the initial metabolism of DHA to 17S-hydroxy-DHA by 15-LO followed by further metabolism by 5-LO to 7S-hydroxy-derivative and then by Cytochrome P450 monooxygenase to 7(S),16(R),17(S)-Resolvin D2 [42]. The observations that the level of the intermediate (17-HDoHE), which is derived from 15-LO, did not change in diabetes together with increased levels of CYP/sEH products, may provide clues that the alteration in RvD2 level observed is most likely due to the increased enzymatic activity of the subsequent metabolic pathways rather than the initial metabolism by 15-LO. These in vivo results were further corroborated by our in vivo data that showed that leukocytes from 12/15-LO−/− mice displayed a similar increase in adhesion to activated endothelial cells as did leukocytes from WT mice. However, because systemic and local bioactive metabolite changes might be very different, it does not necessarily mean that the local change is the only driving force for leukocyte adhesion in DR.
Another important finding of our study is the observation that high glucose per se does not affect TER in mature HREC monolayers; but at the same time it does upregulate ICAM-1 directly and activates HRECs to leukocyte adhesion. This would imply that high glucose affects TER in mature HREC monolayers indirectly through activation of HRECs to leukocyte adhesion. This explains what we have seen in Figure 4; no drop in TER with direct HG-treatment compared to LG-treatment until monocytes were added.
There are two major isoforms of 12/15-LO in mammalian, 12 and 15- LO, with different cellular distributions. To determine the relative contributions of these isoforms towards glucose-induced monocyte-endothelial interactions, the distribution of these isoforms in the two cell types involved in leukostasis; endothelial and monocytes, was determined. An intriguing and important finding described here is the demonstration that 12-LO and 15-LO both are found in retinal microvascular endothelial cells while they were hardly detected in circulating human monocytes. These results are in agreement with prior studies showing a notable 15-LO expression in HRECs [43], but not in agreement with other studies which demonstrated undetectable 15-LO mRNA in basal or stimulated human aortic or umbilical endothelial cells [28, 44]. The discrepancy between those results and our findings may be due to differences in endothelial properties in large vessels, such as aortic artery or umbilical vein, versus microvessels present in the retina [45]. This differential expression of 12/15-LO might explain contradictory findings regarding the contribution of 12/15-LO to vascular dysfunction found in various pathological conditions. For instance, it has been verified in three different mouse models (apoE, LDL-R, and apobec-1/LDL-R deficiency) by at least three research groups the role of 12/15-LO expressing bone marrow–derived cells (e.g. macrophages) in preference to macrovascular endothelial cells in initiating pro-atherogenic inflammatory response [46]. On the contrary, our results suggest a role for 12/15-LO expressing retinal microvascular endothelial cells, and not monocytic/macrophagic cells, in mediating inflammatory responses seen in early DR.
The next important step would be to establish the connection between the enhanced 12/15-LO induction by high glucose and monocyte adhesion. The monocyte adhesion appears to be mediated primarily by intercellular adhesion molecule-1 (ICAM-1). Here, we showed that using 12/15-LO knockdown approach, a dramatic reduction of ICAM-1 expression induced in HRECs by hyperglycemia was observed. Moreover, a lipoxygenase inhibitor, baicalein, mimics the effect obtained by siRNA. Although the current study focused on endothelial and monocytic cells, additional cell types involved in the pathogenesis of DR and expressing 12/15-LO, such as resident macrophages, M2- type macrophages, and epithelial cells, could play a role in mediating endothelial ICAM-1 expression and leukocyte adhesion and their contributions need to be further investigated.
Several potential mechanisms for the regulation of ICAM-1 expression by 12/15-LO can be considered. First, the 12/15-LO product, 12- or 15-HETE, which we have recently demonstrated to be increased in high glucose-treated HRECs [22], has shown to mediate the ICAM-1 induction through RhoA, protein kinase C (PKC), and NF-κB activation pathways [47]. Second, Bolick and colleagues postulated, in the same study [47], that the 12/15-LO product can activate a G protein–coupled receptor (GPCR) to initiate these events. Criteria for making this assumption were based on the activation of the small GTPase by 12/15-LO product as well as the reduction in ICAM-1 expression in response to blocking the G proteins G12 and G13 but not Gi in the endothelium [47]. The isolation of this receptor has proven later to be GPCR-31 for 12-HETE [48]. Nevertheless, no research group has “adopted” one of the orphan members as a 15-HETE receptor. Furthermore, whether 15-HETE can activate the same receptor as does 12-HETE has yet to be clarified. Others have demonstrated the ability of 12/15-LO products to bind an intracellular receptor that associates with other proteins, which is more reminiscent of nuclear receptor signaling [49, 50]. Such observations leave an open question with regard to 12/15-LO products: whether their cellular effects are mediated by a secreted product binding to a receptor or whether they act as second messengers within the cell. Although our studies do not directly address this issue, previous studies from our group showed that exogenous addition of 12/15-LO products in nano-molar concentrations was sufficient to stimulate monocyte binding [22]. Yet, the possibility that 12/15-LO products may function intracellularly cannot be ruled out because it is generally recognized that lipophilic molecules can cross the cell membrane via passive diffusion. In any case, understanding the pathway of HREC activation under hyperglycemia from 12/15-LO induction to monocyte adhesion would be a substantial step forward in translating eicosanoid biology to therapeutics in DR.
In summary, our data have shown that one mechanism by which glucose may mediate monocyte–endothelial cell interaction in the retinal endothelial cells is via the 12/15-LO pathway Furthermore, our current and previously published data [20, 22] indicate a differential role of endothelial 12/15-LO versus the one in monocytic/macrophagic cells in mediating the inflammatory responses during DR. We, therefore, conclude that glucose promotes production of 12- or 15-HETE through retinal endothelial 12/15-LO rather than monocye/macrophage 12/15-LO, which results in autocrine activation of endothelial cells and induction of ICAM-1 on the surface of HRECs that would allow monocytes to firmly adhere to the endothelium (Figure 6). Because the interaction between monocytes and the endothelium is a key early event in the development of DR, targeting this enzyme in endothelial cells may facilitate the development of more precisely targeted treatment strategies for DR.
Figure 6. Proposed cascade of events in the pathogenesis of diabetic retinopathy.
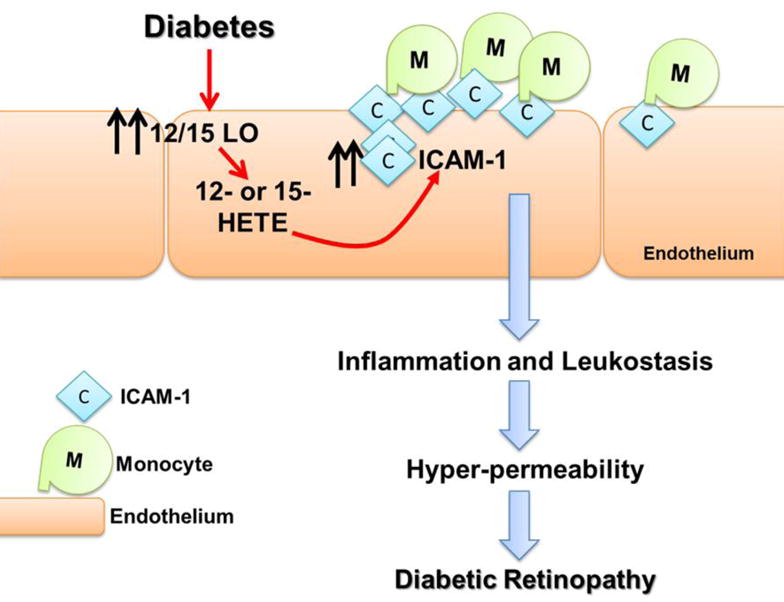
Hyperglycemia activates 12/15-LO to release 12- and 15-hydroxyeicosatetraeanoic acids (HETEs) that in turn activate retinal endothelial cells through various inflammatory signaling system leading to increases in ICAM-1 expression, leukocyte adhesion, followed by hyperpermeability, the cardinal signs of diabetic retinopathy.
Highlights.
Endothelial 12/15-LO rather than the myeloid lineage 12/15-LO has a role in HG-induced leukostasis.
No significant change in plasma levels of metabolites derived from of 12/15-LO in diabetic mice.
12/15-LO−/− Leukocytes have a similar increase in adhesion to endothelial cells as WT Leukocytes.
Inhibition of endothelial 12/15-LO in HRECs reduces HG-induced leukostasis and ICAM-1 expression.
Acknowledgments
This work has been supported by the National Eye Institute (NIH) Grant 5R01EY023315-02 (MA). This study was also supported in part by National Center for Research Resources Grant S10RR027926 for the lipid analysis and by James and Jean Culver Vision Discovery Institute (ASI). The work in NS laboratory is supported in part by non-restricted funds from Research to Prevent Blindness Foundation, Retina Research Foundation, EY016665, EY022883, and EPA 83573701.The authors would like to thank Dr. Bhagelu Achyut for his assistance with heat map making software.
List of Nonstandard Abbreviations
- DR
diabetic retinopathy
- 12/15-LO
12/15-Lipoxygenase
- HETEs
hydroxyeicosatetraenoic acids
- ELISA
Enzyme-linked immunosorbent assay
- ROS
reactive oxygen species
- STZ
Streptozotocin
- HRECs
human retinal endothelial cells
- LC/MS
Liquid chromatography–mass spectrometry
- VEGF
vascular endothelial growth factor
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Congdon NG, Friedman DS, Lietman T. Important causes of visual impairment in the world today. JAMA. 2003;290:2057–2060. doi: 10.1001/jama.290.15.2057. [DOI] [PubMed] [Google Scholar]
- 2.Nguyen TT, Wang JJ, Sharrett AR, Islam FM, Klein R, Klein BE, Cotch MF, Wong TY. Relationship of retinal vascular caliber with diabetes and retinopathy: the Multi-Ethnic Study of Atherosclerosis (MESA) Diabetes Care. 2008;31:544–549. doi: 10.2337/dc07-1528. [DOI] [PubMed] [Google Scholar]
- 3.Wilkinson CP, Ferris FL, 3rd, Klein RE, Lee PP, Agardh CD, Davis M, Dills D, Kampik A, Pararajasegaram R, Verdaguer JT, G Global Diabetic Retinopathy Project Proposed international clinical diabetic retinopathy and diabetic macular edema disease severity scales. Ophthalmology. 2003;110:1677–1682. doi: 10.1016/S0161-6420(03)00475-5. [DOI] [PubMed] [Google Scholar]
- 4.Wong TY, Liew G, Tapp RJ, Schmidt MI, Wang JJ, Mitchell P, Klein R, Klein BE, Zimmet P, Shaw J. Relation between fasting glucose and retinopathy for diagnosis of diabetes: three population-based cross-sectional studies. Lancet. 2008;371:736–743. doi: 10.1016/S0140-6736(08)60343-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cheung N, Mitchell P, Wong TY. Diabetic retinopathy. Lancet. 2010;376:124–136. doi: 10.1016/S0140-6736(09)62124-3. [DOI] [PubMed] [Google Scholar]
- 6.Rangasamy S, McGuire PG, Das A. Diabetic retinopathy and inflammation: novel therapeutic targets. Middle East Afr J Ophthalmol. 2012;19:52–59. doi: 10.4103/0974-9233.92116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Joussen AM, Poulaki V, Le ML, Koizumi K, Esser C, Janicki H, Schraermeyer U, Kociok N, Fauser S, Kirchhof B, Kern TS, Adamis AP. A central role for inflammation in the pathogenesis of diabetic retinopathy. FASEB J. 2004;18:1450–1452. doi: 10.1096/fj.03-1476fje. [DOI] [PubMed] [Google Scholar]
- 8.Miyamoto K, Khosrof S, Bursell SE, Rohan R, Murata T, Clermont AC, Aiello LP, Ogura Y, Adamis AP. Prevention of leukostasis and vascular leakage in streptozotocin-induced diabetic retinopathy via intercellular adhesion molecule-1 inhibition. Proc Natl Acad Sci U S A. 1999;96:10836–10841. doi: 10.1073/pnas.96.19.10836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schroder S, Palinski W, Schmid-Schonbein GW. Activated monocytes and granulocytes, capillary nonperfusion, and neovascularization in diabetic retinopathy. Am J Pathol. 1991;139:81–100. [PMC free article] [PubMed] [Google Scholar]
- 10.McLeod DS, Lefer DJ, Merges C, Lutty GA. Enhanced expression of intracellular adhesion molecule-1 and P-selectin in the diabetic human retina and choroid. Am J Pathol. 1995;147:642–653. [PMC free article] [PubMed] [Google Scholar]
- 11.Joussen AM, Murata T, Tsujikawa A, Kirchhof B, Bursell SE, Adamis AP. Leukocyte-mediated endothelial cell injury and death in the diabetic retina. Am J Pathol. 2001;158:147–152. doi: 10.1016/S0002-9440(10)63952-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bai N, Tang S, Ma J, Luo Y, Lin S. Increased expression of intercellular adhesion molecule-1, vascular cellular adhesion molecule-1 and leukocyte common antigen in diabetic rat retina. Yan Ke Xue Bao. 2003;19:176–183. [PubMed] [Google Scholar]
- 13.Adamis AP, Berman AJ. Immunological mechanisms in the pathogenesis of diabetic retinopathy. Semin Immunopathol. 2008;30:65–84. doi: 10.1007/s00281-008-0111-x. [DOI] [PubMed] [Google Scholar]
- 14.Joussen AM, Poulaki V, Mitsiades N, Kirchhof B, Koizumi K, Dohmen S, Adamis AP. Nonsteroidal anti-inflammatory drugs prevent early diabetic retinopathy via TNF-alpha suppression. FASEB J. 2002;16:438–440. doi: 10.1096/fj.01-0707fje. [DOI] [PubMed] [Google Scholar]
- 15.Hirano Y, Sakurai E, Matsubara A, Ogura Y. Suppression of ICAM-1 in retinal and choroidal endothelial cells by plasmid small-interfering RNAs in vivo. Invest Ophthalmol Vis Sci. 2010;51:508–515. doi: 10.1167/iovs.09-3457. [DOI] [PubMed] [Google Scholar]
- 16.Zhang XL, Wen L, Chen YJ, Zhu Y. Vascular endothelial growth factor up-regulates the expression of intracellular adhesion molecule-1 in retinal endothelial cells via reactive oxygen species, but not nitric oxide. Chin Med J (Engl) 2009;122:338–343. [PubMed] [Google Scholar]
- 17.Nagai N, Izumi-Nagai K, Oike Y, Koto T, Satofuka S, Ozawa Y, Yamashiro K, Inoue M, Tsubota K, Umezawa K, Ishida S. Suppression of diabetes-induced retinal inflammation by blocking the angiotensin II type 1 receptor or its downstream nuclear factor-kappaB pathway. Invest Ophthalmol Vis Sci. 2007;48:4342–4350. doi: 10.1167/iovs.06-1473. [DOI] [PubMed] [Google Scholar]
- 18.Nonaka A, Kiryu J, Tsujikawa A, Yamashiro K, Miyamoto K, Nishiwaki H, Honda Y, Ogura Y. PKC-beta inhibitor (LY333531) attenuates leukocyte entrapment in retinal microcirculation of diabetic rats. Invest Ophthalmol Vis Sci. 2000;41:2702–2706. [PubMed] [Google Scholar]
- 19.Gubitosi-Klug RA, Talahalli R, Du Y, Nadler JL, Kern TS. 5-Lipoxygenase, but not 12/15-lipoxygenase, contributes to degeneration of retinal capillaries in a mouse model of diabetic retinopathy. Diabetes. 2008;57:1387–1393. doi: 10.2337/db07-1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Al-Shabrawey M, Mussell R, Kahook K, Tawfik A, Eladl M, Sarthy V, Nussbaum J, El-Marakby A, Park SY, Gurel Z, Sheibani N, Maddipati KR. Increased expression and activity of 12-lipoxygenase in oxygen-induced ischemic retinopathy and proliferative diabetic retinopathy: implications in retinal neovascularization. Diabetes. 2011;60:614–624. doi: 10.2337/db10-0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Othman A, Ahmad S, Megyerdi S, Mussell R, Choksi K, Maddipati KR, Elmarakby A, Rizk N, Al-Shabrawey M. 12/15-Lipoxygenase-derived lipid metabolites induce retinal endothelial cell barrier dysfunction: contribution of NADPH oxidase. PLoS One. 2013;8:e57254. doi: 10.1371/journal.pone.0057254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ibrahim AS, Elshafey S, Sellak H, Hussein KA, El-Sherbiny M, Abdelsaid M, Rizk N, Beasley S, Tawfik AM, Smith SB, Al-Shabrawey M. A lipidomic screen of hyperglycemia-treated HRECs links 12/15-Lipoxygenase to microvascular dysfunction during diabetic retinopathy via NADPH oxidase. J Lipid Res. 2015;56:599–611. doi: 10.1194/jlr.M056069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ibrahim AS, Tawfik AM, Hussein KA, Elshafey S, Markand S, Rizk N, Duh EJ, Smith SB, Al-Shabrawey M. Pigment epithelium-derived factor inhibits retinal microvascular dysfunction induced by 12/15-lipoxygenase-derived eicosanoids. Biochim Biophys Acta. 2015;1851:290–298. doi: 10.1016/j.bbalip.2014.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kuhn H, Belkner J, Suzuki H, Yamamoto S. Oxidative modification of human lipoproteins by lipoxygenases of different positional specificities. J Lipid Res. 1994;35:1749–1759. [PubMed] [Google Scholar]
- 25.Belkner J, Stender H, Kuhn H. The rabbit 15-lipoxygenase preferentially oxygenates LDL cholesterol esters, and this reaction does not require vitamin E. J Biol Chem. 1998;273:23225–23232. doi: 10.1074/jbc.273.36.23225. [DOI] [PubMed] [Google Scholar]
- 26.Takahashi Y, Glasgow WC, Suzuki H, Taketani Y, Yamamoto S, Anton M, Kuhn H, Brash AR. Investigation of the oxygenation of phospholipids by the porcine leukocyte and human platelet arachidonate 12-lipoxygenases. Eur J Biochem. 1993;218:165–171. doi: 10.1111/j.1432-1033.1993.tb18362.x. [DOI] [PubMed] [Google Scholar]
- 27.Funk CD. The molecular biology of mammalian lipoxygenases and the quest for eicosanoid functions using lipoxygenase-deficient mice. Biochim Biophys Acta. 1996;1304:65–84. doi: 10.1016/s0005-2760(96)00107-5. [DOI] [PubMed] [Google Scholar]
- 28.Kim JA, Gu JL, Natarajan R, Berliner JA, Nadler JL. A leukocyte type of 12-lipoxygenase is expressed in human vascular and mononuclear cells. Evidence for upregulation by angiotensin II. Arterioscler Thromb Vasc Biol. 1995;15:942–948. doi: 10.1161/01.atv.15.7.942. [DOI] [PubMed] [Google Scholar]
- 29.Harats D, Shaish A, George J, Mulkins M, Kurihara H, Levkovitz H, Sigal E. Overexpression of 15-lipoxygenase in vascular endothelium accelerates early atherosclerosis in LDL receptor-deficient mice. Arterioscler Thromb Vasc Biol. 2000;20:2100–2105. doi: 10.1161/01.atv.20.9.2100. [DOI] [PubMed] [Google Scholar]
- 30.Shen J, Herderick E, Cornhill JF, Zsigmond E, Kim HS, Kuhn H, Guevara NV, Chan L. Macrophage-mediated 15-lipoxygenase expression protects against atherosclerosis development. J Clin Invest. 1996;98:2201–2208. doi: 10.1172/JCI119029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Maddipati KR, Romero R, Chaiworapongsa T, Zhou SL, Xu Z, Tarca AL, Kusanovic JP, Munoz H, Honn KV. Eicosanomic profiling reveals dominance of the epoxygenase pathway in human amniotic fluid at term in spontaneous labor. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2014 doi: 10.1096/fj.14-254383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Su X, Sorenson CM, Sheibani N. Isolation and characterization of murine retinal endothelial cells. Mol Vis. 2003;9:171–178. [PubMed] [Google Scholar]
- 33.Ibrahim AS, El-Remessy AB, Matragoon S, Zhang W, Patel Y, Khan S, Al-Gayyar MM, El-Shishtawy MM, Liou GI. Retinal microglial activation and inflammation induced by amadori-glycated albumin in a rat model of diabetes. Diabetes. 2011;60:1122–1133. doi: 10.2337/db10-1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Patricia MK, Kim JA, Harper CM, Shih PT, Berliner JA, Natarajan R, Nadler JL, Hedrick CC. Lipoxygenase products increase monocyte adhesion to human aortic endothelial cells. Arterioscler Thromb Vasc Biol. 1999;19:2615–2622. doi: 10.1161/01.atv.19.11.2615. [DOI] [PubMed] [Google Scholar]
- 35.Natarajan R, Gu JL, Rossi J, Gonzales N, Lanting L, Xu L, Nadler J. Elevated glucose and angiotensin II increase 12-lipoxygenase activity and expression in porcine aortic smooth muscle cells. Proc Natl Acad Sci U S A. 1993;90:4947–4951. doi: 10.1073/pnas.90.11.4947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Antonipillai I, Nadler J, Vu EJ, Bughi S, Natarajan R, Horton R. A 12-lipoxygenase product, 12-hydroxyeicosatetraenoic acid, is increased in diabetics with incipient and early renal disease. J Clin Endocrinol Metab. 1996;81:1940–1945. doi: 10.1210/jcem.81.5.8626861. [DOI] [PubMed] [Google Scholar]
- 37.Setty BN, Stuart MJ. 15-Hydroxy-5,8,11,13-eicosatetraenoic acid inhibits human vascular cyclooxygenase. Potential role in diabetic vascular disease. J Clin Invest. 1986;77:202–211. doi: 10.1172/JCI112277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hatley ME, Srinivasan S, Reilly KB, Bolick DT, Hedrick CC. Increased production of 12/15 lipoxygenase eicosanoids accelerates monocyte/endothelial interactions in diabetic db/db mice. J Biol Chem. 2003;278:25369–25375. doi: 10.1074/jbc.M301175200. [DOI] [PubMed] [Google Scholar]
- 39.Natarajan R, Gerrity RG, Gu JL, Lanting L, Thomas L, Nadler JL. Role of 12-lipoxygenase and oxidant stress in hyperglycaemia-induced acceleration of atherosclerosis in a diabetic pig model. Diabetologia. 2002;45:125–133. doi: 10.1007/s125-002-8253-x. [DOI] [PubMed] [Google Scholar]
- 40.Patricia MK, Natarajan R, Dooley AN, Hernandez F, Gu JL, Berliner JA, Rossi JJ, Nadler JL, Meidell RS, Hedrick CC. Adenoviral delivery of a leukocyte-type 12 lipoxygenase ribozyme inhibits effects of glucose and platelet-derived growth factor in vascular endothelial and smooth muscle cells. Circ Res. 2001;88:659–665. doi: 10.1161/hh0701.088838. [DOI] [PubMed] [Google Scholar]
- 41.Hirata F, Yoshida M, Ogura Y. High glucose exacerbates neutrophil adhesion to human retinal endothelial cells. Exp Eye Res. 2006;82:179–182. doi: 10.1016/j.exer.2005.08.022. [DOI] [PubMed] [Google Scholar]
- 42.Duvall MG, Levy BD. DHA- and EPA-derived resolvins, protectins, and maresins in airway inflammation. Eur J Pharmacol. 2016;785:144–155. doi: 10.1016/j.ejphar.2015.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bajpai AK, Blaskova E, Pakala SB, Zhao T, Glasgow WC, Penn JS, Johnson DA, Rao GN. 15(S)-HETE production in human retinal microvascular endothelial cells by hypoxia: Novel role for MEK1 in 15(S)-HETE induced angiogenesis. Invest Ophthalmol Vis Sci. 2007;48:4930–4938. doi: 10.1167/iovs.07-0617. [DOI] [PubMed] [Google Scholar]
- 44.Lopez S, Vila L, Breviario F, de Castellarnau C. Interleukin-1 increases 15-hydroxyeicosatetraenoic acid formation in cultured human endothelial cells. Biochim Biophys Acta. 1993;1170:17–24. doi: 10.1016/0005-2760(93)90170-e. [DOI] [PubMed] [Google Scholar]
- 45.Zetter BR. The endothelial cells of large and small blood vessels. Diabetes. 1981;30:24–28. doi: 10.2337/diab.30.2.s24. [DOI] [PubMed] [Google Scholar]
- 46.Funk CD. Lipoxygenase pathways as mediators of early inflammatory events in atherosclerosis. Arterioscler Thromb Vasc Biol. 2006;26:1204–1206. doi: 10.1161/01.ATV.0000222960.43792.ff. [DOI] [PubMed] [Google Scholar]
- 47.Bolick DT, Orr AW, Whetzel A, Srinivasan S, Hatley ME, Schwartz MA, Hedrick CC. 12/15-lipoxygenase regulates intercellular adhesion molecule-1 expression and monocyte adhesion to endothelium through activation of RhoA and nuclear factor-kappaB. Arterioscler Thromb Vasc Biol. 2005;25:2301–2307. doi: 10.1161/01.ATV.0000186181.19909.a6. [DOI] [PubMed] [Google Scholar]
- 48.Guo Y, Zhang W, Giroux C, Cai Y, Ekambaram P, Dilly AK, Hsu A, Zhou S, Maddipati KR, Liu J, Joshi S, Tucker SC, Lee MJ, Honn KV. Identification of the orphan G protein-coupled receptor GPR31 as a receptor for 12-(S)-hydroxyeicosatetraenoic acid. J Biol Chem. 2011;286:33832–33840. doi: 10.1074/jbc.M110.216564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Herbertsson H, Kuhme T, Hammarstrom S. The 650–kDa 12(S)-hydroxyeicosatetraenoic acid binding complex: occurrence in human platelets, identification of hsp90 as a constituent, and binding properties of its 50-kDa subunit. Arch Biochem Biophys. 1999;367:33–38. doi: 10.1006/abbi.1999.1233. [DOI] [PubMed] [Google Scholar]
- 50.Zhang L, Ma J, Li Y, Guo L, Ran Y, Liu S, Jiang C, Zhu D. 15-Hydroxyeicosatetraenoic acid (15-HETE) protects pulmonary artery smooth muscle cells against apoptosis via HSP90. Life Sci. 2010;87:223–231. doi: 10.1016/j.lfs.2010.06.019. [DOI] [PubMed] [Google Scholar]