

Abstract
The introduction of calcineurin inhibitors (CNI) into clinical practice in the late 1970s transformed organ transplantation and led to significant improvement in acute rejection episodes. However, despite their significant clinical utility, the use of these agents is hampered by the development of hypertension and nephrotoxicity, which ultimately lead to end-stage kidney disease and overt cardiovascular outcomes. There are currently no effective agents to treat or prevent these complications. Importantly, CNI-free immunosuppressive regimens lack the overall efficacy of CNI-based treatments and put patients at risk of allograft rejection. Cytochrome P-450 epoxygenase metabolites of arachidonic acid, epoxyeicosatrienoic acids (EETs), have potent vasodilator and antihypertensive properties in addition to many cytoprotective effects, but their effects on CNI-induced nephrotoxicity have not been explored. Here, we show that PVPA, a novel, orally active analog of 14,15-EET, effectively prevents the development of hypertension and ameliorates kidney injury in cyclosporine-treated rats. PVPA treatment reduced proteinuria and renal dysfunction induced by cyclosporine. PVPA inhibited inflammatory cell infiltration into the kidney and decreased renal fibrosis. PVPA also reduced tubular epithelial cell apoptosis, attenuated the generation of reactive oxygen species, and modulated the unfolded protein response that is associated with endoplasmic reticulum stress. Consistent with the in vivo data, PVPA attenuated cyclosporine-induced apoptosis of NRK-52E cells in vitro. These data indicate that the cytochrome P-450/EET system offers a novel therapeutic strategy to treat or prevent CNI-induced nephrotoxicity.
Keywords: cyclosporine, transplantation, nephrotoxicity, hypertension, epoxyeicosatrienoic acids
cyclosporine a (csa) is a calcineurin inhibitor (CNI) whose widespread introduction into clinics in the late 1970s transformed organ transplantation and led to significant improvement in acute rejection episodes. Tacrolimus came into the market subsequently (3, 5, 13, 44). To date, the CNIs continue to be a major component of immunosuppressive regimens in solid organ transplantation worldwide. CNIs are also used in the treatment of many autoimmune conditions (1, 23). However, despite the well-documented improvements in acute rejection rates and short-term graft outcomes, CNI use has not been associated with any significant enhancements in long-term allograft survival. Instead, CNI use is limited by the development of hypertension and nephrotoxicity (2, 28, 29, 31, 32). Indeed, CNI-induced end-stage kidney failure is a major complication and is an ever-increasing problem in nonkidney solid organ recipients (36). Unfortunately, efficient treatment to prevent or reduce this devastating outcome of CNI therapy is nonexistent. Rather, this has led to trials of CNI-free regimens that lack the overall efficacy of CNI-based regimens and actually put patients at risk for allograft rejection (9–11, 16, 18). Also of note, control of CNI-induced hypertension per se has not been shown to be adequate in preventing nephrotoxicity (24, 39). The underlying mechanism of CNI-induced nephrotoxicity remains unclear. CNIs activate the intrarenal renin-angiotensin system and also increase the expression of endothelin-1, leading to afferent arteriolar vasoconstriction and reduction in renal blood flow. CNIs have also been reported to cause oxidative stress, which acts variously to cause tissue injury. Inflammatory cell infiltration into the kidney is a common finding, and this is initially associated with augmented expression and release of inflammatory cytokines and chemokines in the kidney. The macrophages subsequently, along with other cells, become activated and elaborate large amounts of extracellular matrix components such as collagen, which leads to tubulointerstitial fibrosis. Tubular epithelial cell (TEC) apoptosis may be due to the direct effect of the CNI or from local hypoxia due to renal vasoconstriction (8, 17, 34, 40, 49). The foregoing suggests that CNI-induced nephrotoxicity occurs via a multifactorial mechanism, and therefore, to be effective in preventing or treating these CNI-induced complications, potential therapeutic agents will need to have broad-based modulating effects on vascular tone, inflammation, oxidative stress, and apoptosis. Such an agent would allow patients to benefit from these useful drugs while preventing overt complications.
Arachidonic acid is metabolized by key enzyme systems to small-molecule mediators with diverse physiological and pathophysiological effects. Specifically, the cytochrome P-450 epoxygenases (mostly CYP2C and CYP2J) metabolize arachidonic acid to four bioactive regioisomeric epoxyeicosatrienoic acids (EETs): 5,6-EET, 8,9-EET, 11,12-EET, and 14,15-EET (6, 52). EETs are mostly produced in the endothelium and are expressed in many tissues including the kidney, heart, lung, and liver. EETs mediate many autocrine and paracrine functions, including the regulation of vascular tone and tubular sodium and water absorption in the kidney. EETs have antihypertensive actions in vivo (4, 19, 51). Also, previous studies have demonstrated that EETs have anti-inflammatory, antifibrotic, and antiapoptotic effects (14, 21, 22, 35, 42, 48). In support of the above, we and others recently reported that EETs attenuated cisplatin-induced nephrotoxicity by reducing oxidative stress, inflammation, endoplasmic reticulum (ER) stress, and apoptosis, without affecting the chemotherapeutic effects of cisplatin (20, 26, 41). In this study, we examined the protective role of the novel, orally active EET analog [N-isopropyl-N-(5-((3-(5-(N-isopropylheptanamido)pentyl)-2-(pivaloylimino)-2,3-dihydrobenzo[d]thiazol-4-yl)oxy)pentyl)heptanamide] (PVPA) in an experimental model of CsA-induced nephrotoxicity. We demonstrate that PVPA administration robustly prevented CsA-induced hypertension and ameliorated histological damage and kidney dysfunction. The mechanism of action seems to be through the suppression of inflammation, oxidative stress, ER stress, and apoptosis.
MATERIALS AND METHODS
Materials.
All chemicals and assay kits were purchased from Sigma-Aldrich (St. Louis, MO) unless otherwise indicated. The novel, orally active 14,15-epoxyeicosatrienoic acid analog PVPA was designed and synthesized in the laboratory of Dr. John R. Falck (Dept. of Biochemistry, Univ. of Texas Southwestern Medical Center, Dallas, TX). PVPA was given in the drinking water, and its concentration was adjusted in a way that the daily intake (dose) was 10 mg/kg body wt. The administered dose was chosen on the basis of the drug's pharmacokinetic properties and is similar to the dose of similar agents used in recent in vivo experiments (20).
Animals, dosing, and experimental design.
Male Sprague-Dawley rats, weighing 180–200 g (Envigo, Indianapolis, IN), were used. Animals were kept in a temperature-controlled environment with a 12:12-h light-dark cycle and were allowed free access to a low-salt diet (0.02% sodium, Teklad Global 2918, Madison, WI) and to tap water, except in the PVPA treatment group, where the drug was put in the drinking water. CsA (supplied at 50 mg/ml in Cremophor EL plus 32.5% vol/vol alcohol by Novartis Pharma, Basel, Switzerland) was diluted in normal saline to 15 mg/ml. Rats were acclimatized for a minimum of 1 wk before experimentation. All animals received humane care in compliance with the National Research Council's Guide for the Care and Use of Laboratory Animals. The animal protocol was approved by the Institutional Animal Care and Use Committee of the Medical College of Wisconsin. The animals were placed into three groups. Group 1 (Control group) received a daily subcutaneous (sc) injection of Cremophor EL only. Group 2 (CsA group) received a daily sc injection of CsA only (20 mg/kg). Group 3 (PVPA group) received a daily sc injection of CsA (20 mg/kg) plus PVPA (10 mg·kg−1·day−1) in the drinking water. PVPA was initiated 5 days before the start of CsA treatment. All the animals were treated for 28 days. At 1 day before euthanasia, rats were maintained in individual metabolic cages, and urine was collected over a 24-h period.
Noninvasive blood pressure measurements.
Noninvasive blood pressure measurements were performed by tail plethysmography (IITC Life Sciences, Woodland Hills, CA). Rats were acclimatized to the apparatus during three sessions over 5–7 days. Following acclimatization, weekly blood pressure measurement was done during the course of the study. Systolic blood pressure (SBP) was measured exactly at the same period of the day (9–10 AM) on every occasion. The average values for SBP were obtained from 10 sequential cuff inflation-deflation cycles.
Biochemical analysis.
The levels of blood urea nitrogen (BUN; BioAssay Systems, Hayward, CA), urinary protein, and creatinine (Cayman Chemical, Ann Arbor, MI) were measured spectrophotometrically using commercial kits. Urinary albumin was measured using ELISA kits (Exocell, Philadelphia, PA). A kidney cortex and medulla homogenate was prepared with a lysis buffer (50 mM HEPES, pH 7.4, with 5 mM CHAPS and 5 mM DTT), centrifuged at 10,000 g for 10 min, and then the resulting supernatant was used for the assay. Caspase 3 activity in the kidney homogenate was determined using a commercial fluorimetric assay kit (Sigma-Aldrich). The caspase 3 fluorimetric assay is based on the hydrolysis of the peptide substrate acetyl-Asp-Glu-Val-Asp-7-amido-4-methylcoumarin (Ac-DEVD-AMC) by caspase 3, resulting in the release of the fluorescent 7-amino-4-methylcoumarin (AMC) moiety. Caspase 3 activity is expressed as nanomoles AMC per minute per microliter. Kidney tissue protein content was measured using a BCA protein assay kit (Thermo Scientific, Rockford, IL). Lipid peroxidation in the renal medulla and cortical homogenate was measured as thiobarbituric acid-reactive substances (TBARS) by a fluorometric assay (Cayman Chemical).
Immunohistochemistry and terminal uridine deoxynucleotidyl transferase-mediated dUTP nick-end labeling assay.
Formalin-fixed kidneys were embedded in paraffin and used for immunohistochemical studies. After routine deparaffinization, heat-induced epitope retrieval was done using a citrate-based buffer. Endogenous peroxidase was quenched by incubation with 3% H2O2 in PBS (pH 7.4) for 5 min. The sections were permeabilized using 0.1% (wt/vol) Triton X-100 in PBS for 15 min. Nonspecific binding was minimized by incubating sections with 5% normal serum from the species in which the secondary antibody was raised (diluted in 5% BSA/PBS) for 60 min at room temperature. Endogenous biotin and avidin binding sites were blocked by sequential incubation with avidin and biotin for 15 min (Vector Laboratories, Burlingame, CA). The primary antibodies used to detect glucose-regulated protein 78 (GRP78)/BiP, C/EBP homologous protein (CHOP) and CD68 were goat anti-rat GRP78 (Santa Cruz Biotechnology, Santa Cruz, CA), rabbit anti-rat CHOP (Santa Cruz Biotechnology), and mouse anti-rat CD68 (Bio-Rad, Hercules, CA), respectively. Biotinylated secondary antibodies were used for development with avidin-biotinylated horseradish peroxidase complex (Vectastain ABC kits; Vector Laboratories). The slides were counterstained with hematoxylin or methyl green and photographed. Apoptosis was examined by terminal uridine deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL) staining with an ApopTag Plus Peroxidase In Situ Apoptosis Kit (EMD Millipore, Billerica, MA), according to the manufacturer's instructions. Quantification of CD68- and TUNEL-positive cells was done by counting the positive cells in 15 randomly selected, nonoverlapping fields (×200 magnification) in the outer medulla and cortex. Quantification of GRP78 and CHOP staining were assessed at ×200 magnification using image analyzing software (NIS Elements AR version 3.0, Nikon Instruments, Melville, NY). To minimize observer bias, the analysis was performed in a blinded fashion without knowledge of the treatment group from which the tissues originated.
Histological analysis.
Formalin (10%)-fixed kidney samples were embedded in paraffin. Sections (4 μm) were prepared and stained with hematoxylin and eosin and Masson's trichrome. The slides were scanned and visualized using a Hamamatsu NanoZoomer HT digital scanner/NDP.view2 viewing software (Hamamatsu Photonics).The hematoxylin- and eosin-stained kidneys were reviewed and assessed for tubular vacuolization. The scoring was done semiquantitatively by counting the number of fields with three or more tubules affected by isometric vacuolization from a total of 20 randomly selected nonoverlapping fields (×200 magnification) observed in the renal cortex. The trichrome-stained slides were assessed for the degree of tubulointerstitial fibrosis using NIS Elements AR version 3.0 software (Nikon Instruments). The renal tissue areas positive for collagen staining were expressed as the percentage area fraction relative to total area analyzed. All examinations and scoring were done in a blinded manner.
Quantitative real-time PCR.
Total RNA was prepared from kidney tissues stored in RNAlater stabilization solution using a TRIzol Plus RNA Purification Kit and then reverse transcribed with a High-Capacity cDNA Reverse Transcription Kit according to the manufacturer's instructions (Thermo Fisher Scientific). Quantitative amplification of the cDNA was performed on an ABI-Prism 7900HT Sequence Detection system and evaluated using SDS and RQ manager software (versions 2.3 and 1.2, respectively; Applied Biosystems, Thermo Fisher Scientific). Results were normalized to GAPDH content by the comparative CT method, and relative mRNA levels are expressed as fold-change compared with vehicle-treated control animals. The following primer and probe sets (Integrated DNA Technologies, Coralville, IA) were used: GAPDH forward: GTAACCAGGCGTCCGATAC, reverse: TCTCTGCTCCTCCCTGTTC, and probe:/56-FAM/CACACCGAC/ZEN/CTTCACCATCTTGTCT/3IABkFQ/; collagen 1a1 forward: CATTGTGTATGCAGCTGACTTC, reverse: CGCAAAGAGTCTACATGTCTAGG, and probe:/56-FAM/CCGGAGGTC/ZEN/CACAAAGCTGAACA/3IABkFQ/.
Cell cultures.
The well-characterized normal rat proximal TEC line NRK-52E was obtained from the American Type Culture Collection (Manassas, VA). Cells were cultured in Dulbecco's modified Eagle's medium (with 4.5 g/l glucose; D5671, Sigma-Aldrich) supplemented with 5% FBS, 1 mM sodium pyruvate, and 4 mM l-glutamine in 5% CO2 at 37°C.
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay.
NRK-52E cells were seeded into 96-well plates in a volume of 200 μl/well (40,000 cells/ml) and allowed to grow to 80% confluence. After washing once with serum-free media, cells were incubated in 200 μl/well of serum-free media containing the indicated amount of PVPA for 24 h. Cell viability was determined by addition of 20 μl of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) at a concentration of 5 mg/ml in PBS. After incubation for 1 h, the medium was removed and 100 μl of DMSO was added to dissolve the formazan crystals. The absorbance was read at 570 nm using a Synergy HT microplate reader (Bio-Tek Instruments, Winooski, VT).
TUNEL staining of NRK-52E cells.
NRK-52E cells were seeded into Lab-Tek II Chamber Slides (8 chambers; Thermo Fisher Scientific) at a density of 25,000 cells/chamber in 0.5 ml complete media and allowed to grow to 80% confluence. After washing once with serum-free media, cells were incubated in 0.5 ml/chamber of serum-free media with or without 10 μM PVPA for 1 h. Cells were then treated with 5 μM CsA or an equivalent amount of vehicle. After 24 h, cells were washed once with PBS before fixation in 4% paraformaldehyde in PBS for 10 min at room temperature. Apoptosis was examined by TUNEL staining with an ApopTag Plus Peroxidase In Situ Apoptosis Kit (EMD Millipore), according to the manufacturer's instructions. Quantification of TUNEL-positive cells was done by counting the positive cells in 10 randomly selected, nonoverlapping fields/chamber (×200 magnification). Six replicate chambers/condition were counted.
Statistical analysis.
Results are reported as means ± SE. In vitro experiments were performed at least twice, and statistical significance between two measurements was determined by a two-tailed unpaired Student's t-test. Differences between multiple groups were determined by ANOVA, using GraphPad Prism Version 4.0 software (GraphPad Software, La Jolla, CA). P values of ≤ 0.05 were considered statistically significant.
RESULTS
PVPA prevents development of hypertension in CsA-treated rats.
Hypertension is a common complication in patients who receive calcineurin inhibitor treatment, and this has been associated with increased cardiovascular morbidity and mortality, as well as graft loss in renal transplant recipients (7, 27, 38). To investigate whether PVPA affected the development of hypertension in an experimental model of CsA-induced nephrotoxicity, we measured SBP in conscious male Sprague-Dawley rats by the noninvasive tail-cuff method. Baseline SBP was not different among the three groups of rats. Consistent with the known effect of CsA on blood pressure, the animals developed robust hypertension by day 7 of CsA treatment with an increase in SBP of 33 ± 4 mmHg (P < 0.001), followed by a steady rise in SBP throughout the remainder of experimentation. By the end of the 28-day study period, the mean SBP in the CsA-only group was significantly elevated compared with the PVPA group (+Δ32 mmHg; P < 0.001; Fig. 1). Indeed, the final mean SBP in the PVPA group remained very close to the baseline value and was not statistically different from that of the control rats that did not receive CsA treatment (+Δ9 mmHg; P = 0.17). These results indicate that PVPA had a profound protective effect on the development of CsA-induced hypertension. The final body weight of the CsA-only animals was not statistically different from that of the PVPA group (316 ± 7 vs. 313 ± 4 g; P = 0.71), and hence the observed differences in SBP are not due to differences in body weight.
Fig. 1.
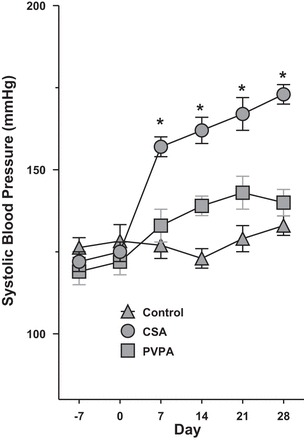
[N-isopropyl-N-(5-((3-(5-(N-isopropylheptanamido)pentyl)-2-(pivaloylimino)-2,3-dihydrobenzo[d]thiazol-4-yl)oxy)pentyl)heptanamide] (PVPA) prevents the development of hypertension in cyclosporin A (CsA)-treated Rats. A rat model of CsA-induced nephrotoxicity was established by daily subcutaneous (sc) injection of CsA for a total of 28 days. The 3 groups of rats studied were treated with either vehicle only, CsA only, or CsA plus PVPA. Systolic blood pressure (SBP) was measured by the tail-cuff method at weekly intervals. Values are means ± SE; n = 5–8/group. *P < 0.001 vs. PVPA group.
PVPA attenuates proteinuria and renal dysfunction in CsA-treated rats.
Prolonged treatment with CsA leads to the development of irreversible parenchymal changes in the kidney which progresses to chronic kidney disease and is a major risk factor for the development of end-stage kidney failure in solid organ transplant recipients. We assessed albuminuria and proteinuria as markers of chronic renal damage. Albuminuria was 3.5-fold higher in the CsA-treated animals at the end of the 28-day treatment period compared with the control group, which only received a vehicle (P < 0.001). Administration of PVPA significantly reduced the CsA-induced albuminuria by ∼90% (P < 0.001; Fig. 2A). Similarly, the increased proteinuria induced by CsA treatment was significantly reduced by coadministration of PVPA (P < 0.05; Fig. 2B). Importantly, at the end of the study, no differences in urinary sodium and potassium excretion rates were noted in the CsA-only group vs. the PVPA group (sodium: 0.60 ± 0.08 vs. 0.79 ± 0.08 mmol/day, P = 0.14; potassium, 1.68 ± 0.12 vs 1.74 ± 0.18 mmol/day, P = 0.8). This indicates that salt intake was similar between the CsA-only and PVPA rats. Altogether, these data imply that the reduction in albuminuria and proteinuria in the PVPA group is not due to differences in salt consumption or natriuresis. Consistent with the albuminuria/proteinuria results, BUN, a measure of renal function was increased more than two-fold in the CsA-only group at the end of the study compared with the vehicle-treated rats while the increase was significantly attenuated in the PVPA group by ∼30% (P < 0.001; Fig. 2C).
Fig. 2.
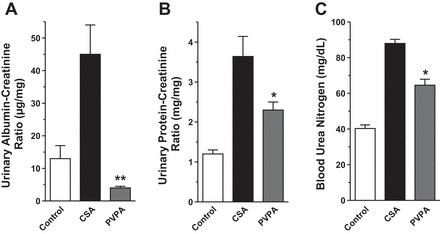
PVPA attenuates proteinuria and renal dysfunction in CsA-treated rats. A 24-h urine sample was collected at the end of the treatment period and was used to measure albumin excretion rate (A) and protein excretion rate (B). C: blood urea nitrogen (BUN) was measured at the end of the treatment. Values are means ± SE; n = 5–8/group. *P < 0.05, **P < 0.001 vs. CsA group.
PVPA ameliorates renal morphological changes induced by CsA.
To assess histological features of CsA-induced tubular and interstitial damage, kidney sections stained with hematoxylin and eosin and Masson's trichrome were examined. Prolonged CsA treatment resulted in tubular injury with swelling and isometric vacuolization, which was evident on the hematoxylin and eosin-stained slides. Cotreatment with PVPA significantly reduced the extent and severity of this injury (Fig. 3, A and B). Chronic damage from CsA results in deposition of extracellular matrix, including collagen within kidney parenchyma that ultimately leads to chronic kidney disease and end-stage kidney failure. CsA treatment resulted in 1.8–2.8 fold increase in Masson's trichrome staining in the cortex and medulla, consistent with enhanced interstitial fibrosis. PVPA administration led to significant improvement in the CsA-induced interstitial fibrosis, reducing the extent of collagen deposition by 25–45% (blue area, Fig. 3, C and D). We further assessed the fibrosis by measurement of the gene expression level of collagen 1a1. As depicted in Fig. 3E, CsA treatment induced collagen 1a1 mRNA expression. In contrast, the increased expression was significantly attenuated by PVPA cotreatment.
Fig. 3.
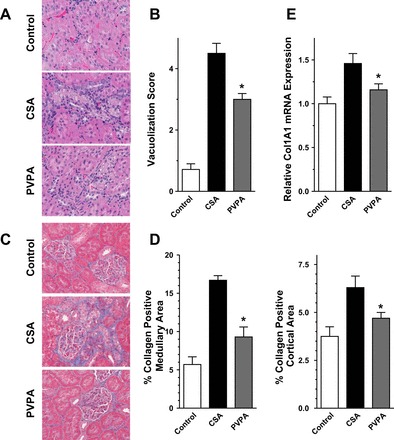
PVPA ameliorates morphological changes induced by CsA. A: representative hematoxylin and eosin staining of kidney sections from the 3 groups of rats showing areas of isometric vacuolization. B: semiquantitative score of vacuolization in the kidney sections. C: representative Masson's trichrome-stained sections from the 3 groups of rats showing areas of fibrosis (blue areas). D: semiquantitative score of renal fibrosis as depicted by collagen deposition. E: real-time RT-PCR quantification of collagen 1 expression in kidney sections. Original magnification of slides, ×400. Values are means ± SE; n = 5–8/group. *P < 0.05 vs. CsA group.
PVPA reduces CsA-induced renal oxidative stress.
Reactive oxygen species (ROS) play key roles in normal renal physiology, regulating many different processes including gluconeogenesis, electrolyte transport, hemodynamics, and gene expression (15, 43, 46). Excessive elaboration of ROS during pathological states, however, leads to oxidative stress, which is associated with cellular apoptosis, tissue inflammation, and fibrosis. Oxidative stress has been shown to be an important mediator of CsA-induced nephrotoxicity. We evaluated whether PVPA administration modifies oxidative stress during CsA treatment. The level of TBARS, a measure of ROS levels, was significantly increased by 60–80% in the cortex and medulla after CsA treatment compared with vehicle treatment. Coadministration of PVPA significantly suppressed the ROS generation in the medulla by ∼30% compared with the CsA-only group. In the cortex, PVPA coadministration reduced TBARs levels by ∼20%, although this did not reach statistical significance (Fig. 4).
Fig. 4.
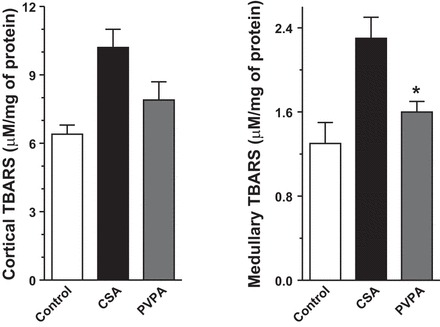
PVPA reduces CsA-induced oxidative stress. Kidney thiobarbituric acid-reactive substances (TBARS) levels in the 3 groups of rats are shown. Values are means ± SE; n = 5–8/group. *P < 0.05 vs. CsA group.
PVPA reduces renal inflammation during CsA treatment.
CsA-induced nephropathy, like other causes of chronic kidney disease, is associated with tubulointerstitial inflammatory response and macrophage influx, which contributes to the establishment of the renal damage and tubulointerstitial fibrosis. To ascertain the effect of PVPA on the CsA-induced inflammatory response, we performed immunohistochemical staining using an antibody against CD68, a marker of macrophages. As shown in Fig. 5, the number of infiltrating macrophages into the kidney increased more than sixfold in CsA-treated rats compared with the control animals. The coadministration of PVPA resulted in a significant reduction in macrophage infiltration by 50%.
Fig. 5.
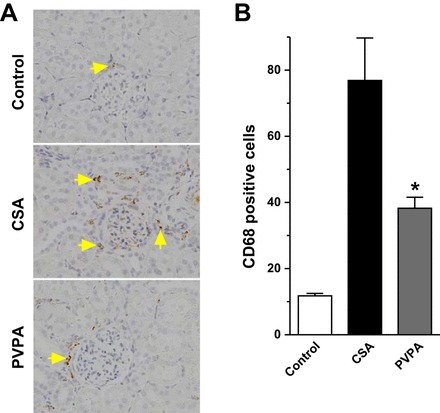
PVPA inhibits inflammation during CsA treatment. A:representative CD68-stained sections of kidney tissues from the 3 groups of rats (original magnification, ×400). Arrows show infiltrated CD-68 positive cells. B: semiquantitative score of CD68-positive cells in kidney sections. Values are means ± SE; n = 5–8/group. *P < 0.05 vs. CsA group.
PVPA reduces renal TEC apoptosis during CsA treatment.
TEC apoptosis could be a direct effect of the CNI or may occur as a result of local hypoxia due to renal vasoconstriction. Apoptosis represents a key feature in the pathogenesis of CsA-induced nephrotoxicity (45, 47). We assessed the protective effects of the PVPA on CsA-induced renal TEC apoptosis by using the TUNEL staining method. The number of TECs containing TUNEL-positive nuclei increased markedly after 28 days of CsA treatment compared with the vehicle-treated animals. Administration of PVPA significantly ameliorated tubular cell apoptosis induced by CsA (P < 0.01, Fig. 6, A and B). Apoptosis or programmed cell death involves the activation of a group of proteases called “caspases.” Caspase 3 is the final effector caspase that mediates apoptotic cell death. Therefore, we examined the effect of PVPA on caspase 3 activity in the kidney. We found that caspase 3 activity increased by more than twofold with CsA treatment. Coadministration of PVPA reduced caspase 3 activity by more than 50% in the medulla of CsA-treated rats (Fig. 6C).
Fig. 6.
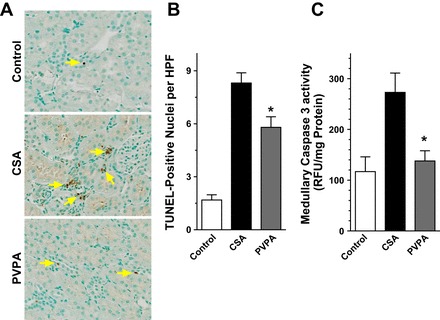
PVPA reduces renal tubular epithelial cell apoptosis during CsA treatment. A: representative light photomicrographs of kidney sections stained by the terminal uridine deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL) method at the completion of 28 days of treatment (original magnification, ×400). Arrows show TUNEL-positive cells. B: semiquantitative score of TUNEL-positive cells in kidney sections. C: caspase 3 activity in kidney tissues. Values are means ± SE; n = 5–8/group. *P < 0.05 vs. CsA group.
PVPA ameliorates ER stress in the kidney during CsA treatment.
Recent evidence indicates that CsA causes ER stress, which has been implicated in the pathophysiology of various renal diseases. Excessive ER stress leads to overexpression of the transcription factor CHOP, which mediates ER-stress induced apoptosis in addition to induction of inflammation (33). To elucidate the effect of PVPA treatment on this process, we examined the expression of the ER stress marker GRP78 and also CHOP in kidney tissues using immunohistochemical analysis. We found more than a twofold increase in GRP78 expression in both the cortex and medulla consistent with increased ER stress following CsA treatment compared with control rats. The coadministration of PVPA to CsA-treated rats downregulated GRP78 expression levels by more than 50% (Fig. 7, A and B). Similarly, the expression level of the proapoptotic transcription factor CHOP was upregulated by 1.6- to 2.8-fold in the cortex and medulla in CsA-treated rats, and cotreatment with PVPA significantly attenuated this increase by ∼80% (Fig. 7, B and C). These results are in support of the other findings showing attenuation of inflammation and apoptosis after coadministration of PVPA and suggest that the protective effect of PVPA during CsA treatment is mediated, to some extent, by modulation of ER stress.
Fig. 7.
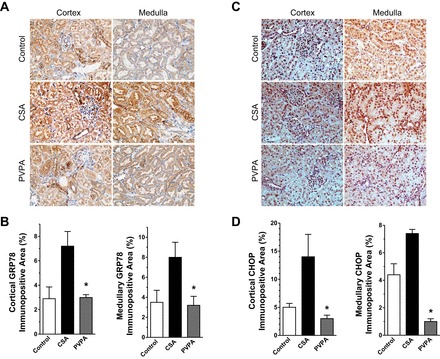
PVPA ameliorates ER stress in the kidney during CsA treatment. A: representative light photomicrographs of kidney sections showing immunopositive staining for GRP78 (original magnification, ×400). B: semiquantitative score of GRP78 immunostaining in kidney sections. C: representative light photomicrographs of kidney sections showing immunopositive staining for C/EBP homologous protein (CHOP). D: semiquantitative score of CHOP immunostaining in kidney sections. Values are means ± SE; n = 5–8/group. *P < 0.05 vs. CsA group.
PVPA reduces CsA-induced apoptosis in vitro.
In the present study, we demonstrated antihypertensive and kidney-protective effects of PVPA in CsA-treated animals, and such kidney-protective effects of PVPA can be attributed to its antihypertensive action. To investigate whether the kidney-protective effects of PVPA in the CsA-treated animals also resulted from its direct cytoprotective effects, we conducted in vitro studies using NRK-52E cells. First, to confirm the safety of PVPA in kidney cells, NRK-52E cells were incubated with various concentrations of PVPA and cell viability was assessed by an MTT assay. PVPA had no deleterious effect on the proliferation or viability of the cells over a wide range of pharmacologically relevant dosing (1–20 μM; Fig. 8A). As in the in vivo system, we found that incubation of NRK-52E cells with CsA resulted in a significant increase in the number of apoptotic cells as determined by TUNEL staining compared with vehicle treatment. Cotreatment of the cells with PVPA significantly reduced apoptosis in the cells (Fig. 8, B and C). These results suggest that PVPA ameliorates CsA-induced nephrotoxicity, at least in part, through direct cytoprotective effects on epithelial cells.
Fig. 8.
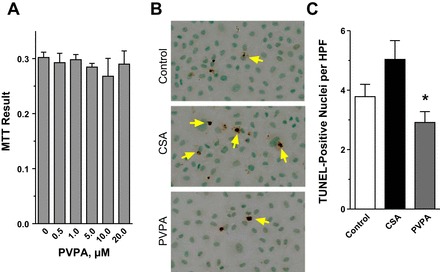
PVPA reduces CsA-induced apoptosis in vitro. A: NRK-52E cells were incubated with various concentrations of PVPA for 24 h. PVPA did not affect the proliferation or viability of the cells as assessed by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. B: representative light photomicrographs of NRK-52E cells stained by the TUNEL method after 24 h of treatment with vehicle (control) or 5 μM CsA ± pretreatment with 10 μM PVPA (original magnification, ×400). C: semiquantitative score of TUNEL-positive cells. *P < 0.05 vs. CsA (cells not pretreated with PVPA).
DISCUSSION
CNIs are a major component of the immunosuppressive regimen used in solid organ transplant recipients and are also used to treat many other immune-mediated diseases. By inhibiting calcineurin, the CNIs exert their immunosuppressive effects by preventing the dephosphorylation and nuclear translocation of NFAT and thus reducing the expression of IL-2 which is important for T cell proliferation and activation. Unfortunately, despite their significant clinical utility, CNI use is hampered by the development of hypertension, nephrotoxicity, and cardiovascular morbidity. The nephrotoxicity is universal and occurs over time. The irreversible kidney changes noted following chronic CNI use include renal arteriolar hyalinosis, glomerular sclerosis, tubular atrophy, and tubulointerstitial fibrosis, which are associated with progressive renal dysfunction (30). Attempts at developing immunosuppressive protocols that avoid, withdraw, or minimize CNI use in solid organ transplantation have mostly been unsuccessful due to the increased risk of acute rejection and therefore the potential for chronic graft failure (9–11, 16, 18). Consequently, effective therapy to prevent or treat CNI-induced nephrotoxicity is urgently needed. Accumulating evidence indicates that the cytochrome P-450/EET system has many organ-protective properties. In the present study, we investigated the protective role of the novel, orally active EET analog PVPA on an experimental model of CsA-induced hypertension and nephrotoxicity. Our results show that exogenous administration of PVPA effectively prevented CsA-induced hypertension and ameliorated interstitial fibrosis and kidney dysfunction. We demonstrate that PVPA administration inhibited inflammatory cell infiltration into the kidney, indicating that PVPA has anti-inflammatory effects in this model. Also, PVPA reduced oxidative stress, modulated the unfolded protein response that is associated with ER stress, and, importantly, attenuated TEC apoptosis. Overall, our data suggest that PVPA decreased CsA-induced renal injury through lowering of blood pressure and also through its direct cytoprotective effects on epithelial cells. To our knowledge, this study is the first to show a role for EETs in this model, and the findings suggest that the cytochrome P-450/EET system may offer a novel, multimechanism therapeutic strategy to prevent CNI-induced nephrotoxicity.
The mechanism of CNI-induced hypertension and nephrotoxicity is complex and remains incompletely understood. CNIs may cause hypertension by enhancing salt and water retention through the activation of sodium channels, including the thiazide-sensitive Na+-Cl− cotransporter (NCC) and epithelial sodium channel (ENaC) (25, 50). CNIs have also been reported to stimulate the intrarenal renin-angiotensin system (RAS) in addition to promoting increased levels of endothelin 1, both of which could contribute to the development of hypertension, tissue hypoxia, and renal fibrosis. Also, CNI administration is associated with increased production of ROS, and there is extensive evidence supporting the involvement of oxidative stress in the pathogenesis of CNI-induced hypertension and nephrotoxicity. Among others, oxidative stress due to excess production of ROS can lead to reduction in nitric oxide levels, resulting in endothelial dysfunction and vasoconstriction and may play a role in the development of hypertension and nephrotoxicity. Furthermore, chronic CNI administration leads to tubular atrophy, at least in part, through TEC apoptosis. CNI-induced TEC apoptosis could be a direct effect of the CNI or from indirect causes. Another common finding with CNI treatment is that of an increased inflammatory response within the kidney. Numerous studies have demonstrated that CNI treatment leads to increased production of inflammatory cytokines, chemokines, and adhesion molecules, and this is associated with enhanced inflammatory cell infiltration into the kidney (30). The infiltrating cells are a source of ROS and may also cause chronic damage through the deposition of excessive amounts of extracellular matrix that results in renal fibrosis. Recent studies have identified ER stress as a potential mechanism for CNI-induced nephrotoxicity (40). Chronic treatment with CNIs has been shown to cause ER stress, an ER dysfunctional state characterized by the accumulation of unfolded (or misfolded) proteins. The adaptive cellular response to ER stress involves the activation of the unfolded protein response (UPR), aimed at restoring homeostasis and maintaining functional integrity of the cells. During excessive or persistent ER stress, the UPR can activate inflammatory and apoptotic pathways, leading to cell death (33).
In the rat model of CNI-induced hypertension and nephrotoxicity, we noted a profound increase in SBP as early as 7 days after initiation of CsA followed by a sustained elevation in the SBP until the end of the experiment. In comparison, the animals that received PVPA in addition to CsA maintained their SBP close to the baseline value, and at the end of the treatment period had SBP that was not statistically different from the control animals. The mechanism of the antihypertensive effect of PVPA in this model was not examined, but most likely involves vasodilatation of the renal microvessels and also through inhibition of sodium channels (4, 19). The mean weight of the CsA-only group at the end of the study was not different from the PVPA group, implying that the observed differences in blood pressure were not driven by weight differences. Renal fibrosis is a hallmark of chronic damage by CNI treatment, and, like other causes of CKD, it is the final common pathway through which CNI-induced renal injury leads to end-stage kidney failure. In this study, we found that CsA caused tubulointerstitial fibrosis and this was associated with renal dysfunction as shown by the increased proteinuria and BUN levels. The CsA-induced collagen deposition and renal dysfunction were significantly ameliorated in animals treated with PVPA. Macrophage infiltration into the kidney was increased by CsA, and this was attenuated by coadministration of PVPA. The reduction in macrophage infiltration due to PVPA treatment could explain the improved fibrosis and renal dysfunction noted in our study. We also observed increased expression of TBARs, a marker of oxidative stress, extensive tubular injury as shown by tubular vacuolization, and also increased apoptosis in the kidneys of animals treated with cyclosporine. All these are precursors to the development of fibrosis during chronic administration of cyclosporine (30), and these changes were all ameliorated by coadministration of PVPA. Following 28 days of CsA administration to the animals, we found evidence of ER stress in tubular epithelial cells as depicted by the marked increase in expression of the master chaperone GRP 78 and also the transcription factor CHOP, which is associated with apoptotic cell death. Cotreatment of the animals with PVPA attenuated ER stress. These findings are consistent with our recent report showing the modulating effects of EET analogs on cisplatin-induced ER stress (20). Future studies will elucidate the molecular mechanisms through which PVPA modulates ER stress.
Under physiological conditions, the cytochrome P-450 epoxygenases metabolize arachidonic acid to bioactive EETs in various tissues, and these compounds act in autocrine and paracrine fashions, mediating vasodilatory and other effects. Recent studies have provided evidence that EETs activate cell surface receptors to increase the levels of cAMP, which activates smooth muscle cell large-conductance calcium-activated K+ channels, resulting in vasodilation. Specifically, the kidney expresses high amounts of epoxygenase activity and produces 14,15-EET as the major epoxide. The renal EETs are expressed in both the microvessels and tubules, and they have been shown to control hemodynamic and epithelial transport functions in the kidney. During pathophysiological states, there is impaired epoxygenase generation of EETs (20), and this may contribute to reduction in renal blood flow and glomerular filtration. The current availability of exogenous EETs implies that we can explore this pathway further to understand the physiological basis for certain diseases, and possibly treat or prevent the occurrence of specific diseases. Accumulating evidence indicates that in addition to its vasodilator properties and, hence, ability to alleviate hypertension, EETs are able to protect tissues through other mechanisms, including anti-inflammatory, antiapoptotic, and antioxidative effects. Indeed, studies by our group and others have shown that EETs provide organ protection in a number of preclinical models of human diseases, including diabetes, hypertension, ischemic cardiac injury, and cisplatin-induced nephrotoxicity (12, 20, 37, 51). Our current findings are in total agreement with the above and demonstrate the renoprotective effect of PVPA on CsA-induced hypertension and nephrotoxicity. The beneficial effect of PVPA in this model is likely due to the suppression of inflammation, oxidative stress, and apoptosis through the modulation of ER stress. The protective effects of PVPA were also seen in the in vitro system, where PVPA reduced cyclosporine-induced apoptosis in rat proximal TEC.
In conclusion, the 14,15-EET analog PVPA effectively prevented CsA-induced hypertension and reduced interstitial fibrosis and kidney dysfunction by ameliorating inflammation, oxidative stress, ER stress, and apoptosis. The results of these experiments have important implications not only for CNI-induced nephropathy but also for the treatment and prevention of other kidney diseases that are mediated by apoptosis, inflammation, and oxidative stress.
GRANTS
This study was funded by Department of Medicine, Medical College of Wisconsin support (to M. M. Yeboah), a Research Starter Grant from the PhRMA Foundation USA (to M. A. Hye Khan), the Robert A. Welch Foundation (I-0011 to J. R. Falck), and the Dr. Ralph and Marian Falk Medical Research Trust, Bank of America, N.A. Trustee grant (to J. D. Imig).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
M.M.Y., M.A.H.K., and J.D.I. provided conception and design of research; M.M.Y., M.A.H.K., M.A.C., and A.S. performed experiments; M.M.Y., M.A.H.K., M.A.C., A.S., M.P.P., J.R.F., and J.D.I. analyzed data; M.M.Y., M.A.H.K., M.A.C., A.S., M.P.P., J.R.F., and J.D.I. interpreted results of experiments; M.M.Y., M.A.H.K., M.A.C., and J.D.I. prepared figures; M.M.Y., M.A.H.K., M.A.C., A.S., and J.D.I. drafted manuscript; M.M.Y., M.A.H.K., M.A.C., A.S., M.P.P., J.R.F., and J.D.I. edited and revised manuscript; M.M.Y., M.A.H.K., M.A.C., A.S., M.P.P., J.R.F., and J.D.I. approved final version of manuscript.
REFERENCES
- 1.Andreoni KA, Brayman KL, Guidinger MK, Sommers CM, Sung RS. Kidney and pancreas transplantation in the United States, 1996–2005. Am J Transplant 7: 1359–1375, 2007. [DOI] [PubMed] [Google Scholar]
- 2.Bennett WM, Pulliam JP. Cyclosporine nephrotoxicity. Ann Intern Med 99: 851–854, 1983. [DOI] [PubMed] [Google Scholar]
- 3.Calne RY, White DJ, Thiru S, Evans DB, McMaster P, Dunn DC, Craddock GN, Pentlow BD, Rolles K. Cyclosporin A in patients receiving renal allografts from cadaver donors. Lancet 2: 1323–1327, 1978. [DOI] [PubMed] [Google Scholar]
- 4.Campbell WB, Harder DR. Endothelium-derived hyperpolarizing factors and vascular cytochrome P450 metabolites of arachidonic acid in the regulation of tone. Circ Res 84: 484–488, 1999. [DOI] [PubMed] [Google Scholar]
- 5.Canadian Multicentre Transplant Study Group. A randomized clinical trial of cyclosporine in cadaveric renal transplantation. N Engl J Med 309: 809–815, 1983. [DOI] [PubMed] [Google Scholar]
- 6.Capdevila JH, Falck JR, Estabrook RW. Cytochrome P450 and the arachidonate cascade. FASEB J 6: 731–736, 1992. [DOI] [PubMed] [Google Scholar]
- 7.Curtis JJ. Cyclosporine and posttransplant hypertension. J Am Soc Nephrol 2: S243–S245, 1992. [DOI] [PubMed] [Google Scholar]
- 8.Djamali A, Reese S, Hafez O, Vidyasagar A, Jacobson L, Swain W, Kolehmainen C, Huang L, Wilson NA, Torrealba JR. Nox2 is a mediator of chronic CsA nephrotoxicity. Am J Transplant 12: 1997–2007, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ekberg H. Calcineurin inhibitor sparing in renal transplantation. Transplantation 86: 761–767, 2008. [DOI] [PubMed] [Google Scholar]
- 10.Ekberg H, Grinyo J, Nashan B, Vanrenterghem Y, Vincenti F, Voulgari A, Truman M, Nasmyth-Miller C, Rashford M. Cyclosporine sparing with mycophenolate mofetil, daclizumab and corticosteroids in renal allograft recipients: the CAESAR Study. Am J Transplant 7: 560–570, 2007. [DOI] [PubMed] [Google Scholar]
- 11.Ekberg H, Tedesco-Silva H, Demirbas A, Vitko S, Nashan B, Gurkan A, Margreiter R, Hugo C, Grinyo JM, Frei U, Vanrenterghem Y, Daloze P, Halloran PF, ELITE Symphony Study. Reduced exposure to calcineurin inhibitors in renal transplantation. N Engl J Med 357: 2562–2575, 2007. [DOI] [PubMed] [Google Scholar]
- 12.Elmarakby AA, Faulkner J, Al-Shabrawey M, Wang MH, Maddipati KR, Imig JD. Deletion of soluble epoxide hydrolase gene improves renal endothelial function and reduces renal inflammation and injury in streptozotocin-induced type 1 diabetes. Am J Physiol Regul Integr Comp Physiol 301: R1307–R1317, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.European Multicentre Trial Group. Cyclosporin in cadaveric renal transplantation: one-year follow-up of a multicentre trial. Lancet 2: 986–989, 1983. [PubMed] [Google Scholar]
- 14.Feng D, Yang C, Geurts AM, Kurth T, Liang M, Lazar J, Mattson DL, O'Connor PM, Cowley AW Jr. Increased expression of NAD(P)H oxidase subunit p67(phox) in the renal medulla contributes to excess oxidative stress and salt-sensitive hypertension. Cell Metab 15: 201–208, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Feng W, Xu X, Zhao G, Li G, Liu T, Zhao J, Dong R, Wang DW, Tu L. EETs and CYP2J2 inhibit TNF-alpha-induced apoptosis in pulmonary artery endothelial cells and TGF-beta1-induced migration in pulmonary artery smooth muscle cells. Int J Mol Med 32: 685–693, 2013. [DOI] [PubMed] [Google Scholar]
- 16.Flechner SM, Kobashigawa J, Klintmalm G. Calcineurin inhibitor-sparing regimens in solid organ transplantation: focus on improving renal function and nephrotoxicity. Clin Transplant 22: 1–15, 2008. [DOI] [PubMed] [Google Scholar]
- 17.Gonzalez-Guerrero C, Ocana-Salceda C, Berzal S, Carrasco S, Fernandez-Fernandez B, Cannata-Ortiz P, Egido J, Ortiz A, Ramos AM. Calcineurin inhibitors recruit protein kinases JAK2 and JNK, TLR signaling and the UPR to activate NF-kappaB-mediated inflammatory responses in kidney tubular cells. Toxicol Appl Pharmacol 272: 825–841, 2013. [DOI] [PubMed] [Google Scholar]
- 18.Hricik DE, Formica RN, Nickerson P, Rush D, Fairchild RL, Poggio ED, Gibson IW, Wiebe C, Tinckam K, Bunnapradist S, Samaniego-Picota M, Brennan DC, Schroppel B, Gaber O, Armstrong B, Ikle D, Diop H, Bridges ND, Heeger PS; Clinical Trials in Organ Transplantation-09 Consortium. Adverse outcomes of tacrolimus withdrawal in immune-quiescent kidney transplant recipients. J Am Soc Nephrol 26: 3114–3122, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Imig JD, Navar LG, Roman RJ, Reddy KK, Falck JR. Actions of epoxygenase metabolites on the preglomerular vasculature. J Am Soc Nephrol 7: 2364–2370, 1996. [DOI] [PubMed] [Google Scholar]
- 20.Khan MA, Liu J, Kumar G, Skapek SX, Falck JR, Imig JD. Novel orally active epoxyeicosatrienoic acid (EET) analogs attenuate cisplatin nephrotoxicity. FASEB J 27: 2946–2956, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim J, Imig JD, Yang J, Hammock BD, Padanilam BJ. Inhibition of soluble epoxide hydrolase prevents renal interstitial fibrosis and inflammation. Am J Physiol Renal Physiol 307: F971–F980, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim J, Yoon SP, Toews ML, Imig JD, Hwang SH, Hammock BD, Padanilam BJ. Pharmacological inhibition of soluble epoxide hydrolase prevents renal interstitial fibrogenesis in obstructive nephropathy. Am J Physiol Renal Physiol 308: F131–F139, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kurita T, Yasuda S, Amengual O, Atsumi T. The efficacy of calcineurin inhibitors for the treatment of interstitial lung disease associated with polymyositis/dermatomyositis. Lupus 24: 3–9, 2015. [DOI] [PubMed] [Google Scholar]
- 24.Lafayette RA, Mayer G, Meyer TW. The effects of blood pressure reduction on cyclosporine nephrotoxicity in the rat. J Am Soc Nephrol 3: 1892–1899, 1993. [DOI] [PubMed] [Google Scholar]
- 25.Lazelle RA, McCully BH, Terker AS, Himmerkus N, Blankenstein K, Mutig K, Bleich M, Bachmann S, Yang CL, Ellison DH. Renal deletion of 12 kDa FK506-binding protein attenuates tacrolimus-induced hypertension. J Am Soc Nephrol 27: 1456–1464, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu Y, Webb HK, Fukushima H, Micheli J, Markova S, Olson JL, Kroetz DL. Attenuation of cisplatin-induced renal injury by inhibition of soluble epoxide hydrolase involves nuclear factor kappaB signaling. J Pharmacol Exp Ther 341: 725–734, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Luke RG. Pathophysiology and treatment of posttransplant hypertension. J Am Soc Nephrol 2: S37–S44, 1991. [DOI] [PubMed] [Google Scholar]
- 28.Meier-Kriesche HU, Schold JD, Srinivas TR, Kaplan B. Lack of improvement in renal allograft survival despite a marked decrease in acute rejection rates over the most recent era. Am J Transplant 4: 378–383, 2004. [DOI] [PubMed] [Google Scholar]
- 29.Myers BD, Ross J, Newton L, Luetscher J, Perlroth M. Cyclosporine-associated chronic nephropathy. N Engl J Med 311: 699–705, 1984. [DOI] [PubMed] [Google Scholar]
- 30.Naesens M, Kuypers DR, Sarwal M. Calcineurin inhibitor nephrotoxicity. Clin J Am Soc Nephrol 4: 481–508, 2009. [DOI] [PubMed] [Google Scholar]
- 31.Nankivell BJ, Borrows RJ, Fung CL, O'Connell PJ, Allen RD, Chapman JR. The natural history of chronic allograft nephropathy. N Engl J Med 349: 2326–2333, 2003. [DOI] [PubMed] [Google Scholar]
- 32.Nankivell BJ, Borrows RJ, Fung CL, O'Connell PJ, Chapman JR, Allen RD. Calcineurin inhibitor nephrotoxicity: longitudinal assessment by protocol histology. Transplantation 78: 557–565, 2004. [DOI] [PubMed] [Google Scholar]
- 33.Nishitoh H. CHOP is a multifunctional transcription factor in the ER stress response. J Biochem 151: 217–219, 2012. [DOI] [PubMed] [Google Scholar]
- 34.Nishiyama A, Kobori H, Fukui T, Zhang GX, Yao L, Rahman M, Hitomi H, Kiyomoto H, Shokoji T, Kimura S, Kohno M, Abe Y. Role of angiotensin II and reactive oxygen species in cyclosporine A-dependent hypertension. Hypertension 42: 754–760, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Node K, Huo Y, Ruan X, Yang B, Spiecker M, Ley K, Zeldin DC, Liao JK. Anti-inflammatory properties of cytochrome P450 epoxygenase-derived eicosanoids. Science 285: 1276–1279, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ojo AO. Renal disease in recipients of nonrenal solid organ transplantation. Semin Nephrol 27: 498–507, 2007. [DOI] [PubMed] [Google Scholar]
- 37.Olearczyk JJ, Quigley JE, Mitchell BC, Yamamoto T, Kim IH, Newman JW, Luria A, Hammock BD, Imig JD. Administration of a substituted adamantyl urea inhibitor of soluble epoxide hydrolase protects the kidney from damage in hypertensive Goto-Kakizaki rats. Clin Sci (Lond) 116: 61–70, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Opelz G, Wujciak T, Ritz E. Association of chronic kidney graft failure with recipient blood pressure. Collaborative Transplant Study. Kidney Int 53: 217–222, 1998. [DOI] [PubMed] [Google Scholar]
- 39.Padi SS, Chopra K. Salvage of cyclosporine A-induced oxidative stress and renal dysfunction by carvedilol. Nephron 92: 685–692, 2002. [DOI] [PubMed] [Google Scholar]
- 40.Pallet N, Bouvier N, Bendjallabah A, Rabant M, Flinois JP, Hertig A, Legendre C, Beaune P, Thervet E, Anglicheau D. Cyclosporine-induced endoplasmic reticulum stress triggers tubular phenotypic changes and death. Am J Transplant 8: 2283–2296, 2008. [DOI] [PubMed] [Google Scholar]
- 41.Parrish AR, Chen G, Burghardt RC, Watanabe T, Morisseau C, Hammock BD. Attenuation of cisplatin nephrotoxicity by inhibition of soluble epoxide hydrolase. Cell Biol Toxicol 25: 217–225, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sanders WG, Morisseau C, Hammock BD, Cheung AK, Terry CM. Soluble epoxide hydrolase expression in a porcine model of arteriovenous graft stenosis and anti-inflammatory effects of a soluble epoxide hydrolase inhibitor. Am J Physiol Cell Physiol 303: C278–C290, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sedeek M, Nasrallah R, Touyz RM, Hebert RL. NADPH oxidases, reactive oxygen species, and the kidney: friend and foe. J Am Soc Nephrol 24: 1512–1518, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shevach EM. The effects of cyclosporin A on the immune system. Annu Rev Immunol 3: 397–423, 1985. [DOI] [PubMed] [Google Scholar]
- 45.Shihab FS, Bennett WM, Yi H, Andoh TF. Effect of pirfenidone on apoptosis-regulatory genes in chronic cyclosporine nephrotoxicity. Transplantation 79: 419–426, 2005. [DOI] [PubMed] [Google Scholar]
- 46.Taylor NE, Glocka P, Liang M, Cowley AW Jr. NADPH oxidase in the renal medulla causes oxidative stress and contributes to salt-sensitive hypertension in Dahl S rats. Hypertension 47: 692–698, 2006. [DOI] [PubMed] [Google Scholar]
- 47.Thomas SE, Andoh TF, Pichler RH, Shankland SJ, Couser WG, Bennett WM, Johnson RJ. Accelerated apoptosis characterizes cyclosporine-associated interstitial fibrosis. Kidney Int 53: 897–908, 1998. [DOI] [PubMed] [Google Scholar]
- 48.Thomson SJ, Askari A, Bishop-Bailey D. Anti-inflammatory effects of epoxyeicosatrienoic acids. Int J Vasc Med 2012: 605101, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang C, Salahudeen AK. Lipid peroxidation accompanies cyclosporine nephrotoxicity: effects of vitamin E. Kidney Int 47: 927–934, 1995. [DOI] [PubMed] [Google Scholar]
- 50.Wang J, Zhang ZR, Chou CF, Liang YY, Gu Y, Ma HP. Cyclosporine stimulates the renal epithelial sodium channel by elevating cholesterol. Am J Physiol Renal Physiol 296: F284–F290, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yang L, Maki-Petaja K, Cheriyan J, McEniery C, Wilkinson IB. The role of epoxyeicosatrienoic acids in the cardiovascular system. Br J Clin Pharmacol 80: 28–44, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yu Z, Xu F, Huse LM, Morisseau C, Draper AJ, Newman JW, Parker C, Graham L, Engler MM, Hammock BD, Zeldin DC, Kroetz DL. Soluble epoxide hydrolase regulates hydrolysis of vasoactive epoxyeicosatrienoic acids. Circ Res 87: 992–998, 2000. [DOI] [PubMed] [Google Scholar]