

Abstract
Calcineurin dephosphorylates nuclear factor of activated T cells transcription factors, thereby facilitating T cell-mediated immune responses. Calcineurin inhibitors are instrumental for immunosuppression after organ transplantation but may cause side effects, including hypertension and electrolyte disorders. Kidneys were recently shown to display activation of the furosemide-sensitive Na-K-2Cl cotransporter (NKCC2) of the thick ascending limb and the thiazide-sensitive Na-Cl cotransporter (NCC) of the distal convoluted tubule upon calcineurin inhibition using cyclosporin A (CsA). An involvement of major hormones like angiotensin II or arginine vasopressin (AVP) has been proposed. To resolve this issue, the effects of CsA treatment in normal Wistar rats, AVP-deficient Brattleboro rats, and cultured renal epithelial cells endogenously expressing either NKCC2 or NCC were studied. Acute administration of CsA to Wistar rats rapidly augmented phosphorylation levels of NKCC2, NCC, and their activating kinases suggesting intraepithelial activating effects. Chronic CsA administration caused salt retention and hypertension, along with stimulation of renin and suppression of renal cyclooxygenase 2, pointing to a contribution of endocrine and paracrine mechanisms at long term. In Brattleboro rats, CsA induced activation of NCC, but not NKCC2, and parallel effects were obtained in cultured cells in the absence of AVP. Stimulation of cultured thick ascending limb cells with AVP agonist restored their responsiveness to CsA. Our results suggest that the direct epithelial action of calcineurin inhibition is sufficient for the activation of NCC, whereas its effect on NKCC2 is more complex and requires concomitant stimulation by AVP.
Keywords: hypertension, sodium-chloride cotransporter, sodium-potassium-chloride cotransporter, salt transport, vasopressin
calcineurin inhibitors (CNI) like cyclosporine A (CsA) and tacrolimus are broadly used for immunosuppression after organ transplantation to prolong survival of allografts. In spite of their undoubted benefit, however, CNI cause adverse side effects, which include renal functional impairment leading to hypertension and electrolyte disorders (13, 14, 45). To improve the benefit-to-risk ratio of CNI therapy, recent research has been focused on pathogenetic mechanisms underlying their renal side effects. Studies from our and other groups have suggested that inhibition of calcineurin in the distal nephron results in an activation of the crucial cation-coupled chloride cotransporters (CCC), the furosemide-sensitive Na-K-2Cl cotransporter type 2 (NKCC2) of the thick ascending limb (TAL) and the thiazide-sensitive Na-Cl cotransporter (NCC) of the distal convoluted tubule (DCT). As a result, salt retention, volume expansion, and hypertension have been reported (2, 13, 36). NKCC2 and NCC share several conserved NH2-terminal serine/threonine phosphoacceptor sites (7, 8, 10). Phosphorylation of these residues by a kinase cascade comprising members of the with-no-lysine [K] kinase family (WNK) and two homologous Ste20-related kinases, Ste20-related proline/alanine-rich kinase (SPAK) and oxidative stress-responsive kinase 1 (OSR1), is critical to the transport activity of NKCC2 and NCC (2, 35). Calcineurin appears to be involved in dephosphorylation and deactivation of the transporters via its action on these kinases. CNI have been reported to increase the abundance of phosphorylated CCC and their activating kinases along the distal nephron (2, 13). In line with this, application of diuretic drugs acting in the distal nephron efficiently alleviates CNI-induced salt retention and hypertension both in mouse models and in patients (13, 20). However, the mechanisms of CNI-induced hypertension are more complex, encompassing not only intraepithelial but also paracrine and systemic variables that affect renal salt handling, and individual susceptibility of patients also plays a role (12, 14, 19, 25). With respect to the systemic action of CsA, it has been suggested that stimulated arginine vasopressin (AVP) signaling may contribute to CsA-induced hypertension through vascular effects (23, 24). Other studies demonstrated activation of the renin-angiotensin-aldosterone system (RAAS) upon CsA administration (19). CNI may therefore affect renal homeostasis systemically via modulation of major endocrine signals with the result of an aggravation of their renal side effects (19, 26). Endocrine stimuli also affect activity of the CCC critically, as elucidated by substantially decreased NKCC2 function in AVP-deficient Brattleboro rats or reduced NCC function when RAAS activity is suppressed (5, 29, 30, 41, 42). Despite their structural similarity, NKCC2 and NCC react differentially upon endocrine stimuli, with the TAL being more sensitive to AVP than the DCT, and the DCT more responsive to RAAS components (27, 30, 33, 38). CNI may also modulate renal salt handling via their effects on relevant paracrine mechanisms, including the effects on cyclooxygenase-2 (COX-2) and local prostaglandin pathways. Notably, the function of renal COX-2 has been linked with AVP signaling (12, 18). Despite previous effort, the mechanisms of CNI-induced direct activation of NKCC2 and NCC as well as the contribution of CNI-dependent systemic effects have so far not been sufficiently clarified. To gain better insight, we have evaluated the effects of CsA in normal Wistar rats, AVP-deficient Brattleboro rats, and cultured TAL and DCT cells. Our results suggest that local calcineurin inhibition in DCT cells is sufficient to induce NCC activation, whereas activation of NKCC2 requires additional systemic stimulation by AVP.
MATERIALS AND METHODS
Animals, tissues, treatments.
Adult (10–12 wk) male Wistar and AVP-deficient Brattleboro rats were kept on standard diet and tap water. For evaluation of short-term CsA effects in wild-type rats, animals were divided into groups (n = 5 for biochemical evaluation and at least n = 4 for morphology) receiving CsA (30 mg/kg body wt sandimmune; Novartis, Nürnberg, Germany) or vehicle (chremophor; Sigma-Aldrich, Munich, Germany) for 1 or 4 h, respectively, by intraperitoneal injection. For biochemical analysis, rats were killed, and the kidneys were removed. For morphological evaluation, rats were anesthetized (ketamine/xylazine; Sigma-Aldrich) and perfusion fixed retrogradely via the aorta abdominalis using 3% paraformaldehyde (39). For long-term CsA studies, wild-type rats were divided into groups (n = 10 for physiological analysis and n = 4 for morphology) receiving CsA (30 mg/kg body wt) or vehicle (chremophor) for 14 days by daily subcutaneous injection. For physiological analysis, animals were individually placed in metabolic cages. Rats received normal food and tap water ad libitum, and 24-h urine was collected. Urine sodium and creatinine concentrations as well as plasma renin activity were determined. Evaluation of systolic blood pressure was performed by the tail-cuff method on anesthetized rats. For furosemide tests, rats were injected intraperitoneally with one dose of furosemide (40 mg/kg body wt; Sigma-Aldrich), and urine was collected 4 h following injection. Blood was collected via the tail vein and during euthanization concomitantly with organ collection. For morphological evaluation, rats were anesthetized and perfusion fixed retrogradely via the aorta abdominalis using 3% paraformaldehyde. For evaluation of the effects of calcineurin inhibition in rats lacking endogenous AVP, Brattleboro rats (n = 6/group) received a single dose of CsA (30 mg/kg body wt ip) or vehicle (chremophor) and were killed 4 h after injection; kidneys were removed. All experiments were approved by the Regional Office for Health and Social Affairs Berlin (LAGESO permission G0220/12).
Cell culture.
SV40-transformed rat TAL cells (raTAL) were cultured as previously described (44). Mouse DCT (mDCT) cells (9) were cultivated in 75-cm2 cell culture flasks in RPMI medium (Biochrom, Berlin, Germany) with 10% fetal calf serum and 1% penicillin/streptavidin at 37°C, 95% humidity, and 5% CO2. Cells were grown to confluent monolayers, stimulated with 1 µM CsA (Santa Cruz Biotechnology, Heidelberg, Germany), 10 µM desmopressin (DDAVP; Sigma-Aldrich), or both agents simultaneously in culture medium for 4 h, harvested in Igepal lysis buffer (Sigma-Aldrich), and whole cell lysates were prepared for immunoblotting.
Primary antibodies.
Antibodies recognizing NKCC2 (29), phospho-T96/T101-NKCC2 (29), NCC (29), phospho-S71-NCC (29), renal outer medullary K channel (ROMK; Sigma-Aldrich), ATP-sensitive inward-rectifier K channel (Kir4.1; Alomone Laboratories, Duisburg, Germany), SPAK (COOH-terminal) (21), OSR1 (University of Dundee) and phosphorylated SPAK/OSR1 species [pT243-SPAK/pT185-OSR1 (catalytic domain) and pS383-SPAK/pS325-OSR1 (regulatory domain); University of Dundee], COX-2 (Santa Cruz Biotechnology), β-actin (Sigma-Aldrich), and GAPDH (Santa Cruz Biotechnology) were applied as primary antibodies.
Immunofluorescence.
Cryo (7 µm)- or paraffin (4 µm) sections from rat kidneys were incubated with blocking medium (30 min) before incubation with primary antibodies diluted in blocking medium (1 h, overnight); multiple stainings were separated by washing steps. Fluorescent Cy2-, Cy3-, or Cy5-conjugated antibodies (DIANOVA, Hamburg, Germany) were applied for detection. A Zeiss confocal microscope (LSM 5 Exciter) was used for evaluation of sections. To identify TAL and DCT, kidney sections were double stained with NKCC2 or NCC, respectively. For evaluation of phosphorylated (p)-NKCC2, pNCC, and pSPAK/OSR1 fluorescent signal intensities, sections were labeled with antibodies to their phosphorylated species followed by labeling with antibodies recognizing the respective proteins independently on their phosphorylation state. Micrographs were obtained using ZEN2008 software (Zeiss), and signal intensities in individual tubular profiles were analyzed with ImageJ software (40). Phosphosignals were normalized to respective total signals. Four to six animals with at least 20 similar tubular profiles per individual were evaluated per group in a blind fashion. Mean signal values were obtained within the depth of 2 µm to the apical membrane, and normalization to background signal over the cell nuclei was performed. Quantification of COX-2 signal at the macula densa and adjacent TAL portions was performed by counting cells histochemically positive for COX-2.
Immunoblotting.
Whole kidneys were homogenized in buffer containing 250 mM sucrose, 10 mM triethanolamine, and protease inhibitors (Complete; Roche Diagnostics, Berlin, Germany) and sonicated for 10 s, and nuclei were removed by centrifugation (1,000 g for 10 min). Cell lysates were homogenized, sonicated, and centrifuged likewise for protein extraction. Polyacrylamide minigels (10%) were used to electrophoretically separate proteins, which were subsequently transferred to polyvinylidene fluoride membranes, followed by blocking (30 min) and incubation with primary antibody (1 h at room temperature or overnight at 4°C). Detection was performed using horseradish peroxidase-conjugated secondary antibodies (DAKO, Hamburg, Germany) followed by chemiluminescence exposure of the membranes. Signals were detected using the ChemoCam Imager ECL (Intas, Göttingen, Germany) and evaluated densitometrically using ImageJ software.
Quantitative PCR.
RNA was extracted from whole kidney lysates using an RNA extraction kit (Stratec Biomedical, Birkenfeld, Germany) and reverse transcribed into cDNA using Tetro Reverse Transcriptase (Promega, Mannheim, Germany). Quantitative PCR was performed using HOT FIREPol EvaGreen qPCR Mix (Solis BioDyne, Tallinn, Estonia) and primers specific for COX-2 (forward: 5′-TGACAGCCCACCAACTTACA-3′, reverse: 5′-TCCTTATTTCCTTTCACACCCA-3′), NKCC2 (forward: 5′-TGTGAAGTTTGGATGGGT-3′, reverse: 5′-CCGCTTCTCCTACAATCC-3′), NCC (forward: 5′-TGATCATCCTTACCTTGCCCA-3′, reverse: 5′-ACGTTCTCCTGGTTACCTCG-3′), Kir4.1 (forward: 5′-CAGCCACTCCACCTCTGTG-3′, reverse: 5′-GACGTATTCCTGGAGCCACT-3′), ROMK (forward: 5′-CAAGCACCACTTGCTTGC-3′, reverse: 5′-TGAACATCCTTTCTGTCAGTGC-3′), OSR1 (forward: 5′-AAAGACGTTTGTTGGCACCC-3′, reverse: 5′-GCCCCTGTGGCTAGTTCAAT-3′), SPAK (forward: 5′-TGCCAGACGAGTATGGATGA-3′, reverse: 5′-CCACAGCTCATCTTTGACCA-3′), WNK1 (forward: 5′-AAGTATGCCTCAGTCCGTGG-3′, reverse: 5′-ACTTTCGGTGGACAGGTAGG-3′), and WNK3 (forward: 5′-GAGCTACAGGACCGCAAATTA-3′, reverse: 5′-TCGAACTATATTGGGATGCTGGA-3′). Gene expression analysis was performed applying the ΔΔCt method. Expression was normalized against β‐actin.
Ultrastructural analysis.
For evaluation of NKCC2 and NCC distribution, electron microscopy was performed on perfusion-fixed ultrathin LR-White sections. Primary antibodies and 5-nm nanogold-labeled secondary antibodies (Amersham) were applied for detection and visualized using transmission electron microscopy (Tecnai; FEI). Immunogold signals in TAL and DCT profiles were quantified according to an established protocol (38, 39). Signals within a 20-nm distance to the plasma membrane were defined as membrane bound, whereas signals detected below 20 nm until the nuclear envelope were defined as cytoplasmic. At least 10 profiles and 4 cells per profile were evaluated per individual animal.
In situ hybridization.
For evaluation of renin mRNA, in situ hybridization was performed on perfusion-fixed paraffin-embedded kidney sections. Antisense RNA probes labeled with digoxygenin (DIG) were hybridized and recognized with secondary anti-DIG-alkaline phosphatase-conjugated antibody (DAKO) (1). Detection was performed by incubating the sections with nitro blue tetrazolium and 5-bromo-4-chloro-3-indolyl phosphate (Roche Diagnostics). Renin mRNA-expressing arteriolar sites were quantified as described previously (3). Sections were analyzed with a Leica DMRB microscope (Leitz).
Statistical analysis.
Statistical significance was determined applying parametric Student’s t-test or nonparametric Mann-Whitney test. Two-way ANOVA with Bonferroni correction was applied for evaluation of physiological data received from metabolic cage experiments. P values of <0.05 were considered significant. All data are expressed as means ± SE.
RESULTS
Acute administration of CsA induces NKCC2 and NCC activation in vivo.
Wistar rats were treated with CsA or vehicle for 1 or 4 h, respectively, to evaluate short-term effects of CsA on renal CCC and their activating kinases. The 1-h period was chosen to assess fast changes on posttranslational level such as protein phosphorylation or trafficking events, whereas the 4-h period aimed at the detection of potential early changes in mRNA or protein abundance. Phosphorylation of NKCC2 and NCC was increased after both 1h [+131% for pT96/pT101-NKCC2 (pNKCC2) and +190% for pS71-NCC (pNCC), P < 0.05; Fig. 1, A and B] and 4h (+197% for pNKCC2 and +410% for pNCC, P < 0.05; Fig. 2, A and B) of CsA administration compared with the vehicle control groups, whereas their total abundance was unchanged (Figs. 1, A and B, and 2, A and B). Because CNI are known to induce hyperkalemia in patients and animal models (31), we also evaluated the relevant distal potassium channels, ROMK and Kir4.1, by immunoblotting. Protein abundance of ROMK and Kir4.1 was not altered upon 4 h CsA (Fig. 2, A and B). Surface expression of NKCC2 and NCC, as assessed by immunogold electron microscopy, was unchanged after 1 h of CsA (not shown), whereas a moderate increase was detected after 4 h of CsA treatment compared with vehicle treatment (+20% for NKCC2 and +13% for NCC, P < 0.05; Fig. 3, A and B). Confocal microscopic evaluation of phosphorylated SPAK/OSR1 species revealed no significant CsA-induced changes in the TAL but substantially increased apical signals in the DCT (Fig. 4B) upon CsA administration [+49% for pS383-SPAK/pS325-OSR1; regulatory domain (pS-SPAK/OSR1) after 1 h and +94% for pS-SPAK/OSR1 after 4 h, P < 0.05; Fig. 4, C and D]. Total abundance of the kinases was not affected as shown by immunoblots (Fig. 5, A–D). At the mRNA level, expression profiles of WNKs, SPAK, OSR1, NKCC2, and NCC showed no changes upon 4 h CsA (Fig. 6). COX-2, which is regulated by the calcineurin-nuclear factor of activated T cells pathway (12), was expectedly suppressed after 4 h of CsA treatment (−41%, P < 0.05; Fig. 6). These results suggest that short-term calcineurin inhibition using CsA activates the WNK-SPAK/OSR1 cascade and their renal substrates, NKCC2 and NCC, by increasing their apical abundance and phosphorylation levels. Parallel suppression of COX-2 may further contribute to the activation of the transporters via paracrine mechanisms (33).
Fig. 1.
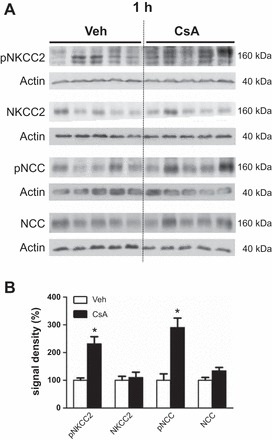
Short-term (1-h) effects of cyclosporine A on the abundance and phosphorylation of the distal Na-K-Cl cotransporters. A: representative immunoblots of kidney lysates from vehicle-treated (Veh) and cyclosporine A-treated (CsA) rats showing immunoreactive signals for total Na-K-2Cl cotransporter (NKCC2), phosphorylated NKCC2 [pT96/pT101-NKCC2 (pNKCC2)], total Na-Cl cotransporter (NCC), and phosphorylated NCC [pS71-NCC (pNCC)], all ~160 kDa; β-actin served as loading control (~40 kDa). B: graphs showing densitometric evaluation of immunoreactive signals (A) normalized to loading controls. Data are means ± SE; *P < 0.05.
Fig. 2.
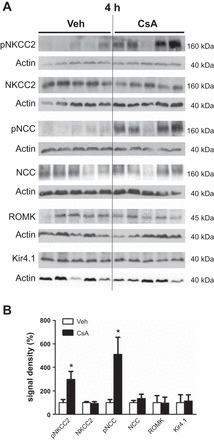
Short-term (4-h) effects of cyclosporine A on the distal Na-K-Cl cotransporters and potassium channels [renal outer medullary K channel (ROMK) and ATP-sensitive inward-rectifier K channel (Kir4.1)]. A: representative immunoblots of kidney lysates from vehicle-treated and cyclosporine A-treated rats showing immunoreactive signals for NKCC2, pNKCC2, NCC, pNCC (all ~160 kDa), ROMK (~45 kDa), and Kir4.1 (~40 kDa); β-actin served as loading control (~40 kDa). B: graphs showing densitometric evaluation of immunoreactive signals (A) normalized to loading controls. Data are means ± SE; *P < 0.05.
Fig. 3.
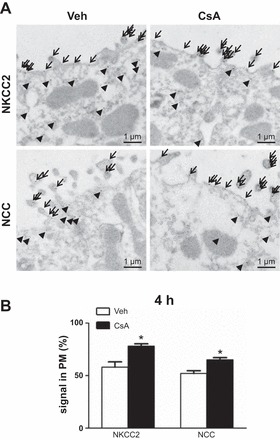
Short-term (4-h) effects of cyclosporine A on surface expression of the distal Na-K-Cl cotransporters. A: representative immunoelectron microscopic images showing cellular distribution of NKCC2 and NCC in plasma membrane (PM, arrows) vs. cytoplasm (arrowheads) in kidneys from vehicle-treated and cyclosporine A-treated rats; 5 nm gold grain labeling. B: numerical quantification of NKCC2 and NCC signals in plasma membrane/respective total cellular signals. Data are means ± SE; *P < 0.05.
Fig. 4.
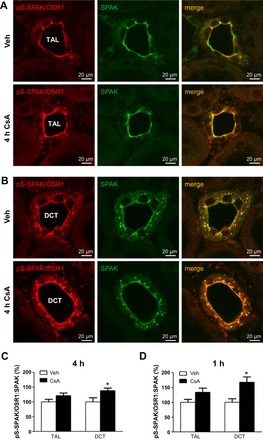
Short-term (1- and 4-h) effects of cyclosporine A on apical abundance of phosphorylated Ste20-related proline/alanine-rich kinase (SPAK)/oxidative stress-responsive kinase 1 (OSR1) kinases as evaluated by confocal microscopy. A and B: representative images of kidney sections from vehicle- or cyclosporine A-treated rats (4 h) showing immunofluorescent labeling of phosphorylated regulatory SPAK/OSR1 domain [pS383/pS325 (pS-SPAK/OSR1)] and double labeling for SPAK in thick ascending limb (TAL, A) and distal convoluted tubule (DCT, B). TAL and DCT were identified according to morphological criteria and specific SPAK signal patterns (predominant apical signal in TAL vs. apical and punctate cytoplasmic signal in DCT). C: graphs showing pS-SPAK/OSR1-to-SPAK signal ratio in TAL and DCT of rats treated with vehicle or cyclosporine A for 4 h. D: graphs showing evaluation of pS-SPAK/OSR1-to-SPAK signal ratio in TAL and DCT of rats treated with vehicle or cyclosporine A for 1 h; respective immunofluorescent images are not shown. Data are means ± SE; *P < 0.05.
Fig. 5.
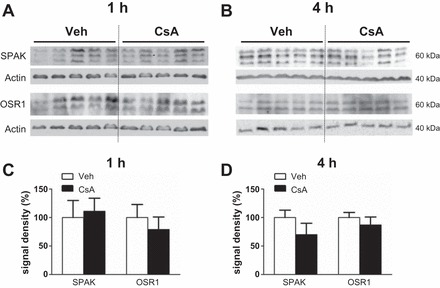
Short-term (1- and 4-h) effects of cyclosporine A on whole kidney protein levels of SPAK and OSR1. A and B: representative immunoblots of kidney lysates obtained from rats after vehicle or cyclosporine A administration for 1h (A and C) and 4h (B and D) showing total SPAK (~60 kDa) or total OSR1 signals (~60 kDa); β-actin served as loading control (~40 kDa). C and D: graphs showing densitometric evaluation of immunoreactive signals in A and B, normalized to loading controls. Data are means ± SE.
Fig. 6.
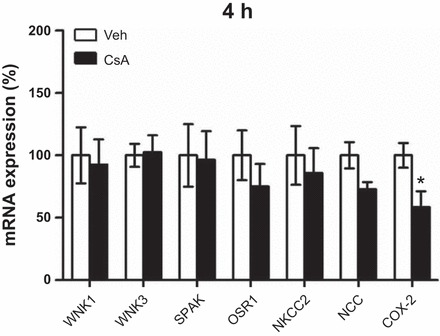
Short-term (4-h) effects of cyclosporine A on with-no-lysine [K] kinase (WNK) 1, WNK3, SPAK, OSR1, NKCC2, NCC, and cyclooxygenase (COX)-2 mRNA expression in kidney lysates from rats treated with vehicle or cyclosporine A for 4 h. Quantitative PCR; all results were normalized to GAPDH expression. Data are means ± SE; *P < 0.05.
Chronic administration of CsA activates renal CCC, modulates juxtaglomerular COX-2 and renin expression, and induces salt retention and hypertension.
Long-term effects of CsA were analyzed in Wistar rats treated with vehicle or CsA for 14 days. At the mRNA level, expression of WNKs, SPAK, NKCC2, and NCC was not altered upon 14 days of CsA treatment, but OSR1 expression was moderately increased (+24%, P < 0.05; Fig. 7). Expression of COX-2 mRNA was suppressed following 14 days of CsA treatment (Fig. 7). Confocal microscopic evaluation of NKCC2, NCC, and SPAK/OSR1 showed CsA-induced increases in abundance of their phosphorylated species without concomitant changes in their total protein levels [pNKCC2: +46%; pNCC: +19%; Fig. 8, A–C; pT243-SPAK/pT185-OSR1; catalytic domain (pT-SPAK/OSR1): +123% in DCT; pS-SPAK/OSR1: +36% in DCT; P < 0.05; Fig. 9, A–C]. Because CNI have been reported to modulate the function of the juxtaglomerular apparatus with effects on distal salt handling, we analyzed local COX-2 and renin synthesis as well as the systemic activity of renin. Chronic administration of CsA markedly suppressed the juxtaglomerular expression of COX-2 (−93%, P < 0.05; Fig. 10, A and C) but augmented the expression of renin mRNA (+72%, P < 0.05; Fig. 10, B and D) and strongly increased the plasma renin activity (+966%, P < 0.05; Fig. 11A). These results suggest that CsA may stimulate renal salt reabsorption via local intraepithelial, paracrine, and systemic mechanisms. In line with this, physiological evaluation in metabolic cages revealed marked CsA-induced decreases of urinary salt excretion on day 1 (−31%, P < 0.05; Fig. 11B) and day 10 (−51%, P < 0.05; Fig. 11B) of treatment as well as a moderate but significant increase in blood pressure on day 7 of treatment compared with vehicle [113 (vehicle) vs. 121 (CsA) mmHg; P < 0.05; Fig. 11C]. Previous studies propose that CNI-induced salt retention results chiefly from the activation of NCC. To demonstrate the respective contribution of NKCC2, we performed a furosemide response test, which revealed stronger furosemide-induced salt loss in rats receiving CsA compared with vehicle-treated animals, indicating increased NKCC2 function upon CsA treatment (+104%, P < 0.05; Fig. 11D).
Fig. 7.
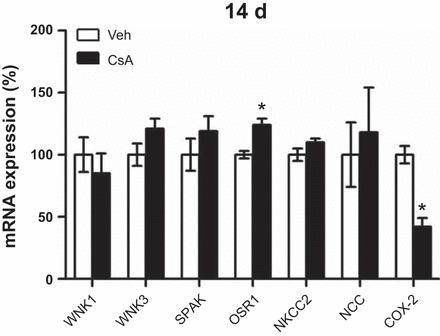
Long-term (14-days) effects of cyclosporine A on mRNA expression levels of the distal Na-K-Cl cotransporters, WNK-SPAK/OSR1 kinases, and COX-2. Graphs show results of quantitative PCR evaluation of WNK1, WNK3, SPAK, OSR1, NKCC2, NCC, and COX-2 mRNA levels in kidney lysates from rats treated with vehicle or cyclosporine A for 14 days; all results were normalized to GAPDH expression. Data are means ± SE; *P < 0.05.
Fig. 8.
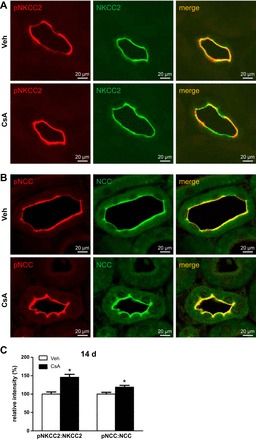
Long-term (14-days) effects of cyclosporine A on the abundance and phosphorylation levels of the distal Na-K-Cl cotransporters as evaluated by confocal microscopy. A and B: representative images of kidney sections from vehicle-treated and cyclosporine A-treated (14 days) rats showing immunofluorescent labeling of pNKCC2 and double labeling for NKCC2 in TAL (A) or immunofluorescent labeling of pS71-NCC and double labeling for NCC in DCT (B). C: graphs showing relative signal intensities of pNKCC2 and pNCC signals normalized to colocalized total NKCC2 or NCC signals as evaluated using ZEN and ImageJ software. Data are means ± SE; *P < 0.05.
Fig. 9.
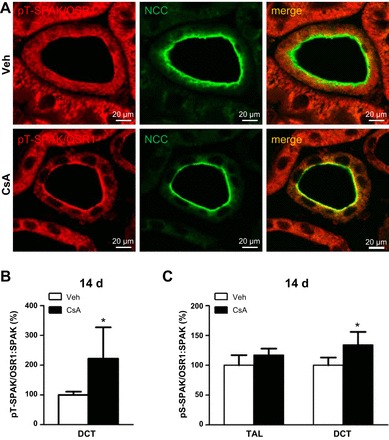
Long-term (14-days) effects of cyclosporine A on apical phosphorylation levels of SPAK/OSR1 kinases as evaluated by confocal microscopy. A: representative images of DCT in kidney sections from vehicle-treated and cyclosporine A-treated (14 days) rats showing immunofluorescent labeling of phosphorylated catalytic SPAK/OSR1 domain (pT243/pT185; pT-SPAK/OSR1) and double labeling for NCC to identify DCT. B and C: relative intensities of apical pT-SPAK/OSR1 in DCT (B) or pS-SPAK/OSR1 in TAL and DCT (C; the respective immunofluorescent images are not shown) after normalization to colocalized SPAK signals, as evaluated using ZEN2008 and ImageJ software. Data are means ± SE; *P < 0.05.
Fig. 10.
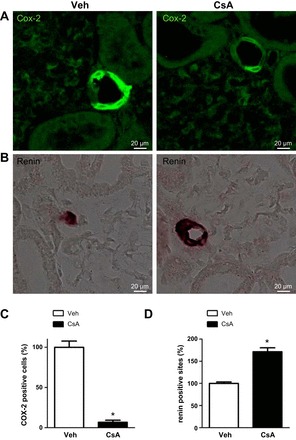
Long-term (14-days) effects of cyclosporine A on juxtaglomerular COX-2 abundance and renin expression. A: representative images of macula densa regions in kidney sections from vehicle-treated and cyclosporine A-treated (14 days) rats showing immunofluorescent labeling of COX-2. B: representative images of afferent arterioles in kidney sections from vehicle- and cyclosporine A-treated rats showing renin mRNA signal detected by nonradioactive in situ hybridization (brown signal). C and D: numerical quantification of COX-2-positive cells (C) or renin-positive sites (D) normalized for respective glomeruli numbers. Data are means ± SE; *P < 0.05.
Fig. 11.
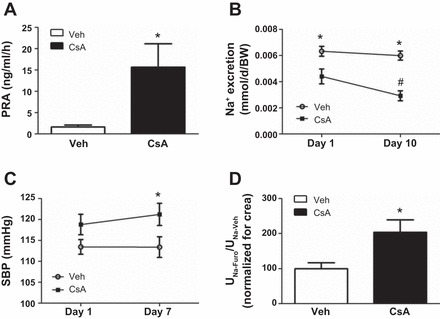
Long-term (14-days) effects of cyclosporine A on renin activity, arterial blood pressure, and sodium handling. A: plasma renin activity in vehicle-treated and cyclosporine A-treated (14 days) rats. B: 24-h urine sodium excretion in vehicle- and cyclosporine A-treated rats on treatment days 1 and 10. C: systolic blood pressure (SBP) in anesthetized vehicle- and cyclosporine A-treated rats measured by noninvasive tail cuff measurement. D: furosemide test in vehicle- and cyclosporine A-treated rats, as evaluated by the ratio of urinary sodium excretion after (UNa-Furo) and before (UNa-Veh) furosemide application; data were normalized to creatinine excretion. Data are means ± SE; *P < 0.05 for vehicle vs. cyclosporine A; #P < 0.05 for day 1 vs. day 10 (B).
AVP is required for CsA-induced NKCC2 activation but dispensable for activation of NCC.
To test whether AVP contributes to CNI-induced activation of NKCC2 or NCC, we treated AVP-deficient Brattleboro rats with CsA or vehicle for 4 h and compared immunoblot profiles of the distal transporters and kinases between groups. CsA did not alter the abundance of NKCC2, NCC, or pNKCC2 but significantly increased the level of pNCC (+68%, P < 0.05; Fig. 12, A and B). These results suggest that CNI-induced activation of NKCC2 requires at least baseline AVP levels, whereas activation of NCC can occur independent of AVP signaling.
Fig. 12.
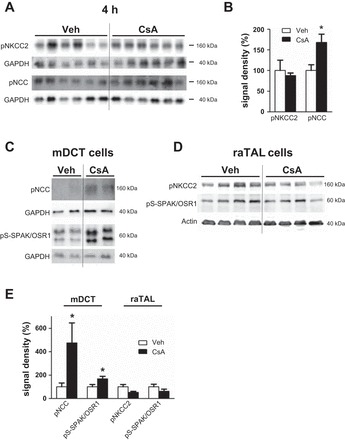
Short-term (4-h) effects of cyclosporine A on phosphorylation levels of the distal Na-K-Cl cotransporters and SPAK/OSR1 kinases in vasopressin-deficient Brattleboro rats and in cultured cells. A: representative immunoblots of kidney lysates from vehicle-treated and cyclosporine A-treated (4 h) Brattleboro rats showing immunoreactive signals for pNKCC2 and pNCC (both ~160 kDa); GAPDH served as loading control (~40 kDa). B: densitometric evaluation of immunoreactive signals (A) normalized to loading controls. C: representative immunoblots of cell lysates from vehicle- and cyclosporine A-treated DCT cells showing immunoreactive signals for pNCC (~160 kDa) and pS-SPAK/OSR1 (~60 kDa); GAPDH served as loading control (~40 kDa). D: representative immunoblots of cell lysates from vehicle- and cyclosporine A-treated TAL cells showing immunoreactive signals for pNKCC2 (~160 kDa) and pS-SPAK/OSR1 (~60 kDa); β-actin served as loading control (~40 kDa). E: densitometric evaluation of immunoreactive signals (C and D) normalized to loading controls. Data are means ± SE; *P < 0.05.
To provide further support for the idea that local inhibition of calcineurin in DCT cells is sufficient for NCC activation, we examined the effects of CsA in cultured mDCT cells (9). Compared with vehicle, application of CsA for 4 h increased the levels of phosphorylated NCC (+375% for pS71-NCC, P < 0.05; Fig. 12, C and E) and SPAK/OSR1 (+68% for pS-SPAK/OSR1, P < 0.05; Fig. 12, C and E) as detected by immunoblotting. In contrast, the same CsA treatment protocol did not induce any significant changes in SPAK/OSR1 or NKCC2 phosphorylation in cultured raTAL cells in the absence of AVP (Fig. 12, D and E). However, in the presence of the AVP receptor 2 agonist DDAVP in the culture medium, CsA markedly increased phosphorylation levels of NKCC2 (+46% for DDAVP and +97% for DDAVP + CsA, P < 0.05; Fig. 13, A and B) and SPAK/OSR1 (+65% for DDAVP, not significant; +92% for DDAVP + CsA, P < 0.05; Fig. 13, A and B) in raTAL cells. These results corroborate the dominant role of local calcineurin-dependent regulation of NCC in DCT cells and suggest a permissive role of AVP in TAL.
Fig. 13.
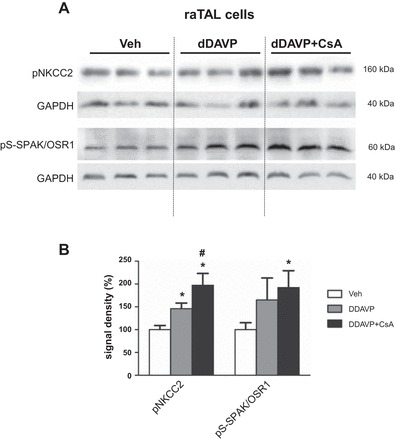
Short-term (4-h) effects of cyclosporine A on phosphorylation levels of NKCC2 and SPAK/OSR1 kinases in cultured TAL cells stimulated by desmopressin (DDAVP). A: representative immunoblots of cell lysates from TAL cells treated with vehicle, DDAVP, or DDAVP and cyclosporine A simultaneously showing immunoreactive signals for pNKCC2 (~160 kDa) and pS-SPAK/OSR1 (~60 kDa); GAPDH served as loading control (~40 kDa). B: densitometric evaluation of immunoreactive signals normalized to loading controls. Data are means ± SE; *P < 0.05 for DDAVP or DDAVP + cyclosporine A vs. vehicle; #P < 0.05 for DDAVP + cyclosporine A vs. DDAVP.
DISCUSSION
The presented data provide new insight into the effects of CsA on salt handling along the distal nephron and the contribution of endocrine stimulation by AVP. With respect to the local effects of calcineurin inhibition in TAL or DCT cells, our results on short- and long-term effects of CsA corroborate and extend previous reports on the role of posttranslational modification of the CCC by phosphorylation (2, 10). The observation of a parallel increase in surface expression of both NKCC2 and NCC upon CsA treatment provides additional evidence for their activated state and supports previous hypotheses that phosphorylation stabilizes the transporters within the plasma membrane (4, 37). Increased abundance of phosphorylated NKCC2 and NCC may thus result either from inhibition of relevant phosphatases or activation of WNK/SPAK-OSR1 kinases (21, 34). Earlier studies have documented increased abundance of WNK and SPAK kinases following chronic CsA or tacrolimus administration (13, 22). Although no substantial changes in expression or protein abundance of WNKs or SPAK/OSR1 were detected throughout the CsA treatment protocols used in this study, increases in their phosphorylation levels clearly support the idea that effects of CsA on CCC are, at least in part, mediated via activation of the WNK-SPAK/OSR1 pathway (13, 22). Our results thus underline the role of posttranslational regulation by phosphorylation/dephosphorylation reactions. Previously, we suggested that calcineurin may interact with renal CCC or their activating kinases via scaffolding mechanisms rather than direct interactions because neither the transporters nor the kinases possess known calcineurin-binding motifs but show multiple interactions by co-immunoprecipitation (2, 39). Alternatively, effects of calcineurin may be mediated by protein phosphatase 1 (34).
Apart from the direct effects in distal renal epithelia, CsA may facilitate renal salt reabsorption via endocrine and paracrine mechanisms, or by stimulation of the renal sympathetic innervation (12, 19, 26). Renin expression and activity were clearly increased upon CsA administration in our rat model, likely reflecting augmented sympathetic activity and local effects of calcineurin inhibition in renin-producing juxtaglomerular cells (19). Stimulation of renin activity was probably unrelated to paracrine mechanisms within the juxtaglomerular apparatus because COX-2 expression was suppressed in CsA-treated rats. Transcriptional control of COX-2 is mediated by nuclear factor of activated T cells transcription factors and, therefore, depends on calcineurin (12, 15). COX-2 is a critical enzyme for the biosynthesis of prostaglandin E2 and I2, which have been implicated in the modulation of glomerular filtration rate and NKCC2 function (16, 32). Although effects of renal COX-2 suppression on salt handling are reportedly complex (33), global inhibition of COX-2 has been shown to induce or aggravate salt-sensitive hypertension via cross talk between hematopoietic cells and the kidney (46). Therefore, CsA-induced stimulation of RAAS and inhibition of COX-2 may synergistically enhance salt reabsorption along the distal nephron, thus contributing to the development of CNI-induced hypertension (14, 46).
Another principal component of this study was the role of AVP during CsA-induced activation of NKCC2 and NCC. Treatment of AVP-deficient Brattleboro rats with CsA for 4 h induced an increase of NCC phosphorylation but no concomitant stimulation of NKCC2, suggesting that activation of the latter depends on the presence of AVP. In line with this, application of CsA in cell culture induced activation of SPAK and NCC in mDCT cells but did not affect SPAK or NKCC2 in raTAL cells. Although robust expression of the vasopressin V2 receptor (V2R) was documented along the entire distal nephron (6, 27, 29), the impact of AVP signaling on transport function may differ between the TAL and the DCT (17). The TAL in particular appears to strongly depend on AVP, which is compatible with its major role in the urinary concentration process (5, 27). In line with this idea, previous studies in Brattleboro rats documented almost complete absence of phosphorylated NKCC2 in their kidneys, whereas NCC phosphorylation was less affected in this model (29, 30). Along the same line, our recent evaluation of V2R function in the TAL using transgenic rats with segment-specific overexpression of a dominant-negative V2R mutant showed substantial deficits in NKCC2 phosphorylation and function (27). Overall, previous and present results suggest that AVP signaling is indispensable for proper NKCC2 phosphorylation, most likely because of its facilitating effects on relevant kinases such as WNK-SPAK/OSR1 kinases, protein kinase A (PKA), or AMP-activated protein kinase (11, 39). Some of these effects, such as activation of PKA, may depend on AVP-induced intracellular cAMP release (11), whereas the molecular components activating WNK-SPAK/OSR1 kinases still remain to be found (39). Because the sensing of intracellular chloride concentration has been increasingly recognized as a unique property and major function of WNK kinases (43), it is tempting to speculate that AVP may affect intracellular chloride levels in TAL cells to regulate the WNK-SPAK/OSR1 pathway. In this context, we have previously shown that uromodulin contributes to adjust baseline and AVP-induced NKCC2 phosphorylation via effects on intracellular chloride (28). Therefore, diminished activity of NKCC2-phosphorylating kinases in the absence of AVP may explain the failure of CsA to augment its phosphorylation in Brattleboro rats or cultured raTAL cells lacking AVP. This idea is further supported by the fact that, in DDAVP-treated raTAL cells, CsA produced clear increases of SPAK/OSR1 and NKCC2 phosphorylation levels in the present study. Therefore, in the presence of AVP, NKCC2 activity appears to contribute to CsA-induced salt retention, as has been demonstrated by the furosemide test in the present study. In contrast to the TAL, local inhibition of calcineurin in DCT cells by CsA induced a substantial increase of NCC phosphorylation. Likewise, our recent work demonstrated that tacrolimus-induced activation of NCC chiefly relies on local calcineurin inhibition in DCT cells because renal deletion of the respective immunophilin, FK506-binding protein 12, abolished effects of tacrolimus on NCC phosphorylation in mice (20).
In summary, the results of the present study demonstrate that CsA-induced activation of NKCC2 and NCC occurs chiefly at the posttranslational level via increased phosphorylation of the transporters and their activating kinases. Local calcineurin inhibition in DCT cells is sufficient to induce NCC activation, whereas activation of NKCC2 appears to require additional stimulation by AVP. Our data provide further support for the use of thiazides in patients with CNI-induced hypertension and suggest that furosemide or V2R-antagonists may be useful as well. In view of the broad use of CNI our data have clinical implications in posttransplantational immunosuppression.
GRANTS
This study was supported by the Deutsche Forschungsgemeinschaft.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
K.I.B. and K.M. conceived and designed research; K.I.B., A.B., C.B., M.B., and K.M. performed experiments; K.I.B., A.B., R.L., A.P., M.B., and K.M. analyzed data; K.I.B. and K.M. interpreted results of experiments; K.I.B. and A.B. prepared figures; K.I.B. and K.M. drafted manuscript; K.I.B., R.L., A.P., J.A.M., D.H.E., S.B., and K.M. edited and revised manuscript; K.I.B., D.H.E., S.B., and K.M. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Kerstin Riskowsky, Frauke Grams, Julia Skodowski, John Horn, and Petra Schrade for technical assistance.
REFERENCES
- 1.Bachmann S, Mutig K, Bates J, Welker P, Geist B, Gross V, Luft FC, Alenina N, Bader M, Thiele BJ, Prasadan K, Raffi HS, Kumar S. Renal effects of Tamm-Horsfall protein (uromodulin) deficiency in mice. Am J Physiol Renal Physiol 288: F559–F567, 2005. doi: 10.1152/ajprenal.00143.2004. [DOI] [PubMed] [Google Scholar]
- 2.Borschewski A, Himmerkus N, Boldt C, Blankenstein KI, McCormick JA, Lazelle R, Willnow TE, Jankowski V, Plain A, Bleich M, Ellison DH, Bachmann S, Mutig K. Calcineurin and Sorting-Related Receptor with A-Type Repeats Interact to Regulate the Renal Na+-K+-2Cl− Cotransporter. J Am Soc Nephrol 27: 107–119, 2016. doi: 10.1681/ASN.2014070728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bosse HM, Böhm R, Resch S, Bachmann S. Parallel regulation of constitutive NO synthase and renin at JGA of rat kidney under various stimuli. Am J Physiol Renal Physiol 269: F793–F805, 1995. [DOI] [PubMed] [Google Scholar]
- 4.Dathe C, Daigeler A-L, Seifert W, Jankowski V, Mrowka R, Kalis R, Wanker E, Mutig K, Bachmann S, Paliege A. Annexin A2 mediates apical trafficking of renal Na+-K+-2Cl− cotransporter. J Biol Chem 289: 9983–9997, 2014. doi: 10.1074/jbc.M113.540948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ecelbarger CA, Kim GH, Wade JB, Knepper MA. Regulation of the abundance of renal sodium transporters and channels by vasopressin. Exp Neurol 171: 227–234, 2001. doi: 10.1006/exnr.2001.7775. [DOI] [PubMed] [Google Scholar]
- 6.Fenton RA, Brønd L, Nielsen S, Praetorius J. Cellular and subcellular distribution of the type-2 vasopressin receptor in the kidney. Am J Physiol Renal Physiol 293: F748–F760, 2007. doi: 10.1152/ajprenal.00316.2006. [DOI] [PubMed] [Google Scholar]
- 7.Gamba G. Molecular physiology and pathophysiology of electroneutral cation-chloride cotransporters. Physiol Rev 85: 423–493, 2005. doi: 10.1152/physrev.00011.2004. [DOI] [PubMed] [Google Scholar]
- 8.Gamba G. Regulation of the renal Na+-Cl- cotransporter by phosphorylation and ubiquitylation. Am J Physiol Renal Physiol 303: F1573–F1583, 2012. doi: 10.1152/ajprenal.00508.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gesek FA, Friedman PA. Mechanism of calcium transport stimulated by chlorothiazide in mouse distal convoluted tubule cells. J Clin Invest 90: 429–438, 1992. doi: 10.1172/JCI115878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Giménez I, Forbush B. Short-term stimulation of the renal Na-K-Cl cotransporter (NKCC2) by vasopressin involves phosphorylation and membrane translocation of the protein. J Biol Chem 278: 26946–26951, 2003. doi: 10.1074/jbc.M303435200. [DOI] [PubMed] [Google Scholar]
- 11.Gunaratne R, Braucht DWW, Rinschen MM, Chou C-L, Hoffert JD, Pisitkun T, Knepper MA. Quantitative phosphoproteomic analysis reveals cAMP/vasopressin-dependent signaling pathways in native renal thick ascending limb cells. Proc Natl Acad Sci USA 107: 15653–15658, 2010. doi: 10.1073/pnas.1007424107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Höcherl K, Dreher F, Vitzthum H, Köhler J, Kurtz A. Cyclosporine A suppresses cyclooxygenase-2 expression in the rat kidney. J Am Soc Nephrol 13: 2427–2436, 2002. doi: 10.1097/01.ASN.0000031702.86799.B9. [DOI] [PubMed] [Google Scholar]
- 13.Hoorn EJ, Walsh SB, McCormick JA, Fürstenberg A, Yang C-L, Roeschel T, Paliege A, Howie AJ, Conley J, Bachmann S, Unwin RJ, Ellison DH. The calcineurin inhibitor tacrolimus activates the renal sodium chloride cotransporter to cause hypertension. Nat Med 17: 1304–1309, 2011. doi: 10.1038/nm.2497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hoorn EJ, Walsh SB, McCormick JA, Zietse R, Unwin RJ, Ellison DH. Pathogenesis of calcineurin inhibitor-induced hypertension. J Nephrol 25: 269–275, 2012. doi: 10.5301/jn.5000174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Iñiguez MA, Martinez-Martinez S, Punzón C, Redondo JM, Fresno M. An essential role of the nuclear factor of activated T cells in the regulation of the expression of the cyclooxygenase-2 gene in human T lymphocytes. J Biol Chem 275: 23627–23635, 2000. doi: 10.1074/jbc.M001381200. [DOI] [PubMed] [Google Scholar]
- 16.Kim G-H. Renal effects of prostaglandins and cyclooxygenase-2 inhibitors. Electrolyte Blood Press 6: 35–41, 2008. doi: 10.5049/EBP.2008.6.1.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kortenoeven MLA, Pedersen NB, Rosenbaek LL, Fenton RA. Vasopressin regulation of sodium transport in the distal nephron and collecting duct. Am J Physiol Renal Physiol 309: F280–F299, 2015. doi: 10.1152/ajprenal.00093.2015. [DOI] [PubMed] [Google Scholar]
- 18.Kotnik P, Nielsen J, Kwon T-H, Krzisnik C, Frøkiaer J, Nielsen S. Altered expression of COX-1, COX-2, and mPGES in rats with nephrogenic and central diabetes insipidus. Am J Physiol Renal Physiol 288: F1053–F1068, 2005. doi: 10.1152/ajprenal.00114.2004. [DOI] [PubMed] [Google Scholar]
- 19.Kurtz A, Della Bruna R, Kühn K. Cyclosporine A enhances renin secretion and production in isolated juxtaglomerular cells. Kidney Int 33: 947–953, 1988. doi: 10.1038/ki.1988.92. [DOI] [PubMed] [Google Scholar]
- 20.Lazelle RA, McCully BH, Terker AS, Himmerkus N, Blankenstein KI, Mutig K, Bleich M, Bachmann S, Yang C-L, Ellison DH. Renal Deletion of 12 kDa FK506-Binding Protein Attenuates Tacrolimus-Induced Hypertension. J Am Soc Nephrol 27: 1456–1464, 2016. doi: 10.1681/ASN.2015040466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McCormick JA, Mutig K, Nelson JH, Saritas T, Hoorn EJ, Yang C-L, Rogers S, Curry J, Delpire E, Bachmann S, Ellison DH. A SPAK isoform switch modulates renal salt transport and blood pressure. Cell Metab 14: 352–364, 2011. doi: 10.1016/j.cmet.2011.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Melnikov S, Mayan H, Uchida S, Holtzman EJ, Farfel Z. Cyclosporine metabolic side effects: association with the WNK4 system. Eur J Clin Invest 41: 1113–1120, 2011. doi: 10.1111/j.1365-2362.2011.02517.x. [DOI] [PubMed] [Google Scholar]
- 23.Meyer-Lehnert H, Schrier RW. Cyclosporine A enhances vasopressin-induced Ca2+ mobilization and contraction in mesangial cells. Kidney Int 34: 89–97, 1988. doi: 10.1038/ki.1988.149. [DOI] [PubMed] [Google Scholar]
- 24.Meyer-Lehnert H, Schrier RW. Potential mechanism of cyclosporine A-induced vascular smooth muscle contraction. Hypertension 13: 352–360, 1989. doi: 10.1161/01.HYP.13.4.352. [DOI] [PubMed] [Google Scholar]
- 25.Moes AD, Hesselink DA, Zietse R, van Schaik RHN, van Gelder T, Hoorn EJ. Calcineurin inhibitors and hypertension: a role for pharmacogenetics? Pharmacogenomics 15: 1243–1251, 2014. doi: 10.2217/pgs.14.87. [DOI] [PubMed] [Google Scholar]
- 26.Moss NG, Powell SL, Falk RJ. Intravenous cyclosporine activates afferent and efferent renal nerves and causes sodium retention in innervated kidneys in rats. Proc Natl Acad Sci USA 82: 8222–8226, 1985. doi: 10.1073/pnas.82.23.8222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mutig K, Borowski T, Boldt C, Borschewski A, Paliege A, Popova E, Bader M, Bachmann S. Demonstration of the functional impact of vasopressin signaling in the thick ascending limb by a targeted transgenic rat approach. Am J Physiol Renal Physiol 311: F411–F423, 2016. doi: 10.1152/ajprenal.00126.2016. [DOI] [PubMed] [Google Scholar]
- 28.Mutig K, Kahl T, Saritas T, Godes M, Persson P, Bates J, Raffi H, Rampoldi L, Uchida S, Hille C, Dosche C, Kumar S, Castañeda-Bueno M, Gamba G, Bachmann S. Activation of the bumetanide-sensitive Na+,K+,2Cl- cotransporter (NKCC2) is facilitated by Tamm-Horsfall protein in a chloride-sensitive manner. J Biol Chem 286: 30200–30210, 2011. doi: 10.1074/jbc.M111.222968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mutig K, Paliege A, Kahl T, Jöns T, Müller-Esterl W, Bachmann S. Vasopressin V2 receptor expression along rat, mouse, and human renal epithelia with focus on TAL. Am J Physiol Renal Physiol 293: F1166–F1177, 2007. doi: 10.1152/ajprenal.00196.2007. [DOI] [PubMed] [Google Scholar]
- 30.Mutig K, Saritas T, Uchida S, Kahl T, Borowski T, Paliege A, Böhlick A, Bleich M, Shan Q, Bachmann S. Short-term stimulation of the thiazide-sensitive Na+-Cl- cotransporter by vasopressin involves phosphorylation and membrane translocation. Am J Physiol Renal Physiol 298: F502–F509, 2010. doi: 10.1152/ajprenal.00476.2009. [DOI] [PubMed] [Google Scholar]
- 31.Naesens M, Kuypers DRJ, Sarwal M. Calcineurin inhibitor nephrotoxicity. Clin J Am Soc Nephrol 4: 481–508, 2009. doi: 10.2215/CJN.04800908. [DOI] [PubMed] [Google Scholar]
- 32.Paliege A, Mizel D, Medina C, Pasumarthy A, Huang YG, Bachmann S, Briggs JP, Schnermann JB, Yang T. Inhibition of nNOS expression in the macula densa by COX-2-derived prostaglandin E(2). Am J Physiol Renal Physiol 287: F152–F159, 2004. doi: 10.1152/ajprenal.00287.2003. [DOI] [PubMed] [Google Scholar]
- 33.Palmer LG, Schnermann J. Integrated control of Na transport along the nephron. Clin J Am Soc Nephrol 10: 676–687, 2015. doi: 10.2215/CJN.12391213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Picard N, Trompf K, Yang C-L, Miller RL, Carrel M, Loffing-Cueni D, Fenton RA, Ellison DH, Loffing J. Protein phosphatase 1 inhibitor-1 deficiency reduces phosphorylation of renal NaCl cotransporter and causes arterial hypotension. J Am Soc Nephrol 25: 511–522, 2014. doi: 10.1681/ASN.2012121202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rafiqi FH, Zuber AM, Glover M, Richardson C, Fleming S, Jovanović S, Jovanović A, O’Shaughnessy KM, Alessi DR. Role of the WNK-activated SPAK kinase in regulating blood pressure. EMBO Mol Med 2: 63–75, 2010. doi: 10.1002/emmm.200900058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rojas-Vega L, Jiménez-Vega AR, Bazúa-Valenti S, Arroyo-Garza I, Jiménez JV, Gómez-Ocádiz R, Carrillo-Pérez DL, Moreno E, Morales-Buenrostro LE, Alberú J, Gamba G. Increased phosphorylation of the renal Na+-Cl- cotransporter in male kidney transplant recipient patients with hypertension: a prospective cohort. Am J Physiol Renal Physiol 309: F836–F842, 2015. doi: 10.1152/ajprenal.00326.2015. [DOI] [PubMed] [Google Scholar]
- 37.Rosenbaek LL, Kortenoeven MLA, Aroankins TS, Fenton RA. Phosphorylation decreases ubiquitylation of the thiazide-sensitive cotransporter NCC and subsequent clathrin-mediated endocytosis. J Biol Chem 289: 13347–13361, 2014. doi: 10.1074/jbc.M113.543710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sandberg MB, Riquier ADM, Pihakaski-Maunsbach K, McDonough AA, Maunsbach AB. ANG II provokes acute trafficking of distal tubule Na+-Cl(-) cotransporter to apical membrane. Am J Physiol Renal Physiol 293: F662–F669, 2007. doi: 10.1152/ajprenal.00064.2007. [DOI] [PubMed] [Google Scholar]
- 39.Saritas T, Borschewski A, McCormick JA, Paliege A, Dathe C, Uchida S, Terker A, Himmerkus N, Bleich M, Demaretz S, Laghmani K, Delpire E, Ellison DH, Bachmann S, Mutig K. SPAK differentially mediates vasopressin effects on sodium cotransporters. J Am Soc Nephrol 24: 407–418, 2013. doi: 10.1681/ASN.2012040404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9: 671–675, 2012. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sevá Pessôa B, van der Lubbe N, Verdonk K, Roks AJM, Hoorn EJ, Danser AHJ. Key developments in renin-angiotensin-aldosterone system inhibition. Nat Rev Nephrol 9: 26–36, 2013. doi: 10.1038/nrneph.2012.249. [DOI] [PubMed] [Google Scholar]
- 42.Terker AS, Yarbrough B, Ferdaus MZ, Lazelle RA, Erspamer KJ, Meermeier NP, Park HJ, McCormick JA, Yang C-L, Ellison DH. Direct and Indirect Mineralocorticoid Effects Determine Distal Salt Transport. J Am Soc Nephrol 27: 2436–2445, 2016. doi: 10.1681/ASN.2015070815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Terker AS, Zhang C, Erspamer KJ, Gamba G, Yang C-L, Ellison DH. Unique chloride-sensing properties of WNK4 permit the distal nephron to modulate potassium homeostasis. Kidney Int 89: 127–134, 2016. doi: 10.1038/ki.2015.289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang D, Pedraza PL, Abdullah HI, McGiff JC, Ferreri NR. Calcium-sensing receptor-mediated TNF production in medullary thick ascending limb cells. Am J Physiol Renal Physiol 283: F963–F970, 2002. doi: 10.1152/ajprenal.00108.2002. [DOI] [PubMed] [Google Scholar]
- 45.Williams CR, Gooch JL. Calcineurin inhibitors and immunosuppression - a tale of two isoforms. Expert Rev Mol Med 14: e14, 2012. doi: 10.1017/erm.2012.8. [DOI] [PubMed] [Google Scholar]
- 46.Zhang M-Z, Yao B, Wang Y, Yang S, Wang S, Fan X, Harris RC. Inhibition of cyclooxygenase-2 in hematopoietic cells results in salt-sensitive hypertension. J Clin Invest 125: 4281–4294, 2015. doi: 10.1172/JCI81550. [DOI] [PMC free article] [PubMed] [Google Scholar]