

Abstract
Endothelial cells are essential for normal lung function: they sense and respond to circulating factors and hemodynamic alterations. In inflammatory lung diseases such as acute respiratory distress syndrome, endothelial cell apoptosis is an inciting event in pathogenesis and a prominent pathological feature. Endothelial cell apoptosis is mediated by circulating inflammatory factors, which bind to receptors on the cell surface, activating signal transduction pathways, leading to caspase-3-mediated apoptosis. We hypothesized that yes and src have differential effects on caspase-3 activation in human pulmonary microvascular endothelial cells (hPMVEC) due to differential downstream signaling effects. To test this hypothesis, hPMVEC were treated with siRNA against src (siRNAsrc), siRNA against yes (siRNAyes), or their respective scramble controls. After recovery, the hPMVEC were treated with cytomix (LPS, IL-1β, TNF-α, and IFN-γ). Treatment with cytomix induced activation of the extracellular signal-regulated kinase (ERK) pathway and caspase-3-mediated apoptosis. Treatment with siRNAsrc blunted cytomix-induced ERK activation and enhanced cleaved caspase-3 levels, while treatment with siRNAyes enhanced cytomix-induced ERK activation and attenuated levels of cleaved caspase-3. Inhibition of the ERK pathway using U0126 enhanced cytomix-induced caspase-3 activity. Treatment of hPMVEC with cytomix induced Akt activation, which was inhibited by siRNAsrc. Inhibition of the phosphatidylinositol 3-kinase/Akt pathway using LY294002 prevented cytomix-induced ERK activation and augmented cytomix-induced caspase-3 cleavage. Together, our data demonstrate that, in hPMVEC, yes activation blunts the ERK cascade in response to cytomix, resulting in greater apoptosis, while cytomix-induced src activation induces the phosphatidylinositol 3-kinase pathway, which leads to activation of Akt and ERK and attenuation of apoptosis.
Keywords: caspase-3, extracellular signal-regulated kinase, cytokines
inflammation is involved in the pathogenesis of almost all lung diseases. As one important example, inflammation underlies the acute lung injury (ALI) that leads to acute respiratory distress syndrome (ARDS) (34, 36). ALI/ARDS usually develops in hospitalized patients within 24–72 h of the onset of their illness and is secondary to inflammatory conditions such as sepsis, pneumonia, and trauma (34, 36). Treatments for ALI/ARDS are largely supportive, consisting of treatment of the underlying symptoms, as well as judicious mechanical ventilation and fluid management (27). There is a real need for the development of pharmacotherapies directed at ALI/ARDS (4), particularly pharmacotherapies that could be given within 24–72 h of onset to prevent or attenuate the development of full-blown ARDS (25). Another example of an inflammatory lung disease is bronchopulmonary dysplasia (BPD). Much like ALI/ARDS, BPD develops in preterm infants secondary to ALI caused by inflammatory conditions such as surfactant deficiency, pneumonia, and sepsis (11, 35).
Apoptosis is central to the pathogenesis of the lung injury leading to ALI/ARDS and BPD. In ALI/ARDS, endothelial cell apoptosis is well established as a key step in the formation of pulmonary edema and inflammatory cell infiltration, which are the hallmarks of the disease (10, 36). Hashimoto et al. (13) found significant mRNA and protein markers for apoptotic mediators in bronchoalveolar lavage fluid from patients with early ALI/ARDS. In an autopsy study of BPD patients, endothelial apoptosis was substantially greater in the lungs from patients with BPD than in age-matched controls (22). Animal models of BPD also showed that apoptosis is central to the pathogenesis of the disease (1, 8, 23).
The Src family of tyrosine kinases (STK) represents a group of closely related evolutionally conserved nonreceptor protein tyrosine kinases defined by a common molecular structure (3, 17) with distinct structural motifs termed Src homology (SH) domains. The STK consist of an NH2-terminal sequence (the SH4 domain) that directs myristoylation, resulting in membrane localization, followed by a nonconserved unique domain, an SH3 domain that directs binding to polyproline-rich sequences, an SH2 domain that binds phosphotyrosine, a catalytic domain (the SH1 domain), which includes the positive regulator site tyrosine 416, and a short COOH-terminal tail that contains a negative regulator site, tyrosine 527 (3, 17). We previously showed in human pulmonary microvascular endothelial cells (hPMVEC) that activation of the STK is necessary for LPS-induced inducible nitric oxide (NO) synthase expression (5). The STK include at least nine members: src, fyn, yes, lyn, lck, hck, blk, fgr, and yrk; however, yrk has not been found in humans. We previously reported that hPMVEC express yes, src, and fyn (5). The signal transduction pathways that mediate apoptosis and caspase-3 activation in endothelial cells are not well understood. We previously showed in hPMVEC that the individual STK could be knocked down using specific siRNA and demonstrated that cytomix-induced urea production was differentially regulated by the various STK family members (5). Moreover, studies using hPMVEC have revealed differential effects of src, fyn, and yes on TNF-α-induced permeability (2). Therefore, we hypothesize that the STK src and yes will have opposing effects on cytokine-induced apoptosis in hPMVEC and that these opposing effects will be due to differential downstream signaling effects.
METHODS
Human pulmonary microvascular endothelial cells.
hPMVEC (Lonza, Allendale, NJ) were grown in six-well plates according to the manufacturer's recommendations using endothelial cell basal medium (EBM2, Lonza) supplemented with an EGM-2 bullet kit (Lonza). On the day of study, the hPMVEC were washed three times with 2 ml of Hanks' balanced salt solution (HBSS). Then 1 ml of medium was placed on the hPMVEC (control), and the cells were cultured in an incubator for 24 h at 37°C in 5% CO2-balance air. Included in the medium used to culture the cytomix-treated hPMVEC were 1.5 μg/ml LPS, 3 ng/ml TNF-α, 2 ng/ml IL-1β, and 30 ng/ml INF-γ (all from Sigma Chemical, St. Louis, MO), as previously described (5). After 24 h, the medium was removed and frozen at −70°C. The hPMVEC were washed three times with 4 ml of HBSS and treated with lysis buffer for protein extraction. A transfection reagent provided by the manufacturer (Dharmacon) was used to transfect the hPMVEC with the siRNA of interest (Dharmacon) or a scramble siRNA as a control, as previously described (5, 33). We previously showed that the siRNAs against the STK (src, fyn, yes, and lyn) are specific for their targeted family member and do not alter expression of the other family members in the hPMVEC (see Fig. 4D in Ref. 5). After 48 h, the hPMVEC were washed three times, and medium containing cytomix or vehicle was added to the cell culture wells. After an additional 24 h of incubation, the medium was harvested, and the cells were lysed for protein extraction.
Fig. 4.
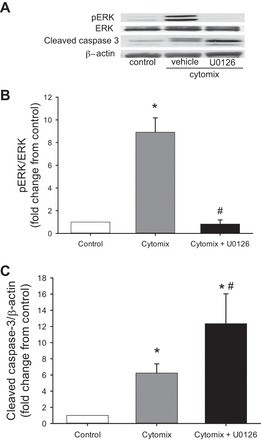
Inhibition of ERK signaling augmented cytomix-induced cleaved caspase-3 expression. Cells were untreated (controls) or were treated with vehicle or 10 μM U0126; cells treated with vehicle or U0126 were also given cytomix (n = 3 for each condition). After 24 h, protein was harvested for Western blotting of pERK, and membranes were stripped and reprobed for total amounts of ERK. Western blotting was also done for cleaved caspase-3, and these membranes were stripped and reprobed for β-actin. A: representative Western blots for pERK, cleaved caspase-3, and β-actin from control cells and vehicle- and U0126-treated cytomix-stimulated cells. B: densitometry data for pERK demonstrating that U0126 prevented cytomix-induced pERK expression. C: densitometry data for cleaved caspase-3 demonstrating substantially greater cytomix-induced cleaved caspase-3 expression in U0126- than vehicle-treated hPMVEC. *P < 0.05, cytomix vs. control. #P < 0.05, U0126 + cytomix vs. cytomix.
Protein isolation.
Protein was isolated from the hPMVEC as previously described (5, 7, 33). Briefly, the cells were washed with HBSS, and lysis buffer (0.2 M NaOH and 0.2% SDS) was added. At 30 min before the hPMVEC were used, the following protease inhibitors were added to each milliliter of lysis buffer: 0.2 μl of aprotinin (10 mg/ml double-distilled H2O), 0.5 μl of leupeptin (10 mg/ml double-distilled H2O), 0.14 μl of pepstatin A (5 mg/ml methanol), and 5 μl of phenylmethylsulfonyl fluoride (34.8 mg/ml methanol). The cells were scraped and placed in sterile centrifuge tubes on ice. The supernatant was stored in 1-ml tubes at −70°C for Western blot analysis. Total protein concentration was determined by the Bradford method using a commercially available assay (Bio-Rad, Hercules, CA).
Western blotting.
Cell lysates were assayed for cleaved caspase-3, fyn, src, yes, β-actin, phosphorylated and total amounts of extracellular signal-regulated kinase (ERK), c-Jun NH2-terminal kinase (JNK), and p38, as well as phosphorylated and total Akt, using Western blot analysis, as previously described (5, 16). Aliquots of cell lysate were diluted 1:1 with SDS sample buffer, heated to 80°C for 15 min, and then centrifuged at 10,000 g at room temperature for 2 min. Aliquots of the supernatant were used for SDS-polyacrylamide gel electrophoresis. The proteins were transferred to polyvinylidene difluoride membranes and blocked overnight in PBS with 0.1% Tween containing 5% nonfat dry milk and 3% albumin. The membranes were then incubated with the primary antibody of interest (cleaved caspase-3, 1:1,000 dilution; Cell Signaling Technology, Danvers, MA; all others, 1:1,000 dilution; Transduction Laboratories, Lexington, KY). The blots were washed with PBS with 0.1% Tween and 1% nonfat dry milk. The membranes were then incubated with the biotinylated IgG secondary antibody (1:5,000 dilution; Vector Laboratories, Burlingame, CA) for 1 h, washed, and incubated with streptavidin-horseradish peroxidase conjugate (1:1,500 dilution; Bio-Rad) for 30 min. The bands of interest were visualized using enhanced chemiluminescence (Amersham, Piscataway, NJ) and quantified using densitometry (Sigma Gel, Jandel Scientific, San Rafael, CA). To control for protein loading, the blots were stripped using a stripping buffer (62.5 mM Tris·HCl, pH 6.8, 2% SDS, and 100 mM β-mercaptoethanol). The blots for cleaved caspase-3, fyn, src, and yes were reprobed for β-actin (1:10,000 dilution; Abcam, Cambridge, MA), while the blots for the phosphorylated proteins were reprobed for the total amounts of the protein of interest [e.g., ERK for phosphorylated ERK (pERK) and Akt for phosphorylated Akt (pAkt)], as described above. The quantified densitometry data are shown as the protein of interest over the appropriate protein loading control normalized to the values for control such that control values are 1.
Caspase-3 activity.
Caspase-3 activity was measured in cell lysates using a fluorescence-based assay (BioVision, Mountain View, CA).
Proliferation assay.
Proliferation of the hPMVEC was determined in six-well plates, as previously described (33). Cultured hPMVEC, 5 × 104 cells, were plated into each well of six-well plates. The appropriate reagents were included in the medium, and the cells were incubated for 48 h. At the end of the experimental protocol, the cells were removed from the incubator and the plates were washed three times with HBSS. After the final wash, 1 ml of trypsin was added to each well, and the plates were incubated for 3 min; then the plates were incubated with 2 ml of trypsin-neutralizing solution. The cells from each well were placed in 15-ml conical tubes and centrifuged for 5 min at 1,220 g at 4°C. The supernatant was discarded, and the cells were resuspended in 1 ml of EGM. The cells were mixed 1:1 with Trypan blue and viable cells; in other words, cells that were not blue, were counted using a hemocytometer.
Experimental protocols.
To examine the effect of src or yes knockdown on caspase-3 activity, we utilized Western blotting and activity assays for caspase-3. The hPMVEC were not transfected (control) or were transfected with a scramble siRNA or a siRNA against src (siRNAsrc) for 48 h. In parallel studies, the hPMVEC were not transfected or were transfected with a scramble siRNA or a siRNA against yes (siRNAyes) for 48 h. Then the cells were treated with cytomix for 24 h, and protein was harvested for Western blotting and activity assays for caspase-3.
To examine the effect of selective knockdown of src or yes on viable cell numbers, we used siRNA-induced selective knockdown of src or yes and Trypan blue exclusion to quantify viable cell numbers. The cells were not treated (controls) or were treated with a scramble siRNA, siRNAsrc, or siRNAyes. After 48 h, the cells were washed and trypsinized, and 5 × 104 cells were plated in each well of a six-well plate. The siRNA-transfected cells were treated with cytomix, while controls were left untreated; after 48 h, the number of viable cells was counted using Trypan blue exclusion.
To investigate the role of src and yes in cytomix-induced activation of the mitogen-activated protein kinases (MAPK), the hPMVEC were treated with siRNAsrc or a scramble siRNA; in parallel experiments, the hPMVEC were treated with siRNAyes or a scramble siRNA. After 48 h, the cells were washed and then treated with cytomix for an additional 24 h. Protein was harvested for Western blotting for total and phosphorylated ERK, p38, and JNK.
Next we looked at the effect of an ERK pathway inhibitor, U0126, on levels of cleaved caspase-3 in cytomix-treated hPMVEC. The cells were untreated (controls) or were treated with vehicle or 10 μM U0126 and also given cytomix. After 24 h, protein was harvested for Western blotting of phosphorylated and total ERK, JNK, and p38.
To determine if cytomix-induced src and/or yes signaling activates the phosphatidylinositol 3-kinase (PI3K) pathway, we examined the activation of Akt following treatment with scramble siRNA, siRNAsrc, or siRNAyes. The cells were transfected with scramble RNA, siRNAsrc, or siRNAyes for 48 h or were not transfected (controls). The cells were then washed and treated with cytomix for an additional 24 h, and the protein was subsequently harvested for Western blotting for phosphorylated and total Akt.
To determine if inhibition of PI3K activation would affect cytomix-induced cleaved caspase-3 protein levels, we utilized the putative PI3K inhibitor LY294002. The cells were not treated (controls) or were treated with cytomix together with vehicle or 10 μM LY294002; after 24 h, protein was harvested for Western blotting to detect cleaved caspase-3.
To determine if ERK plays a role in the inhibitory effect of PI3K on cytomix-induced cleaved caspase-3 protein levels, the hPMVEC were not treated (controls) or were treated with cytomix in the presence or absence of 10 μM LY294002. After 24 h, protein was harvested for Western blotting for phosphorylated and total ERK, JNK, and p38, respectively.
Statistical analysis.
Values are means ± SE. One-way analysis of variance was used to compare the densitometry data between control and treated cells or cellular proliferation. Significant differences were identified using a Newman-Keuls post hoc test (SigmaStat, Jandel Scientific, Carlsbad, CA). Differences were considered significant when P < 0.05.
RESULTS
Knockdown of src augmented cytomix-induced caspase-3 activation, while knockdown of yes attenuated cytomix-induced caspase-3 activation.
Transfection of the hPMVEC with a scramble siRNA and cytomix had little effect on total src protein levels, while transfection with the siRNAsrc led to a dramatic src knockdown (Fig. 1A). Levels of cleaved caspase-3 protein were greater in the scramble siRNA-treated hPMVEC stimulated with cytomix. Levels of cleaved caspase-3 protein were greater in the hPMVEC treated with siRNAsrc and cytomix than in those treated with the scramble siRNA and cytomix (Fig. 1B). This observation was confirmed with the caspase-3 activity assays. Caspase-3 activity was significantly greater in scramble siRNA-transfected hPMVEC following cytomix treatment than in control cells, and cytomix-induced caspase-3 activity was substantially greater in siRNAsrc-treated hPMVEC than in controls or scramble-treated hPMVEC (Fig. 1C). Similar results were found when an siRNA against fyn was substituted for siRNAsrc (data not shown).
Fig. 1.
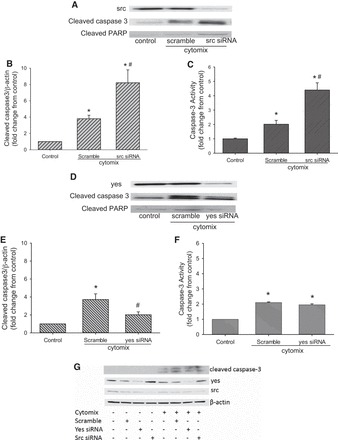
Knockdown of src augmented cytomix-induced caspase-3 activation, while knockdown of yes had less effect on cytomix-induced caspase-3 activation. A–C: human pulmonary microvascular endothelial cells (hPMVEC) were treated with a scramble siRNA (scramble) or siRNAsrc for 48 h, and scramble- and siRNAsrc-treated cells were stimulated with cytomix for 24 h. A: representative Western blot showing knockdown of src, as well as cleaved caspase-3 and cleaved poly(ADP-ribose) polymerase (PARP). B: densitometry data for cleaved caspase-3 normalized to β-actin (membranes were stripped and reprobed for β-actin; n = 3 for each condition). C: caspase-3 activity in cell lysates (n = 3 for each condition). D–F: hPMVEC were treated with scramble or siRNAyes for 48 h and stimulated with cytomix for 24 h. D: representative Western blot showing knockdown of yes, as well as attenuation of cleaved caspase-3 and cleaved PARP expression. E: densitometry data for cleaved caspase-3 normalized to β-actin (n = 7 for each condition). F: caspase-3 activity in cell lysates (n = 5 for each condition). *P < 0.01, cytomix vs. control. #P < 0.05, siRNA vs. scramble. G: hPMVEC were not treated or were treated with scramble, siRNAyes, or siRNAsrc for 48 h and then treated with vehicle or cytomix for 24 h. Experiment was repeated 3 times. Protein was harvested for Western blotting for cleaved caspase-3, total yes, total src, and β-actin. Representative Western blot demonstrates that transfection in the absence of cytomix had no effect on cleaved caspase-3 levels in hPMVEC. Furthermore, representative yes and src blots demonstrate that the siRNA were specific for their targeted member of the Src family of tyrosine kinases (STK).
We previously found that selective knockdown of yes had little effect on urea and NO production in cytomix-treated hPMVEC, while selective knockdown of fyn prevented cytomix-induced urea and NO production in hPMVEC (5). As expected, siRNAyes effectively knocked down cytomix-induced yes protein expression (Fig. 1D). Cytomix-induced levels of cleaved caspase-3 were somewhat lower in the hPMVEC transfected with siRNAyes than in scramble-transfected cytomix-stimulated hPMVEC (Fig. 1E). However, the levels of cytomix-induced caspase-3 activity in cells transfected with siRNAyes were not significantly different from those in scramble-transfected hPMVEC (Fig. 1F).
To determine if transfection with the scramble siRNAs had an effect on cleaved caspase-3 expression, the hPMVEC were treated as described above with an additional group of scramble-transfected, vehicle-treated cells included. A representative Western blot, shown in Fig. 1G, demonstrates that transfection in the absence of cytomix had no effect on cleaved caspase-3 levels in the hPMVEC. Furthermore, the representative yes and src Western blots (Fig. 1G) demonstrate that the siRNAs were specific for their targeted STK, as we showed previously (5).
Knockdown of src, but not yes, leads to decreased viable cell numbers.
There were significantly fewer viable scramble siRNA-transfected cells treated with cytomix than control cells in all experiments (Fig. 2). Selective knockdown of src resulted in substantially fewer viable cells than scramble siRNA transfection following cytomix treatment (Fig. 2). In contrast, selective knockdown of yes resulted in similar numbers of viable cells, as seen in scramble-transfected cells following cytomix treatment, such that, following cytomix stimulation, there were significantly more viable siRNAyes- than siRNAsrc-transfected hPMVEC (Fig. 2).
Fig. 2.
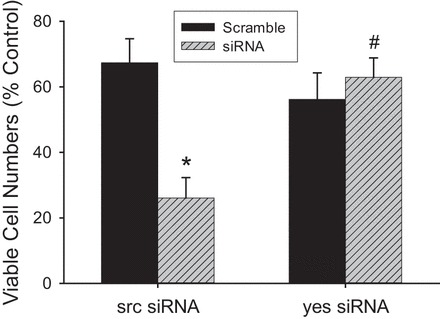
Knockdown of src resulted in substantially fewer viable cells following cytomix treatment. hPMVEC (n = 3 for each condition) were not treated (controls) or were treated with scramble, siRNAsrc, or siRNAyes for 48 h. Cells were then washed and trypsinized, and 5 × 104 cells were plated in each well of a 6-well plate. siRNA-transfected cells were treated with cytomix, while controls were left untreated; after 48 h, viable cells were counted using Trypan blue exclusion. There were significantly fewer viable cells among those treated with cytomix (i.e., scramble and siRNA; P < 0.01) than among the untreated (control) cells. There were substantially fewer viable siRNAsrc-treated (P < 0.005) than scramble-treated cells, while siRNAyes had a negligible effect on the number of viable cells following cytomix treatment. The effect of siRNAyes on viable cell number was significantly different from the effect of siRNAsrc. *P < 0.01, siRNA vs. scramble. #P < 0.01, siRNAyes vs. siRNAsrc.
Knockdown of src resulted in lower levels of cytomix-induced phosphorylated ERK levels, while knockdown of yes augmented cytomix-induced pERK levels, in the hPMVEC.
Levels of pERK were lower in the hPMVEC treated with cytomix and siRNAsrc than in cytomix-stimulated scramble siRNA-transfected cells (Fig. 3A). siRNAsrc did not affect levels of phosphorylated JNK (pJNK) or phosphorylated p38 (pp38) (Fig. 3, C and D). Levels of pERK were greater in cytomix-stimulated hPMVEC treated with siRNAyes than in scramble-treated cells (Fig. 3B). However, knockdown of yes had little effect on pJNK or pp38 levels in cytomix-treated hPMVEC (Fig. 3, D and F).
Fig. 3.
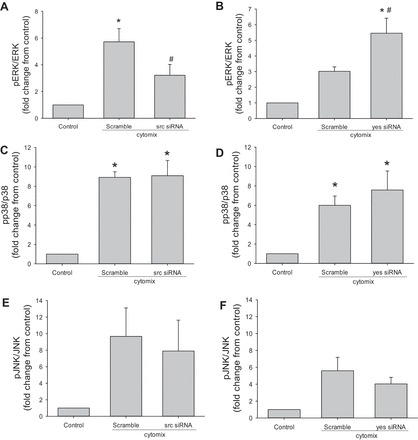
Of the MAPK, only cytomix-induced ERK activation was affected by src or yes knockdown. hPMVEC (n = 4 for each condition) were treated with siRNAsrc or scramble for 48 h, washed, and then treated with cytomix. After 24 h, protein was harvested for Western blotting of phosphorylated ERK (pERK), JNK (pJNK), or p38 (pp38); then the membranes were then stripped and reprobed for total amounts of ERK, JNK, or p38, respectively. Intensities of the bands were quantified by densitometry normalized to the respective total protein level. Experiment was repeated using siRNAyes and scramble, as described above. A: pERK levels were lower in siRNAsrc- than scramble-treated cells. B: pERK levels were augmented in siRNAyes- compared with scramble-treated cells. C and E: knockdown of src had little effect on cytomix-induced levels of pp38 or pJNK. D and F: knockdown of yes had little effect on cytomix-induced levels of pp38 or pJNK. *P < 0.05, cytomix vs. control. #P < 0.05, siRNA vs. scramble.
Inhibition of ERK signaling augmented cytomix-induced caspase-3 activity.
pERK levels were greater in the hPMVEC treated with cytomix alone than in controls (Fig. 4, A and B). pERK levels in the hPMVEC treated with cytomix and U0126 were similar to those in the control cells (Fig. 4, A and B). Cleaved caspase-3 protein levels were greater in the cells treated with cytomix than in control cells, and inhibition of ERK signaling with U0126 significantly augmented the cytomix-induced levels of cleaved caspase-3 (Fig. 4C).
Knockdown of src, but not yes, resulted in lower levels of pAkt in cytomix-treated hPMVEC.
Cytomix treatment resulted in greater levels of pAkt in scramble siRNA-treated cells (Fig. 5). siRNAsrc prevented the cytomix-induced increase in pAkt levels in the hPMVEC (Fig. 5A), while siRNAyes had no discernible effect on levels of pAkt protein in the hPMVEC (Fig. 5, C and D).
Fig. 5.

Knockdown of src, but not yes, resulted in lower levels of phosphorylated Akt (pAkt) in cytomix-treated hPMVEC. hPMVEC were not treated (controls) or were treated with scramble, siRNAsrc, or siRNAyes for 48 h (n = 5 for each condition). Cells were then washed, and scramble- and siRNA-treated cells were treated with cytomix for an additional 24 h. Protein was harvested for Western blotting for pAkt and total Akt. A: representative Western blot for control cells or cells treated with scramble or siRNAsrc. B: densitometry data using pAkt levels normalized to total Akt levels. pAkt levels were substantially greater in cytomix-treated than control cells, and siRNAsrc prevented the cytomix-induced phosphorylation of Akt. C: representative Western blot for control cells or cells treated with scramble or siRNAyes. D: densitometry data showing that cytomix-induced phosphorylation of Akt was essentially unaffected by siRNAyes. *P < 0.01, cytomix vs. control. #P < 0.01, siRNA vs. scramble.
Inhibition of PI3K augmented cytomix-induced cleaved caspase-3 expression.
While cytomix treatment resulted in an increase in cleaved caspase-3 protein levels in cells treated with vehicle relative to control cells, treatment with LY294002 resulted in a substantial increase in cleaved caspase-3 protein levels relative to cytomix-stimulated cells treated with vehicle (Fig. 6).
Fig. 6.
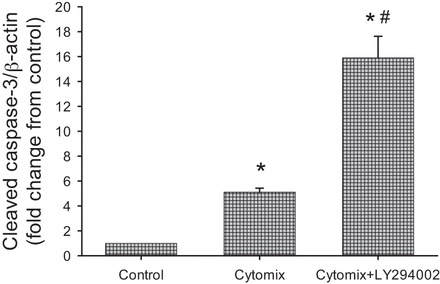
Inhibition of Akt augmented cytomix-induced cleaved caspase-3 expression. hPMVEC were not treated (controls) or were treated with cytomix and vehicle or 10 μM LY294002 (a putative inhibitor of the phosphatidylinositol 3-kinase/Akt pathway; n = 9 for each condition). After 24 h, protein was harvested for cleaved caspase-3 Western blotting. Levels of cytomix-induced cleaved caspase-3 protein were substantially higher in LY294002- than vehicle-treated hPMVEC. *P < 0.01, cytomix vs. control. #P < 0.01, LY294002 vs. vehicle.
Inhibition of PI3K activation prevented cytomix-induced ERK activation.
Levels of cytomix-induced pERK protein were significantly lower in LY294002- than vehicle-treated cells (Fig. 7A). Treatment of the hPMVEC with LY294002 had little effect on cytomix-induced levels of pp38 protein (Fig. 7B) or pJNK (Fig. 7C). These results suggest that PI3K inhibits apoptosis in cytomix-treated cells via the Akt and ERK pathways.
Fig. 7.
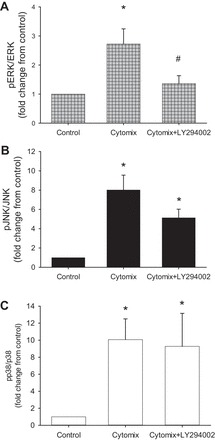
Inhibition of Akt activation prevented cytokine-induced pERK expression. hPMVEC were not treated (controls) or were treated with cytomix in the presence or absence of 10 μM LY294002 (n = 7 for each condition). After 24 h, protein was harvested for Western blotting for pERK, pJNK, or pp38, and blots were stripped and reprobed for total ERK, JNK, or p38, respectively. Treatment with LY294002 prevented only cytomix-induced pERK protein expression (A), with no significant effect on pJNK (B) or pp38 (C) expression. *P < 0.05, cytomix vs. control. #P < 0.05, LY294002 vs. vehicle.
DISCUSSION
In the present study we demonstrate that the specific STK family members src and yes have differing effects on cytomix-induced apoptosis in hPMVEC. Inhibition of src using an siRNA resulted in a substantial augmentation of cytomix-induced cleaved caspase-3 protein expression and activity. In contrast, inhibition of yes with siRNAyes resulted in a small decrease in cleaved caspase-3 protein expression. The changes in cleaved caspase-3 expression paralleled viable cell numbers, such that siRNAsrc resulted in substantially fewer viable cells following cytomix treatment, while siRNAyes had little effect on the number of viable cells following cytomix treatment. When exploring downstream signaling cascades activated by src and yes, we found that knockdown of src resulted in lower cytomix-induced pERK expression with little effect on pp38 or pJNK levels, while knockdown of yes augmented pERK expression with little effect on pp38 or pJNK levels. A small-molecule inhibitor of the ERK pathway resulted in augmentation of cytomix-induced cleaved caspase-3 protein expression in hPMVEC. Knockdown of src resulted in substantially lower levels of pAkt following cytomix treatment, while knockdown of yes had little effect on cytomix-induced pAkt levels in hPMVEC. Use of a small-molecule inhibitor of the PI3K/Akt pathway resulted in substantially increased levels of cleaved caspase-3 and decreased levels of pERK following cytomix treatment in hPMVEC, with no significant effect on cytomix-induced pp38 or pJNK levels. Together, our data demonstrate that src and yes differentially affect cytomix-induced apoptosis in hPMVEC, wherein src activates PI3K/Akt, which in turn activates ERK to prevent caspase-3 activation, while yes inhibits ERK activation, thereby augmenting caspase-3 activation (Fig. 8). Thus our findings support our hypothesis that src and yes have opposing effects on cytomix-induced caspase-3 activation, and this is due to the effects of src and yes on Akt/ERK signaling in hPMVEC. Further studies are needed to determine if these effects of src and yes on caspase-3 activation in hPMVEC correlate with disease pathogenesis or progression in vivo.
Fig. 8.
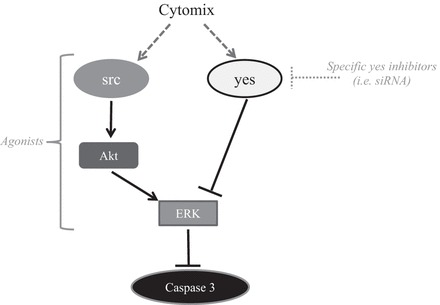
Proposed model for opposing effects of src and yes on caspase-3 activation. Antiapoptotic effects of src were through downstream activation of Akt, which in turn led to activation of ERK and, subsequently, attenuated caspase-3 activation. Proapoptotic effects of yes were through blunting of ERK activation, which dampened the inhibitory effect of ERK on caspase-3, resulting in augmentation of caspase-3 activation. Agonists of src and/or Akt and antagonists of yes may be potential therapeutic targets in preventing inflammatory lung diseases.
The STK have been implicated in the pathogenesis of lung injury in animal models. In BALB/c mice (18, 30), it has been shown that LPS, administered intraperitoneally or intratracheally, leads to lung injury associated with STK activation and that when the STK are inhibited using small-molecule inhibitors, LPS-induced lung injury is attenuated. Oyaizu et al. (26) found that ischemia-reperfusion lung injury in Sprague-Dawley rats resulted in STK phosphorylation and that treatment of the mice with a small-molecule inhibitor of the STK attenuated lung injury and reduced the number of terminal deoxynucleotidyl transferase-mediated dUTP nick-end label-positive cells in the lung. Zhao et al. (38) found that high-tidal-volume ventilation in Wistar rats resulted in lung injury and activation of STK and that the lung injury was significantly attenuated when STK activation was prevented by treatment with PP2, a STK inhibitor. Using a hyperoxia-and-high-tidal-volume model of ventilator-induced lung injury, Liu et al. (21) found that lung injury was attenuated in mice heterozygous for Src (Src+/−) compared with mice homozygous for Src (Src+/+). Han et al. (12) found greater susceptibility to LPS-induced lung injury in lyn-deficient than wild-type mice. These results highlight the importance of STK signaling in lung injury models and demonstrate that individual STK have a role in these lung injury models, although their role may depend on the model used to induce lung injury.
Our data support the concept that src is antiapoptotic, while yes is proapoptotic, in hPMVEC following cytomix exposure. This is consistent with findings in hepatocytes, where, in response to cell swelling, src is proproliferative, while yes is proapoptotic (28). On the other hand, in mesothelioma cell lines, knockdown of yes, but not src, using siRNA led to cell growth suppression due in part to enhanced apoptosis (29). In a neuroblastoma cell line, siRNA against either src or fyn resulted in decreased apoptosis following oxygen/glucose deprivation or amyloid-β peptide treatment (9). Similarly, in Hepa-1 cells (a mouse hepatoma cell line), overexpression of src or yes led to greater oxidant-induced apoptosis (24). Together, these findings support a differential role for the STK in apoptosis, although the effect of the individual STK family member likely depends on the specific cell type and/or the apoptotic stimulus. Indeed, Lewis-Tuffin et al. (19) recently reported that knockdown of the individual STK had highly variable effects in glioblastoma cell lines and xenograft models in mice, depending on the cell line. However, to the best of our knowledge, this is the first demonstration of a differential role of the STK in inflammation-induced apoptosis in hPMVEC.
We found that src, but not yes, was necessary for cytomix-induced Akt activation in hPMVEC. This finding is consistent with previous work showing that inhibition of STK signaling with PP2 inhibits phosphorylation of Akt (15). A specific role for src in Akt activation was demonstrated in mutant embryonic fibroblasts when knockout of src, fyn, and yes prevented estrogen-induced pAkt protein expression; if only yes and fyn were knocked out, there was an estrogen-induced increase in pAkt protein levels (14). We also found that Akt activity was necessary for attenuation of cytomix-induced apoptosis, given that inhibition of the PI3K/Akt pathway led to augmented cytomix-induced caspase-3 cleavage in hPMVEC. This finding is consistent with a report that inhibition of the PI3K/Akt pathway using LY294002 in human umbilical vein endothelial cells resulted in greater numbers of terminal deoxynucleotidyl transferase-mediated dUTP nick-end label-positive cells following TNF-α/cycloheximide treatment (37). Similarly, in bovine pulmonary artery endothelial cells, inhibition of Akt resulted in augmentation of LPS-induced caspase-3 activation (31). Thus, src mediates the Akt cascade in response to cytomix stimulation, and this src/Akt pathway is necessary to attenuate cytomix-induced apoptosis in hPMVEC.
We found that ERK activation was necessary to attenuate cytomix-induced apoptosis in hPMVEC. When ERK activity was inhibited using U0126, cytomix-induced cleaved caspase-3 levels were considerably increased. Furthermore, the effect of siRNAsrc on enhancement of caspase-3 activation was associated with lower levels of pERK protein, while the effect of siRNAyes in decreasing cleaved caspase-3 expression following cytomix was associated with greater levels of pERK. Finally, the effect of Akt inhibition on enhancement of cytomix-induced cleaved caspase-3 levels was associated with decreased pERK protein levels. These results demonstrate a central role for ERK in attenuation of cytomix-induced apoptosis in hPMVEC (Fig. 8). This is consistent with studies in endothelial cells. Taraseviciene-Stewart et al. (32) found that simvastatin suppressed pERK expression and induced caspase-3 activation in rat pulmonary microvascular endothelial cells. Chen et al. (6) reported that treatment of human umbilical vein endothelial cells with urotensin II resulted in ERK phosphorylation and protection of cells from doxorubicin-induced caspase-3 activation and apoptosis and that when ERK phosphorylation was prevented with U0126, urotensin II had no effect on doxorubicin-induced caspase-3 activation and apoptosis. Recently, using rat pulmonary microvascular endothelial cells, Li et al. (20) reported that inhibition of ERK augmented LPS-induced apoptosis, while inhibition of JNK protected cells from LPS-induced apoptosis, consistent with the notion that ERK is involved in attenuation of inflammation-induced apoptosis. However, their findings also suggest that the other MAPK may have a proapoptotic role in pulmonary endothelial cells. Our present findings that knockdown of src or yes resulted in no differences in p38 or JNK activation following cytomix treatment suggest that src and yes are not involved in p38 or JNK activation in hPMVEC.
In summary, we found opposing effects of two STK family members on caspase-3 activation in hPMVEC following cytomix treatment: src was antiapoptotic, while yes was proapoptotic (Fig. 8). The antiapoptotic effects of src were through downstream activation of Akt, which in turn led to activation of ERK and subsequent attenuation of caspase-3 activation and apoptosis. The proapoptotic effects of yes were through blunting of ERK activation, which dampened the inhibitory effect of ERK on caspase-3, thereby resulting in augmentation of caspase-3 activation. These findings may suggest novel therapeutic targets for inflammatory lung diseases, such as ARDS, wherein agonists of src or antagonists of yes may act to prevent endothelial cell apoptosis, which is one of the inciting events in the development of ARDS and BPD.
GRANTS
This study was supported in part by National Institutes of Health Grants K08 HL-105677 (B. Chen) and R21 AI-113930 (Y. Liu).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
L.D.N., B.C., and Y.L. developed the concept and designed the research; L.D.N., H.A.W., and Y.L. analyzed the data; L.D.N., Y.J., J.K.T., and Y.L. interpreted the results of the experiments; L.D.N. and Y.J. prepared the figures; L.D.N., H.A.W., Y.J., J.K.T., and B.C. drafted the manuscript; L.D.N., H.A.W., J.K.T., B.C., and Y.L. edited and revised the manuscript; L.D.N., H.A.W., Y.J., J.K.T., B.C., and Y.L. approved the final version of the manuscript; Y.J. performed the experiments.
REFERENCES
- 1.Alphonse RS, Vadivel A, Coltan L, Eaton F, Barr AJ, Dyck JR, Thébaud B. Activation of Akt protects alveoli from neonatal oxygen-induced lung injury. Am J Respir Cell Mol Biol 44: 146–154, 2011. [DOI] [PubMed] [Google Scholar]
- 2.Angelini DJ, Hyun SW, Grigoryev DN, Garg P, Gong P, Singh IS, Passaniti A, Hasday JD, Goldblum SE. TNF-α increases tyrosine phosphorylation of vascular endothelial cadherin and opens the paracellular pathway through fyn activation in human lung endothelia. Am J Physiol Lung Cell Mol Physiol 291: L1232–L1245, 2006. [DOI] [PubMed] [Google Scholar]
- 3.Byeon SE, Yi YS, Oh J, Yoo BC, Hong S, Cho JY. The role of Src kinases in macrophage-mediated inflammatory responses. Med Inf (Lond) 2012: 512926, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cepkova M, Matthay MA. Pharmacotherapy of acute lung injury and the acute respiratory distress syndrome. J Intensive Care Med 21: 119–143, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chang R, Chicoine LG, Cui H, Kanagy NL, Walker BR, Liu Y, English BK, Nelin LD. Cytokine-induced arginase activity in pulmonary endothelial cells depends on Src-family tyrosine kinase activity. Am J Physiol Lung Cell Mol Physiol 295: L688–L697, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen YL, Tsai YT, Lee CY, Lee CH, Chen CY, Liu CM, Chen JJ, Loh SH, Tsai CS. Urotensin II inhibits doxorubicin-induced human umbilical vein endothelial cell death by modulating ATF expression and via the ERK and Akt pathway. PLos One 9: e106812, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cui H, Chen B, Chicoine LG, Nelin LD. Over-expression of cationic amino acid transporter-1 increases nitric oxide production in human pulmonary microvascular endothelial cells. Clin Exp Pharmacol Physiol 38: 796–803, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Das KC, Ravi D, Holland W. Increased apoptosis and expression of p21 and p53 in premature infant baboon model of bronchopulmonary dysplasia. Antioxid Redox Signal 6: 109–116, 2004. [DOI] [PubMed] [Google Scholar]
- 9.Du CP, Tan R, Hou XY. Fyn kinases play a critical role in neuronal apoptosis induced by oxygen and glucose deprivation or amyloid-β peptide treatment. CNS Neurosci Ther 18: 754–761, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Galani V, Tatsaki E, Bai M, Kitsoulis P, Lekka M, Nakos G, Kanavaros P. The role of apoptosis in the pathophysiology of acute respiratory distress syndrome: an up-to-date cell-specific review. Pathol Res Pract 206: 145–150, 2010. [DOI] [PubMed] [Google Scholar]
- 11.Gien J, Kinsella JP. Pathogenesis and treatment of bronchopulmonary dysplasia. Curr Opin Pediatr 23: 305–313, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Han J, Zhang G, Welch EJ, Liang Y, Fu J, Vogel SM, Lowell CA, Du X, Cheresh DA, Malik AB, Li Z. A critical role for Lyn kinase in strengthening endothelial integrity and barrier function. Blood 122: 4140–4149, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hashimoto S, Kobayashi A, Kooguchi K, Kitamura Y, Onodera H, Nakajima H. Upregulation of two death pathways of perforin/granzyme and FasL/Fas in septic acute respiratory distress syndrome. Am J Respir Crit Care Med 161: 237–243, 2000. [DOI] [PubMed] [Google Scholar]
- 14.Haynes MP, Li L, Sinha D, Russell KS, Hisamoto K, Baron R, Collinge M, Sessa WC, Bender JR. Src kinase mediates phosphatidylinositol 3-kinase/Akt-dependent rapid endothelial nitric-oxide synthase activation by estrogen. J Biol Chem 278: 2118–2123, 2003. [DOI] [PubMed] [Google Scholar]
- 15.Ikeda Y, Tajima S, Izawa-Ishizawa Y, Kihira Y, Ishizawa K, Yoshida S, Aihara K, Tsuchiya K, Tamaki T. Bovine milk-derived lactoferrin exerts proangiogenic effects in an Src-Akt-eNOS-dependent manner in response to ischemia. J Cardiovasc Pharmacol 61: 423–429, 2013. [DOI] [PubMed] [Google Scholar]
- 16.Jin Y, Liu Y, Nelin LD. Extracellular signal-regulated kinase mediates expression of arginase II but not inducible nitric oxide synthase in lipopolysaccharide-stimulated macrophages. J Biol Chem 290: 2099–2111, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kim MP, Park SI, Kopetz S, Gallick GE. Src family kinases as mediators of endothelial permeability: effects on inflammation and metastasis. Cell Tissue Res 335: 249–259, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee HS, Moon C, Lee HW, Park EM, Cho MS, Kang JL. Src tyrosine kinases mediate activations of NF-κB and integrin signal during lipopolysaccharide-induced acute lung injury. J Immunol 179: 7001–7011, 2007. [DOI] [PubMed] [Google Scholar]
- 19.Lewis-Tuffin LJ, Feathers R, Hari P, Durand N, Li Z, Rodriguez FJ, Bakken K, Carlson B, Schroeder M, Sarkaria JN, Anastasiadis PZ. Src family kinases differentially influence glioma growth and motility. Mol Oncol 9: 1783–1798, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li Y, Cao Y, Zeng Z, Liang M, Xue Y, Xi C, Zhou M, Jiang W. Angiotensin-converting enzyme 2/angiotensin-(1–7)/Mas axis prevents lipopolysaccharide-induced apoptosis of pulmonary microvascular endothelial cells by inhibiting JNK/NF-κB pathways. Sci Rep 5: 8209, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu YY, Li LF, Fu JY, Kao KC, Huang CC, Chien Y, Liao YW, Chiou SH, Chang YL. Induced pluripotent stem cell therapy ameliorates hyperoxia-augmented ventilator-induced lung injury through suppressing the Src pathway. PLos One 9: e109953, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.May M, Strobel P, Preisshofen T, Seidenspinner S, Marx A, Speer CP. Apoptosis and proliferation in lungs of ventilated and oxygen-treated preterm infants. Eur Respir J 23: 113–121, 2004. [DOI] [PubMed] [Google Scholar]
- 23.Mokres LM, Parai K, Hilgendorff A, Ertsey R, Alvira CM, Rabinovitch M, Bland RD. Prolonged mechanical ventilation with air induces apoptosis and causes failure of alveolar septation and angiogenesis in lungs of newborn mice. Am J Physiol Lung Cell Mol Physiol 298: L23–L35, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Niture SK, Jain AK, Shelton PM, Jaiswal AK. Src subfamily kinases regulate nuclear export and degradation of transcription factor Nrf2 to switch off Nrf2-mediated antioxidant activation of cytoprotective gene expression. J Biol Chem 286: 28821–28832, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 25.Ortiz-Diaz E, Festic E, Gajic O, Levitt JE. Emerging pharmacological therapies for prevention and early treatment of acute lung injury. Semin Respir Crit Care Med 34: 448–458, 2013. [DOI] [PubMed] [Google Scholar]
- 26.Oyaizu T, Fung SY, Shiozaki A, Guan Z, Zhang Q, dos Santos CC, Han B, Mura M, Keshavjee S, Liu M. Src tyrosine kinase inhibition prevents pulmonary ischemia-reperfusion-induced acute lung injury. Intensive Care Med 38: 894–905, 2012. [DOI] [PubMed] [Google Scholar]
- 27.Raghavendran K, Pryhuber GS, Chess PR, Davidson BA, Knight PR, Notter RH. Pharmacotherapy of acute lung injury and acute respiratory distress syndrome. Curr Med Chem 15: 1911–1924, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Reinehr R, Sommerfeld A, Häussinger D. The Src family kinases: distinct functions of c-Src, Yes, and Fyn in the liver. Biomol Concepts 4: 129–142, 2013. [DOI] [PubMed] [Google Scholar]
- 29.Sato A, Sekine M, Virgona N, Ota M, Yano T. Yes is a central mediator of cell growth in malignant mesothelioma cells. Oncol Rep 28: 1889–1893, 2012. [DOI] [PubMed] [Google Scholar]
- 30.Severgnini M, Takahashi S, Tu P, Perides G, Homer RJ, Jhung JW, Bhavsar D, Cochran BH, Simon AR. Inhibition of the Src and Jak kinases protects against lipopolysaccharide-induced acute lung injury. Am J Respir Crit Care Med 171: 858–867, 2005. [DOI] [PubMed] [Google Scholar]
- 31.Sylte MJ, Kuckleburg CJ, Atapattu D, Leite FP, McClenahan D, Inzana TJ, Czuprynski CJ. Signaling through interleukin-1 type 1 receptor diminishes Haemophilus somnus lipooligoshaccharide-mediated apoptosis of endothelial cells. Microb Pathog 39: 121–130, 2005. [DOI] [PubMed] [Google Scholar]
- 32.Taraseviciene-Stewart L, Scerbavicius R, Choe KH, Cool C, Wood K, Tuder RM, Burns N, Kasper M, Voelkel NF. Simvastatin causes endothelial cell apoptosis and attenuates severe pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 291: L668–L676, 2006. [DOI] [PubMed] [Google Scholar]
- 33.Toby I, Chicoine LG, Cui H, Chen B, Nelin LD. Hypoxia-induced proliferation of human pulmonary microvascular endothelial cells depends on epidermal growth factor receptor tyrosine kinase activation. Am J Physiol Lung Cell Mol Physiol 298: L600–L606, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Uhling U, Uhling S. Ventilation-induced lung injury. Compr Physiol 1: 635–661, 2011. [DOI] [PubMed] [Google Scholar]
- 35.Viscardi RM. Perinatal inflammation and lung injury. Semin Fetal Neonatal Med 17: 30–35, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ware LB, Matthay MA. The acute respiratory distress syndrome. N Engl J Med 342: 1334–1349, 2000. [DOI] [PubMed] [Google Scholar]
- 37.Watanabe T, Suzuki J, Yamawaki H, Sharma VK, Sheu SS, Berk BC. Losartan metabolite EXP3179 activates Akt and endothelial nitric oxide synthase via vascular endothelial growth factor receptor-2 in endothelial cells: angiotensin II type 1 receptor-independent effects of EXP3179. Circulation 112: 1798–1805, 2005. [DOI] [PubMed] [Google Scholar]
- 38.Zhao T, Liu M, Gu C, Wang X, Wang Y. Activation of c-Src tyrosine kinase mediated the degradation of occludin in ventilator-induced lung injury. Respir Res 15: 158, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]