

Abstract
Respiratory syncytial virus (RSV) represents a threat to infants, the elderly, and the immunocompromised. RSV entry blockers are in clinical trials, but escape mutations challenge their potential. In search of RSV inhibitors, we have integrated a signature resistance mutation into are combinant RSV virus and applied the strain to high-throughput screening. Counter screening of candidates returned 14 confirmed hits with activity in the nano- to low-micromolar range. All blocked RSV polymerase activity in minigenome assays. Compound 1a (GRP-74915) was selected for development based on activity (EC50=0.21 µM, selectivity index (SI) 40), and scaffold. Resynthesis confirmed potency of the compound, which suppressed viral RNA synthesis in infected cells. However, metabolic testing revealed a short half-life in the presence of mouse hepatocyte fractions. Metabolite tracking and chemical elaboration combined with 3D-quantitative structure-activity relationship modeling yielded analogs (i.e. 8n: EC50=0.06 µM, SI 500) that establish a platform for the development of a therapeutic candidate.
Keywords: respiratory syncytial virus, RNA-dependent RNA polymerase inhibitor, pneumovirus, antiviral
INTRODUCTION
A member of the recently established pneumovirus family of negative stranded non-segmented RNA viruses1, RSV is the leading cause of infant hospitalization due to viral infections in the United States and is responsible for the majority of fatalities due to respiratory infectious diseases of children less than one year of age2. The risk of progressing to severe RSV disease is highest during primary infection of infants, but re-infections with RSV can occur throughout life3. Whereas disease is typically less severe at re-infection, RSV constitutes a major clinical threat to the elderly and the immunocompromised4.
RSV is a highly contagious airborne pathogen that spreads through the respiratory route. Initial infection of airway epithelia cells is followed by viral spread from the nasopharynx to the lower airways that can impair respiratory function5, 6. Frequently observed with acute virus infections are immunopathogenic effects, such that disease symptoms are predominantly a consequence of the host immune response to infection rather than induced by the pathogen directly7. Since viral titers are typically already immune-controlled when the infection becomes fully symptomatic, the available time window is often too narrow for efficacious therapeutic intervention with acute viral disease. However, severe and life-threatening RSV disease is characterized by the spread of infection to the lower respiratory tract. Although the connection between initial viral load and progression to lower respiratory infection remains poorly understood, clinical studies have identified viral load as correlated with increased disease severity in adult volunteers8. In RSV-infected hospitalized children under the age of two, higher viral loads on the third day after hospitalization were linked to an increased requirement for intensive care and progression to respiratory failure9. These studies suggest that efficacious therapeutics given early to hospitalized children may improve downstream morbidity and reduce immunopathology, opening a time window for therapeutic disease management.
Ribavirin is currently the only licensed drug approved for the treatment of RSV infection, but toxicity liabilities and limited efficacy undermine its clinical importance10, 11. Recent anti-RSV drug discovery campaigns have predominantly focused on two targets areas, the viral entry machinery and the RSV RNA-dependent RNA polymerase (RdRp) complex. Of eight small molecule drug candidates that were advanced to testing in primate models of RSV infection and/or clinical trials, six target the viral fusion protein, preventing viral entry, while only two block viral RNA synthesis12. Currently most advanced developments are the nucleoside analog inhibitor ALS-8176 (2)13 and the entry inhibitorGS-5806 (3)14, which are both orally available and decreased viral load and disease symptoms in phase II clinical trials.
Despite a high structural variety among the different small-molecule RSV entry inhibitors that were subjected to advanced development, point mutations in either one of two microdomains of the F protein, residues 392–401 and 486–489, respectively, induce robust viral escape from inhibition by all of these compounds12, 15. This unusual RSV pan-resistance phenotype to entry inhibition does not necessarily coincide with viral attenuation, since we noted that a pan-resistant RSV recombinant remained as pathogenic in a mouse model of RSV disease as the corresponding parent strain15. If this finding equally extends to the human host, broad clinical implementation may result in the induction of preexisting resistance in circulating strains as observed with the adamantes used for the treatment of influenza A virus (IAV) infections. Due to the development of widespread resistance from 2002–2006, the CDC advises against the use of adamantanes for influenza therapy16, 17.
Guided by the past experience that pan-resistance-sensitive RSV entry inhibitors emerge readily in HTS campaigns, we have in recent work developed an innovative HTS design that counterselects against identifying additional chemical scaffolds sensitive to pan-resistance by including a signature resistance mutation into a recombinant RSV reporter strain18 that also features a fusion protein derived from the line19 RSV isolate19. For automated detection of replication, this recombinant recRSV-A2-L19FD489E-fireSMASh strain expresses a firefly luciferase reporter from an additional transcription unit that is linked to an engineered small-molecule assisted shut off (SMASh) domain20, which significantly boosts the dynamic range of the assay, allowing effective assay miniaturization to 384-well plate format. Used in a co-infection approach with a recombinant IAV reporter strain harboring nano luciferase, this strategy supports the simultaneous identification of RSV-specific, IAV-specific, and broad-spectrum hit candidates in a single well setting18.
Here, we describe the implementation of this screening protocol against a 50,640-entry, open discovery library with the goal to identify novel scaffolds with anti-RSV activity. Based on the design of the infection protocol, we anticipated identifying predominantly inhibitors of early and intermediate stages of the viral life cycle, such as a new entry-inhibitor class insensitive to pan-resistance and RdRp blockers, while the discovery of compounds targeting late stages of viral replication such as virion assembly and egress was less favored. Having validated hit candidates through direct and orthogonal counterscreens and initial mechanistic characterization, we tested the developmental potential of a confirmed novel hit class through synthetic exploration of the scaffold and 3D-QSAR development. This work establishes a foundation for our program to develop much-needed next generation RSV therapeutics.
RESULTS
SCREENING AND HIT IDENTIFICATION
The primary HTS screen was carried out in single replicate in 384-well plate format, using a final compound concentration in the assay plates of 5 µM. Target viruses were IAV-WSN-nanoLuc and recRSV-A2-L19FD489E-fireSMASh, which carried the signature D489E mutation mediating pan-resistance against currently developed RSV entry inhibitors15.
For automated identification of primary hit candidates, we applied two complementary statistical methods: i) raw values imported into the MScreen package21 were automatically normalized for positive and negative controls present in 32 replicates each on every test plate and % inhibition by plate, scaled across all plates present in the campaign, calculated; and ii) we determined control-independent robust z scores by plate22, followed by scaling of values derived from each plate against the full campaign. Hit candidate cut-off values were arbitrarily set to ≥75% inhibition and robust z scores ≥4.5, respectively. Plotting of robust z scores as a function of % inhibition identified 180 compounds that met both hit identification targets and were selected for confirmation retesting (Figure 2A).
Figure 2.

Primary HTS and counterscreening for hit discovery. (A) Primary hit identification. Assay plates were analyzed using both control-dependent (% inhibition by plate) and independent (robust z-score) statistical approaches. Dashed lines represent hit cutoffs (75% inhibition and a z-score of >4.5, respectively), and red symbols denote hit candidates (180 total) selected for counterscreening. (B) Direct counter and cytotoxicity screens of the selected hit candidates against recRSV-A2-L19FD489E-fireSMASh. Confirmation plates were screened twice, error bars depict range. (C) Orthogonal dose-response counterscreens of 26 hit candidates selected from (B) against recRSV-A2-L19FD489E-fireSMASh and recRSV-A2-L19F-renilla. EC50 concentrations were determined through regression modeling. The solid line marks parity of active concentrations against either target virus, the dashed line specifies the cut-off for further consideration (15-fold less potency against recRSV-A2-L19F-renilla than recRSV-A2-L19FD489E-fireSMASh. (D) RSV minigenome dose-response assay of the 14 confirmed compounds from (C), red symbols. Regression curves are shown for each candidate, symbols represent averages of relative minireplicon activities ± SEM.
To verify hit candidates, we implemented a two-stage counterscreening strategy consisting of single concentration direct counter- and cytotoxicity screens in 384-well format and dose-response orthogonal counterscreens in 96-well format after sourcing of shortlisted compounds. Of the 180 compounds selected for the first stage of counterscreens, only 39 failed the direct counterscreen, corresponding to a false positive rate of 21.7% of the primary hit identification strategy (Figure 2B). We noted a relatively high frequency of cytotoxic compounds (defined as metabolic activity of cells <70% of vehicle-exposed control) in this candidate pool of 41% when metabolic activity of uninfected cells was assessed after 48-hour exposure to the different hit candidates. This distribution likely reflected that the primary hit pool included both RSV-specific and broad-spectrum myxovirus blockers, since a large proportion of the cytotoxic candidates originated not surprisingly from the broad-spectrum pool.
Of the remaining 67 candidates, a total of 26 with acceptable first-pass toxicity profile were sourced after visual inspection of the chemical scaffolds and advanced to orthogonal dose-response counterscreens to triage reporter-interfering candidates. We tested each compound intriplicate against the original screening strain and a recRSV-A2-L19F-renilla luciferase recombinant that we have previously generated23, capitalizing on the different protein structures and substrate chemistry between firefly and renilla luciferase24 that make simultaneous interference of a candidate compound with both reporters unlikely. Active concentrations against each target RSV strain were calculated independently through four-parameter variable slope regression modeling and plotted as a function of each other (Figure 2C). Fourteen compounds returned EC50 concentrations with <15-fold difference of each other against either target virus, indicating potency against both RSV reporter strains and thus either suggesting inhibition of RSV replication rather than reporter interference or, in an inversion of the desired activity profile, dependence on the D489E-substitution for antiviral activity. This subset of 14 hit candidates was subsequently tested for inhibitory activity in a firefly luciferase reporter-based RSV minigenome assay that we have previously developed15. Remarkably, we observed in all cases dose-dependent inhibition of reporter expression, indicating interference of the confirmed hit scaffolds with activity of the RSV RdRp complex (Figure 2D).
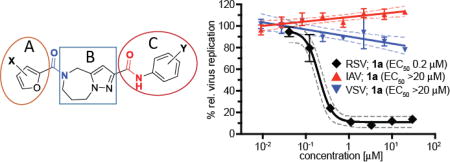
In an independent second orthogonal counterscreen, we tested the ability of five selected candidates (Figure 3) to reduce virus load, using standard recRSV-A2-L19F and progeny virus titers as the assay readout (Figure 4A). Combined, this panel of primary and counterscreens was highly effective, returning five confirmed hits with active concentrations below 1 µM against a standard RSV target strain. Based on structure and amenability for synthetic modification, we next selected one of the confirmed hits, compound 1a, for chemical hit-to-lead development. The compound was first resynthesized in-house using the procedure outlined in Scheme 1 to confirm chemical structure, purity and bioactivity. Testing against the original screening strain recRSV-A2-L19FD489E-fireSMASh demonstrated active concentrations of the sourced and resynthesized material within a 4-fold range of each other (EC50= 0.84 µM (sourced) vs. 0.21 µM (resynthesized), respectively), supporting the structural integrity of the original hit candidate (Figure 4B). The observed potency difference is likely due to higher purity of the resynthesized (98.2%) compared to the sourced (≥90%) material. Parallel testing of resynthesized 1a against two additional RNA viruses, vesicular stomatitis virus (VSV), which is like RSV a member of the order mononegavirales, and influenza A virus A/WSN/1933 (H1N1), revealed RSV specificity of the compound, since neither of these pathogens was inhibited by 1a (Figure 4B). These results underscore that anti-RSV activity of the compound is not due to underlying unspecific cytotoxicity and/or interference of the 1a with host cell polymerases.
Figure 3.

Structures of confirmed hits.
Figure 4.

(A) Progeny virus yield-based dose-response assay of five selected candidates from the subpanel shown in figure 2C (red symbols) against recRSV-A2-L19F. Virus titers were determined through TCID50 titration. Symbols represent averages of two repeats ± range, regression curves and calculated EC50 values are shown. Compounds tested were 1a, 4, 5, 6, and 7. (B) Dose-response reporter assays against the recRSV-A2-L19FD489E-fireSMASh screening strain to compare potency of resynthesized 1a and the sourced material. Only resynthesized 1a was tested against two distinct RNA viruses, vesicular stomatitis virus (VSV) and influenza A virus A/WSN/1933 (H1N1) (IAV). Symbols represent averages of three repeats ± SEM, nonlinear (RSV) and linear (VSV, IAV) regression curves, each with 95% confidence intervals, and EC50 values are shown. N/A, not applicable. (C) RT-qPCR assay to determine relative RSV N mRNA levels. Cells were infected with recRSV-A2-L19F (MOI 3.0) and incubated in the presence of a range of 1a concentrations as specified, previously reported 2 (30 µM), or volume-equivalent of vehicle (DMSO) for 20 hours. Values represent averages of three experiments, determined in duplicate each ± SD. Statistical variation was assessed through one-way ANOVA with Sidak’s multiple comparison post test (***: P <0.001, **: P <0.01, NS not significant).
Scheme 1.

General synthesis of compounds 1a–1s and syntheses of compound 1t and 1u
aReagents and conditions (a) EDCI, HOBt, aniline, DMF, RT, 65%. (b) PhSH, Cs2CO3, CH3CN, RT, 50%. (c) EDCI, HOBt, R1COOH, DMF, RT, 22–87%. (d) 3-methyl-2-furaldehyde, NaBH(OAc)3, AcOH, DMA, 60°C, 13%. (e) furan-2-sulfonyl chloride, DIPEA, CH2Cl2, 0°C to RT, 58%
To furthermore assess consistency of the MOA profile, we employed reverse transcription qPCR as an independent alternative approach to minigenome assays to compare relative levels of viral mRNA synthesized by the RSV polymerase complex in the presence or absence of inhibitor as exemplified by reference compound 2 (Figure 4C). Consistent with the minireplicon data obtained with the sourced material, resynthesized 1a reduced the relative amount of RSV N protein-encoding mRNA in infected cells after 20-hour exposure to the compound. The inhibitory effect was dose-dependent, and statistically significant reductions were observed when 1a concentrations exceeded 1 µM. Taken together, these results demonstrate suitability of the recently developed pan-resistant recRSV-A2-L19FD489E-fireSMASh strain for automated drug discovery. The presence of the resistance mutation has shifted the hit pool of the drug discovery campaign described here towards inhibitors of the viral polymerase complex, illuminating that the pan-resistance mechanism apparently is challenging to overcome by small molecule entry inhibitors.
STRUCTURE-ACTIVITY RELATIONSHIP DEVELOPMENT
Upon selection of 1a as a starting point for hit-to-lead development, we sought to synthetically explore this scaffold with a primary goal of establishing a structure-activity relationship (SAR) as a platform for pharmacophore-informed lead development. In addition to maximizing antiviral potency and selectivity, a thorough understanding of the underlying SAR will be essential for meaningful attempts to synthetically address pharmacologic liabilities that may emerge during development. A second goal of the initial synthetic campaign was to eliminate potential metabolically labile groups wherever possible at the earliest stage of hit-to-lead development.
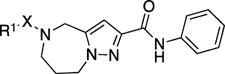
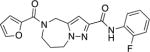
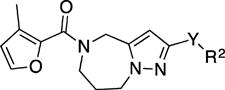
Before initiating in-house synthesis, we used the ChemNavigator similarity search application and SciFinder’s similarity search function25 to search for commercially available analogs of 1a. Disappointingly, however, the commercial source compounds showed little promise to yield interpretable information due to the presence of multiple scaffold modifications in each available analog. We therefore opted to launch a traditional SAR campaign that divided the scaffold into three specific regions (Figure 5, A–C) and concentrated synthetic efforts on probing each of these sections individually through the introduction of singular modifications. Bioactivity results of our serial query of each of these regions are summarized in Tables 1–3 for molecule section A, B, and C, respectively.
Figure 5.

SAR strategy of hit compound 1a.
Table 1.
RSV inhibitory (EC50) and cytotoxic (CC50) concentrations and selectivity indices of 1a and analogs featuring modifications of section A, tested on BEAS-2B human respiratory cells.a
| |||||
|---|---|---|---|---|---|
| Compound No. | R1 | X | EC50 [µM] | CC50 [µM] | SI |
| 1a |
|
C=O | 0.21 (0.14 to 0.3) | 8.3 | 40 |
|
| |||||
| 1b |
|
C=O | 7.1 (6.1 to undef) | ND | ND |
|
| |||||
| 1c |
|
C=O | >10 | ND | ND |
|
| |||||
| 1d |
|
C=O | >10 | ND | ND |
|
| |||||
| 1e |
|
C=O | >10 | ND | ND |
|
| |||||
| 1f |
|
C=O | 0.76 (0.63 to 0.9) | 30 | 39 |
|
| |||||
| 1g |
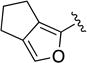
|
C=O | ~10 | 10 | ND |
|
| |||||
| 1h |
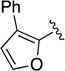
|
C=O | >10 | ND | ND |
|
| |||||
| 1i |
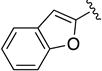
|
C=O | ~10 | ND | ND |
|
| |||||
| 1j |
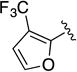
|
C=O | 5.4 (3.3 to 11.6) | 30 | 6.0 |
|
| |||||
| 1k |
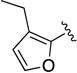
|
C=O | 0.73 (0.61 to 0.88) | 10 | 14 |
|
| |||||
| 1l |
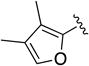
|
C=O | 2.25 (1.13 to 4.5) | 30 | 13 |
|
| |||||
| 1m |
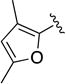
|
C=O | 0.96 (0.64 to 1.45) | 2.6 | 37 |
|
| |||||
| 1n |
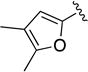
|
C=O | >10 | ND | ND |
|
| |||||
| 1o |
|
C=O | >10 | ND | ND |
|
| |||||
| 1p |
|
C=O | >10 | ND | ND |
|
| |||||
| 1q |
|
C=O | >10 | ND | ND |
|
| |||||
| 1r |
|
C=O | >10 | ND | ND |
|
| |||||
| 1s |
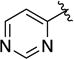
|
C=O | >10 | ND | ND |
|
| |||||
| 1t |
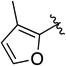
|
CH2 | >10 | ND | ND |
|
| |||||
| 1u |
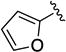
|
SO2 | >10 | ND | ND |
Highest concentration assessed for potency calculation was 10 µM, and for cytotoxicity assessment 90 µM. Active concentrations were calculated through four-parameter variable slope regression modeling, values in parentheses specify 95% confidence intervals.
ND: not determined.
Table 3.
RSV inhibitory (EC50) and cytotoxic (CC50) concentrations and selectivity indices (SI) of 1a analogs featuring modifications of section B or modifications of multiple sections.a
| Compound No. | Structure | EC50[µM] | CC50[µM] | SI |
|---|---|---|---|---|
| 9a |
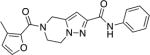
|
>10 | ND | ND |
| 9b |
|
3.19 (2.4 to 4.2) | >90 | >28 |
| 9c |
|
0.32 (0.21 to 0.5) | 3.7 | 12 |
Testing and analysis were carried out as specified for table 1.
As outlined in Scheme 1, coupling of compound 1026 with aniline, followed by removal of the o-nosyl protecting group with thiophenol and cesium carbonate provided amine 12. Coupling this compound with the corresponding carboxylic acids gave access to final compounds 1a–1s in low to good yields. Compound 1t was prepared via reductive amination of 3-methyl-2-furaldehyde with amine 12. The synthesis of compound 1u was achieved through reaction of amine 12 and furan-2-sulfonyl chloride under standard sulfonylation conditions.
Compounds 8a–8o were prepared according to the general synthesis outlined in Scheme 2. Amine 14 was prepared by removing of the o-nosyl group from compound 1326. Coupling of 14 and 3-methylfuran-2-carboxylic acid,27 followed by hydrolysis of the ethyl ester moiety with sodium hydroxide afforded carboxylic acid 16. Coupling 16 with the corresponding amines gave access to final compounds 8a–8o in low to moderate yields.
Scheme 2.

General synthesis of compounds 8a–8oa
aReagents and conditions (a) PhSH, Cs2CO3, CH3CN, RT, 66%. (b) EDCI, HOBt, 3-methylfuran-2-carboxylic acid, DMF, RT, 90%. (c) 3N NaOH, MeOH/THF, then aq. HCl, RT, 95%. (d) EDCI, HOBt, R3R4NH, DMF or HATU, DIPEA, DMF, RT, 12–74%.
As described in Scheme 3, conversion of carboxylic acid 16 into the corresponding Weinreb amide 17, followed by treatment with benzylmagnesium chloride provided compound 8p. Conversion of carboxylic acid 16 into a mixed carbonic acid anhydride, followed by reduction with sodium borohydride afforded alcohol 18.28 Treatment with mesyl chloride and displacement of the resulting mesylate group with aniline provided compound 8q.29
Scheme 3.

Syntheses of compound 8p and 8qa
aReagents and conditions (a) MeNH(OMe)•HCl, EDCI, Et3N, CH2Cl2, 41% (b) BnMgCl, THF, 0 °C to RT, 25%. (c) IBCF, NMM, THF, 0 °C, then aq. NaBH4, 0 °C, 41%. (d) MsCl, Et3N, THF, 0 °C. (e) aniline, Et3N, THF, 20% (2 steps).
Sulfonamide 8r was prepared in six steps from compound 1026. Carbamate 19 was prepared via a Curtius rearrangement starting from carboxylic acid 10. Boc deprotection provided amine 20, which in turn was converted into sulfonyl chloride 21 through a modified Sandmeyer reaction.30,31 Formation of sulfonanilide 22 under standard sulfonylation conditions, followed by deprotection of the o-nosyl group with thiophenol and cesium carbonate, provided amine 23. This compound was then coupled with 3-methylfuran-2-carboxylic acid to give final compound 8r.
Compounds 8s and 8t were prepared in three steps from compound 20. Treatment of this compound with benzoyl chloride and benzenesulfonyl chloride afforded the corresponding benzamide 24 and sulfonamide 26, which were treated with thiophenol and cesium carbonate to remove the o-nosyl groups. The resulting unprotected azepines 25 and 27 were coupled with 3-methylfuran-2-carboxylic acid to provide the final compounds 8s and 8t, respectively.
As shown in Schemes 6 and 7, compounds 9a and 9b were prepared in three steps from known compounds 2826,32 and 3126. Coupling with 3-methylfuran-2-carboxylic acid, followed by hydrolysis of the ethyl esters provided carboxylic acids 30 and 33, respectively. These compounds were coupled with aniline to provide the corresponding final compounds 9a and 9b.
Scheme 6.

Synthesis of compound 9aa
aReagents and conditions (a) EDCI, HOBt, 3-methylfuran-2-carboxylic acid, DMF, RT, 65%. (b) NaOH, H2O/MeOH/THF, then aq. HCl, RT. (c) EDCI, HOBt, aniline, DMF, RT, 54% (2 steps)
Scheme 7.

Synthesis of compound 9ba
aReagents and conditions (a) HATU, Et3N, 3-methylfuran-2-carboxylic acid, CH2Cl2, RT, 22%. (b) LiOH, H2O/MeOH/THF, then aq. HCl, RT. (c) HATU, DIPEA, aniline, DMF, RT, 15% (2 steps).
As shown in Scheme 8, compound 9c was prepared in a similar manner to that described above for compounds 1a–1s. Coupling of carboxylic acid 1026 with o-fluoroaniline afforded anilide 34. Removal of the o-nosyl group, followed by coupling with furan-2-carboxylic acid gave final compound 9c.
Scheme 8.

Synthesis of compound 9ca
aReagents and conditions (a) EDCI, HOBt, o-fluoroaniline, DMF, RT, 65%. (b) PhSH, Cs2CO3, CH3CN, RT, 55%. (c) EDCI, HOBt, furan-2-carboxylic acid, DMF, RT, 59%.
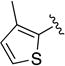
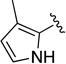
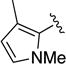
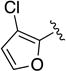
Given the known potential for metabolic liabilities associated with furan groups33, we first turned our attention to section A of the scaffold with a goal of finding a suitable alternative (see Table 1). Direct replacement with alternate heterocycles such as thiophene 1c, pyrroles 1d and 1e, oxazole 1q, isoxazole 1r, benzofuran 1i and 6-membered pyrimidine 1s, resulted in complete loss of potency. We therefore narrowed the scope of modification and concentrated on alteration of substituents on the furan ring that might offer increases in potency, selectivity index, and/or metabolic stability. We found, however, that this region of the molecule was very sensitive to manipulation, since all of these changes substantially reduced bioactivity. For example, removal of the furanyl-C3-methyl group (1b) resulted in a drastic loss of activity (EC50= 7.1 µM). Similarly, relocation of the methyl group to the 4-furanyl (1o) and 5-furanyl (1p) positions led to loss of activity, suggesting that substitution at the 3 position is required for activity. This was reinforced by the bioactivity results obtained for the 3,4-dimethyl (1l), 3,5-dimethyl (1m) and 4,5-dimethyl (1n) compounds, of which only 1n lacked antiviral activity. We next sought to define the scope of modification and functional diversity that could be accommodated at the C3 position. Replacement of the 3-methyl with a 3-chloro substituent, gave slightly less active 1f (EC50 = 0.76 µM and CC50 = 30 µM). We also replaced the methyl with phenyl (1h) and incorporated the C3 carbon into a fused5-membered ring (1g). However, the EC50 of 1g was reduced to approximately 10 µM and 1h lacked appreciable inhibitory activity. Finally, the trifluoromethyl (1j) and ethyl (1k) analogs both had negative impacts on the potency, revealing a very narrow tolerance range to C3 substitutions in section A. Turning our attention to the carboxamide group in section A, we obtained comparable results. We substituted the carbonyl bond for either CH2 (1t) or SO2 (1u), but in both cases the resulting compounds experienced a complete loss of bioactivity. Motivated by this limited window of opportunity for modifications in section A, we next examined section C of the scaffold.
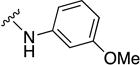
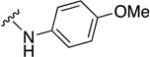
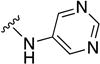
In parallel with our initial strategy towards probing section A, we again sampled the effect on bioactivity of different heteroaromatic rings, altered substitutions on the phenyl ring, and modified linkers other than the amide bond (Table 2). Two pyrimidine ring analogs were synthesized and tested (8m and 8o). However, 8m lacked inhibitory activity (EC50>10 µM) and 8o likewise experienced a drop in potency, albeit less pronounced than that seen for 8m (EC50 = 1.5 µM). We then substituted the aromatic ring with different groups on a variety of positions, such as o-F & p-F (8c), o-Me (8d), o-F (8g), o-OMe (8j), m-Me (8e), m-F (8h), m-OMe (8k), p-Me (8f), p-F (8i), p-OMe (8l) and o-Cl & p-F (8n). Several compounds of this panel showed improved inhibitory activity compared to hit compound 1a; for instance, compounds 8c (EC50 = 0.060 µM), 8g (EC50 = 0.050 µM) and 8n (EC50 = 0.060 µM) all returned an approximately 4-fold improvement in antiviral potency. The SI was acceptable (SI > 20) in most cases with some values well above 100; however, there was no apparent correlation with structure that allowed for predictive design around this parameter.
Table 2.
RSV inhibitory (EC50) and cytotoxic (CC50) concentrations and selectivity indices (SI) of 1a analogs featuring modifications of section C.a
| |||||
|---|---|---|---|---|---|
| Compound No. | Y | R2 | EC50 [µM] | CC50 [µM] | SI |
| 8a | C=O |
|
10 | >90 | >9 |
|
| |||||
| 8b | C=O |
|
1.2 (0.79 to 1.84) | 50 | 42 |
|
| |||||
| 8c | C=O |
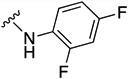
|
0.06 (0.04 to 0.1) | 5 | 83 |
|
| |||||
| 8d | C=O |
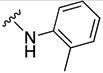
|
0.14 (0.08 to 0.2) | 1.1 | 8 |
|
| |||||
| 8e | C=O |
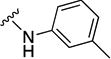
|
0.28 (0.21 to undef) | 3.3 | 12 |
|
| |||||
| 8f | C=O |
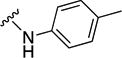
|
0.41 (0.35 to 0.51) | 30 | 73 |
|
| |||||
| 8g | C=O |
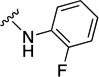
|
0.05 (0.03 to 0.08) | 1.1 | 22 |
|
| |||||
| 8h | C=O |
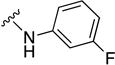
|
0.21 (0.15 to 0.3) | 30 | 143 |
|
| |||||
| 8i | C=O |
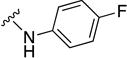
|
0.33 (0.26 to 0.4) | 90 | 273 |
|
| |||||
| 8j | C=O |
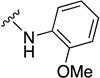
|
0.31 (0.16 to 0.6) | 3.3 | 11 |
|
| |||||
| 8k | C=O |
|
0.54 (0.17 to 1.7) | 30 | 56 |
|
| |||||
| 8l | C=O |
|
0.32 (0.26 to 0.4) | 90 | 281 |
|
| |||||
| 8m | C=O |
|
>10 | ND | ND |
|
| |||||
| 8n | C=O |
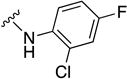
|
0.06 (0.002 to 1.7) | 30 | 500 |
|
| |||||
| 8o | C=O |
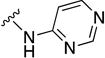
|
1.53 (0.71–3.33) | 16.6 | 11 |
|
| |||||
| 8p | C=O |
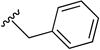
|
0.07 (0.06 to 0.09) | 5.5 | 79 |
|
| |||||
| 8q | CH2 |
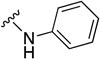
|
0.5 (0.39 to 0.6) | 10 | 20 |
|
| |||||
| 8r | SO2 |
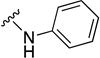
|
>10 | ND | ND |
|
| |||||
| 8s | NH |
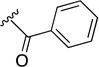
|
>10 | ND | ND |
|
| |||||
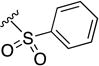
| 8t | NH |
|
>10 | ND | ND |
Testing and analysis were carried out as specified for table 1.
Encouraged by these promising results, we assessed the sensitivity of 8g to phase I liver oxidation by incubating them with mouse S9 hepatocyte subcellular fractions to determine if the metabolic stability was suitable to support efficacy testing in the mouse model of RSV infection15, 19. Unfortunately, the half-life of compound 8g co-incubated with mouse S9 fractions was only 11 min, and the compound hydrolyzed rapidly even in the absence of cofactors (Table 4 and Supporting Information). Interestingly, the half-life against human S9 liver fractions was much higher (7 h) suggesting that metabolic stability of this compound, and possibly others in the series, in humans may be sufficient to support advancement toward clinical develoment.
Table 4.
Aqueous solubility for selected compounds at various pH valuesa
| Compound No. |
Structure | Aq. Solubility (µg/mL) |
Metabolic Stability (t1/2) | |||||
|---|---|---|---|---|---|---|---|---|
| pH | Mouse | Human | ||||||
| 7.4 | 5.0 | 3.0 | liver S9 | plasma | liver micro somes |
plasm a |
||
| 1a |
|
63 | 75 | 74 | 15 min | - | 4.7 h | - |
| 8g |
|
30 | 51 | 75 | 11 min | 2.4 h | 5.3 h | >24 h |
| 8p |
|
18 | 18 | <15 | 20 min | - | 42 min | - |
| 8n |
|
4 minb | 3.6 h | |||||
| 8a |
|
58 | 25 | 20 | ||||
| 8j |
|
28 | 43 | 48 | ||||
Detailed solubility and metabolism procedures in Experimental Section
Tested against mouse liver microsomes
In order to develop a rational plan to address the observed metabolic liability in mice, we assessed the metabolites that arose from the exposure of 1a to mouse liver microsomes (see Supporting Information for detailed protocols and results). The half-life was 15 min both with and without the presence of cofactors. Given that amidases do not require cofactors for activity, amide hydrolysis was postulated to be the primary metabolic pathway. Analytical standards of the two most likely metabolites, (Scheme 1, compound 12; Scheme 2, compound 16) were then prepared, and 16 was the only compound identified as a metabolic product of 1a by mouse liver microsomes; thus, implicating the anilinic amide as the primary site for hydrolysis (amide bond b in Figure 6). The evidence also suggests that amide bond a is stable and not labile to hydrolytic cleavage.
Figure 6.

Amide bonds in compound 1a.
As a follow-up experiment, the anilinic nitrogen in 1a was replaced with a methylene to provide 8p and eliminate the possibility of amide hydrolysis of amide bond b The calculated half-life remained short (20 min) in the presence of cofactors, but the parent remained intact after 2 hours when incubated in the presence of mouse liver microsomes without cofactors. CYP-mediated oxidative metabolism was therefore implicated as a competing metabolic pathway. We then incubated 8p at a higher concentration (80 µM) in order to elucidate the primary metabolites and determine potential sites for oxidative metabolism. Proposed metabolites and biotransformations are listed in Table 5 (see Supporting Information for a detailed rationale for this analysis). The core ring system and the phenyl ring were implicated as primary sites for oxidative metabolism, and much of our SAR strategy was accordingly directed toward modifications at these sites.
Table 5.
Major metabolites of 8p by mouse liver microsomes and proposed biotransformationsa
| Metabolit e |
Retention Time (min) |
m/z (Δ Da) | Proposed Biotransformation | Proposed Site of Biotransformation |
|---|---|---|---|---|
| M1 | 7.0 | 556 (+192) | Glucuronide of M3 | Phenyl ring hydroxyl |
| M2 | 9.7 | 396 (+32) | (di)hydroxylation/N-oxide(s) | Pyrazole ring |
| M3 | 10.1 | 380 (+16) | Monohydroxylation | Phenyl ring |
| M4 | 10.6 | 380 (+16) | Monohydroxylation or N-oxide | Pyrazole ring |
| Parent | 12.9 | 364 (MH+) | Parent | N/A |
Detailed procedure in Supporting Information
Two additional analogs were designed specifically to substitute the amide bond b for a metabolically more stable alternative. This effort produced compounds that feature a CH2 substitution (8q), and an SO2 replacement (8r). Encouragingly, compounds 8p and, albeit to a lesser degree, 8q maintained antiviral activity (EC50= 0.070 µM and 0.5 µM, respectively). Despite the successful replacement of amide bond b, however, compound 8p yielded only a 2-fold improvement in half-life (20 min) compared to 8g when incubated with mouse S9 subcellular fractions. Several other compounds were assessed with modifications on the aromatic ring, but none were successful in addressing the high rate of metabolism by mouse liver S9 fractions, likely rendering them unsuitable for use in mouse models of RSV infection. However, the longer half-life in human liver S9 fractions and the acceptable aqueous solubility observed with several compounds tested (Table 4), supports further investigation with this series for possible advancement toward use in humans.
Our third target for SAR development was section B of the hit scaffold, the central 5–7 fused ring system (Table 3). To explore amenability of this section to substitution, we altered the size and nature of the system. Replacing the 7-membered with a 6-membered ring (compound 9a) completely eliminated antiviral activity (EC50 > 10 µM). In addition, substitution of the pyrazole ring for a pyrrole moiety (compound 9b) reduced potency, although modest antiviral activity was still observed (EC50 = 3.19 µM).
Several representatives of each analogs set were tested for aqueous solubility (Table 4) including 1a, 8g, 8p and 8n. Poor solubility at concentrations exceeding approximately 50 µM interfered with cytotoxicity testing in several cases. The solubility showed minimal dependence on pH. We did not focus on aqueous solubility as a selection criterion because the observed solubilities are on the lower end of what are typically considered acceptable. However, we recognize that detailed preformulation studies may be required before compounds of this type can be advanced in development.
QSAR MODEL BUILDING
To extract a graphical summary of the nascent SAR that may serve as a framework for future chemical development of the scaffold, we generated 3D-QSAR models using the AutoGPA module34 embedded in the MOE software package35.
Ranking of 147 automatically generated initial pharmacophore models identified a superior representation that showed acceptable predictive capacity (q2 = 0.7552) when evaluated against a test set of six compounds selected from the pool of bioactives (Figure 7). This top-scoring modelfully reflected our initial impression that both the central region B and in particular section A of the scaffold are sterically highly controlled and unaccepting of bulkier substitutions. The predicted allowable space contour map is most relaxed around section C, which accurately represents our experience that a number of analogs of the second series demonstrate consistent or improved inhibitory activity. Highlighted by the red and blue contours in the model, the presence of electron donating groups in section A and electron acceptance by the C region is predicted to be a key feature of ligand docking.
Figure 7.

Pharmacophore modeling. (A) AutoGPA-based 3D-QSAR model, shown from two rotational angels. Orange and green spheres represent aromatic and hydrophobic centers, respectively. The allowable space model is shown in grey, and electrostatically active portions are in red and blue, respectively. Areas of steric constrain (yellow) and freedom (solid green) are marked. (B) Correlation between the predicted and observed pIC50 values for the training (19 compounds) and test (6 compounds) sets. Model statistics are shown in the graph.
DISCUSSION AND CONCLUSIONS
In this study, we have implemented an open screening campaign against a novel RSV reporter virus carrying a signature pan-resistance mutation that mediates viral escape from known RSV entry inhibitors. A panel of automated direct and orthogonal counterscreens centered on recombinant RSV strains with distinct reporter technology expedited the identification of 14 confirmed hits, corresponding to an overall hit discovery rate in this campaign of approximately 0.028%. Mechanistic characterization through minigenome reporter assays monitoring RSV RdRp activity suggested that all of these confirmed compounds interfere with activity of the RSV polymerase machinery. In the case of 1a, this observation was corroborated by quantitation of the relative viral N mRNA level in virus infected cells. Although this particular screen was limited to a moderate size library of 50,640 compounds, an entirely RdRp-centric hit activity profile is in our experience unusual for drug screens against RSV since small-molecule entry inhibitors emerge readily in anti-RSV campaigns. Based on the available dataset alone, we cannot definitely address whether or not it may be feasible in principle to overcome RSV pan-resistance through a new generation of small molecule entry blockers. However, the noticeable absence of this usually prominent hit class from our pool of confirmed candidates indicates that the threshold against efficient inhibition of the entry machinery of this mutant RSV strain must be considerably increased. This finding is consistent with the notion that pan-resistance likely represents a composite phenotype, arising from a blend of primary resistance and kinetic, secondary escape from entry inhibition15, 36. It remains to be seen whether structure-guided optimization of currently existing, pan-resistance-sensitive scaffolds will illuminate a path towards overcoming viral escape, but the outcome of our HTS campaign provides little encouragement. Ultimately, therapeutic targeting of the viral RdRp complex may represent the more fruitful strategy towards developing anti-RSV therapeutics with a lasting clinical impact.
Compound 1a was selected for SAR development based on initial potency, absence of obvious chemical liabilities, and potential for chemical elaboration. Although resistance profiles have not yet been generated for this compound class, quantitation of relative viral RNA levels synthesized in the presence of compound in a one-step replication cycle confirmed inhibition of RdRp complex activity. The 3D-QSAR model best summarizes the insight gained from our initial chemical elaboration efforts. Without knowledge of the binding site or docking pose of the hit compound, it was the primary goal of the initial synthetic efforts to establish a basic framework of the underlying structure-activity relationship (SAR) profile. Consequently, synthesis was directed towards appreciating the basic impact of structural modifications on antiviral potency and cytotoxicity. Three major conclusions can be drawn from the resulting dataset:
The section A portion of the molecule, bearing the furanyl amide, presented highly sensitive to modification. A diverse set of substitutions were made that contributed modest electronic and/or steric alterations, and all resulted in moderate to catastrophic losses in antiviral potency, suggesting that this region likely stands in close-range interaction with the RdRp target. Fortunately, data generated from in vitro metabolism studies showed that this portion of the molecule was not sensitive to either amide hydrolysis or oxidative metabolism. We therefore refrained from further modification of this substructure and turned our attention to section B, bearing the phenyl amide moiety, which was found sensitive to both types of metabolic attack.
Importantly, the central amide linker presented as partially amenable to modification, providing a basis to probe its contribution to metabolic instability. While several modifications designed to address hydrolytic cleavage of the amide linker resulted in loss of activity including N-methylation (8a), inversion of the amide group (8s) and isosteric substitution as a sulfonamide (8r and 8t), others retained sufficient activity to remain viable as potential tools for the development of more metabolically stable analogs. These included conversion of the amide to a ketone (8p) and alkylamine (8q). However, a detailed analysis of the metabolites emerging from the exposure of 8p to mouse microsomes revealed that even though the hydrolytic cleavage issue had been circumvented, oxidative metabolism was occurring both on the core ring structure and on the aromatic ring. Initial attempts to modify the core bicyclic ring structure compromised antiviral potency.
Substitutions on the aromatic ring were well-tolerated, indicating a considerable degree of structural freedom at this position, which argues against a direct engagement of the target by this substructure. Accordingly, we explored several iterations at the ortho- and para-positions in order to block the most common sites of oxidative metabolism. This exercise yielded compounds 8i, 8l and 8n, which revealed that both positions accept substituents that can block oxidative metabolism, retain nanomolar potency, and provide acceptable SI’s.
Based on this QSAR-driven insight, we intend to next launch a comprehensive lead optimization program to identify an analog that is suitable for efficacy testing in the mouse model of RSV infection before ultimate advancing to formal development.
EXPERIMENTAL SECTION
CELLS AND VIRUSES
Human carcinoma (HEp-2, ATCC CCL-23), human bronchial epithelial (BEAS-2B, ATCC CRL-9609), human embryonic kidney (293T, ATCC CRL-3216), and Madin Darby canine kidney (MDCK, ATCC CCL-34) cells were maintained at 37°C and 5% CO2 in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 7.5% fetal bovine serum. Cell lines were obtained directly from the ATCC. Cell lines are replaced with cryo-preserved low-passage stocks after approximately 20–30 passages in culture. All cell lines in culture in the laboratory are routinely screened for mycoplasma contaminations on a biannual basis. GeneJuice (EMD) was used for all transient transfection reactions. IAV stocks were grown on MDCK cells, while all RSV stocks were propagated on HEp-2 cells inoculated at a multiplicity of infection (MOI) of 0.01 pfu/cell. Infected cells were kept for 16 hours at 37°C, followed by incubation at 32°C for five to seven days. Cell-associated progeny virus was released through one freeze/thaw cycle and titers determined by TCID50 titration on HEp-2 cells. Asunaprevir (ASV) was obtained from Santa Cruz Biotechnology. When included, ASV was added at the time of virus infection. For purification of virus screening stocks, progeny virions in culture supernatants (IAV stocks) or released through one freeze/thaw cycle from infected cells (RSV stocks) were cleared (4,000×g for 20 minutes at 4°C), then pelleted (60,000×g for 30 minutes at 4°C). The pelleted material was resuspended in TNE buffer (50 mM Tris/Cl pH 7.2, 10 mM EDTA) and purified through a 20/60% one-step sucrose gradient in TNE buffer (100,000×g for 90 minutes at 4°C). Virions were harvested from the gradient intersection.
HIGH-THROUGHPUT SCREENING
Screening libraries were sourced from the ChemBridge collections and dissolved in DMSO to 10 mM concentration in 96-well master plates and stored at −80°C. The MScreen software package21 was used for electronic compound management, HTS data storage and data analysis. Inventoried libraries were reformatted into barcoded 384-well daughter plates using a Nimbus liquid handler (Hamilton Robotics) with multichannel pipetting head. Sixty-four wells on each 384-well stock plate were reserved for positive and negative controls, located in the outermost columns and arranged in a checkerboard pattern. For screening, BEAS-2B cells (3.5 × 103/well) were injected in 30 µL/well into barcoded white wall/clear bottom 384-well plates using a MultiFlo automated dispenser (BioTek) equipped with dual 10-µL peristaltic pump manifolds, collected (150×g for 90 seconds at 25°C), and incubated for 14 hours at 37°C and 5% CO2. Compound was added to a final concentration of 5 µM (20 nL/well) using a high-density pin tool (V&P Scientific) attached to the pipetting head of the Nimbus liquid handler, followed by co-infection with recRSV A2-L19FD489E-fireSMASh (MOI = 0.1) and recIAV WSN-NanoLuc (MOI = 0.02) in 10 µL/well using the MultiFlo dispenser unit, spin collection (150×g for 90 seconds at 25°C), and incubation for 40 hours at 37°C and 5% CO2. Final vehicle (DMSO) concentration was 0.05%. Barcodes of source and assay plates were automatically detected and recorded by the Nimbus unit at the time of stamping. Using a stacker unit with integrated barcode reader (Biotek) attached to an H1 synergy plate reader, plates were automatically loaded, dual-Glo substrates (15 µL/well each) injected, and bioluminescence recorded after a three minute lag time for each well and substrate. Readouts were automatically saved by plate barcode and imported into the MScreen IT environment.
DATA ANALYSIS AND HIT IDENTIFICATION
Normalized relative values were automatically calculated in the MScreen package by subtracting from each value the average of the plate vehicle controls, followed by dividing the results by the difference between the means of plate vehicle positive and negative controls. Robust z-scores were calculated as follows: and MAD (Sall) = 1.4826 × median (|Si − median (Sall)|). Hit candidates were defined as compounds showing ≥75% inhibition of normalized signal intensity against either viral target or both and robust z-score ≥4.5.
COUNTERSCREENING CAMPAIGNS
For single concentrations direct counter- and cytotoxicity screens, hit candidates were automatically picked into a single 384-well plate and stamped against recRSV-L19FD489E-fireSMASh grown on BEAS-2B cells. Reporter signals were recorded as outlined above, but only control well-dependent % inhibition calculated due to the high number of positives present on the confirmation plates. All confirmation plates were tested twice in independent repeats. To determine cell viability, PrestoBlue substrate (life technologies) was added after 48 hours of incubation of uninfected but compound treated cells at 37°C (5 µL/well) and top-read fluorescence (excitation at 560 nm, emission at 590 nm, instrument gain of 85) recorded after incubation for 90 min at 37°C using the H1 synergy plate reader. For dose-response counterscreens, serial 3-fold compound dilutions were prepared in three repeats in 96-well plates using the Nimbus liquid handler. BEAS-2B cells (1.5×104 cells/well) were plated in 96-well plates, serial dilutions transferred to the cell plates using the liquid handler, and cells infected with recRSV-A2-L19FD489E-fireSMASh (MOI = 0.1) or recRSV-A2-L19F-renilla (MOI = 0.1). Each plate contained negative and positive control wells in four replicates each, and raw data of all dose-response screens were analyzed according to the formula % inhibition = (XSample−XMin)/(XMax−XMin)×100 with XMin representing the average of the positive and XMax the average of the negative control wells. Four-parameter variable slope regression was applied to determine 50% active (EC50) concentrations. For virus yield assays, cells were infected in a 12-well plate format with recRSV-A2-L19F-mKate expressing a far-red fluorescent protein37 at an MOI of 0.05 particles/cell in the presence of serial compound dilutions and incubated at 37°C. Cell-associated progeny virions were harvested 48 hours post-infection, released as described, and virus titers in each sample determined through TCID50 titration.
MINIGENOME REPORTER ASSAYS
For minireplicon assays, an RSV firefly luciferase minigenome construct under the control of the constitutive RNA pol I promoter (pHH-RSV-repl-firefly) was used that we have previously described15. 293T cells were co-transfected with this minigenome and plasmids pRSV-L, pRSV-M2-1, pRSV-N and pRSV-P, respectively, under CMV promoter control. Test compounds were added in serial dilutions and luciferase reporter activities determined 40–44 hours post-transfection.
REVERSE TRANSCRIPTION QPCR
Cells were infected with recRSV-A2-L19F-mKate (MOI = 3 particles/cell) and incubated in the presence of different 1a concentrations ranging from 0.1 to 10 µM, 30 µM of the nucleoside-analog RSV RdRp inhibitor 213, or vehicle (DMSO) for control at 37°C. Twenty hours post-infection, total RNA was prepared from all wells using a QIAcube automated extractor and the RNeasy Mini Kit (Qiagen), and subjected to reverse transcription using Superscript III Reverse Transcriptase and oligo-dT primer of first strand synthesis. Real-time reactions were carried out using an Applied Biosystems 7500 Fast real-time PCR system, PowerUp Sybr Green Master mix (Thermo-Fisher scientific), and primer pairs specific for a fragment in the RSV N open reading frame or human GAPDH, respectively. Melting curves were generated for each primer pair to verify amplification of a single product. To calculate ΔΔCT values, CT values obtained for each sample were normalized for GAPDH as reference and then ΔCT values of inhibitor treated samples normalized for the DMSO-treated controls. Final quantification was based on three independent experiments in which each treatment condition and RT primer setting were assessed in duplicate.
QSAR MODEL BUILDING
All energy minimization, conformation searches, and model building were performed by MOE 2015.1035. The AutoGPA module34 embedded in MOE was used to develop 3D-QSAR models. For model building, 25 structures were chosen that showed various inhibitory activities, ranging in EC50 concentration from 0.01 to 30 µM. This panel was divided into training set (19 entries compounds 1a, 1c, 1f, 1g, 1l, 1m, 8a, 8b, 8c, 8d, 8f, 8g, 8h, 8i, 8j, 8k, 8l, 8o, and 8q) and a test set (6 entries; compounds 1b, 8e, 8n, 8p, 9b, and 9c). Prior to input, EC50 values were converted to pIC50 (−log10(EC50)), compounds then drawn and energy minimized under the Merck molecular force field 94X (MMFF94X). Conformational alternatives of each compound were generated using the conformational search function of the AutoGPA package. A total of 983 and 351 conformations were generated for the training and test sets, respectively, which were then aligned and assigned pharmacophore properties. AutoGPA identified common features and created 147 initial pharmacophore models based on the training set. Employing the CoMFA algorithm38, all molecules were placed in a 3D grid box with 2Å separation, and electrostatic and steric interaction energies between each molecule were calculated. An sp3 carbon atom probe evaluated the allowable space and molecular field surrounding the compounds. Subsequently, a partial-least squares analysis was performed to analyze the relationship between grid potential fields and inhibitory activity to create the AutoGPA-based 3D-QSAR. The test set subsequently provided analogs with known bioactivity to evaluate the predictive capacity of the model.
STATISTICAL ANALYSIS
The MScreen, Excel and Prism 7 (GraphPad) software packages were used for data analysis. To determine antiviral potency, EC50 concentrations were determined from dose-response data sets through four parameter variably slope regression modeling using the Prism package. The statistical significance of differences between groups was assessed through one-way analysis of variance (ANOVA) in combination with Sidak’s multiple comparison post-test. Experimental uncertainties are identified by error bars, representing standard deviations (SD) or standard error of the mean (SEM) as specified in figure legends.
PROCEDURE FOR AQUEOUS SOLUBILITY
The compounds and an internal control were dissolved in 100% DMSO to obtain a stock concentration of 30 mg/mL. The 30 mg/mL stock solution was serially diluted (concentration profile: 30, 20, 15, 10, 7.5, 5, 2.5, 1.25, 0.63, 0.31, and 0.15 mg/mL) in test tubes with 100% DMSO. The concentration profile was transferred to 96 well microplates and serially diluted to a final DMSO (EMD) concentration of 1% and a final drug concentration 300, 200, 150, 100, 75, 50, 25, 12, 6, 3 and 1.5 µg/mL with phosphate buffered saline, pH 7.4, (Sigma). Compounds that are pH dependent were assayed at pH 3 and 5 in addition to pH 7.4. Citrate buffer was the diluent used for pH 3 and 5. Microplates were incubated for 90 minutes at ambient temperature. Laser nephelometry (NEPHELOstar, BMG Lab Technologies), measuring forward scattered light, was used to determine the point at which the solute began to precipitate out of solution.
METABOLIC STABILITY STUDY
Compounds were incubated in triplicate with mouse or human S9 hepatocyte subcellular fractions or microsomes (XenoTech/BioreclamationIVT, 0.5 mg total protein) at final concentrations of 1 µM for metabolic stability or 80 µM for metabolite ID, with the necessary cofactors for Phase I oxidations and Phase II glucuronide conjugation. Incubations were performed in 13 × 100 mm glass culture tubes. Samples were placed in a water bath shaker set at 37°C and shaken at 150 rpm. Positive controls consisted of 1 µM verapamil (Phase I) and 175 µM 7-hydroxycoumarin (Phase II) and were run in parallel to test system for competency. Negative controls (without cofactors) were also run to access non-cytochrome-P450 metabolism. A negative control containing no microsomes was run to assess chemical stability. Aliquots of 100 µL were taken at the following time-points: 0, 15, 30, 60 and 120 minutes; and only at 0 and 120 minutes for the negative controls. These aliquots were mixed with 400 µL of a suitable organic internal standard solution (ISTD) in 1.7 mL conical polypropylene microcentrifuge tubes. Sub-samples plus ISTD from each time point were vortexed for about 10 seconds and then centrifuged for 2 minutes at 13,000 rpm. Supernatants were decanted into 2 mL LC vials and analyzed by LC-MS full-scan and/or LC-MS. For metabolite ID samples, after 2 hours of incubation the whole sample was quenched with acidified acetonitrile and the organic solvent was evaporated under a gentle nitrogen stream prior analysis. Half-life (t1/2) was estimated by plotting the natural logarithm of % parent vs. time and obtaining the slope of the line. Assuming first-order kinetics, the elimination rate constant, k, is the negative (−) of the slope of the plot (ln % parent vs. time). Half-life (t1/2) (min) = −0.693/ (slope).
CHEMISTRY
Unless otherwise noted, all materials were obtained from commercial suppliers and used without purification. Dry organic solvents, packaged under nitrogen in septum sealed bottles, were purchased from EMD Millipore and Sigma-Aldrich Co. Reactions were monitored using EMD silica gel 60 F254 TLC plates or using an Agilent 1200 series HPLC system with a diode array detector and an Agilent 6120 quadrupole MS detector. Compound purification was accomplished by liquid chromatography on a Teledyne Isco CombiFlash RF+ flash chromatography system. 1H NMR and 19F spectra were recorded on an Agilent NMR spectrometer (400 MHz) at room temperature. Chemical shifts are reported in ppm relative to CDCl3 (7.26) or CD3OD (3.31). The residual shifts were taken as internal references and reported in parts per million (ppm). All target compounds in this manuscript appear as two or more distinct rotamers on the NMR time scale; accordingly, the 13C NMR spectra were complex and not interpretable. Data was therefore not included. Purities of all compounds were determined by high-performance liquid chromatography (HPLC), using an Agilent 1200 HPLC system with a Zorbax Eclipse XDB C18 column (4.6 × 50 mm, 3.5 µm) at 254 nm, and 0.1% formic acid in MeOH (solvent A) and 0.1% formic acid in water (solvent B) solvent mixtures, at 1 mL/min. Method A: 5 min, 60% solvent A/40% solvent B to 95% solvent A/5% solvent B; Method B: 8 min, 60% solvent A/40% solvent B to 95% solvent A/5% solvent B; Method C: 5 min, 70% solvent A/30% solvent B to 95% solvent A/5% solvent B. The HRMS experiments were performed with a Thermo LTQ-FTMS, using a nanospray source.
5-((2-Nitrophenyl)sulfonyl)-N-phenyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (11)
To a solution of HOBt (367 mg, 2.72 mmol), EDCI (521 mg, 2.72 mmol) and aniline (207 µL, 2.27 mmol) in DMF (11 mL) was added 5-((2-nitrophenyl)sulfonyl)-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxylic acid (10)26(830 mg, 2.27 mmol) in one portion. The reaction was stirred for 72 h, diluted with water (100 mL), and the aqueous phase was extracted with EtOAc (3 × 75 mL). The combined organic phases were washed with brine (50 mL), dried (Na2SO4), filtered and concentrated. The residue was purified via flash chromatography (SiO2, 25 to 70% EtOAc in hexanes) to give 11 (654 mg, 1.48 mmol, 65% yield) as a light yellow solid. 1H NMR (400 MHz, CDCl3) δ 8.58 (s, 1H), 8.03-7.96 (m, 1H), 7.74-7.59 (m, 5H), 7.40-7.32 (m, 2H), 7.15-7.07 (m, 1H), 6.81 (s, 1H), 4.60 (s, 2H), 4.49-4.43 (m, 2H), 3.80-3.72 (m, 2H), 2.17-2.05 (m, 2H). MS (ESI) [M+H]+: 442.0.
N-phenyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (12)
To a suspension of 5-((2-nitrophenyl)sulfonyl)-N-phenyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide 11 (0.87 g, 1.971 mmol) and cesium carbonate (1.284 g, 3.94 mmol) in acetonitrile (20 mL) was added a solution of thiophenol (0.405 mL, 3.94 mmol) in acetonitrile (1 mL). The reaction was stirred at RT for 4h, and concentrated under vacuum. The residue was suspended in CH2Cl2, filtered through a cotton plug and purified via flash chromatography (SiO2, 0 to 4% MeOH in CH2Cl2, Rf = 0.25, 5% MeOH in CH2Cl2) to give 12 (250 mg, 0.975 mmol, 50% yield) as a white crystalline solid. 1H NMR (400 MHz, CDCl3) δ 8.66 (s, 1H), 7.69-7.64 (m, 2H), 7.37-7.30 (m, 2H), 7.12-7.06 (m, 1H), 6.66 (s, 1H), 4.45-4.35 (m, 2H), 3.92 (s, 2H), 3.24-3.17 (m, 2H), 1.92-1.81 (m, 2H). MS (ESI) [M+H]+: 257.1.
Synthesis of 5-(3-methylfuran-2-carbonyl)-N-phenyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (1a)
To a solution of HOBt (89 mg, 0.66 mmol), EDCI (126 mg, 0.66 mmol) and N-phenyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (12) (140 mg, 0.55 mmol) in DMF (5.4 mL) was added 3-methylfuran-2-carboxylic acid (69 mg, 0.55 mmol) in one portion. The reaction was stirred for 24 h. The reaction mixture was partitioned between EtOAc (50 mL) and water (50 mL) and the aqueous phase was extracted with EtOAc (2 × 50 mL). The combined organic phases were washed with brine (50 mL), dried (Na2SO4), filtered and purified via flash chromatography (SiO2, 40 to 60% EtOAc in hexanes, Rf = 0.2, 60% EtOAc in hexanes) to give 1a (111 mg, 0.30 mmol, 56% yield) as a white solid. 1H NMR (400 MHz, CDCl3) δ 8.64 (s, 1H), 7.66 (d, J = 7.8 Hz, 2H), 7.41-7.30 (br s, 1H, overlapped), 7.34 (t, J = 7.9 Hz, 2H), 7.10 (t, J = 7.4 Hz, 1H), 6.77 (br s, 1H), 6.34 (d, 1H), 4.84 (s, 2H), 4.55-4.45 (m, 2H), 4.05-3.93 (m, 2H), 2.27 (s, 3H), 2.13 (br s, 2H). HRMS–ESI (m/z): [M + H]+ calcd for C20H21N4O3, 365.16082; found, 365.16077. HPLC purity: 98.2% (method A).
Synthesis of compounds 1b–1s
Compounds 1b–1s were prepared by employing a similar procedure to that described for compound 1a, with N-phenyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (12) and the corresponding carboxylic acid.
5-(Furan-2-carbonyl)-N-phenyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (1b)
Compound 1b was obtained as a white solid in 66% yield. 1H NMR (400 MHz, CDCl3) δ 8.63 (s, 1H), 7.66 (m, 2H), 7.54 (br s, 1H), 7.37-7.31 (m, 2H), 7.14-7.08 (m, 2H), 6.81 (br s, 1H), 6.50 (dd, J = 3.5, 1.8 Hz, 1H), 4.93 (br s, 2H), 4.57-4.47 (m, 2H), 4.07 (br s, 2H), 2.19-2.09 (br m, 2H). HRMS–ESI (m/z): [M + H]+ calcd for C19H19N4O3, 351.14517; found, 351.14511. HPLC purity: 96.9% (method A).
5-(3-Methylthiophene-2-carbonyl)-N-phenyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (1c)
Compound 1c was obtained as a white solid in 57% yield. 1H NMR (400 MHz, CDCl3) δ 8.63 (s, 1H), 7.74-7.62 (m, 2H), 7.39-7.31 (m, 2H), 7.28 (d, J = 5.0 Hz, 1H), 7.15-7.07 (m, 1H), 6.85 (d, J = 5.0 Hz, 1H), 6.67 (br s, 1H), 4.68 (br s, 2H), 4.55-4.47 (m, 2H), 3.94 (br s, 2H), 2.15 (s, 3H), 2.13-2.01 (br m, 2H). HRMS–ESI (m/z): [M + H]+ calcd for C19H19N4O3, 351.14517; found, 351.14511. HPLC purity: 98.8% (method A).
5-(3-Methyl-1H-pyrrole-2-carbonyl)-N-phenyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (1d)
Compound 1d was obtained as a white solid in 87% yield. 1H NMR (400 MHz, CD3OD/CDCl3 1:1) δ 7.67-7.59 (m, 2H), 7.37-7.27 (m, 2H), 7.13-7.07 (m, 1H), 6.74 (d, J = 2.6 Hz, 1H), 6.64 (br s, 1H), 6.00-5.97 (m, 1H), 4.72 (br s, 2H, overlapped with HOD signal), 4.57-4.48 (m, 1H), 3.94 (t, J = 5.4 Hz, 2H), 2.10 (s, 3H), 2.06-1.98 (m, 2H). HRMS–ESI (m/z): [M + H]+ calcd for C19H22N5O2, 364.17680; found, 364.17609. HPLC purity: 95.7% (method B).
5-(1,3-Dimethyl-1H-pyrrole-2-carbonyl)-N-phenyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (1e)
Compound 1e was obtained as a white solid in 68% yield. 1H NMR (400 MHz, CDCl3) δ 8.62 (s, 1H), 7.66 (d, J = 7.9 Hz, 2H), 7.34 (t, J = 7.6 Hz, 3H), 7.10 (t, J = 7.2 Hz, 2H), 6.56 (s, 1H), 5.91 (s, 1H), 4.95 (br s, 1H), 4.68-4.14 (br m, 4H), 3.34 (s, 3H), 2.16-1.85 (br m, 2H, overlapped), 2.06 (s, 3H). HRMS–ESI (m/z): [M + Na]+ calcd for C21H23N5O2Na, 400.17440; found, 400.17435. HPLC purity: 99.2% (method A).
5-(3-Chlorofuran-2-carbonyl)-N-phenyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (1f)
Compound 1f was obtained as a white solid in 62% yield. 1H NMR (400 MHz, CDCl3) δ 8.63 (s, 1H), 7.70-7.63 (m, 2H), 7.52-7.39 (br m, 1H), 7.34 (t, J = 7.9 Hz, 2H), 7.10 (t, J = 7.4 Hz, 1H), 6.95-6.58 (br m, 1H), 6.51 (br s, 1H), 4.78 (s, 2H), 4.54-4.46 (m, 2H), 3.97 (br s, 2H), 2.16-2.08 (br m, 2H). HRMS–ESI (m/z): [M + H]+ calcd for C19H18N4O3Cl, 385.10619; found, 385.10609. HPLC purity: 98.6% (method A).
5-(5,6-Dihydro-4H-cyclopenta[c]furan-1-carbonyl)-N-phenyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (1g)
Compound 1g was obtained as a white solid in 59% yield. 1H NMR (400 MHz, CDCl3) δ 8.62 (s, 1H), 7.75-7.58 (m, 2H), 7.38-7.28 (m, 2H), 7.17-7.04 (m, 2H), 6.81 (br s, 1H), 4.96 (br s, 2H), 4.60-4.42 (m, 2H), 4.07 (br s, 2H), 2.82 (t, J = 7.3 Hz, 2H), 2.62 (td, J = 7.3, 1.4 Hz, 2H), 2.36 (p, J = 7.4 Hz, 2H), 2.16-2.06 (br m, 2H). HRMS–ESI (m/z): [M + H]+ calcd for C22H23N4O3Cl, 391.17647; found, 391.17641. HPLC purity: 99.2% (method A).
N-phenyl-5-(3-phenylfuran-2-carbonyl)-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (1h)
Compound 1h was obtained as a white solid in 86% yield. 1H NMR (400 MHz, CDCl3) δ 8.59 (br s, 1H), 7.67 (br d, J = 7.9 Hz, 2H), 7.51 (br s, 1H), 7.42 (d, J = 7.5 Hz, 2H ), 7.36-7.20 (m, 5H), 7.15-7.06 (m, 1H), 6.91 (br s, 0.5 H), 6.66 (br s, 1H), 6.45 (br s, 0.5H), 4.81 (br s, 1H), 4.49 (br s, overlapped, 1H), 4.42 (br s, 2H), 3.99 (br s, 1H), 3.63 (br s, 1H), 2.12 (br s, 1H), 1.78 (br s, 1H), 1.62 (s, 3H). MS (ESI) [M+H]+: HRMS–ESI (m/z): [M + H]+ calcd for C25H23N4O3, 427.17647; found, 427.17677. HPLC purity: 98.9% (method A).
5-(3-Methylbenzofuran-2-carbonyl)-N-phenyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (1i)
Compound 1i was obtained as a white solid in 79% yield. 1H NMR (400 MHz, CDCl3) δ 8.64 (br s, 1H), 7.70-7.65 (m, 2H), 7.63-7.58 (m, 1H), 7.48-7.38 (br m, 1H), 7.38-7.28 (m, 3H), 7.14-7.08 (m, 1H), 6.98-6.69 (br m, 1H), 4.87 (s, 2H), 4.60-4.50 (m, 2H), 4.13-3.99 (br m, 2H), 2.46 (s, 3H), 2.25-2.15 (br m, 2H). HRMS–ESI (m/z): [M + Na]+ calcd for C24H22N4O3Na, 437.15841; found, 437.15927. HPLC purity: 98.6% (method C).
N-phenyl-5-(3-(trifluoromethyl)furan-2-carbonyl)-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (1j)
Compound 1j was obtained as a white solid in 22% yield. 1H NMR (400 MHz, CDCl3) δ 8.62 (br s, 1H), 7.70-7.62 (m, 2H), 7.55 (br s, 0.5H, overlapped), 7.46 (br s, 0.5H, overlapped), 7.40-7.28 (m, 2H), 7.15-7.06 (m, 1H), 6.90 (br s, 0.5H), 6.71 (br s, 0.5H, overlapped), 6.67 (br s, 0.5H, overlapped), 6.58 (br s, 0.5H), 4.82 (br s, 1H), 4.60 (br s, 1H), 4.55-4.46 (m, 2H), 4.03 (br s, 1H), 3.74 (br s, 1H), 2.12 (br s, 2H). 19F NMR (376 MHz, CDCl3) δ −58.3 (s), −58.4 (s). Note: The1H and 19F NMR spectra reveal the presence of two rotamers. HRMS–ESI (m/z): [M + H]+ calcd for C20H18N4O3F3, 419.13255; found, 419.13254. HPLC purity: 89.0% (method B).
5-(3-Ethylfuran-2-carbonyl)-N-phenyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (1k)
Compound 1k was obtained as a white solid in 77% yield. 1H NMR (400 MHz, CDCl3) δ 8.62 (s, 1H), 7.72-7.62 (m, 2H), 7.39 (br s, 1H, overlapped), 7.40-7.29 (m, 2H), 7.16-7.05 (m, 1H), 6.76 (br s, 1H), 6.41 (d, J = 1.8 Hz, 1H), 4.83 (s, 2H), 4.56-4.47 (m, 2H), 4.04-3.94 (m, 2H), 2.71 (q, J = 7.6 Hz, 2H), 2.21-2.07 (m, 2H), 1.15 (t, J = 7.6 Hz, 3H). HRMS–ESI (m/z): [M + H]+ calcd for C21H23N4O3, 379.17646; found, 379.17636. HPLC purity: 99.5% (method B).
5-(3,4-Dimethylfuran-2-carbonyl)-N-phenyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (1l)
Compound 1l was obtained as a white solid in 45% yield. 1H NMR (400 MHz, CDCl3) δ 8.63 (s, 1H), 7.68-7.64 (m, 2H), 7.38-7.29 (m, 2H), 7.19 (br s, 1H), 7.13-7.06 (m, 1H), 6.75 (br s, 1H), 4.84 (br s, 2H), 4.55-4.44 (m, 2H), 4.06-3.92 (m, 2H), 2.18 (s, 3H), 2.15-2.08 (br m, 2H), 1.95 (d, J = 1.0 Hz, 3H). HRMS–ESI (m/z): [M + H]+ calcd for C21H23N4O3, 379.17647; found, 379.17634. HPLC purity: 98.9% (method C).
5-(3,5-Dimethylfuran-2-carbonyl)-N-phenyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (1m)
Compound 1m was obtained as a white solid in 68% yield. 1H NMR (400 MHz, CDCl3) δ 8.63 (s, 1H), 7.69-7.63 (m, 2H), 7.38-7.29 (m, 2H), 7.13-7.06 (m, 1H), 6.76 (br s, 1H), 5.95 (s, 1H), 4.84 (br s, 1H), 4.54-4.44 (m, 1H), 4.04-3.93 (m, 2H), 2.32 (br s, 3H), 2.22 (s, 3H), 2.16-2.07 (m, 2H). HRMS–ESI (m/z): [M + H]+ calcd for C21H23N4O3, 379.17647; found, 379.17648. HPLC purity: 97.4% (method C).
5-(4,5-Dimethylfuran-2-carbonyl)-N-phenyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (1n)
Compound 1n was obtained as a white solid in 54% yield. 1H NMR (400 MHz, CDCl3) δ 8.63 (s, 1H), 7.69-7.63 (m, 2H), 7.37-7.31 (m, 2H), 7.14-7.06 (m, 1H), 6.88 (br s, 1H), 6.79 (br s, 1H), 4.92 (br s, 2H), 4.53-4.48 (m, 2H), 4.04 (br s, 2H), 2.30 (s, 1H), 2.16-2.07 (br m, 1H), 1.96 (br s, 1H). HRMS–ESI (m/z): [M + H]+ calcd for C21H23N4O3, 379.17647; found, 379.17645. HPLC purity: 97.9% (method C).
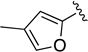
5-(4-Methylfuran-2-carbonyl)-N-phenyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (1o)
Compound 1o was obtained as a white solid in 76% yield. 1H NMR (400 MHz, CDCl3) δ 8.63 (s, 1H), 7.68-7.64 (m, 2H), 7.37-7.29 (m, 2H), 7.28 (br s, overlapped, 1H), 7.13-7.07 (m, 1H), 6.95 (s, 1H), 6.79 (br s, 1H), 4.92 (br s, 2H), 4.54-4.47 (m, 2H), 4.05 (br s, 2H), 2.15-2.07 (br m, 2H), 2.05 (s, 3H). HRMS–ESI (m/z): [M + H]+ calcd for C20H21N4O3, 365.16082; found, 365.16147. HPLC purity: 96.4% (method A).
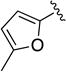
5-(5-Methylfuran-2-carbonyl)-N-phenyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (1p)
Compound 1p was obtained as a white solid in 54% yield. 1H NMR (400 MHz, CDCl3) δ 8.63 (s, 1H), 7.68-7.62 (m, 2H), 7.37-7.30 (m, 2H), 7.12-7.07 (m, 1H), 6.99 (d, J = 3.3 Hz, 1H), 6.79 (br s, 1H), 6.11-6.07 (m, 1H), 4.90 (br s, 2H), 4.56-4.41 (m, 2H), 4.04 (br s, 2H), 2.39 (s, 3H), 2.18-2.04 (br m, 2H). HRMS–ESI (m/z): [M + H]+ calcd for C20H21N4O3, 365.16082; found, 365.16074. HPLC purity: 98.7% (method A).
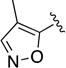
5-(4-Methylisoxazole-5-carbonyl)-N-phenyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (1q)
Compound 1q was obtained as a white solid in 65% yield. 1H NMR (400 MHz, CDCl3) δ 8.61 (s, 1H), 8.18 (s, 0.44 H), 8.16 (s, 0.56H), 7.69-7.63 (m, 2H), 7.40-7.29 (m, 2H), 7.14-7.08 (m, 1H), 6.91 (br s, 0.56H), 6.68 (br s, 0.44H), 4.85 (d, J = 8.6 Hz, 1H), 4.58-4.50 (br m, 2H), 4.10-3.98 (m, 0.88H), 4.02-3.92 (m, 1.12H), 2.21 (s, 3H), 2.13 (s, 2H). HRMS–ESI (m/z): [M + H]+ calcd for C19H20N5O3, 366.15607; found, 366.15601. HPLC purity: 98.7% (method A).
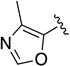
5-(4-Methyloxazole-5-carbonyl)-N-phenyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (1r)
Compound 1r was obtained as a white solid in 81% yield. 1H NMR (400 MHz, CDCl3) δ 8.63 (s, 1H), 7.94-7.74 (br m, 1H), 7.68-7.59 (m, 2H), 7.36-7.26 (m, 2H), 7.13-7.03 (m, 1H), 6.91-6.60 (br m, 1H), 4.78 (br s, 2H), 4.52-4.43 (m, 2H), 4.05-3.89 (m, 2H), 2.40 (s, 3H), 2.04-2.04 (br m, 2H). HRMS–ESI (m/z): [M + H]+ calcd for C19H20N5O3, 366.15607; found, 366.15582. HPLC purity: 98.5% (method A).
N-phenyl-5-(pyrimidine-4-carbonyl)-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (1s)
Compound 1s was obtained as a light yellow solid in 64% yield. 1H NMR (400 MHz, CDCl3) δ 9.32 (d, J = 1.4 Hz, 0.5H), 9.23 (d, J = 1.4 Hz, 0.5H), 8.93 (d, J = 5.1 Hz, 0.5H), 8.90 (d, J = 5.1 Hz, 0.5H), 8.62 (br s, 1H, overlapped), 8.61 (br s, 1H, overlapped), 7.70-7.62 (m, 2.5H), 7.60 (dd, J = 5.1, 1.4 Hz, 0.5H), 7.39-7.31 (m, 2H), 7.15-7.07 (m, 1H), 6.94 (s, 0.5H), 6.45 (s, 0.5H), 4.93 (s, 1H), 4.87 (s, 1H), 4.60-4.49 (m, 2H), 4.14-4.05 (m, 1H), 3.97-3.88 (m, 1H), 2.21-2.12 (br m, 2H). HRMS–ESI (m/z): [M + H]+ calcd for C19H19N6O2, 363.15640; found, 363.15688. HPLC purity: 99.0% (method A).
5-((3-Methylfuran-2-yl)methyl)-N-phenyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (1t)
To a solution of N-phenyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (12) (100 mg, 0.39 mmol) in DMA (2.6 mL) were added 3-methylfuran-2-carbaldehyde (107 mg, 0.98 mmol) and acetic acid (67 µL, 1.2 mmol). The mixture was stirred for 10 minutes and then NaBH(OAc)3 (273 mg, 1.3 mmol) was added in one portion. The reaction was heated at 60 °C for 8 h, allowed to cool down to RT and diluted with sat. aq. NaHCO3 (50 mL). The aqueous phase was extracted with EtOAc (3 × 50 mL) and the combined organic phases were washed with brine (50 mL), dried (Na2SO4), filtered and concentrated under vacuum. The residue was purified via flash chromatography (SiO2, 20 to 60% EtOAc in hexanes, Rf = 0.20, 60% EtOAc in hexanes) to afford 1t (18 mg, 0.051 mmol, 13% yield) as a light yellow oil.1H NMR (400 MHz, CDCl3) δ 8.66 (s, 1H), 7.68 (d, J = 7.7 Hz, 3H), 7.38-7.32 (m, 2H), 7.31 (d, J = 1.8 Hz, 1H), 7.13-7.07 (m, 1H), 6.68 (s, 1H), 6.20 (d, J = 1.8 Hz, 1H), 4.44-4.33 (m, 2H), 3.85 (s, 2H), 3.54 (s, 2H), 3.18-3.09 (m, 2H), 1.95 (s, 3H), 1.95-1.89 (m, 2H, overlapped). HRMS–ESI (m/z): [M + H]+ calcd for C20H23N4O2, 351.18155; found, 351.18135. HPLC purity: 99.5% (method A).
5-(Furan-2-ylsulfonyl)-N-phenyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (1u)
To a solution of N-phenyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (12) (50 mg, 0.20 mmol) in CH2Cl2 (2 mL) at 0 °C, under argon, were added DIPEA (51 µL, 0.20 mmol) and furan-2-sulfonyl chloride (36 mg, 0.22 mmol). The reaction was stirred overnight and quenched with sat. aq. NaHCO3 (50 mL). The aqueous phase was extracted with EtOAc (3 × 50 mL) and the combined organic phases were washed with brine (50 mL), dried (Na2SO4), filtered and concentrated under vacuum. The residue was purified via flash chromatography (SiO2, 20 to 60% EtOAc in hexanes, Rf = 0.2, 60% EtOAc in hexanes) to afford 1u (44 mg, 0.11 mmol, 58% yield) as a white solid. 1H NMR (400 MHz, CDCl3) δ 8.57 (s, 1H), 7.72-7.60 (m, 2H), 7.41-7.29 (m, 3H), 7.16-7.06 (m, 1H), 6.96-6.93 (m, 1H), 6.68 (s, 1H), 6.43-6.40 (m, 1H), 4.56 (s, 2H), 4.45-4.36 (m, 2H), 3.85-3.73 (m, 2H), 1.95-2.05 (br m, 2H). HRMS–ESI (m/z): [M + H]+ calcd for C18H19N4O4S, 387.11215; found, 387.11227. HPLC purity: 99.2% (method A).
Ethyl 5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxylate (14)
Compound 14 was prepared from 5-((2-nitrophenyl)sulfonyl)-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxylate (13)26, by employing a similar procedure to that described for compound 12. It was isolated in 66% yield as a colorless oil.1H NMR (400 MHz, CDCl3) δ 6.60 (s, 1H), 4.49-4.44 (m, 1H), 4.37 (q, J = 7.1 Hz, 2H), 3.91 (s, 2H), 3.23-3.17 (m, 1H), 1.90-1.79 (m, 2H), 1.37 (t, J = 7.1 Hz, 2H). MS (ESI) [M+H]+: 210.1.
Ethyl 5-(3-methylfuran-2-carbonyl)-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxylate (15)
A solution of HOBt (275 mg, 2.04 mmol), EDCI (390 mg, 2.04 mmol) and ethyl 5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxylate 14 (355 mg, 1.70 mmol) in DMF (8.5 mL) was added 3-methylfuran-2-carboxylic acid (214 mg, 1.70 mmol) in one portion. The reaction was stirred for 24 h. The reaction mixture was partitioned between EtOAc (50 mL) and water (50 mL), and the aqueous phase was extracted with EtOAc (2 × 50 mL). The combined organic phases were washed with brine (50 mL), dried (Na2SO4), filtered and concentrated. The residue was purified via flash chromatography (SiO2, 20 to 60% EtOAc in hexanes, Rf= 0.1, 50% EtOAc in hexanes) to give ethyl 5-(3-methylfuran-2-carbonyl)-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxylate (15) (485 mg, 1.53 mmol, 90% yield) as a colorless oil.1H NMR (400 MHz, CDCl3) δ 7.33 (br s, 1H), 6.71 (br s, 0H), 6.32 (br s, 1H), 4.80 (br s, 2H), 4.59-4.49 (m, 2H), 4.35 (q, J = 7.1 Hz, 2H), 4.03-3.92 (m, 2H), 2.24 (s, 3H), 2.14-2.04 (m, 2H), 1.35 (t, J = 7.1 Hz, 3H). MS (ESI) [M+H]+: 318.1.
5-(3-methylfuran-2-carbonyl)-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxylic acid (16)
To a solution of ethyl 5-(3-methylfuran-2-carbonyl)-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxylate (15) (0.444 g, 1.40 mmol) in 4:1 THF/MeOH (14 mL) was added 3N NaOH (1.87 mL, 5.60 mmol) and the resulting solution was stirred for 2h, concentrated to dryness, and suspended in water (25 mL). The aqueous phase was extracted with diethyl ether (3 × 10 mL), acidified to pH 3 with 1N HCl, and extracted with EtOAc (3 ×10 mL). The combined EtOAclayers were rinsed with brine (10 mL), dried (Na2SO4), filtered and concentrated to provide 16 (385 mg, 1.33 mmol, 95% yield) as an amorphous white solid. The material was used in the following step without further purification.1H NMR (400 MHz, CDCl3) δ 7.36 (br s, 1H), 6.78 (br s, 1H), 6.34 (s, 1H), 4.83 (br s, 2H), 4.64-4.49 (br m, 2H), 4.07-3.92 (br m, 2H), 2.26 (s, 3H), 2.17-2.05 (br m, 2H). MS (ESI) [M+H]+: 290.1.
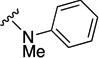
N-methyl-5-(3-methylfuran-2-carbonyl)-N-phenyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (8a)
To a solution of 5-(3-methylfuran-2-carbonyl)-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxylic acid (16) (75 mg, 0.26 mmol), HOBt (42 mg, 0.31 mmol), EDCI (60 mg, 0.31 mmol) in DMF (1.3 mL) was added N-methylaniline (28 µL, 0.26 mmol) in one portion. The reaction was stirred for 24 h and partitioned between EtOAc (50 mL) and water (50 mL), and the aqueous phase was extracted with EtOAc (2 × 50 mL). The combined organic phases were washed with brine (50 mL), dried (Na2SO4), filtered and purified via flash chromatography (SiO2, 35 to 100% EtOAc in hexanes, Rf = 0.05, 60% EtOAc in hexanes) to give 8a (30 mg, 0.079 mmol, 31% yield) as a white solid. 1H NMR (400 MHz, CDCl3) δ 7.37-7.22 (m, 4H), 7.17-7.07 (m, 2H), 6.30 (d, J = 1.8 Hz, 1H), 5.54 (br s, 1H), 4.57 (br s, 2H), 4.46-4.34 (br m, 2H), 3.93-3.85 (m, 2H), 3.44 (br s, 3H), 2.22 (s, 3H), 1.98 (br s, 2H). HRMS–ESI (m/z): [M + H]+ calcd for C21H23N4O3, 379.17647; found, 379.17617. HPLC purity: 97.9% (method A).
Synthesis of compounds 8b–8n
Compounds 8b–8n were prepared by employing a similar procedure to that described for compound 8a, with 5-(3-methylfuran-2-carbonyl)-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxylic acid (16) and the corresponding amine.
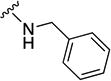
N-benzyl-5-(3-methylfuran-2-carbonyl)-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (8b)
Compound 8b was obtained as a white solid in 15% yield. 1H NMR (400 MHz, CDCl3) δ 7.47-7.20 (m, 5H), 7.19-7.05 (br m, 1H), 6.71 (br s, 1H), 6.33 (d, J = 1.7 Hz, 1H), 4.81 (br s, 2H), 4.60 (s, 1H), 4.58 (s, 1H), 4.46-4.38 (m, 2H), 4.00-3.92 (m, 2H), 2.26 (s, 3H), 2.12-2.03 (br m, 2H). HRMS–ESI (m/z): [M + H]+ calcd for C21H23N4O3, 379.17647; found, 379.17626. HPLC purity: 94.7% (method A).
N-(2,4-difluorophenyl)-5-(3-methylfuran-2-carbonyl)-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (8c)
Compound 8c was obtained as a white solid in 55% yield. 1H NMR (400 MHz, CDCl3) δ 8.78 (s, 1H), 8.45-8.36 (m, 1H), 7.36 (br s, 1H), 6.92-6.84 (m, 2H), 6.77 (br s, overlapped, 1H), 6.34 (d, J = 1.4 Hz, 1H), 4.84 (s, 2H), 4.55-4.48 (m, 2H), 4.05-3.94 (m, 2H), 2.27 (s, 3H), 2.18-2.09 (br m, 2H). HRMS–ESI (m/z): [M + H]+ calcd for C21H23N4O3, 379.17647; found, 379.17626. HPLC purity: 96.5% (method C).
5-(3-Methylfuran-2-carbonyl)-N-(o-tolyl)-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (8d)
Compound 8d was obtained as a white solid in 52% yield. 1H NMR (400 MHz, CDCl3) δ 8.59 (s, 1H), 7.52 (br m, 1H), 7.45 (d, J = 8.4 Hz, 1H), 7.37 (br s, 1H), 7.22 (t, J = 7.8 Hz, 1H), 6.92 (d, J = 7.5 Hz, 1H), 6.76 (br s, 1H), 6.34 (d, J = 1.7 Hz, 1H), 4.84 (br s, 2H), 4.55-4.45 (m, 2H), 4.04-3.94 (m, 2H), 2.35 (s, 3H), 2.27 (s, 3H), 2.21-2.04 (br m, 2H). HRMS–ESI (m/z): [M + H]+ calcd for C21H23N4O3, 379.17647; found, 379.17638. HPLC purity: 99.2% (method A).
5-(3-Methylfuran-2-carbonyl)-N-(m-tolyl)-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (8e)
Compound 8e was obtained as a light yellow solid in 62% yield. 1H NMR (400 MHz, CDCl3) δ 8.60 (s, 1H), 8.10 (d, J = 8.0 Hz, 1H), 7.38 (br s, 1H), 7.25-7.16 (m, 2H), 7.05 (td, J = 7.4, 1.3 Hz, 1H), 6.78 (br s, 1H), 6.35 (d, J = 1.8 Hz, 1H), 4.85 (s, 2H), 4.59-4.46 (m, 2H), 4.06-3.95 (m, 2H), 2.35 (s, 3H), 2.28 (s, 3H), 2.20-2.09 (br m, 1H). HRMS–ESI (m/z): [M + H]+ calcd for C21H23N4O3, 379.17647; found, 379.17633. HPLC purity: 97.4% (method A).
5-(3-Methylfuran-2-carbonyl)-N-(p-tolyl)-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (8f)
Compound 8f was obtained as a white solid in 64% yield. 1H NMR (400 MHz, CDCl3) δ 8.57 (s, 1H), 7.59-7.49 (m, 2H), 7.37 (br s, 1H), 7.14 (d, J = 8.2 Hz, 2H), 6.76 (br s, 1H), 6.34 (d, J = 1.7 Hz, 1H), 4.84 (br s, 2H), 4.55-4.44 (m, 2H), 4.05-3.92 (br m, 2H), 2.32 (s, 3H), 2.27 (s, 3H), 2.18-2.08 (br m, 2H). HRMS–ESI (m/z): [M + H]+ calcd for C21H23N4O3, 379.17647; found, 379.17689. HPLC purity: 97.5% (method A).
N-(2-fluorophenyl)-5-(3-methylfuran-2-carbonyl)-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (8g)
Compound 8g was obtained as a white solid in 52% yield. 1H NMR (400 MHz, CDCl3) δ 8.91 (s, 1H), 8.47 (td, J = 8.1, 1.7 Hz, 1H), 7.38 (br s, 1H), 7.20-6.97 (m, 3H), 6.78 (br s, 1H), 6.35 (d, J = 1.4 Hz, 1H), 4.85 (br s, 2H), 4.59-4.48 (m, 2H), 4.14-3.89 (m, 2H), 2.28 (s, 3H), 2.21-2.08 (br m, 2H). 19F NMR (376 MHz, CDCl3) δ −131.3 (s). HRMS–ESI (m/z): [M + H]+ calcd for C20H20N4O3F, 379.15140; found, 379.15114. HPLC purity: 97.7% (method A).
N-(3-fluorophenyl)-5-(3-methylfuran-2-carbonyl)-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (8h)
Compound 8h was obtained as a yellow solid in 62% yield. 1H NMR (400 MHz, CDCl3) δ 8.68 (s, 1H), 7.67-7.60 (m, 1H), 7.37 (br s, 1H), 7.32-7.27 (m, 2H), 6.90-6.69 (m, 2H), 6.35 (d, J = 1.7 Hz, 1H), 4.85 (s, 2H), 4.59-4.47 (m, 2H), 4.09-3.94 (m, 2H), 2.27 (s, 3H), 2.18-2.09 (br m, 2H). 19F NMR (376 MHz, CDCl3) δ −111.61 (s). HRMS–ESI (m/z): [M + H]+ calcd for C20H20N4O3F, 379.15140; found, 379.15150. HPLC purity: 97.0% (method A).
N-(4-fluorophenyl)-5-(3-methylfuran-2-carbonyl)-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (8i)
Compound 8i was obtained as a white solid in 53% yield. 1H NMR (400 MHz, CDCl3) δ 8.61 (s, 1H), 7.68-7.56 (m, 2H), 7.37 (br s, 1H), 7.09-6.96 (m, 2H), 6.77 (s, 1H), 6.35 (d, J = 1.7 Hz, 1H), 4.85 (s, 2H), 4.62-4.41 (m, 2H), 4.07-3.96 (m, 2H), 2.27 (s, 3H), 2.20-2.08 (br m, 2H). 19F NMR (376 MHz, CDCl3) δ −118.6 (s). HRMS–ESI (m/z): [M + H]+ calcd for C20H20N4O3F, 379.15140; found, 379.15204. HPLC purity: 98.7% (method A).
N-(2-methoxyphenyl)-5-(3-methylfuran-2-carbonyl)-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (8j)
Compound 8j was obtained as a white solid in 66% yield. 1H NMR (400 MHz, CDCl3) δ 9.25 (br s, 1H), 8.50 (dd, J = 7.9, 1.7 Hz, 1H), 7.38 (br s, 1H), 7.05 (td, J = 7.8, 1.7 Hz, 1H), 6.98 (td, J = 7.7, 1.4 Hz, 1H), 6.90 (dd, J = 8.0, 1.4 Hz, 1H), 6.76 (br s, 1H), 6.34 (d, J = 1.4 Hz, 1H), 4.85 (br s, 2H), 4.58-4.46 (m, 2H), 4.04-3.97 (br m, 2H), 3.93 (s, 3H), 2.27 (s, 3H), 2.20-2.05 (br m, 2H). HRMS–ESI (m/z): [M + H]+ calcd for C21H23N4O4, 395.17138; found, 395.17109. HPLC purity: 99.1% (method A).
N-(3-methoxyphenyl)-5-(3-methylfuran-2-carbonyl)-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (8k)
Compound 8k was obtained as a yellow solid in 62% yield. 1H NMR (400 MHz, CDCl3) δ 8.63 (s, 1H), 7.47 (t, J = 2.2 Hz, 1H), 7.37 (br s, 1H), 7.22 (t, J = 8.1 Hz, 1H), 7.10 (ddd, J = 8.0, 2.0, 0.9 Hz, 1H), 6.77 (br s, 1H), 6.66 (ddd, J = 8.3, 2.5, 0.9 Hz, 1H), 6.34 (d, J = 1.8 Hz, 1H), 4.84 (s, 2H), 4.58-4.42 (m, 2H), 4.05-3.93 (m, 2H), 3.81 (s, 3H), 2.26 (s, 3H), 2.18-2.06 (br m, 2H). HRMS–ESI (m/z): [M + H]+ calcd for C21H23N4O4, 395.17138; found, 395.17125. HPLC purity: 98.6% (method A).
N-(4-methoxyphenyl)-5-(3-methylfuran-2-carbonyl)-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (8l)
Compound 8l was obtained as a white solid in 74% yield. 1H NMR (400 MHz, CDCl3) δ 8.53 (br s, 1H), 7.63-7.50 (m, 2H), 7.37 (br s, 1H), 6.94-6.84 (m, 2H), 6.76 (br s, 1H), 6.35 (d, J = 1.7 Hz, 1H), 4.85 (br s, 2H), 4.59-4.43 (m, 2H), 4.06-3.93 (m, 2H), 3.80 (s, 3H), 2.27 (s, 3H), 2.21-2.06 (m, 2H). HRMS–ESI (m/z): [M + H]+ calcd for C20H23N4O4, 395.17138; found, 395.17141. HPLC purity: 98.7% (method A).
5-(3-Methylfuran-2-carbonyl)-N-(pyrimidin-5-yl)-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (8m)
Compound 8m was obtained as a light yellow solid in 15% yield. 1H NMR (400 MHz, CDCl3) δ 9.15 (s, 2H), 8.98 (s, 1H), 8.68 (br s, 1H), 7.38 (br s, 1H), 6.81 (br s, 1H), 6.36 (d, J = 1.7 Hz, 1H), 4.86 (s, 2H), 4.64-4.43 (m, 2H), 4.11-3.95 (br m, 2H), 2.28 (s, 3H), 2.21.2.09 (br m, 1H). HRMS–ESI (m/z): [M + H]+ calcd for C18H19N6O3, 367.15131; found, 367.15111. HPLC purity: 97.2% (method A).
N-(2-chloro-4-fluorophenyl)-5-(3-methylfuran-2-carbonyl)-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (8n)
Compound 8n was obtained as a white solid in 29% yield. 1H NMR (400 MHz, CDCl3) δ 9.15 (s, 1H), 8.49 (dd, J = 9.2, 5.7 Hz, 1H), 7.37 (br s, 1H), 7.15 (dd, J = 8.1, 2.9 Hz, 1H), 7.02 (ddd, J = 9.2, 7.9, 2.9 Hz, 1H), 6.78 (br s, 1H), 6.35 (d, J = 1.5 Hz, 1H), 4.85 (s, 2H), 4.61-4.47 (m, 2H), 4.09-3.92 (br m, 2H), 2.27 (s, 3H), 2.21-2.08 (br m, 2H). 19F NMR (376 MHz, CDCl3) δ -117.1 (s). HRMS–ESI (m/z): [M + H]+ calcd for C20H19N4O3ClF, 417.11242; found, 417.11213. HPLC purity: 97.8% (method C).
5-(3-Methylfuran-2-carbonyl)-N-(pyrimidin-4-yl)-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (8o)
To a solution of 5-(3-methylfuran-2-carbonyl)-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxylic acid 16 (122 mg, 0.42 mmol), HATU (192 mg, 0.51 mmol) and DIPEA (176 µL, 1.01 mmol) in DMF (2.1 mL) was added pyrimidin-4-amine (120 mg, 1.265 mmol) in one portion. The reaction was stirred for 72 h, diluted with water (50 mL), and the aqueous phase was extracted with EtOAc (3 × 50 mL). The combined organic phases were washed with brine (50 mL), dried (Na2SO4), filtered and concentrated under vacuum. The residue was purified via flash chromatography (SiO2, 30 to 100% EtOAc in hexanes, Rf = 0.05, 60% EtOAc in hexanes) to afford 8o (19 mg, 0.052 mmol, 12% yield) as a light yellow solid. 1H NMR (400 MHz, CDCl3) δ 9.37 (s, 1H), 8.90 (s, 1H), 8.64 (br d, J = 5.8 Hz, 1H), 8.29 (d, J = 5.5 Hz, 1H), 7.37 (br s, 1H), 6.81 (br s, 1H), 6.35 (d, J = 1.7 Hz, 1H), 4.85 (br s, 2H), 4.62-4.41 (m, 2H), 4.07-3.98 (m, 2H), 2.27 (s, 3H), 2.20-2.08 (br m, 2H). HRMS–ESI (m/z): [M + H]+ calcd for C18H19N6O3, 367.15131; found, 367.15112. HPLC purity: 96.1% (method A).
N-methoxy-N-methyl-5-(3-methylfuran-2-carbonyl)-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (17)
To a solution of 5-(3-methylfuran-2-carbonyl)-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxylic acid (16)(85 mg, 0.29 mmol) in CH2Cl2 (1 mL) were added EDCI (56 mg, 0.29 mmol), N,O-dimethylhydroxylamine hydrochloride (32 mg, 0.32 mmol) and triethylamine (41 µL, 0.29 mmol). The reaction was stirred overnight, diluted with water (100 mL) and the aqueous phase was extracted with EtOAc (3 × 50 mL). The combined organic layers were washed with brine (50 mL), dried (Na2SO4), filtered and concentrated. The residue was purified via flash chromatography (SiO2, 1 to 5% MeOH in CH2Cl2, Rf = 0.1, 5% MeOH in CH2Cl2), to afford 17 (40 mg, 0.120 mmol, 41% yield) as a colorless oil. 1H NMR (400 MHz, CDCl3) δ 7.31 (s, 1H), 6.63 (br s, 1H), 6.29 (d, J = 1.7 Hz, 1H), 4.78 (s, 2H), 4.58-4.40 (m, 2H), 3.98-3.90 (m, 2H), 3.67 (s, 3H), 3.35 (s, 3H), 2.21 (s, 3H), 2.13-1.99 (m, 2H). MS (ESI) [M+H]+: 333.1.
1-(5-(3-Methylfuran-2-carbonyl)-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepin-2-yl)-2-phenylethanone (8p)
To a solution of N-methoxy-N-methyl-5-(3-methylfuran-2-carbonyl)-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (17) (40 mg, 0.12 mmol) in THF (1.2 mL), under argon, at 0 °C, was added benzylmagnesium chloride (963 µL, 0.963 mmol) (1.0 M in 2-methyltetrahydrofuran), and the reaction was allowed to warm to RT and stirred for 1 h at this temperature. The reaction was quenched with sat. aq. NH4Cl (10 mL), diluted with water (40 mL), and the aqueous phase was extracted with EtOAc (3 × 50 mL). The combined organic layers were washed with brine (50 mL), dried (Na2SO4), filtered and concentrated, and the residue was purified via flash chromatography (SiO2,20 to 60% EtOAc in hexanes, Rf = 0.25, 60% EtOAc in hexanes) to provide 8p (11 mg, 0.030 mmol, 25% yield) as a light yellow oil. 1H NMR (400 MHz, CDCl3) δ 7.39-7.17 (m, 6H), 6.68 (br s, 1H), 6.33 (d, J = 1.7 Hz, 1H), 4.80 (s, 2H), 4.59-4.49 (m, 2H), 4.26 (s, 2H), 4.04-3.95 (m, 2H), 2.26 (s, 3H), 2.18-2.10 (br m, 2H). HRMS–ESI (m/z): [M + H]+ calcd for C21H22N3O3, 364.16557; found, 364.16580. HPLC purity: 95.4% (method C).
(2-(Hydroxymethyl)-7,8-dihydro-4H-pyrazolo[1,5-a][1,4]diazepin-5(6H)-yl)(3-methylfuran-2-yl)methanone (18)
To a solution of 5-(3-methylfuran-2-carbonyl)-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxylic acid (16) (200 mg, 0.69 mmol) and 4-methylmorpholine (76 µL, 0.69 mmol) in THF (14 mL) under argon, at 0 °C, was added isobutyl chloroformate (90 µL, 0.69 mmol). The reaction was stirred for 30 min, filtered through Celite into a flask containing a solution of NaBH4 (262 mg, 6.9 mmol) in water (14 mL) at 0°C, and stirred for 2 h at this temperature. The reaction was quenched with AcOH (1.6 mL, 28 mmol) and allowed to warm to RT. The aqueous phase was extracted with EtOAc (3 × 25 mL), and the combined organic phases were washed with brine (25 mL), dried (Na2SO4), filtered and concentrated. The residue was purified via flash chromatography (SiO2, 40 to 100% EtOAc in hexanes) to provide 18 (78 mg, 0.28 mmol, 41% yield) as a colorless oil. 1H NMR (400 MHz, CDCl3) δ 7.34 (br s, 1H), 6.34 (d, J = 1.9 Hz, 1H), 6.20 (br s, 1H), 4.78 (s, 2H), 4.62 (s, 2H), 4.48-4.40 (m, 2H), 4.02-3.92 (m, 2H), 2.27 (s, 3H), 2.13-2.06 (br m, 2H). MS (ESI) [M+H]+: 276.1.
(3-Methylfuran-2-yl)(2-((phenylamino)methyl)-7,8-dihydro-4H-pyrazolo[1,5-a][1,4]diazepin-5(6H)-yl)methanone (8q)
To a solution of (2-(hydroxymethyl)-7,8-dihydro-4H-pyrazolo[1,5-a][1,4]diazepin-5(6H)-yl)(3-methylfuran-2-yl)methanone 18 (72 mg, 0.26 mmol) and triethylamine (73 µL, 0.52 mmol) in THF (1.3 mL), under argon, at 0 °C was added methanesulfonyl chloride (30 µL, 0.39 mmol) in one portion. The reaction was stirred for 2 h, diluted with MeOH (2 mL) and concentrated to dryness. The residue was taken up in dry toluene (2 mL) and concentrated to dryness (2×). According to 1H NMR the residue contained a 4:1 mixture of (5-(3-methylfuran-2-carbonyl)-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepin-2-yl)methyl methanesulfonate and the starting material. The mixture was dissolved in dry THF and used in the next step without further purification. To the resulting solution, under argon, at RT, were added aniline (72 µL, 0.78 mmol) and triethylamine (109 µL, 0.78 mmol). The reaction was stirred overnight at RT and diluted with water (50 mL). The aqueous phase was extracted with EtOAc (3 × 25 mL), and the combined organic phases were washed with brine (50 mL), dried (Na2SO4), filtered and concentrated under vacuum. The residue was separated via flash chromatography (SiO2,0 to 5% MeOH in CH2Cl2) to provide a partially purified product, which was purified via flash chromatography (SiO2,20 to 70% EtOAc in hexanes, Rf = 0.25, 2% MeOH in CH2Cl2) to give 8q (18 mg, 0.051 mmol, 20% yield) as anamorphous white solid. 1H NMR (400 MHz, CDCl3) δ 7.32-7.29 (m, 1H), 7.21-7.13 (m, 2H), 6.73-6.69 (m, overlapped, 1H), 6.69-6.64 (m, overlapped, 2H), 6.33-6.32 (m, 1H), 6.28-5.97 (br m, 1H), 4.75 (s, 2H), 4.47-4.40 (m, 2H), 4.25 (s, 2H), 4.00-3.91 (m, 2H), 2.26 (s, 3H), 2.14-2.06 (br m, 2H). HRMS–ESI (m/z): [M + H]+ calcd for C20H23N4O2, 351.18155; found, 351.18195. HPLC purity: 98.9% (method A).
Tert-butyl (5-((2-nitrophenyl)sulfonyl)-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepin-2-yl)carbamate (19)
To a solution of 5-((2-nitrophenyl)sulfonyl)-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxylic acid (10) (121 mg, 0.33 mmol) in toluene/tert-butanol/1,2-dichloroethane 3:3:2 (1.6 mL) were added triethylamine (92 µL, 0.66 mmol) and diphenyl phosphorazidate (93 µL, 0.43 mmol). The reaction was stirred overnight at RT, and then refluxed for 8 h and allowed to warm to RT. The reaction was diluted with water (25 mL), extracted with EtOAc (3 × 25 mL), the combined organic phases were washed with brine (25 mL), dried (Na2SO4), filtered and concentrated. The residue was purified via flash chromatography (SiO2, 0 to 50% EtOAc in hexanes) to afford 19 (41 mg, 0.094 mmol, 32% yield) as a yellow solid. 1H NMR (400 MHz, CDCl3) δ 8.00-7.95 (m, 1H), 7.73-7.57 (m, 3H), 6.98 (s, 1H), 6.45 (br s, 1H), 4.51 (s, 2H), 4.29-4.20 (m, 2H), 3.75-3.62 (m, 2H), 2.04-1.96 (br m, 2H), 1.50 (s, 9H). MS (ESI) [M+H]+: 438.1.
5-((2-Nitrophenyl)sulfonyl)-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepin-2-amine (20)
Tert-butyl (5-((2-nitrophenyl)sulfonyl)-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepin-2-yl)carbamate 19 (600 mg, 1.37 mmol) was suspended in a 4M solution of HCl in dioxane (8.6 mL, 34 mmol) at RT. The reaction was stirred overnight, diluted with water (50 mL), and sat. aq. NaHCO3 was added to adjust the pH to 8. The aqueous phase was extracted with EtOAc (3 × 50 mL), and the combined organic phases were washed with brine (50 mL), dried (Na2SO4), filtered and concentrated under vacuum. The residue was purified via flash chromatography (SiO2, 0 to 5% MeOH in CH2Cl2) to afford 20 (457 mg, 1.36 mmol, 99% yield) as a yellow oil. 1H NMR (400 MHz, CDCl3) δ 8.00-7.92 (m, 1H), 7.74-7.58 (m, 3H), 5.62 (s, 1H), 4.46 (s, 2H), 4.28-4.07 (m, 2H), 3.66-3.61 (m, 2H), 2.61 (br s, 2H), 2.05-1.91 (m, 2H). MS (ESI) [M+H]+: 338.1.
5-((2-Nitrophenyl)sulfonyl)-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-sulfonyl chloride (21)
A solution of sodium nitrite (98 mg, 1.4 mmol) in water (1 mL) was added to a solution of 5-((2-nitrophenyl)sulfonyl)-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepin-2-amine (20) (457 mg, 1.35 mmol) in AcOH/conc. HCl/water 2:1:1 (6 mL) at −10 °C. The mixture was stirred for 30 min and then added to a suspension of copper (II) sulfate (43 mg, 0.27 mmol) in AcOH (5 mL) saturated with sulfur dioxide. The reaction was stirred for 1 h at −10 °C, allowed to warm to room temperature and diluted with water (50 mL). The pH of was adjusted to 7 with LiOH and the mixture was diluted with water (50 mL). The aqueous phase was extracted with EtOAc (3 × 50 mL), and the combined organic phases were washed with water (25 mL) and brine (50 mL), dried (Na2SO4), filtered and concentrated under vacuum. The residue was purified via flash chromatography (SiO2, 20 to 60% EtOAc in hexanes) to afford 21 (205 mg, 0.487 mmol, 36% yield) as a white solid. 1H NMR (400 MHz, CDCl3) δ 8.05-8.00 (m, 1H), 7.79-7.61 (m, 3H), 6.85 (s, 1H), 4.62 (s, 2H), 4.60-4.54 (m, 2H), 3.77-3.70 (m, 2H), 2.20-2.12 (m, 1H). MS (ESI) [M+H]+: 421.0.
5-((2-Nitrophenyl)sulfonyl)-N-phenyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-sulfonamide (22)
To a solution of 5-((2-nitrophenyl)sulfonyl)-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-sulfonyl chloride 21 (100 mg, 0.24 mmol) and aniline (27 µL, 0.29 mmol) in THF (1.2 mL) was added triethylamine (132 µL, 0.95 mmol) in one portion. The reaction was stirred under argon for 16 h and diluted with water (50 mL). The aqueous phase was extracted with EtOAc (3 × 25 mL) and the combined organic phases were washed with brine (25 mL), dried (Na2SO4), filtered and concentrated under vacuum. The residue was purified via flash chromatography (SiO2, 25 to 70% EtOAc in hexanes) to afford 22 (95 mg, 0.20 mmol, 84% yield) as a white solid. 1H NMR (400 MHz, CDCl3) δ 7.94-7.89 (m, 1H), 7.69-7.65 (m, 3H), 7.63-7.56 (m, 2H), 7.31-7.25 (m, 2H), 7.17-7.11 (m, 3H), 6.53 (s, 1H), 4.51 (s, 2H), 4.46-4.39 (m, 2H), 3.72-3.64 (m, 2H), 2.06-1.99 (m, 2H). MS (ESI) [M+H]+: 478.0.
N-phenyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-sulfonamide (23)
Compound 23 was prepared from 5-((2-nitrophenyl)sulfonyl)-N-phenyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-sulfonamide 22, by employing a similar procedure to that described for compound 12. It was obtained as a colorless oil in 79% yield. 1H NMR (400 MHz, CDCl3) δ 7.67 (s, 1H), 7.28-7.15 (m, 3H), 7.12-7.02 (m, 3H), 6.47 (br s, 1H, overlapped), 6.36 (s, 1H), 4.45-4.25 (m, 2H), 3.82 (s, 2H), 3.24-3.06 (m, 1H), 1.87-1.68 (m, 2H). MS (ESI) [M+H]+: 293.1.
5-(3-Methylfuran-2-carbonyl)-N-phenyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-sulfonamide (8r)
Compound 8r was prepared by employing a similar procedure to that for compound 1a, with 3-methylfuran-2-carboxylic acid and N-phenyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-sulfonamide (23). It was obtained as a white solid in 76% yield. 1H NMR (400 MHz, CDCl3) δ 7.28 (br s, 1H), 7.26-7.20 (m, 2H), 7.17-7.12 (m, 1H), 7.12-7.06 (m, 1H), 6.83 (s, 1H), 6.49 (br s, 1H), 6.34-6.33 (m, 1H), 4.73 (s, 2H), 4.54-4.44 (m, 2H), 4.02-3.92 (m, 2H), 2.25 (s, 3H), 2.13-2.02 (br m, 2H). HRMS–ESI (m/z): [M + H]+ calcd for C19H21N4O4S, 401.12780; found, 401.12790. HPLC purity: 98.2% (method A).
N-(5-((2-nitrophenyl)sulfonyl)-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepin-2-yl)benzamide (24)
To a solution of 5-((2-nitrophenyl)sulfonyl)-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepin-2-amine (20) (136 mg, 0.403 mmol) and DIPEA (351 µL, 2.02 mmol) in THF (4.03 mL) at 0 °C, was added a solution of benzoyl chloride (70 µL, 0.60 mmol) in THF (4.03 mL). The reaction was stirred overnight, quenched with water (50 mL), and the aqueous phase was extracted with EtOAc (3 × 33 mL). The combined organic layerswere washed with brine (33 mL), dried (sodium sulfate), filtered and concentrated under vacuum. The residue was purified via flash chromatography (SiO2, 0 to 100% EtOAc in hexanes, to give 24 (161 mg, 0.365 mmol, 90% yield) as a colorless oil.1H NMR (400 MHz, CDCl3) δ 8.93-8.58 (br m, 1H), 8.05-7.99 (m, 1H), 7.89-7.80 (m, 2H), 7.74-7.51 (m, 4H), 7.51-7.39 (m, 2H), 6.85 (s, 1H), 4.59-4.52 (br m, 1H), 4.26-4.15 (m, 2H), 3.78-3.62 (br m, 2H), 1.99 (brs, 2H). MS (ESI) [M+H]+: 442.0.
N-(5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepin-2-yl)benzamide (25)
Compound 25 was prepared from N-(5-((2-nitrophenyl)sulfonyl)-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepin-2-yl)benzamide (24), by employing a similar procedure to that described for compound 12. It was obtained as colorless oil in quantitative yield. 1H NMR (400 MHz, CDCl3) δ 9.11-8.95 (br m, 1H), 7.88-7.82 (m, 2H), 7.57-7.49 (m, 1H), 7.48-7.40 (m, 2H), 6.71 (s, 1H), 4.17-4.09 (m, 2H), 3.89 (s, 2H), 3.21-3.12 (m, 2H), 1.83-1.64 (m, 2H, overlapped with HOD signal). MS (ESI) [M+H]+: 257.1.
N-(5-(3-methylfuran-2-carbonyl)-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepin-2-yl)benzamide (8s)
Compound 8s was prepared by employing a similar procedure to that for compound 1a, with 3-methylfuran-2-carboxylic acid and N-(5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepin-2-yl)benzamide (25). It was obtained as a white solid in 29% yield. 1H NMR (400 MHz, CDCl3) δ 8.76 (s, 1H), 7.90-7.83 (m, 2H), 7.59-7.51 (m, 1H), 7.50-7.41 (m, 2H), 7.39 (br s, 1H), 6.82 (br s, 1H), 6.35 (d, J = 1.7 Hz, 1H), 4.82 (s, 2H), 4.36-4.27 (m, 2H), 4.00-3.93 (m, 2H), 2.28 (s, 3H), 2.13-2.03 (br m, 2H). HRMS–ESI (m/z): [M + H]+ calcd for C20H21N4O3, 365.16082; found, 365.16051. HPLC purity: 98.6% (method B).
N-(5-((2-nitrophenyl)sulfonyl)-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepin-2-yl)benzenesulfonamide (26)
To a solution of 5-((2-nitrophenyl)sulfonyl)-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepin-2-amine (20) (105 mg, 0.31 mmol) and DIPEA (270 µL, 1.55 mmol) in THF (3 mL) at 0 °C, under Ar, was added benzenesulfonyl chloride (60 µL, 0.47 mmol). The reaction was stirred overnight, quenched with sat. aq. NH4Cl (25 mL) and the aqueous phase extracted with EtOAc (3 × 25 mL). The combined organic phases were washed with brine (25 mL), dried (Na2SO4), filtered and concentrated. The residue was purified via flash chromatography (SiO2, 50 to 100% EtOAc in hexanes, Rf = 0.1 in 80% EtOAc in hexanes), to afford 26 (148 mg, 0.31 mmol, quantitative yield) as a colorless oil.1H NMR (400 MHz, CDCl3) δ 8.56 (br s, 1H), 8.01-7.93 (m, 1H), 7.74-7.67 (m, 3H), 7.67-7.60 (m, 2H), 7.57-7.50 (m, 1H), 7.45-7.38 (m, 2H), 6.24 (s, 1H), 4.47 (s, 2H), 4.35-4.25 (m, 2H), 3.68-3.59 (m, 2H), 2.04-1.91 (m, 2H). MS (ESI) [M+H]+: 478.0.
N-(5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepin-2-yl)benzenesulfonamide (27)
Compound 27 was prepared from N-(5-((2-nitrophenyl)sulfonyl)-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepin-2-yl)benzenesulfonamide (26), by employing a similar procedure to that described for compound 12. It was obtained as a colorless oil in 74% yield. 1H NMR (400 MHz, CDCl3) δ 7.73-7.67 (m, 2H), 7.55-7.49 (m, 1H), 7.38 (t, J = 7.7 Hz, 2H), 4.37-4.29 (m, 2H), 3.82 (s, 2H), 3.18-3.09 (m, 2H), 1.85-1.72 (m, 2H). MS (ESI) [M+H]+: 293.1.
N-(5-(3-methylfuran-2-carbonyl)-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepin-2-yl)benzenesulfonamide (8t)
Compound 8t was prepared by employing a similar procedure to that for compound 1a, with 3-methylfuran-2-carboxylic acid and N-(5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepin-2-yl)benzenesulfonamide (27). It was obtained as a white solid in 13% yield. 1H NMR (400 MHz, CDCl3) δ 9.00 (br s, 1H), 7.75-7.60 (br m, 2H), 7.53-7.47 (m, 1H), 7.36 (t, J = 7.6 Hz, 2H), 6.35 (s, 1H), 6.19 (br s, 1H), 4.72 (s, 2H), 4.42-4.32 (m, 2H), 3.96-3.84 (m, 2H).2.27 (s, 3H), 2.02 (br s, 2H). HRMS–ESI (m/z): [M + H]+ calcd for C19H21N4O4S, 401.12780; found, 401.12781. HPLC purity: 96.3% (method B).
Ethyl 5-(3-methylfuran-2-carbonyl)-4,5,6,7-tetrahydropyrazolo[1,5-a]pyrazine-2-carboxylate (29)
Compound 29 was prepared by employing a similar procedure to that for compound 15, with 3-methylfuran-2-carboxylic acid and ethyl 4,5,6,7-tetrahydropyrazolo[1,5-a]pyrazine-2-carboxylate (28)26,32. It was obtained as a white solid in 65% yield.1H NMR (400 MHz, CDCl3) δ 7.39 (dd, J = 1.8, 0.5 Hz, 1H), 6.65 (s, 1H), 6.38 (dd, J = 1.8, 0.5 Hz, 1H), 4.40 (q, J = 7.1 Hz, 3H), 4.21-4.15 (m, 2H), 2.42 (s, 9H), 2.32 (s, 3H), 1.39 (t, J = 7.1 Hz, 3H). MS (ESI) [M+H]+: 304.1.
5-(3-Methylfuran-2-carbonyl)-N-phenyl-4,5,6,7-tetrahydropyrazolo[1,5-a]pyrazine-2-carboxamide (9a)
To a solution of ethyl 5-(3-methylfuran-2-carbonyl)-4,5,6,7-tetrahydropyrazolo[1,5-a]pyrazine-2-carboxylate (29) (150 mg, 0.495 mmol) in THF/MeOH 4:1 (4.95 mL) was added 3N NaOH (660 µL, 1.98 mmol) and the resulting solution was stirred for 2h, concentrated to dryness, suspended in water (25 mL). The aqueous phase was extracted with diethyl ether (3 × 10 mL), acidified to pH 3 with 1N HCl, and extracted with EtOAc (3 ×10 mL). The combined organic phases were washed with brine (10 mL), dried (Na2SO4), filtered and concentrated to provide 5-(3-methylfuran-2-carbonyl)-4,5,6,7-tetrahydropyrazolo[1,5-a]pyrazine-2-carboxylic acid (30) (116 mg, 0.421 mmol, 85% yield) as an amorphous white solid, which was used without further purification. MS (ESI) [M+H]+: 276.1. To a solution of the crude carboxylic acid 30, HOBt (68 mg, 0.51 mmol), EDCI (97 mg, 0.51 mmol) in DMF (2.10 mL) was added aniline (38 µL, 0.42 mmol) in one portion. The reaction was stirred for 24h and partitioned between EtOAc (50 mL) and water (50 mL). The aqueous phase was extracted with EtOAc (2 × 50 mL), and the combined organic phases were washed with brine (50 mL), dried (Na2SO4), filtered and purified via flash chromatography (SiO2, 20 to 50% EtOAc in hexanes, Rf= 0.30, 60% EtOAc in hexanes) to give 9a (96 mg, 0.27 mmol, 54% yield from 29) as a white solid.1H NMR (400 MHz, CDCl3) δ 8.66 (s, 1H), 7.72-7.64 (m, 2H), 7.40 (d, J = 1.7 Hz, 1H), 7.39-7.30 (m, 2H), 7.16-7.07 (m, 1H), 6.70 (s, 1H), 6.39 (d, J = 1.7 Hz, 1H), 5.00 (br s, 2H), 4.38-4.31 (br m, 2H), 4.24-4.16 (br m, 2H), 2.33 (s, 3H). HRMS–ESI (m/z): [M + H]+ calcd for C19H19N4O3, 351.14517; found, 351.14597. HPLC purity: 96.0% (method A).
Ethyl 2-(3-methylfuran-2-carbonyl)-2,3,4,5-tetrahydro-1H-pyrrolo[1,2-a][1,4]diazepine-8-carboxylate (32)
To a solution of ethyl 2,3,4,5-tetrahydro-1H-pyrrolo[1,2-a][1,4]diazepine-8-carboxylate (31)26 (320 mg, 1.54 mmol), HATU (701 mg, 1.84 mmol), 3-methylfuran-2-carboxylic acid (233 mg, 1.84 mmol) in CH2Cl2 (15.4 mL) was added triethylamine (1.03 mL, 7.38 mmol). The reaction was stirred for 24h and diluted with water (50 mL). The aqueous phase was extracted with EtOAc (3 × 33 mL) and the combined organic phases were washed with brine (50 mL), dried (Na2SO4), filtered and concentrated. The residue was purified via flash chromatography (SiO2, 0 to 70% EtOAc in hexanes Rf = 0.4, 50% EtOAc in hexanes) to afford 32 (106 mg, 0.335 mmol, 22% yield) as a colorless oil. 1H NMR (400 MHz, CDCl3) δ 7.48-7.29 (br m, 1H), 7.24-7.21 (m, 1H), 6.66-6.39 (br m, 2H), 6.33 (s, 1H), 4.74 (br s, 2H), 4.23 (q, J = 7.1 Hz, 2H), 4.19-4.14 (m, 2H), 3.90 (s, 2H), 2.26 (s, 3H), 2.10-2.00 (br m, 2H), 1.30 (t, J = 7.1 Hz, 3H). MS (ESI) [M+H]+: 317.1.
2-(3-Methylfuran-2-carbonyl)-N-phenyl-2,3,4,5-tetrahydro-1H-pyrrolo[1,2-a][1,4]diazepine-8-carboxamide (9b)
To a solution of ethyl 2-(3-methylfuran-2-carbonyl)-2,3,4,5-tetrahydro-1H-pyrrolo[1,2-a][1,4]diazepine-8-carboxylate (32) (0.100 g, 0.316 mmol) in THF/MeOH4:1 (3.16 mL) was added 3N LiOH (420 µL, 1.26 mmol), and the resulting solution was stirred for 6 days, concentrated to dryness, and the residue was suspended in water (25 mL). The aqueous phase was extracted with diethyl ether (3 × 10 mL), acidified to pH 3 with 1N HCl, and extracted with EtOAc (3 ×10 mL). The combined EtOAc were washed with brine (10 mL), dried (Na2SO4), filtered and concentrated to provide 2-(3-methylfuran-2-carbonyl)-2,3,4,5-tetrahydro-1H-pyrrolo[1,2-a][1,4]diazepine-8-carboxylic acid (33) (35 mg, 0.121 mmol, 38% yield) as an amorphous white solid, which was used without further purification. MS (ESI) [M+H]+: 289.1. To a solution of crude carboxylic acid 33, HOBt (20 mg, 0.15 mmol), EDCI (28 mg, 0.15 mmol) in DMF (607 µL) was added aniline (33 µL, 0.36 mmol) in one portion, and the reaction was stirred overnight. The reaction mixture was partitioned between EtOAc (50 mL) and water (50 mL), the aqueous phase was extracted with EtOAc (2 × 50 mL). The combined organic phases were rinsed with brine (50 mL), dried (Na2SO4), filtered and separated via flash chromatography (SiO2, 0 to 5% MeOH in CH2Cl2, Rf= 0.1, 2% MeOH in CH2Cl2) to afford 9b (17 mg, 0.047 mmol, 15% yield from 32) as a light tan solid.1H NMR (400 MHz, CDCl3) δ 7.64-7.54 (m, 2H), 7.47-7.37 (br m, 1H, overlapped), 7.38-7.28 (m, 2H), 7.13-7.03 (m, 1H), 6.50-6.19 (br m, overlapped 1H), 6.34 (s, 1H), 4.76 (br s, 2H), 4.27-4.13 (m, 2H), 3.95 (br s, 2H), 2.27 (s, 3H), 2.12-2.01 (br m, 2H). HRMS–ESI (m/z): [M + H]+ calcd for C21H22N3O3, 364.16557; found, 364.16620. HPLC purity: 97.3% (method A).
N-(2-fluorophenyl)-5-((2-nitrophenyl)sulfonyl)-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (34)
Compound 34 was prepared from 5-((2-nitrophenyl)sulfonyl)-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxylic acid (10) and o-fluoroaniline, by employing a similar procedure to that described for compound 11. It was isolated in 65% yield as a white solid.1H NMR (400 MHz, CDCl3) δ 8.92-8.81 (m, 1H), 8.47 (td, J = 8.1, 1.7 Hz, 1H), 8.02-7.95 (m, 1H), 7.76-7.59 (m, 3H), 7.21-6.98 (m, 3H), 6.82 (d, J = 0.6 Hz, 1H), 4.61 (s, 2H), 4.54-4.41 (m, 2H), 3.83-3.65 (m, 2H), 2.18-2.05 (m, 2H). 19F NMR (376 MHz, CDCl3) δ −131.38- −131.49 (m). MS (ESI) [M+H]+: 460.0.
N-(2-fluorophenyl)-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (35)
Compound 35 was prepared from N-(2-fluorophenyl)-5-((2-nitrophenyl)sulfonyl)-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (34), by employing a similar procedure to that described for compound 12. It was isolated in 56% yield as a white solid. 1H NMR (400 MHz, Chloroform-d) δ 8.93 (s, 1H), 8.49 (td, J = 8.1, 1.7 Hz, 1H), 7.20-6.95 (m, 4H), 6.68 (s, 1H), 4.49-4.40 (m, 2H), 3.95 (s, 2H), 3.28-3.18 (m, 2H), 1.96-1.84 (m, 2H). 19F NMR (376 MHz, CDCl3) δ −131.4- −131.5 (m). MS (ESI) [M+H]+: 275.1.
N-(2-fluorophenyl)-5-(furan-2-carbonyl)-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (9c)
Compound 9c was prepared by employing a similar procedure to that for compound 1a, with furan-2-carboxylic acid and N-(2-fluorophenyl)-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (35). It was obtained as a white solid in 59% yield.1H NMR (400 MHz, CDCl3) δ 8.88 (s, 1H), 8.44 (td, J = 8.1, 1.7 Hz, 1H), 7.51 (br s, 1H), 7.17-6.97 (m, 4H), 6.79 (br s, 1H), 6.53-6.41 (m, 1H), 4.89 (br s, 2H), 4.59-4.37 (m, 2H), 4.04 (br s, 2H), 2.16-2.04 (br m, 2H). 19F NMR (376 MHz, CDCl3) δ −131.0- −131.6 (s). HRMS–ESI (m/z): [M + H]+ calcd for C19H18N4O3F, 369.13575; found, 369.13572. HPLC purity: 99.2% (method A).
Supplementary Material
Figure 1.

RSV inhibitors currently in Phase II clinical trials
Scheme 4.

Synthesis of compound 8ra
aReagents and conditions (a) DPPA, Et3N, t-BuOH/toluene/1,2-dichloroethane 1.5:1.5:1, RT to reflux, 32%. (b) HCl/dioxane, then NaHCO3, RT, 99%. (c) NaNO2/aq. HCl, then CuSO4, SO2, AcOH/water, −10 °C to RT, 36%. (d) PhNH2, Et3N, THF, RT, 84%. (e) PhSH, Cs2CO3, CH3CN, RT, 79%. (f) EDCI, HOBt, 3-methylfuran-2-carboxylic acid, DMF, RT, 76%.
Scheme 5.

Syntheses of compounds 8s and 8ta
aReagents and conditions (a) BzCl, DIPEA, THF, 0 °C to RT, 90%. (b) PhSO2Cl, DIPEA, THF, 0 °C to RT, quant. (c) PhSH, Cs2CO3, CH3CN, RT, 75%–quant. (d) EDCI, HOBt, 3-methylfuran-2-carboxylic acid, DMF, RT, 29–68%.
Acknowledgments
We thank R.T. Jacob for IT support. The MScreen software package was kindly provided by the Center for Chemical Genomics of the University of Michigan under a license agreement by the University of Michigan Office of Technology Transfer, JChem was used for structure database management, search and prediction (JChem 6.2, 2014, ChemAxon (http://www.chemaxon.com)), and Marvin was employed for drawing, displaying and characterizing chemical structures, substructures and reactions (Marvin 14.9.22.0, 2014, ChemAxon (http://www.chemaxon.com)).
Funding Sources
This work was supported, in part, by public health service grants AI071002 and HD079327 from the NIH/NIAID and NIH/NICHD (to R.K.P.).
ABBREVIATIONS USED
- ATCC
American Type Culture Collection
- ANOVA
analysis of variance
- CC50
50% cytotoxic concentration
- Ct value
cycle threshold value
- DIPEA
N,N-diisopropylethylamine
- DMEM
Dulbecco’s modified Eagle’s medium
- DMSO
dimethyl sulfoxide
- DPPA
diphenyl phosphorazidate
- EC50
half-maximal effective concentration
- EDCI
N-(3-dimethylaminopropyl)-N’-ethylcarbodiimide hydrochloride
- GAPDH
glyceraldehyde 3-phosphate dehydrogenase
- HATU
N-[(dimethylamino)-1H-1,2,3-triazolo-[4,5-b]pyridin-1-ylmethylene]-N-methylmethanaminium hexafluorophosphate N-oxide
- HOBt
1-Hydroxybenzotriazole
- IAV
influenza A virus
- IBCF
iso-butyl chloroformate
- ITSD
internal standard
- NMM
N-methylmorpholine
- Ns
2-nitrobenzenesulfonyl
- qPCR
real-time polymerase chain reaction
- RdRp
RNA-dependent RNA polymerase
- RSV
respiratory syncytial virus
- RSV N
RSV nucleocapsid protein
- RSV P
RSV phosphoprotein
- RSV M2-1
RSV matrix protein 2-1
- RSV L
RSV large protein
- SD
standard deviation
- SEM
standard error of the mean
- SI
selectivity index
- TCID50
median tissue culture infective dose
- VSV
vesicular stomatitis virus
Footnotes
ASSOCIATED CONTENT
Supporting Information
The supporting information is available free of charge on the ACS Publications website at DOI: Detailed procedure for metabolism study of 1a and 8p.
Molecular Formula Strings (MFS) for compounds presented in the manuscript.
The authors declare no competing financial interest.
References
- 1.Afonso CL, Amarasinghe GK, Banyai K, Bao Y, Basler CF, Bavari S, Bejerman N, Blasdell KR, Briand FX, Briese T, Bukreyev A, Calisher CH, Chandran K, Cheng J, Clawson AN, Collins PL, Dietzgen RG, Dolnik O, Domier LL, Durrwald R, Dye JM, Easton AJ, Ebihara H, Farkas SL, Freitas-Astua J, Formenty P, Fouchier RA, Fu Y, Ghedin E, Goodin MM, Hewson R, Horie M, Hyndman TH, Jiang D, Kitajima EW, Kobinger GP, Kondo H, Kurath G, Lamb RA, Lenardon S, Leroy EM, Li CX, Lin XD, Liu L, Longdon B, Marton S, Maisner A, Muhlberger E, Netesov SV, Nowotny N, Patterson JL, Payne SL, Paweska JT, Randall RE, Rima BK, Rota P, Rubbenstroth D, Schwemmle M, Shi M, Smither SJ, Stenglein MD, Stone DM, Takada A, Terregino C, Tesh RB, Tian JH, Tomonaga K, Tordo N, Towner JS, Vasilakis N, Verbeek M, Volchkov VE, Wahl-Jensen V, Walsh JA, Walker PJ, Wang D, Wang LF, Wetzel T, Whitfield AE, Xie JT, Yuen KY, Zhang YZ, Kuhn JH. Taxonomy of the order mononegavirales: Update 2016. Arch. Virol. 2016;161:2351–2360. doi: 10.1007/s00705-016-2880-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Thompson WW, Shay DK, Weintraub E, Brammer L, Cox N, Anderson LJ, Fukuda K. Mortality associated with influenza and respiratory syncytial virus in the United States. J. Am. Med. Assoc. 2003;289:179–186. doi: 10.1001/jama.289.2.179. [DOI] [PubMed] [Google Scholar]
- 3.Collins PL, Crowe JE., Jr . Respiratory Syncytial Virus and Metapneumoviruses. In: Knipe DM, Howley PM, editors. Fields Virology. 5. Vol. 2. Lippincott, Williams, & Wilkins; Philadelphia: 2007. pp. 1601–1645. [Google Scholar]
- 4.Elliot AJ, Fleming DM. Influenza and respiratory syncytial virus in the elderly. Expert Rev. Vaccines. 2008;7:249–258. doi: 10.1586/14760584.7.2.249. [DOI] [PubMed] [Google Scholar]
- 5.Aherne W, Bird T, Court SD, Gardner PS, McQuillin J. Pathological changes in virus infections of the lower respiratory tract in children. J. Clin. Pathol. 1970;23:7–18. doi: 10.1136/jcp.23.1.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lugo RA, Nahata MC. Pathogenesis and treatment of bronchiolitis. Clin. Pharm. 1993;12:95–116. [PubMed] [Google Scholar]
- 7.Domachowske JB, Rosenberg HF. Respiratory syncytial virus infection: Immune response, immunopathogenesis, and treatment. Clin. Microbiol. Rev. 1999;12:298–309. doi: 10.1128/cmr.12.2.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.DeVincenzo JP, Wilkinson T, Vaishnaw A, Cehelsky J, Meyers R, Nochur S, Harrison L, Meeking P, Mann A, Moane E, Oxford J, Pareek R, Moore R, Walsh E, Studholme R, Dorsett P, Alvarez R, Lambkin-Williams R. Viral load drives disease in humans experimentally infected with respiratory syncytial virus. Am. J. Respir. Crit. Care Med. 2010;182:1305–1314. doi: 10.1164/rccm.201002-0221OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.El Saleeby CM, Bush AJ, Harrison LM, Aitken JA, Devincenzo JP. Respiratory syncytial virus load, viral dynamics, and disease severity in previously healthy naturally infected children. J. Infect. Dis. 2011;204:996–1002. doi: 10.1093/infdis/jir494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Anderson LJ, Parker RA, Strikas RL. Association between respiratory syncytial virus outbreaks and lower respiratory tract deaths of infants and young children. J. Infect. Dis. 1990;161:640–646. doi: 10.1093/infdis/161.4.640. [DOI] [PubMed] [Google Scholar]
- 11.Groothuis JR, Woodin KA, Katz R, Robertson AD, McBride JT, Hall CB, McWilliams BC, Lauer BA. Early ribavirin treatment of respiratory syncytial viral infection in high-risk children. J. Pediatr. 1990;117:792–798. doi: 10.1016/s0022-3476(05)83347-5. [DOI] [PubMed] [Google Scholar]
- 12.Cox R, Plemper RK. Structure-guided design of small-molecule therapeutics against RSV disease. Expert Opin. Drug Discovery. 2016;11:543–556. doi: 10.1517/17460441.2016.1174212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang G, Deval J, Hong J, Dyatkina N, Prhavc M, Taylor J, Fung A, Jin Z, Stevens SK, Serebryany V, Liu J, Zhang Q, Tam Y, Chanda SM, Smith DB, Symons JA, Blatt LM, Beigelman L. Discovery of 4'-chloromethyl-2'-deoxy-3',5'-di-o-isobutyryl-2'-fluorocytidine (ALS-8176), a first-in-class RSV polymerase inhibitor for treatment of human respiratory syncytial virus infection. J. Med. Chem. 2015;58:1862–1878. doi: 10.1021/jm5017279. [DOI] [PubMed] [Google Scholar]
- 14.DeVincenzo JP, Whitley RJ, Mackman RL, Scaglioni-Weinlich C, Harrison L, Farrell E, McBride S, Lambkin-Williams R, Jordan R, Xin Y, Ramanathan S, O'Riordan T, Lewis SA, Li X, Toback SL, Lin SL, Chien JW. Oral GS-5806 activity in a respiratory syncytial virus challenge study. N. Engl. J. Med. 2014;371:711–722. doi: 10.1056/NEJMoa1401184. [DOI] [PubMed] [Google Scholar]
- 15.Yan D, Lee S, Thakkar VD, Luo M, Moore ML, Plemper RK. Cross-resistance mechanism of respiratory syncytial virus against structurally diverse entry inhibitors. Proc. Natl. Acad. Sci. U. S. A. 2014;111:E3441–3449. doi: 10.1073/pnas.1405198111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bright RA, Medina MJ, Xu X, Perez-Oronoz G, Wallis TR, Davis XM, Povinelli L, Cox NJ, Klimov AI. Incidence of adamantane resistance among influenza a (H3N2) viruses isolated worldwide from 1994 to 2005: A cause for concern. Lancet. 2005;366:1175–1181. doi: 10.1016/S0140-6736(05)67338-2. [DOI] [PubMed] [Google Scholar]
- 17.Bright RA, Shay D, Bresee J, Klimov A, Cox N, Ortiz J, Ctr WC CDC. High levels of adamantane resistance among influenza a (H3N2) viruses and interim guidelines for use of antiviral agents - United States, 2005–06 influenza season (reprinted from mmwr, vol 55, pg 44–46, 2006) J. Am. Med. Assoc. 2006;295:881–882. [Google Scholar]
- 18.Yan D, Weisshaar M, Lamb K, Chung HK, Lin MZ, Plemper RK. Replication-competent influenza virus and respiratory syncytial virus luciferase reporter strains engineered for co-infections identify antiviral compounds in combination screens. Biochemistry. 2015;54:5589–5604. doi: 10.1021/acs.biochem.5b00623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Moore ML, Chi MH, Luongo C, Lukacs NW, Polosukhin VV, Huckabee MM, Newcomb DC, Buchholz UJ, Crowe JE, Jr, Goleniewska K, Williams JV, Collins PL, Peebles RS., Jr A chimeric A2 strain of respiratory syncytial virus (RSV) with the fusion protein of RSV strain line 19 exhibits enhanced viral load, mucus, and airway dysfunction. J. Virol. 2009;83:4185–4194. doi: 10.1128/JVI.01853-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chung HK, Jacobs CL, Huo Y, Yang J, Krumm SA, Plemper RK, Tsien RY, Lin MZ. Tunable and reversible drug control of protein production via a self-excising degron. Nat. Chem. Biol. 2015;11:713–720. doi: 10.1038/nchembio.1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jacob RT, Larsen MJ, Larsen SD, Kirchhoff PD, Sherman DH, Neubig RR. Mscreen: An integrated compound management and high-throughput screening data storage and analysis system. J. Biomol. Screen. 2012;17:1080–1087. doi: 10.1177/1087057112450186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brideau C, Gunter B, Pikounis B, Liaw A. Improved statistical methods for hit selection in high-throughput screening. J. Biomol. Screen. 2003;8:634–647. doi: 10.1177/1087057103258285. [DOI] [PubMed] [Google Scholar]
- 23.Hotard AL, Lee S, Currier MG, Crowe JE, Jr, Sakamoto K, Newcomb DC, Peebles RS, Jr, Plemper RK, Moore ML. Identification of residues in the human respiratory syncytial virus fusion protein that modulate fusion activity and pathogenesis. J. Virol. 2015;89:512–522. doi: 10.1128/JVI.02472-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hawkins E, Butler B, Beck M, O'Grady M, Orr L, Wood K. Promega Notes. Vol. 81. Promega Corporation; 2002. Dual-glo™ luciferase assay system: Convenient dual-reporter measurements in 96- and 384-well plates; pp. 22–26. [Google Scholar]
- 25.Scifinder. Chemical Abstracts Service; Columbus, OH: 2015. [Google Scholar]
- 26.Blackburn C, Chin J, Gigstad KM, Xu H. Substituted hydroxamic acids and uses thereof. 8,946,223. [February 3, 2015];U.S. Patent No.
- 27.Moussa IA, Banister SD, Manoli M, Doddareddy MR, Cui J, MacH RH, Kassiou M. Exploration of ring size in a series of cyclic vicinal diamines with sigma1 receptor affinity. Bioorg. Med. Chem. Lett. 2012;22:5493–5497. doi: 10.1016/j.bmcl.2012.07.026. [DOI] [PubMed] [Google Scholar]
- 28.Lane JW, Chen Y, Williams RM. Asymmetric total syntheses of (−)-jorumycin, (−)-renieramycin g, 3-epi-jorumycin, and 3-epi-renieramycin g. J. Am. Chem. Soc. 2005;127:12684–12690. doi: 10.1021/ja0535918. [DOI] [PubMed] [Google Scholar]
- 29.Tomita N, Hayashi Y, Suzuki S, Oomori Y, Aramaki Y, Matsushita Y, Iwatani M, Iwata H, Okabe A, Awazu Y, Isono O, Skene RJ, Hosfield DJ, Miki H, Kawamoto T, Hori A, Baba A. Structure-based discovery of cellular-active allosteric inhibitors of FAK. Bioorg. Med. Chem. Lett. 2013;23:1779–1785. doi: 10.1016/j.bmcl.2013.01.047. [DOI] [PubMed] [Google Scholar]
- 30.Peng HX Zhili, Zhang Lei, Sun Lihong, Kumaravel Gnanasambandam, Taveras Art, Wang Deping. S1P and/or ATX modulating agents. 14/772481. [February 11, 2016];US. Patent Application No.
- 31.Hoffman RV. Meta-trifluoromethylbenzenesulfonyl chloride. Org. Synth. 1981;60:121–126. [Google Scholar]
- 32.Blackburn C, Barrett C, Brunson M, Chin J, England D, Garcia K, Gigstad K, Gould A, Gutierrez J, Hoar K, Rowland RS, Tsu C, Ringeling J, Wager K, Xu H. Histone deacetylase inhibitors derived from 1,2,3,4-tetrahydropyrrolo[1,2-a]pyrazine and related heterocycles selective for the HDAC6 isoform. Bioorg. Med. Chem. Lett. 2014;24:5450–5454. doi: 10.1016/j.bmcl.2014.10.022. [DOI] [PubMed] [Google Scholar]
- 33.Andaloussi M, Lim HD, van der Meer T, Sijm M, Poulie CB, de Esch IJ, Leurs R, Smits RA. A novel series of histamine h4 receptor antagonists based on the pyrido[3,2-d]pyrimidine scaffold: Comparison of hERG binding and target residence time with PF-3893787. Bioorg. Med. Chem. Lett. 2013;23:2663–2670. doi: 10.1016/j.bmcl.2013.02.091. [DOI] [PubMed] [Google Scholar]
- 34.Asakawa N, Kobayashi S, Goto J, Hirayama N. Autogpa: An automated 3D-QSAR method based on pharmacophore alignment and grid potential analysis. Int. J. Med. Chem. 2012;2012:498931. doi: 10.1155/2012/498931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Molecular operating environment (MOE), 2013.08. Chemical Computing Group Inc.; 1010 Sherbooke St. West, Suite #910, Montreal, QC, Canada, H3a 2r7: 2017. [Google Scholar]
- 36.Battles MB, Langedijk JP, Furmanova-Hollenstein P, Chaiwatpongsakorn S, Costello HM, Kwanten L, Vranckx L, Vink P, Jaensch S, Jonckers TH, Koul A, Arnoult E, Peeples ME, Roymans D, McLellan JS. Molecular mechanism of respiratory syncytial virus fusion inhibitors. Nat. Chem. Biol. 2016;12:87–93. doi: 10.1038/nchembio.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hotard AL, Shaikh FY, Lee S, Yan D, Teng MN, Plemper RK, Crowe JE, Jr, Moore ML. A stabilized respiratory syncytial virus reverse genetics system amenable to recombination-mediated mutagenesis. Virology. 2012;434:129–136. doi: 10.1016/j.virol.2012.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cramer RD, Patterson DE, Bunce JD. Comparative molecular field analysis (CoMFA). 1. Effect of shape on binding of steroids to carrier proteins. J. Am. Chem. Soc. 1988;110:5959–5967. doi: 10.1021/ja00226a005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.