

Abstract
AIM
To investigate alterations in the fecal microbiome using 16S rRNA amplicon sequencing in couples in the same cohabitation environment.
METHODS
Fecal samples were collected from eight ulcerative colitis (UC) patients and their healthy partners at Lishui People’s Hospital, Zhejiang Province, China. DNA was extracted and the variable regions V3 and V4 of the 16S rRNA genes were PCR amplified using a two-step protocol. Clear reads were clustered into operational taxonomic units (OTUs) at the 97% sequence similarity level using UCLUST v1.2.22. The Wilcoxon rank-sum test (R v3.1.2) was used to compare inter-individual differences. Differences with a P value < 0.05 were considered statistically significant.
RESULTS
Fecal microbial communities were more similar among UC patients than their healthy partners (P = 0.024). UC individuals had a lower relative abundance of bacteria belonging to the Firmicutes, especially Blautia, Clostridium, Coprococcus and Roseburia (P < 0.05). Microbiota dysbiosis was detected in UC patients and their healthy partners. Relevant genera included Akkermansiam, Bacteroides, Escherichia, Lactobacillales, Klebsiella and Parabacteroides. The enriched pathways in fecal samples of UC patients were related to lipid and nucleotide metabolism. Additionally, the pathways involved in membrane transport and metabolism of cofactors and vitamins were more abundant in the healthy partners.
CONCLUSION
Our results suggested that the microbial composition might be affected in healthy partners cohabiting with UC patients, especially in terms of microbiota dysbiosis.
Keywords: Ulcerative colitis, Patients, Healthy partner, Fecal microbial communities, Microbiota dysbiosis
Core tip: To identify the influence on the gut microbial community between ulcerative colitis (UC) patients and their healthy partners, we investigated the gut bacterial community using 16S rRNA amplicon sequencing. The results showed that fecal microbial communities were more similar in UC patients, which had a lower relative abundance of Firmicutes bacteria. Microbiota dysbiosis was also founded in healthy partners. The pathways involved in lipid and nucleotide metabolism were more abundant in the UC patients. The membrane transport and metabolism of cofactors and vitamins pathways were significantly enriched in the healthy partners. Microbial composition might be affected in healthy partners cohabiting with UC patients.
INTRODUCTION
Ulcerative colitis (UC), one of the main types of inflammatory bowel disease (IBD), has become increasingly prevalent in developed countries over the past two decades[1]. However, according to the latest Asian epidemiological investigation, the incidence of the IBD has expanded dramatically into developing countries with the increased westernization of lifestyles[1,2].
UC is a multi-faceted disorder associated with a germline genetic background, an aberrant immune system response, and environmental factors[3]. Recently, the gut microbial community has attracted substantial research attention, especially in terms of its influence in healthy and IBD patients[4-6]. Some alterations in the microbial community are shared in Crohn’s disease (CD) and UC patients relative to healthy people, including reduced gut microbiota diversity (particularly Firmicutes), the presence of non-commensals, and increased abundance of pathogenic Proteobacteria strains[7,8]. However, some alterations in the bacterial community are specific to UC. For example, an increased presence of Escherichia coli and Fusobacterium spp group[6], as well as a reduction in Clostridium coccoides group, has been reported in UC patients[9].
Previous studies have demonstrated that environmental factors, including dietary age[9], habits[10], and obesity[11], impact the composition of the gut microbiota. Consequently, we wondered whether the cohabitation environment can influence the microbial community. In 1994, an investigation of 10 couples showed that individuals with IBD symptoms before marriage influenced their partners, resulting in similar symptoms in couples[12]. However, these results mainly focused on clinical symptoms and did not involve the gut microbial community.
Profiling the fecal microbiome using methods based on analysis of the 16S ribosomal RNA gene is less biased than cultivation-based approaches. In recent years, bacterial 16S rRNA amplicon sequencing, referred to as “16S rRNA gene sequencing”, has been used widely for the metagenomic analysis of the environment, including analysis of the composition of human and animal gut and fecal microbiota[13,14]. In particular, compared with bacterial culture, 16S rRNA gene sequencing has a huge advantage in identifying new pathogens or difficult to culture bacteria compared with culture-independent methods. Compared with other high-throughput sequencing platforms (pyrosequencing, Life Technologies platform), the MiSeq platform generates the highest base sequence accuracy with little limitation on the DNA input[15,16]. Although the cohabitation environment has been studied using 16S rRNA gene sequencing, many studies on the gut microbial community of IBD patients focused on comparisons between twins and siblings[11,17].
This study aimed to explore deeply the composition of the UC gut bacterial community using 16S rRNA sequencing to identify the influence on the gut microbial community between couples. Additionally, the current study investigates the predominant fecal microbiota of UC patients compared with their healthy cohabiting partners.
MATERIALS AND METHODS
Patient selection
The diagnosis of UC was determined by endoscopic and pathological findings, and patients diagnosed with UC for longer than 3 mo were recruited from Lishui People’s Hospital at Zhejiang Province. Healthy couples composing the control group, were recruited from a common living environment cohabiting with the UC patients. All participants in this study were divided into two groups (n = 8 each) according to their disease status. Group A consisted of UC patients, identified as UC1-UC8. Group B consisted of their healthy partners, termed HF1-HF8. The study was approved by the Ethics Committee of the hospital and all participants provided written informed consent upon enrolment.
Sample collection and DNA extraction
Fecal samples were collected from all participants and were immediately stored at -80 °C until further processing. The total DNA was extracted from 200 mg of fecal sample using the QIAamp DNA Stool Mini Kit (Qiagen, Inc., Valencia, CA, United States), according to the manufacturer’s instructions. The DNA concentration was quantified using a Qubit 2.0 Fluorometer and diluted appropriately based on the total DNA concentration.
16S amplicon-sequencing
The variable regions V3 and V4 of the bacterial 16S rRNA gene were amplified using a two-step PCR protocol. In brief, the first PCR was performed with universal primers (356F 5’-CCTACGGGNGGCWGCAG-3’ and 803R 5’-GACTACHVGGGTATCTAATCC-3’) and an attached overhang adapter (forward primer overhang adapter 5’-TCGTCGGCAGCGTCAGATGTGTATAAGAGACAG-3’ and reverse primer overhang adapter 5’-GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAG-3’). The first PCR reaction mixture (20 μL) contained 10 ng of DNA template, 10 μL of 2 × High-Fidelity PCR Master Mix with HF Buffer (New England Biolabs, Ipswich, MA, United States), each primer at 5 μmol/L, and reagent-grade water (Sigma Aldrich, St. Louis, MO, United States). The first PCR program for V3 and V4 comprised an initial denaturation of 95 °C for 3 min; followed by 25 cycles of denaturation at 95 °C for 30 s, annealing at 60 °C for 30 s, extension at 72 °C for 30 s; and a final extension at 72 °C for 5 min. Then, the PCR product was cleaned using AMPure XP DNA purification beads (Beckman Coulter, Danvers, MA, United States), according to the manufacturer’s instructions. The second PCR reaction mixture (25 μL) contained 2 μL of cleaned PCR products; 12.5 μL of 2 × High-Fidelity PCR Master Mix and HF Buffer; P5/P7 primers, including adapters and sample barcodes (Illumina); and reagent-grade water. The second PCR program was similar to the first PCR program except the annealing temperature was at 50 °C and the program comprised 10 cycles. Finally, the PCR product was purified using AMPure XP beads to remove primer dimers and was then quantified on an Agilent Bioanalyzer 2100 using High Sensitivity DNA chips (Agilent Technologies, Santa Clara, CA, United States). The library of each sample was pooled at an equimolar concentration, and sequencing was performed on the Illumina MiSeq platform to generate 2 × 250 bp paired-end reads.
Taxonomic classification and pathway profiles of 16S rRNA gene sequencing data
We merged the paired-end reads using FLASH v1.2.11 and obtained approximately 460 bp V3-V4 16S sequences. Merged reads were processed with QIIME v1.8.0, which removed reads with N bases and trimmed reads with more than three consecutive low-quality bases (Q < 20). The reads the passed the high quality filters were aligned to the Greengenes Database (Aug, 2013 version), and USEARCH v6.1 was applied for chimera checking. Then, UCLUST v1.2.22 was used for operational taxonomic unit (OTU) clustering at the 97% sequence similarity level. Each OTU was classified according to an assignment of the taxonomic rank using the reference dataset from the Ribosomal Database Project (version 2.2). Phylogenetic Investigation of Communities by Reconstruction of Unobserved States (PICRUSt) analysis was performed to generate Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway profiles as previously described[18,19].
Statistical analysis
Correlations between the overlapping genera of the 16S rRNA gene sequencing were identified using Pearson’s correlation in the R language. The Wilcoxon rank-sum test (R v3.1.2) was employed to detect interindividual differences. Under the condition of multiple comparisons, P values were corrected to control for the false-discovery rate. Differences with P value < 0.05 were considered statistically significant.
RESULTS
Study subjects and 16S rRNA sequencing
To measure the compositional and functional differences in the common living environment between the gut microbiota of the UC patients and healthy individuals, 16 fecal samples were collected from eight families, including eight UC patients and eight healthy control partners. All patients in this study were cohabiting with their partners. The demographics and characteristics of 8 UC patients and their partners are shown in Table 1.
Table 1.
Information and clinical characteristics of ulcerative colitis patients and their healthy partners n (%)
| UC patients (n = 8) | Healthy partners (n = 8) | |
| mean age (SD) | 53 (9.57) | 42.25 (9.41) |
| Male | 6 (75.0) | 3 (37.5) |
| Age at diagnosis | ||
| 16-40 yr | 3 (37.5) | |
| Above 40 yr | 5 (62.5) | |
| Disease location | ||
| Colonic | 5 (62.5) | |
| Rectal | 3 (37.5) | |
| Concomitant upper GI disease | 4 (50.0) | |
| CRP | ||
| Normal (0-10 mg/L) | 5 (62.5) | |
| Abnormal | 3 (37.5) |
GI: Gastrointestinal; CRP: C-reactive protein; UC: Ulcerative colitis.
16S rRNA amplicon-sequencing of fecal DNA samples was performed using next-generation sequencing (NGS) technology. Low quality reads and chimera sequences were filtered from the raw data, eventually producing an average of 47469 reads per sample. These reads corresponded to 1137 operational taxonomic units (OTUs) at the 97% sequence similarity level and identified 167 genera. There was no significant difference between the number of OTUs in UC patients and their partners (947 ± 298.4 for UC patients and 1327.1 ± 419.9 for their partners, P = 0.071).
Microbiota diversity and similarity analyses
We used three metrics to characterize the fecal microbiota diversity between UC patients and their partners. Microbial alpha diversity was not significantly different by Chao1 analysis (P = 0.573). Additionally, there were no significant differences observed by Shannon-Wiener (P = 0.505) or Simpson’s indices (P = 0.574). The distribution of fecal microbiota was determined using Bray Curtis similarities analysis. The entire microbiota of UC patients was not significantly divergent from those of their cohabiting partners (P = 0.449). However, using un-weighted analysis, the microbiota composition was more similar among UC patients than it was to that of their partners (Figure 1, P = 0.024). These results indicated that cohabitation with UC patients might influence the fecal microbiota composition of healthy people; however, higher dissimilarity of the microbiota was observed among the healthy partners.
Figure 1.
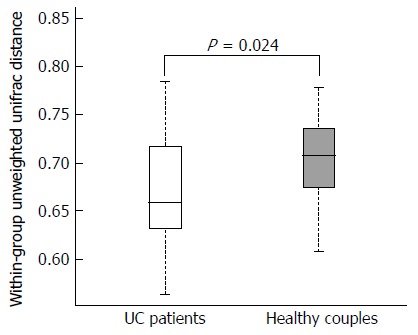
Distributions of the unweighted unifrac distance within the ulcerative colitis patients and healthy partners groups. Orange and red boxplots denote the distributions of 16S rRNA gene sequencing of fecal samples in ulcerative colitis patients and healthy partners, respectively.
Microbial composition
Although there was no significant difference in overall microbial diversity, the Wilcoxon test indicated that some bacteria were significantly different between UC patients and their partners. As shown in Figure 2, at the genus level, 10 genera were less abundant in the fecal microbiota of UC patients compared with that in their healthy partners, and only one bacterium was more abundant in the UC patients (P < 0.05). At the phylum level, nine significantly different bacteria species belonged to the Firmicutes, and the other bacteria were classified as Actinobacteria and Bacteroidetes.
Figure 2.

Significant differences in the fecal microbiota composition between ulcerative colitis patients and their healthy partners. Comparisons for each sample were calculated using the Wilcoxon test. The down arrows indicate less abundant microbiota in ulcerative colitis (UC) patients, and the up arrow represents the more abundant microbiota in UC patients compared with their healthy partners.
In this study, the fecal microbiota in the UC patients showed a decrease in the Firmicutes bacteria, especially Blautia, Clostridium, Coprococcus, and Roseburia. Blautia was detected at 5.81% in the healthy partners, and this percentage was lower in the UC patients. Clostridium and Coprococcus accounted for 6.49% and 3.4% of the fecal microbiota of the healthy partners, whereas these genera were less abundant in the UC patients (0.96% and 0.45%, respectively). A similar trend was observed for Roseburia, which was present at 1.48% in the healthy partners and 0.15% in the UC patients. Other Firmicutes genera, such as Anaerostipes Lachnospira, Megasphaera, and Turicibacter, were resent at a very low abundance in the UC patients. Although they were not detected in all healthy partners, these rare species were detected at significantly different levels in the healthy partners compared with the UC patients. For example, Anaerostipes was detected in UC patients at a percentage of 0.006%, but at 0.68% in the healthy partners. Additionally, Alistipes, belonging to the Bacteroidetes, and an unclassified bacterium of Actinobacteria, did not appear in UC patients, but were detected in the healthy partners at low abundances of 0.14% and 0.23%, respectively.
Microbiota dysbiosis
An unclassified bacterium belonging to the Lactobacillales was detected in a UC patient (UC1) with a higher relative abundance than in their partner (Figure 2, P = 0.032). The proportion of Lactobacillus genus showed large dysbiosis in patient UC1, accounting for 81.8% (Figure 3). Microbiota dysbiosis was also observed in other UC patients and their partners. In patient UC2, the first and second most abundant genera were Bacteroides and Parabacteroides, which were present in percentages of 58.63% and 28.93%, respectively (Figure 3). Bacteroides was also the dominant genus, accounting for 70.24% in patient UC6. In addition, the Escherichia genus accounted for 44.39% in patient UC5. For the healthy partners, microbiota dysbiosis was also found in a few individuals. For example, in HF6, the relative abundance of the Klebsiella genus reached 63.3%. Akkermansia, belonging to the Verrucomicrobia phylum, was only detected in patient UC4 and his partner, accounting for 18.46% and 68.74%, respectively (Figure 3). Microbiota dysbiosis destroys the gut microbiota composition and influences its normal function, which might accelerate the occurrence of intestinal disease.
Figure 3.
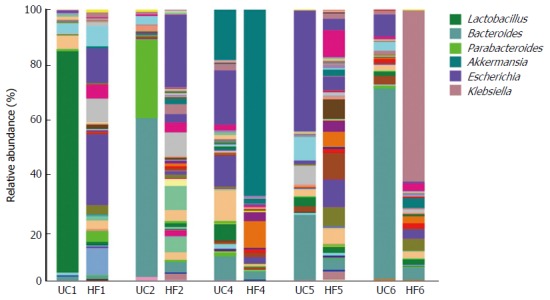
Relative abundance at the genus level, as shown by 16S rRNA gene sequencing, from ulcerative colitis patients and their partners showing a microbiota imbalance. Each column represents one fecal sample and different colors indicate different genera in the microbiota composition. Microbiota with a substantial proportional imbalance are listed. UC: UC patient; HF: Healthy partners.
Microbial metabolic pathways
Combining the microbial composition with a genome database from KEGG (Kyoto Encyclopedia of Genes and Genomes), 328 pathways were identified. We continued our analysis using the Wilcoxon test, and 20 of 328 (6.09%) total metabolic pathways were differentially abundant at Q < 0.05 between the UC patients and their healthy partners. In the UC patients, we observed that 10 pathways were increased significantly, especially those concerning lipid metabolism and nucleotide metabolism. Another eight pathways, such as those involving membrane transport and metabolism of cofactors and vitamins, were more abundant in the healthy partners (Table 2).
Table 2.
Microbial metabolism differentially abundant in ulcerative colitis patients and their healthy partners
| Pathway |
UC patients |
Healthy partners |
P value | ||
| Mean abundance | Standard error | Mean abundance | Standard error | ||
| Membrane transport | |||||
| ABC transporters1 | 3.0271 | 0.5695 | 3.7117 | 0.7130 | 0.04149 |
| Signal transduction | |||||
| Phosphatidylinositol signaling system | 0.1022 | 0.0155 | 0.0845 | 0.0182 | 0.018959 |
| Signaling molecules and interaction | |||||
| G protein-coupled receptors1 | 1.76 × 10-5 | 3.63 × 10-5 | 6.54 × 10-5 | 1.02 × 10-5 | 0.02515 |
| Infectious diseases | |||||
| Staphylococcus aureus infection | 0.295 | 0.582 | 0.00545 | 0.00442 | 0.041492 |
| Biosynthesis of other secondary | |||||
| Metabolites; isoflavonoid biosynthesis | 8.97 × 10-6 | 8.03 × 10-6 | 9.75 × 10-7 | 1.39 × 10-7 | 0.003511 |
| Novobiocin biosynthesis1 | 0.1161 | 0.010 | 0.1277 | 0.0122 | 0.02494 |
| Carbohydrate metabolism | |||||
| C5-Branched dibasic acid metabolism1 | 0.287 | 0.0536 | 0.3214 | 0.0218 | 0.04149 |
| Glycan biosynthesis and metabolism | |||||
| Glycosphingolipid biosynthesis | 3.74 × 10-4 | 3.95 × 10-4 | 1.37 × 10-4 | 1.91× 10-4 | 0.032476 |
| Lipid metabolism | |||||
| Linoleic acid metabolism | 0.0851 | 0.0505 | 0.0518 | 0.0197 | 0.024942 |
| Primary bile acid biosynthesis | 0.0416 | 0.0186 | 0.0257 | 0.0120 | 0.018959 |
| Secondary bile acid biosynthesis | 0.0414 | 0.0187 | 0.0255 | 0.0120 | 0.018959 |
| Metabolism of cofactors and vitamins | |||||
| Porphyrin and chlorophy II metabolism1 | 0.7225 | 0.1723 | 0.8785 | 0.1079 | 0.03248 |
| Nucleotide metabolism | |||||
| Purine metabolism | 2.2171 | 0.2176 | 2.0283 | 0.1561 | 0.018959 |
| Xenobiotics biodegradation and metabolism | |||||
| Atrazine degradation1 | 0.0227 | 0.0252 | 0.0441 | 0.0329 | 0.03248 |
| Ethylbenzene degradation | 0.0769 | 0.0643 | 0.0429 | 0.00795 | 0.018959 |
| Naphthalene degradation | 0.1827 | 0.0823 | 0.1300 | 0.0261 | 0.018959 |
| Nitrotoluene degradation1 | 0.0651 | 0.0346 | 0.1022 | 0.0198 | 0.02494 |
| Styrene degradation1 | 0.0155 | 0.0131 | 0.0329 | 0.0231 | 0.02494 |
Indicated higher abundance in the healthy partners (P < 0.05). UC: Ulcerative colitis.
The lipid metabolic pathway, including primary bile acid biosynthesis, secondary bile acid biosynthesis (P = 0.019), and linoleic acid (P = 0.025), was enriched significantly in the UC patients. The pathway involved in purine metabolism was also enriched in the UC patients (P = 0.019). In contrast, there was a decrease in porphyrin and chlorophyll metabolism in the UC patients (P = 0.032). In addition, the microbiota of the UC patients had fewer ABC transporters for membrane transport (P = 0.041).
DISCUSSION
Alteration in the composition of the gut microbiota and decreases in community diversity are associated with the pathogenesis of UC[20]. However, our understanding of the interaction between couples cohabiting a shared environment in terms of their microbiota is poor. In this study, we analyzed the differences in the bacterial profiles and metabolic pathways between UC patients and their cohabiting partners. Marked microbiota dysbiosis and reduction of the diversity of Firmicutes were observed. Furthermore, to understand the functions of the different bacteria, we compared the differences in microbial metabolic pathways. Lipid metabolism and the biosynthesis of bile acids were significantly upregulated in the microbiota of the UC patients.
Unlike previous reports, we did not observe a significant difference in the OTU distribution associated with the disease state[21]. Additionally, we did not identify significant differences between UC patients and their partners in terms of the microbial alpha diversity using the Chao1, Shannon, and Simpson’s indices[20,22]. In contrast, we confirmed a compositional similarity between UC patients and their partners. Although there was no direct evidence that gut microbiota could spread between IBD patients and normal individuals, the results of this study suggested that fecal microbiota likely influence each other during long-term cohabitation with UC patients. Remarkably, our results indicated that the fecal microbiota composition was more similar among UC patients than among healthy individuals (Figure 1). These findings further demonstrated that the gut microbiota composition, and alterations to it, plays a crucial role in the occurrence of UC.
Although the cause of UC has many uncertain factors, gut microbiota dysbiosis has been considered a major trigger of inflammation[23]. Consistent with previous studies[22,24], we also found that the fecal microbiota in the UC patients in the present study had a decrease in Firmicutes bacteria, especially in Blautia, Clostridium, Coprococcus, and Roseburia bacteria (Figure 2). Although we realized that the gut microbial composition and its interaction with the host likely play an important role in IBD, the relationship between these has remained a mystery. Our research further confirmed that Firmicutes plays a crucial role in UC patients. The reduction in Clostridium likely decreased the utilization of short chain fatty acids and butyric acid salt in intestinal epithelial cells and induced inflammation. The depletion of Actinobacteria and Bacteroidetes is controversial, and we found that these bacteria were greatly depleted[21,25,26].
Interestingly, comparison of the relative abundance at the genus level using 16S rRNA gene sequencing from UC patients and their partners demonstrated that the composition of the fecal microbiota was dominated by Bacteroides in patients UC2 and UC6. By contrast, other samples, including those from healthy partners, did not show this situation. Additionally, among the healthy partners, HF4 and HF6 were dominated by Akkermansia and Klebsiella, respectively. We speculated that this microbiota dysbiosis is likely a consequence of interaction in the same environment.
In agreement with previous studies, we observed that lipid metabolism was remarkably increased[27]. Previous studies have suggested that commensal bacteria might increase or decrease certain specific metabolic pathways to participate in competition for limited energy resources while living in the host intestinal environment. Davenport et al[27] posited that because of a lack of carbohydrates in the inflamed regions, such as in the case of mucin production dysfunction, gut commensal bacteria start metabolizing lipids and amino acids for necessary nutrients. This hypothesis was supported by the decrease in Firmicutes, which are unable to utilize amino acids for energy[27,28].
Furthermore, one study found that patients with IBD may also suffer from the co-occurrence of primary biliary cirrhosis[29]. In the present study, we observed a significant increase in both primary bile acid biosynthesis and secondary bile acid biosynthesis of lipids (P = 0.019) in the UC patients (Table 2). In healthy individuals, it is difficult to detect bile acid in the blood. However, when liver cells are damaged, bile acid biosynthesis is abnormal and its concentration increases. We speculated that the increase in bile acid biosynthesis in UC patients might be associated with frequent liver disease, such as chronic liver disease or alloimmune liver disease. Of course, liver cell damage was also related to drug toxicity produced by long term medication and immune deficiency in UC patients. This would lead to oxidative stress, and some bacteria need to maintain homeostasis under oxidative stress. Interestingly, we observed an increase in linoleic acid metabolism in the UC patients (P = 0.025). Linoleic acid is a type of unsaturated fatty acid that is associated with prostaglandin biosynthesis, which could participate in liver injury protection.
In conclusion, this study presents a comprehensive evaluation of the bacterial composition and the differences in the pathways of UC fecal microbiota. Although our results were similar to the results of previous studies, we also identified an increased prevalence of lipid metabolic pathways and bile acid biosynthesis. The gut microbiota of the UC patients and their partners likely influenced each other. Furthermore, we verified that microbiota dysbiosis is more likely a consequence, rather than a cause, of inflammation.
COMMENTS
Background
Ulcerative colitis (UC) is one of the main types of inflammatory bowel disease. Compared with healthy people, some alterations in the microbial community are shared in Crohn’s disease and UC patients. However, some alterations in the bacterial community are specific to UC patients. The authors wondered whether cohabitation could influence the microbial community. Previous studies mainly focused on clinical symptoms and did not involve gut microbial community. Profiling the fecal microbiome using methods based on analysis of the 16S ribosomal RNA gene is less biased than cultivation-based approaches. Therefore, we investigated the gut bacterial community between UC patients and their healthy partners using 16S rRNA amplicon sequencing.
Research frontiers
Recently, the gut microbial community has attracted substantial attention, especially the influence in healthy and inflammatory bowel disease (IBD) patients. Some studies have demonstrated that environmental factors, including dietary age, habits, and obesity, impact the composition of the gut microbiota.
Innovations and breakthroughs
This study is the first to investigate the influence on the gut microbial community between UC patients and their cohabiting healthy partners. This study showed that microbial composition might affect healthy partners cohabiting with UC patients, especially in terms of microbiota dysbiosis.
Applications
In this study, the authors analyzed the differences in the bacterial profiles and metabolic pathways between UC patients and their cohabiting partners. The findings further demonstrated that the gut microbiota composition and alterations play a crucial role in the occurrence of UC. Furthermore, the gut microbiota of the UC patients and their partners likely influence each other.
Terminology
16S rRNA gene sequencing is a non-culture method based on high-throughput sequencing. At present, 16S rRNA gene sequencing has been utilized widely for metagenomic analysis of the environment, including analysis of the composition of the human and animal guts and fecal microbiota.
Peer-review
Gut dysbiosis is considered one of the factors inducing inflammation in chronic IBD; however, its role in the etiology of ulcerative colitis is controversial. There have been many studies on fecal microbiota in recent years. The present study used a very sensitive method to assess bacterial strains and compared the microbiota in patients with inflammatory bowel disease with that of their healthy partners. The study assessed some type of bacteria only found in patients with UC. These bacteria are difficult to detect using less sensitive methods.
Footnotes
Manuscript source: Unsolicited manuscript
Specialty type: Gastroenterology and hepatology
Country of origin: China
Peer-review report classification
Grade A (Excellent): A
Grade B (Very good): 0
Grade C (Good): 0
Grade D (Fair): 0
Grade E (Poor): 0
Institutional review board statement: This study was approved by the Ethics Committee of Lishui People’s Hospital.
Informed consent statement: All study participants, or their legal guardian, provided informed written consent prior to study enrollment.
Conflict-of-interest statement: To the best of our knowledge, no conflicts of interest exist.
Data sharing statement: No additional data are available.
Peer-review started: February 16, 2017
First decision: April 7, 2017
Article in press: May 19, 2017
P- Reviewer: Bordas JM S- Editor: Qi Y L- Editor: Stewart G E- Editor: Zhang FF
References
- 1.Molodecky NA, Soon IS, Rabi DM, Ghali WA, Ferris M, Chernoff G, Benchimol EI, Panaccione R, Ghosh S, Barkema HW, et al. Increasing incidence and prevalence of the inflammatory bowel diseases with time, based on systematic review. Gastroenterology. 2012;142:46–54.e42; quiz e30. doi: 10.1053/j.gastro.2011.10.001. [DOI] [PubMed] [Google Scholar]
- 2.Ng SC. Epidemiology of inflammatory bowel disease: focus on Asia. Best Pract Res Clin Gastroenterol. 2014;28:363–372. doi: 10.1016/j.bpg.2014.04.003. [DOI] [PubMed] [Google Scholar]
- 3.Flint HJ, Scott KP, Louis P, Duncan SH. The role of the gut microbiota in nutrition and health. Nat Rev Gastroenterol Hepatol. 2012;9:577–589. doi: 10.1038/nrgastro.2012.156. [DOI] [PubMed] [Google Scholar]
- 4.Sokol H, Seksik P, Rigottier-Gois L, Lay C, Lepage P, Podglajen I, Marteau P, Doré J. Specificities of the fecal microbiota in inflammatory bowel disease. Inflamm Bowel Dis. 2006;12:106–111. doi: 10.1097/01.MIB.0000200323.38139.c6. [DOI] [PubMed] [Google Scholar]
- 5.Sokol H, Lepage P, Seksik P, Doré J, Marteau P. Temperature gradient gel electrophoresis of fecal 16S rRNA reveals active Escherichia coli in the microbiota of patients with ulcerative colitis. J Clin Microbiol. 2006;44:3172–3177. doi: 10.1128/JCM.02600-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sokol H, Lay C, Seksik P, Tannock GW. Analysis of bacterial bowel communities of IBD patients: what has it revealed? Inflamm Bowel Dis. 2008;14:858–867. doi: 10.1002/ibd.20392. [DOI] [PubMed] [Google Scholar]
- 7.Kostic AD, Xavier RJ, Gevers D. The microbiome in inflammatory bowel disease: current status and the future ahead. Gastroenterology. 2014;146:1489–1499. doi: 10.1053/j.gastro.2014.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ohkusa T, Sato N, Ogihara T, Morita K, Ogawa M, Okayasu I. Fusobacterium varium localized in the colonic mucosa of patients with ulcerative colitis stimulates species-specific antibody. J Gastroenterol Hepatol. 2002;17:849–853. doi: 10.1046/j.1440-1746.2002.02834.x. [DOI] [PubMed] [Google Scholar]
- 9.Martín-de-Carpi J, Rodríguez A, Ramos E, Jiménez S, Martínez-Gómez MJ, Medina E; SPIRIT-IBD Working Group of Sociedad Española de Gastroenterología, Hepatología y Nutricion Pediátrica. Increasing incidence of pediatric inflammatory bowel disease in Spain (1996-2009): the SPIRIT Registry. Inflamm Bowel Dis. 2013;19:73–80. doi: 10.1002/ibd.22980. [DOI] [PubMed] [Google Scholar]
- 10.Wu GD, Chen J, Hoffmann C, Bittinger K, Chen YY, Keilbaugh SA, Bewtra M, Knights D, Walters WA, Knight R, et al. Linking long-term dietary patterns with gut microbial enterotypes. Science. 2011;334:105–108. doi: 10.1126/science.1208344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Turnbaugh PJ, Hamady M, Yatsunenko T, Cantarel BL, Duncan A, Ley RE, Sogin ML, Jones WJ, Roe BA, Affourtit JP, et al. A core gut microbiome in obese and lean twins. Nature. 2009;457:480–484. doi: 10.1038/nature07540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Comes MC, Gower-Rousseau C, Colombel JF, Belaïche J, Van Kruiningen HJ, Nuttens MC, Cortot A. Inflammatory bowel disease in married couples: 10 cases in Nord Pas de Calais region of France and Liège county of Belgium. Gut. 1994;35:1316–1318. doi: 10.1136/gut.35.9.1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shen H, Ye F, Xie L, Yang J, Li Z, Xu P, Meng F, Li L, Chen Y, Bo X, et al. Metagenomic sequencing of bile from gallstone patients to identify different microbial community patterns and novel biliary bacteria. Sci Rep. 2015;5:17450. doi: 10.1038/srep17450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jiang W, Wu N, Wang X, Chi Y, Zhang Y, Qiu X, Hu Y, Li J, Liu Y. Dysbiosis gut microbiota associated with inflammation and impaired mucosal immune function in intestine of humans with non-alcoholic fatty liver disease. Sci Rep. 2015;5:8096. doi: 10.1038/srep08096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Loman NJ, Misra RV, Dallman TJ, Constantinidou C, Gharbia SE, Wain J, Pallen MJ. Performance comparison of benchtop high-throughput sequencing platforms. Nat Biotechnol. 2012;30:434–439. doi: 10.1038/nbt.2198. [DOI] [PubMed] [Google Scholar]
- 16.Quail MA, Smith M, Coupland P, Otto TD, Harris SR, Connor TR, Bertoni A, Swerdlow HP, Gu Y. A tale of three next generation sequencing platforms: comparison of Ion Torrent, Pacific Biosciences and Illumina MiSeq sequencers. BMC Genomics. 2012;13:341. doi: 10.1186/1471-2164-13-341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hedin C, van der Gast CJ, Rogers GB, Cuthbertson L, McCartney S, Stagg AJ, Lindsay JO, Whelan K. Siblings of patients with Crohn’s disease exhibit a biologically relevant dysbiosis in mucosal microbial metacommunities. Gut. 2016;65:944–953. doi: 10.1136/gutjnl-2014-308896. [DOI] [PubMed] [Google Scholar]
- 18.Langille MG, Zaneveld J, Caporaso JG, McDonald D, Knights D, Reyes JA, Clemente JC, Burkepile DE, Vega Thurber RL, Knight R, et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat Biotechnol. 2013;31:814–821. doi: 10.1038/nbt.2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ye F, Shen H, Li Z, Meng F, Li L, Yang J, Chen Y, Bo X, Zhang X, Ni M. Influence of the Biliary System on Biliary Bacteria Revealed by Bacterial Communities of the Human Biliary and Upper Digestive Tracts. PLoS One. 2016;11:e0150519. doi: 10.1371/journal.pone.0150519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Forbes JD, Van Domselaar G, Bernstein CN. Microbiome Survey of the Inflamed and Noninflamed Gut at Different Compartments Within the Gastrointestinal Tract of Inflammatory Bowel Disease Patients. Inflamm Bowel Dis. 2016;22:817–825. doi: 10.1097/MIB.0000000000000684. [DOI] [PubMed] [Google Scholar]
- 21.Frank DN, St Amand AL, Feldman RA, Boedeker EC, Harpaz N, Pace NR. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc Natl Acad Sci USA. 2007;104:13780–13785. doi: 10.1073/pnas.0706625104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Manichanh C, Rigottier-Gois L, Bonnaud E, Gloux K, Pelletier E, Frangeul L, Nalin R, Jarrin C, Chardon P, Marteau P, et al. Reduced diversity of faecal microbiota in Crohn’s disease revealed by a metagenomic approach. Gut. 2006;55:205–211. doi: 10.1136/gut.2005.073817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hijova E, Soltesova A. Effects of probiotics and prebiotics in ulcerative colitis. Bratisl Lek Listy. 2013;114:540–543. doi: 10.4149/bll_2013_113. [DOI] [PubMed] [Google Scholar]
- 24.Gophna U, Sommerfeld K, Gophna S, Doolittle WF, Veldhuyzen van Zanten SJ. Differences between tissue-associated intestinal microfloras of patients with Crohn’s disease and ulcerative colitis. J Clin Microbiol. 2006;44:4136–4141. doi: 10.1128/JCM.01004-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang W, Chen L, Zhou R, Wang X, Song L, Huang S, Wang G, Xia B. Increased proportions of Bifidobacterium and the Lactobacillus group and loss of butyrate-producing bacteria in inflammatory bowel disease. J Clin Microbiol. 2014;52:398–406. doi: 10.1128/JCM.01500-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gevers D, Kugathasan S, Denson LA, Vázquez-Baeza Y, Van Treuren W, Ren B, Schwager E, Knights D, Song SJ, Yassour M, et al. The treatment-naive microbiome in new-onset Crohn’s disease. Cell Host Microbe. 2014;15:382–392. doi: 10.1016/j.chom.2014.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Davenport M, Poles J, Leung JM, Wolff MJ, Abidi WM, Ullman T, Mayer L, Cho I, Loke P. Metabolic alterations to the mucosal microbiota in inflammatory bowel disease. Inflamm Bowel Dis. 2014;20:723–731. doi: 10.1097/MIB.0000000000000011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kamada N, Chen GY, Inohara N, Núñez G. Control of pathogens and pathobionts by the gut microbiota. Nat Immunol. 2013;14:685–690. doi: 10.1038/ni.2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Raszeja-Wyszomirska J, Wunsch E, Krawczyk M, Rigopoulou EI, Kostrzewa K, Norman GL, Bogdanos DP, Milkiewicz P. Assessment of health related quality of life in polish patients with primary biliary cirrhosis. Clin Res Hepatol Gastroenterol. 2016;40:471–479. doi: 10.1016/j.clinre.2015.10.006. [DOI] [PubMed] [Google Scholar]