

The study demonstrates for the first time that the impaired activity of endothelial nitric oxide-dependent dilatation in aging arteries is mediated by a local renin-angiotensin system and pathological signaling of angiotensin II. Inhibition of angiotensin synthesis or activity in isolated arteries rapidly and completely restores normal endothelial dilatation in old arteries.
Keywords: keywords. angiotensin ii, AT1 receptors, aging, endothelium
Abstract
Aging impairs endothelium-dependent NO-mediated dilatation, which results from increased production of reactive oxygen species (ROS). The local generation of angiotensin II (ANG II) is increased in aging arteries and contributes to inflammatory and fibrotic activity of smooth muscle cells and arterial wall remodeling. Although prolonged in vivo ANG II inhibition improves the impaired endothelial dilatation of aging arteries, it is unclear whether this reflects inhibition of intravascular or systemic ANG II systems. Experiments were therefore performed on isolated tail arteries from young (3–4 mo) and old (22–24 mo) F344 rats to determine if a local renin-angiotensin system contributes to the endothelial dilator dysfunction of aging. Aging impaired dilatation to the endothelial agonist acetylcholine but did not influence responses to a nitric oxide (NO) donor (DEA NONOate). Dilatation to acetylcholine was greatly reduced by NO synthase inhibition [nitro-l-arginine methyl ester (l-NAME)] in young and old arteries. In isolated arteries, acute inhibition of angiotensin-converting enzyme (ACE) (perindoprilat), renin (aliskiren), or AT1 receptors (valsartan, losartan) did not influence dilatation to acetylcholine in young arteries but increased responses in old arteries. After ANG II inhibition, the dilator response to acetylcholine was similar in young and old arteries. ROS activity, which was increased in endothelium of aging arteries, was also reduced by inhibiting ANG II (perindoprilat, losartan). Renin expression was increased by 5.6 fold and immunofluorescent levels of ANG II were confirmed to be increased in aging compared with young arteries. Exogenous ANG II inhibited acetylcholine-induced dilatation. Therefore, aging-induced impairment of endothelium-dependent dilatation in aging is caused by a local intravascular renin-angiotensin system.
Listen to this article’s corresponding podcast at http://ajpheart.podbean.com/e/angii-and-aging-induced-endothelial-dysfunction/.
NEW & NOTEWORTHY
The study demonstrates for the first time that the impaired activity of endothelial nitric oxide-dependent dilatation in aging arteries is mediated by a local renin-angiotensin system and pathological signaling of angiotensin II. Inhibition of angiotensin synthesis or activity in isolated arteries rapidly and completely restores normal endothelial dilatation in old arteries.
aging is associated with structural and functional deterioration of the arterial system that precipitates cardiovascular disease, organ dysfunction, and organ injury (19, 20). Endothelial dysfunction is a key contributor to this process of vascular aging, which includes diminished nitric oxide (NO) dilator activity as a result of increased reactive oxygen species (ROS) (8, 17, 31). Aging arteries have intimal fibrous lesions with deposition of poorly distensible proteins, collagen and fibronectin and degradation of highly distensible elastin fibers (19, 35). This arterial stiffening, which is a hallmark feature of vascular aging, is mediated by expression and activation of a local intravascular angiotensin II (ANG II) system (19, 33, 34). Immunofluorescence for ANG II is increased four- to sixfold in the endothelium and intima of aged compared with young arteries from rats, humans, and nonhuman primates and is paralleled by increased expression of ACE and AT1 receptors that are colocalized with ANG II (36–38). Aging-induced arterial wall remodeling is characterized by activation of ANG II signaling cascades and is mimicked in young arteries by ANG II exposure (12, 19, 34, 35, 38). Moreover, the increased activity of inflammatory and fibrotic mediators in aged arteries or smooth muscle cells (SMCs) is reduced by blocking local ANG II activity, achieved by inhibiting AT1 receptors in isolated cultured arteries or SMCs (12, 16, 37, 38). Chronic in vivo inhibition of ANG II activity is also highly beneficial in aging, including reducing arterial wall remodeling, increasing arterial compliance, and extending lifespan (3, 6, 35).
ANG II is a powerful inducer of endothelial dysfunction, including inhibiting NO dilator activity as a result of increased ROS (10, 21). Prolonged in vivo inhibition of ANG II activity reverses the impaired endothelium-dependent dilatation of aging arteries, which is associated with reduced oxidant stress (24, 27). No previous studies have assessed whether such beneficial effects of ANG II inhibition on endothelial dilatation reflect inhibition of the intravascular or the systemic circulating ANG II system. The present experiments were therefore performed to evaluate the role of a local renin-angiotensin system in the endothelial dilator dysfunction of aging.
MATERIALS AND METHODS
Animals.
Young (3–4 mo) and old (22–24 mo old) male F344 rats were obtained from Charles River and their NIA colony and killed by CO2 asphyxiation. Tail arteries were rapidly removed and placed in cold Krebs-Ringer bicarbonate solution (control solution) (28). Animal use was approved by the Johns Hopkins University Institutional Animal Care and Use Committee and complied with the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Vasodilatation.
Tail arteries were cannulated with micropipettes, secured within a microvascular chamber, and maintained at a transmural pressure of 60 mmHg in the absence of flow, i.e., without perfusion (Living Systems, VT) (28). The arteries were superfused with control solution (37°C, pH 7.4, 16% O2-5% CO2-balance N2), and the chamber was placed on the stage of an inverted microscope (28). Arterial diameter was continuously monitored and recorded (BIOPAC, Santa Barbara, CA) (28). Concentration-effect curves to vasodilators were obtained during constriction to phenylephrine, and acetylcholine responses were determined in paired arteries with one segment serving as control and the other treated with [nitro-l-arginine methyl ester (l-NAME)] (100 μM) to inhibit NO synthase, perindoprilat (1 μM) to inhibit angiotensin-converting enzyme (ACE), aliskiren (0.1 μM) to inhibit renin, or valsartan (1 μM) or losartan (3 μM) to inhibit AT1 receptors, as previously described (40). Arteries were incubated with the inhibitors for 60–90 min before and during exposure to acetylcholine.
Real-time PCR.
Arteries were disrupted in RLT lysis buffer using the Bullet Blender (Next Advance) and total RNA isolated using the RNeasy Plus mini kit (Qiagen) following the manufacturer's directions. The quality and quantity of RNA samples were analyzed using a Nanodrop spectrophotometer (ThermoFisher Scientific). Reverse transcription was performed using the iScript Reverse Transcription super mix kit (Bio-Rad), following the manufacturer's directions. Real-Time PCR was performed in duplicate on an Applied Biosystems 7500 Real-Time PCR System. Relative abundance of renin mRNA in young and aging arteries, compared with GAPDH mRNA, was determined by the comparative cycle threshold (Ct) method using TaqMan probes and primers designed and supplied by Applied Biosystems: rat GAPDH (Rn 01775763_g1) and rat renin (Rn02586313_m1).
Immunofluorescence.
Arteries were fixed with paraformaldehyde, permeabilized in Triton-X and then incubated in donkey serum to reduce nonspecific binding, as described previously (13). They were incubated overnight with a rabbit primary antibody to ANG II (1:500; Peninsula Labs), as performed previously (2, 36–38), and then incubated (120 min) with an AlexaFluor 488 donkey anti rabbit antibody (1:200; Jackson ImmunoResearch) and Draq5 (5 μM, nuclear stain; Biostatus). The endothelium was imaged (1024 × 1024 pixels) with a Leica SP5 LSM using a ×63 objective (NA 1.4), pinhole of 1 Airy unit, scan speed of 400 Hz, 6-line averaging, optical zoom of 3.0, and excitation/emission settings for Alexa 488 (488 nm/492–541 nm) and Draq5 (633 nm/659–758 nm). For quantitative comparison, young and old arteries were processed at the same time using the same instrument settings. For each artery, the fluorescence intensity from three Z-stacks of the endothelial layer (five 0.25-μm slices) was averaged to obtain the arterial fluorescence (n = 1) and is expressed as detector units (40).
ROS activity.
Tail arteries were incubated (control solution, 37°C) for 180 min in the absence or presence of the AT1 receptor antagonist losartan or the ACE inhibitor perindoprilat before being incubated with the ROS-sensitive fluorescent probe 5-(and 6)-chloromethyl-29, 79-dichlorodihydro-fluorescein diacetate (DCDHF; 5 μg/ml; Life Technologies) and Draq5 (5 μM) for 30 min (37°C, control solution) (40). They were then placed in cold control solution (4°C) and the endothelium imaged as in Immunofluorescence using laser-scanning microscopy (×20 air objective, 0.7 NA). The endothelium was visualized using an intensity filter, and optical slices were captured at the highest level of DCDHF fluorescence. For each arterial segment, the fluorescence intensity from multiple images was averaged to obtain the arterial fluorescence (n = 1), which is expressed as detector units (40).
Drugs.
Acetylcholine, l-NAME, and ANG II were from Sigma-Aldrich, losartan and valsartan from Tocris Biosciences, DEA-NONOate from Enzo Life Sciences, perindoprilat from Santa Cruz Biotechnology, and aliskiren from Selleck Chemicals.
Data analysis.
Vasomotor responses were expressed as a percent change in baseline diameter. Agonist concentrations causing 50% dilatation of the phenylephrine constriction (EC50) were calculated by regression analysis and compared as −log EC50. Maximum responses were determined as the maximal observed dilatation of the constriction to phenylephrine. Data are expressed as means ± SE, where “n” equals the number of animals from which arteries were studied. Statistical evaluation of the data was performed by Student's t-test for paired or unpaired observations. When more than two means were compared, ANOVA was used. If a significant F value was found, then the Tukey-Kramer test for multiple comparisons was employed to identify differences among groups. Values were considered to be statistically different when P < 0.05.
RESULTS
Endothelial dilator dysfunction in aging arteries.
Dilatation to acetylcholine was reduced in old compared with young arteries, reflecting a decrease in the maximal response and a rightward shift in the concentration-effect curve (maximums of 108.1 ± 1.1 and 89.6 ± 3.0%, −log EC50 of 7.32 ± 0.06 and 6.87 ± 0.07 in young, n = 17, and old, n = 25, respectively, P < 0.001 for each comparison; Fig. 1). Inhibition of NO synthase with l-NAME (100 μM) suppressed responses to acetylcholine in young and old arteries (Fig. 1). After l-NAME, the residual dilatation to acetylcholine was greater in old arteries (1 μM caused 35.9 ± 8.1 and 66.0 ± 10.7% dilatation in young and old arteries, respectively, n = 6, P < 0.05) (Fig. 1). Dilatation to the NO donor NONOate was not significantly different between young and old arteries (maximums of 106.0 ± 2.4 and 106.9 ± 1.4%, −log EC50 of 7.58 ± 0.14 and 7.31 ± 0.10, respectively, n = 6, P = NS) (Fig. 1).
Fig. 1.
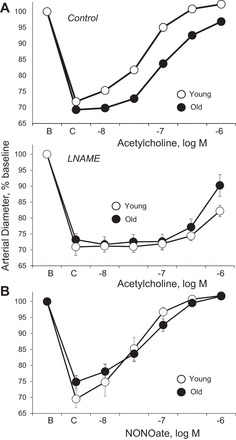
Dilatation of young and old rat isolated tail arteries to the endothelial agonist acetylcholine (A) or the nitric oxide (NO) donor DEA NONOate (B). Responses to acetylcholine (A) were assessed under control conditions (top) and after inhibition of NO synthase with l-NAME (bottom). Dilator responses were analyzed in pressurized arteries during constriction to phenylephrine (C). Data are expressed relative to baseline diameter (B) and presented as means ± SE for A, n = 25 (control old), n = 17 (control young), or n = 6 {[nitro-l-arginine methyl ester (l-NAME)]-treated arteries}; B, n = 6. Aging decreased the maximal dilator response to acetylcholine (P < 0.001) and caused a rightward shift in the concentration-effect curve (P < 0.001) (A). After l-NAME, the residual dilatation to acetylcholine (1 μM) was greater in old compared with young arteries (P < 0.05; B). Aging did not significantly affect the concentration-effect curve to NONOate (C).
Role of ANG II in aging-induced endothelial dilator dysfunction.
Inhibition of endogenous ANG II activity by blocking ACE (perindoprilat), renin (aliskiren), or AT1 receptors (valsartan, losartan) did not affect dilatation to acetylcholine in young arteries but significantly increased responses in old arteries, increasing the maximal dilatation and causing leftward shifts in the concentration-effect curve (Figs. 2 and 3). After ANG II inhibition, there was no longer any significant difference in the concentration-effect curves to acetylcholine between young and old arteries (Figs. 2 and 3). In old arteries treated with perindoprilat to inhibit endogenous production of the peptide, exogenous ANG II (300 nM, 120 min) inhibited dilatation to acetylcholine, decreasing the maximal response and causing a rightward shift in the concentration-effect curve (maximums of 101.4 ± 2.6 and 94.0 ± 4.2%, P < 0.05, and −log EC50 values of −7.13 ± 0.09 and 6.78 ± 0.07, P < 0.01, in perindoprilat and perindoprilat plus ANG II-treated arteries, respectively, n = 6).
Fig. 2.
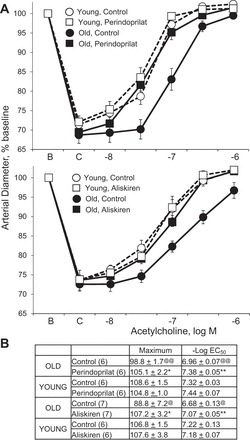
Effects of the ACE inhibitor perindoprilat (top) or the renin inhibitor aliskiren (bottom) on dilatation to acetylcholine in young and old rat isolated tail arteries. Dilatation was analyzed in pressurized arteries during constriction to phenylephrine (C). Data are expressed relative to baseline diameter (B) and presented as means ± SE for n = 6 or 7 (see parenthesis in B). Concentration-effect curves are presented in A and statistical analysis of the curves are presented in B. In B: “Maximum” is maximal dilatation; @P < 0.05, @@P < 0.01, statistically significant difference from the corresponding group in young arteries; *P < 0.05, **P < 0.01, statistically significant difference between the corresponding paired control group. Inhibition of ANG II production augmented dilatation to acetylcholine in old arteries, increasing the maximal response and causing a leftward shift in the concentration-effect curve but had no significant effect in young arteries. After ANG II inhibition, there was no longer any significant difference in the concentration-effect curves to acetylcholine between young and old arteries.
Fig. 3.
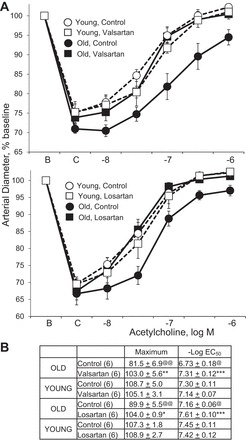
Effects of the AT1 receptor antagonists valsartan (top) or losartan (bottom) on dilatation to acetylcholine in young and old rat isolated tail arteries. Dilatation was analyzed in pressurized arteries during constriction to phenylephrine (C). Data are expressed relative to baseline diameter (B) and presented as means ± SE for n = 6. Concentration-effect curves are presented in A and statistical analysis of the curves are presented in B. In B: “Maximum” is maximal dilatation; @P < 0.05, @@P < 0.01, statistically significant difference from the corresponding group in young arteries; *P < 0.05, **P < 0.01, and ***P < 0.001, statistically significant difference between the corresponding paired control group. Inhibition of AT1 receptors augmented dilatation to acetylcholine in old arteries, increasing the maximal response and causing a leftward shift in the concentration-effect curve, but had no significant effect in young arteries. After ANG II inhibition, there was no longer any significant difference in the concentration-effect curves to acetylcholine between young and old arteries
ROS activity was significantly increased in old compared with young endothelium (Fig. 4). Antagonism of AT1 receptors (losartan) or inhibition of ACE (perindoprilat) significantly reduced ROS activity in old arterial endothelium (Fig. 4). After ANG II inhibition, ROS activity in old endothelium was no longer significantly different from young endothelium.
Fig. 4.
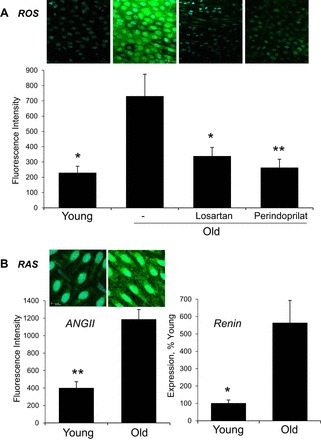
A: reactive oxygen species (ROS) activity, assessed using DCDHF fluorescence (DCDHF, green; Draq5, light blue), in endothelium lining young and old arteries. Data is expressed as fluorescence units and is presented as means ± SE; n = 3. *P < 0.05; **P < 0.01, significant difference from old control arteries. DCDHF fluorescence was increased in old compared with young untreated arterial endothelium, and was reduced by inhibition of AT1 receptors (losartan) or ACE (peridoprilat). B: renin angiotensin system (RAS). Expression levels of ANG II (left) (ANG II, green; Draq5, light blue) and renin (right) in young and old arteries assessed by immunofluorescence and Real-Time PCR, respectively. Fluorescent images are maximal projections of Z-stacks comprising the entire endothelial layer. Data are expressed as fluorescence units (left) or relative to the average value in young arteries (right) and is presented as means ± SE; n = 3. *P < 0.05; **P < 0.01, significant difference from old control arteries. Expression levels of ANG II and renin were increased in old compared with young arteries. Fluorescent images in A and B are presented in their original unprocessed state.
Previous studies demonstrated increased immunofluorescent staining for ANG II in the endothelium and intima of old arteries, which was paralleled by increased expression of ACE and AT1 receptors (36–38). Immunofluorescent levels of ANG II were confirmed to be increased in old compared with young endothelium (Fig. 4). Moreover, expression of renin was increased in old compared with young arteries (Fig. 4).
DISCUSSION
The present study demonstrates that aging-induced impairment of endothelial NO-dependent dilatation is mediated by ANG II generated from a local arterial renin-angiotensin system. Indeed, acute local inhibition of the synthesis (ACE, renin) or activity (AT1 receptors) of ANG II completely restored endothelial dilator activity in aging arteries. Lakatta and colleagues previously demonstrated the presence of an intravascular ANG II signaling system in aging arteries and highlighted its role in the fibrotic and inflammatory activity of intimal SMCs and in arterial remodeling (19, 33, 34). They observed a four to sixfold increase in ANG II immunofluorescence in the endothelium and intimal SMCs of old compared with young arteries from rats, humans and nonhuman primates, which was paralleled by similar increases in ACE and AT1 receptor expression (36–38). The present experiments confirmed an increase in ANG II immunofluorescence levels and further demonstrated a 5.6-fold increase in renin expression in old compared with young arteries. Previous studies demonstrated that prolonged in vivo inhibition of ANG II activity improves endothelium-dependent dilatation of old arteries, which was associated with a decrease in ROS activity (24, 27). The results of the present study indicate that those protective effects of ANG II inhibition may be explained by inhibition of a local rather than systemic renin-angiotensin system.
The results of the present study are consistent with the sequential action of renin and ACE to generate ANG II, which then acts on endothelial AT1 receptors to cause endothelial dilator dysfunction. The expression of chymase, which like ACE can convert ANGI to ANG II, is increased in aortas of old monkeys (36). However, chymase expression was restricted to the adventitia, whereas both ANG II and ACE were localized to the endothelium and intima (36). Therefore, in both of these aging vascular systems, the predominant if not exclusive source of ANG II appears to be ACE (36). Aging is also reported to increase the vascular expression of (pro)renin receptors (39). Aliskiren inhibits ANGI generation by renin and by prorenin bound to the (pro)renin receptor (9). Therefore, both mechanisms could contribute to the local generation of ANG II in aging arteries. Indeed, in addition to the observed increase in vascular expression of renin, capture of (pro)renin from the circulation by (pro)renin receptors could also contribute to increased ANG II production (40). Increased expression of AT1 receptors in the endothelium and intima of aging arteries (37) could potentially increase constitutive ligand-independent activity of the receptors. The similar effects of blocking the synthesis (renin, ACE) and activity (AT1 receptors) of ANG II on endothelial dilatation argue against ligand-independent AT1 receptor activity. Furthermore, although valsartan is a strong inverse agonist and can effectively inhibit ANG II-independent AT1 receptor activity, losartan is considered a poor inverse agonist and is ineffective (40). However, both agents are powerful AT1 receptor antagonists and inhibit responses to ANG II (40).
Other pathological mediators are known to contribute to the endothelial dysfunction of aging. As with prolonged ANG II inhibition, chronic in vivo inhibition of TNF-α increased endothelial dilatation and decreased ROS activity in aging arteries (1, 7, 26). This may reflect the close relationship between TNF-α and ANG II in the aging arterial wall. Indeed, inhibition of TNF-α reduced the heightened expression of ACE, AT1 receptors, and immunofluorescent ANG II in aging arteries (2), suggesting that expression of the local renin-angiotensin system is amplified by TNF-α and the inflammatory activity of aging arteries (19, 32, 33). The results of the present study also explain the parallel mechanisms contributing to endothelial dilator dysfunction in old arteries or in response to ANG II, which include increased expression and activation of arginase, increased production of ROS from NOXs and mitochondria, and uncoupling of NO synthase (5, 8, 11, 17, 30, 31). Cardiovascular effects resulting from chronic administration of ANG II, including vascular remodeling and endothelial dysfunction, can be reduced by mineralocorticoid receptor (MR) antagonism, which may reflect ANG II-induced release of aldosterone from the adrenal gland (4). MR expression is increased in aging arteries and in cultured SMCs derived from them, and an MR antagonist reduced the heightened inflammatory and fibrotic activity of cultured aging SMCs (18). Although ANG II might increase the local vascular production of aldosterone (4, 23, 29), this MR activity in aging SMCs likely results from ANG II and AT1 receptor-dependent transactivation of MRs (15, 18). It is currently unknown if MR activation contributes to the direct effects of ANG II to cause endothelial dilator dysfunction, including in aging arteries.
Aging arterial endothelial cells are severely compromised: their production of protective NO is reduced, their barrier function is impaired, they are highly susceptible to apoptosis, and they have a prominent inflammatory phenotype (19, 32). Aging endothelial cells are therefore caught in a chronic cycle of inflammatory stress, both producing and responding to pathological mediators, and their resulting frailty is considered a key instigator of vascular aging (32, 34). Indeed, specific suppression of endothelial inflammatory activity, by transgenic inhibition of NF-κB, inhibited arterial senescence, increased locomotor activity, and prolonged lifespan in aging mice (14). Similar beneficial effects, including extended lifespan, are observed following systemic inhibition of ANG II (3, 6, 35). The present experiments were restricted to analyzing endothelial NO-mediated dilator function and did not address additional dysfunctional aspects of aging endothelium. However, because of the pleiotropic pathological effects of ANG II and the pleiotropic protective effects of NO (22, 25), ANG II likely contributes to other aspects of the aging endothelial phenotype. Indeed, some of the beneficial effects of inhibiting NF-κB in aging were mediated by increased NO activity (14). Vascular aging represents deterioration of the structure and function of the entire arterial system, from central arteries to the microcirculation (32). Although expression of the intravascular ANG II system occurs in other aging arteries and vascular beds (2, 36–38), the present experiments were restricted to proximal tail arteries and did not analyze the potential role of local ANG II signaling in other systems or in the microcirculation.
In conclusion, we have demonstrated that the aging-induced dysfunction in endothelial NO-dependent dilatation in rat tail arteries is mediated by a local arterial renin-angiotensin system and pathological ANG II activity. Increased understanding of the mechanisms regulating this intravascular system may provide novel therapeutic approaches to alleviate the devastating effects of vascular aging.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
S.F., F.C., and N.A.F. conception and design of research; S.F., F.C., and N.A.F. performed experiments; S.F., F.C., and N.A.F. analyzed data; S.F., F.C., and N.A.F. interpreted results of experiments; S.F., F.C., and N.A.F. edited and revised manuscript; S.F., F.C., and N.A.F. approved final version of manuscript; F.C. and N.A.F. prepared figures; N.A.F. drafted manuscript.
ACKNOWLEDGMENTS
We are grateful to the Biological Resources Branch of the NIA for allowing access to their Aged Rodent Colony.
REFERENCES
- 1.Arenas IA, Armstrong SJ, Xu Y, Davidge ST. Chronic tumor necrosis factor-alpha inhibition enhances NO modulation of vascular function in estrogen-deficient rats. Hypertension 46: 76–81, 2005. [DOI] [PubMed] [Google Scholar]
- 2.Arenas IA, Armstrong SJ, Xu Y, Davidge ST. Tumor necrosis factor-alpha and vascular angiotensin II in estrogen-deficient rats. Hypertension 48: 497–503, 2006. [DOI] [PubMed] [Google Scholar]
- 3.Basso N, Cini R, Pietrelli A, Ferder L, Terragno NA, Inserra F. Protective effect of long-term angiotensin II inhibition. Am J Physiol Heart Circ Physiol 293: H1351–H1358, 2007. [DOI] [PubMed] [Google Scholar]
- 4.Briet M, Schiffrin EL. Vascular actions of aldosterone. J Vasc Res 50: 89–99, 2013. [DOI] [PubMed] [Google Scholar]
- 5.Chalupsky K, Cai H. Endothelial dihydrofolate reductase: critical for nitric oxide bioavailability and role in angiotensin II uncoupling of endothelial nitric oxide synthase. Proc Natl Acad Sci USA 102: 9056–9061, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Conti S, Cassis P, Benigni A. Aging and the renin-angiotensin system. Hypertension 60: 878–883, 2012. [DOI] [PubMed] [Google Scholar]
- 7.Csiszar A, Labinskyy N, Smith K, Rivera A, Orosz Z, Ungvari Z. Vasculoprotective effects of anti-tumor necrosis factor-alpha treatment in aging. Am J Pathol 170: 388–398, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Csiszar A, Ungvari Z, Edwards JG, Kaminski P, Wolin MS, Koller A, Kaley G. Aging-induced phenotypic changes and oxidative stress impair coronary arteriolar function. Circ Res 90: 1159–1166, 2002. [DOI] [PubMed] [Google Scholar]
- 9.Danser AH. (Pro)renin receptors: are they biologically relevant? Curr Opin Nephrol Hypertens 18: 74–78, 2009. [DOI] [PubMed] [Google Scholar]
- 10.Didion SP, Kinzenbaw DA, Faraci FM. Critical role for CuZn-superoxide dismutase in preventing angiotensin II-induced endothelial dysfunction. Hypertension 46: 1147–1153, 2005. [DOI] [PubMed] [Google Scholar]
- 11.Dikalov SI, Nazarewicz RR, Bikineyeva A, Hilenski L, Lassegue B, Griendling KK, Harrison DG, Dikalova AE. Nox2-induced production of mitochondrial superoxide in angiotensin II-mediated endothelial oxidative stress and hypertension. Antioxid Redox Signal 20: 281–294, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fu Z, Wang M, Gucek M, Zhang J, Wu J, Jiang L, Monticone RE, Khazan B, Telljohann R, Mattison J, Sheng S, Cole RN, Spinetti G, Pintus G, Liu L, Kolodgie FD, Virmani R, Spurgeon H, Ingram DK, Everett AD, Lakatta EG, Van Eyk JE. Milk fat globule protein epidermal growth factor-8: a pivotal relay element within the angiotensin II and monocyte chemoattractant protein-1 signaling cascade mediating vascular smooth muscle cells invasion. Circ Res 104: 1337–1346, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Goel A, Su B, Flavahan S, Lowenstein CJ, Berkowitz DE, Flavahan NA. Increased endothelial exocytosis and generation of endothelin-1 contributes to constriction of aged arteries. Circ Res 107: 242–251, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hasegawa Y, Saito T, Ogihara T, Ishigaki Y, Yamada T, Imai J, Uno K, Gao J, Kaneko K, Shimosawa T, Asano T, Fujita T, Oka Y, Katagiri H. Blockade of the nuclear factor-kappaB pathway in the endothelium prevents insulin resistance and prolongs life spans. Circulation 125: 1122–1133, 2012. [DOI] [PubMed] [Google Scholar]
- 15.Jaffe IZ, Mendelsohn ME. Angiotensin II and aldosterone regulate gene transcription via functional mineralocortocoid receptors in human coronary artery smooth muscle cells. Circ Res 96: 643–650, 2005. [DOI] [PubMed] [Google Scholar]
- 16.Jiang L, Wang M, Zhang J, Monticone RE, Telljohann R, Spinetti G, Pintus G, Lakatta EG. Increased aortic calpain-1 activity mediates age-associated angiotensin II signaling of vascular smooth muscle cells. PLoS One 3: e2231, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kim JH, Bugaj LJ, Oh YJ, Bivalacqua TJ, Ryoo S, Soucy KG, Santhanam L, Webb A, Camara A, Sikka G, Nyhan D, Shoukas AA, Ilies M, Christianson DW, Champion HC, Berkowitz DE. Arginase inhibition restores NOS coupling and reverses endothelial dysfunction and vascular stiffness in old rats. J Appl Physiol 107: 1249–1257, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Krug AW, Allenhofer L, Monticone R, Spinetti G, Gekle M, Wang M, Lakatta EG. Elevated mineralocorticoid receptor activity in aged rat vascular smooth muscle cells promotes a proinflammatory phenotype via extracellular signal-regulated kinase 1/2 mitogen-activated protein kinase and epidermal growth factor receptor-dependent pathways. Hypertension 55: 1476–1483, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lakatta EG, Wang M, Najjar SS. Arterial aging and subclinical arterial disease are fundamentally intertwined at macroscopic and molecular levels. Med Clin North Am 93: 583–604, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Laurent S. Defining vascular aging and cardiovascular risk. J Hypertens 30, Suppl: S3–8, 2012. [DOI] [PubMed] [Google Scholar]
- 21.Li JM, Wheatcroft S, Fan LM, Kearney MT, Shah AM. Opposing roles of p47phox in basal versus angiotensin II-stimulated alterations in vascular O2- production, vascular tone, and mitogen-activated protein kinase activation. Circulation 109: 1307–1313, 2004. [DOI] [PubMed] [Google Scholar]
- 22.Michel T, Vanhoutte PM. Cellular signaling and NO production. Pflügers Arch 459: 807–816, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Min LJ, Mogi M, Iwanami J, Li JM, Sakata A, Fujita T, Tsukuda K, Iwai M, Horiuchi M. Cross-talk between aldosterone and angiotensin II in vascular smooth muscle cell senescence. Cardiovasc Res 76: 506–516, 2007. [DOI] [PubMed] [Google Scholar]
- 24.Modrick ML, Didion SP, Sigmund CD, Faraci FM. Role of oxidative stress and AT1 receptors in cerebral vascular dysfunction with aging. Am J Physiol Heart Circ Physiol 296: H1914–H1919, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Montezano AC, Nguyen Dinh Cat A, Rios FJ, Touyz RM. Angiotensin II and vascular injury. Curr Hypertens Rep 16: 431, 2014. [DOI] [PubMed] [Google Scholar]
- 26.Moreau KL, Deane KD, Meditz AL, Kohrt WM. Tumor necrosis factor-alpha inhibition improves endothelial function and decreases arterial stiffness in estrogen-deficient postmenopausal women. Atherosclerosis 230: 390–396, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mukai Y, Shimokawa H, Higashi M, Morikawa K, Matoba T, Hiroki J, Kunihiro I, Talukder HM, Takeshita A. Inhibition of renin-angiotensin system ameliorates endothelial dysfunction associated with aging in rats. Arterioscler Thromb Vasc Biol 22: 1445–1450, 2002. [DOI] [PubMed] [Google Scholar]
- 28.Nowicki PT, Flavahan S, Hassanain H, Mitra S, Holland S, Goldschmidt-Clermont PJ, Flavahan NA. Redox signaling of the arteriolar myogenic response. Circ Res 89: 114–116, 2001. [DOI] [PubMed] [Google Scholar]
- 29.Schiffrin EL. Effects of aldosterone on the vasculature. Hypertension 47: 312–318, 2006. [DOI] [PubMed] [Google Scholar]
- 30.Shatanawi A, Romero MJ, Iddings JA, Chandra S, Umapathy NS, Verin AD, Caldwell RB, Caldwell RW. Angiotensin II-induced vascular endothelial dysfunction through RhoA/Rho kinase/p38 mitogen-activated protein kinase/arginase pathway. Am J Physiol Cell Physiol 300: C1181–C1192, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Trott DW, Seawright JW, Luttrell MJ, Woodman CR. NAD(P)H oxidase-derived reactive oxygen species contribute to age-related impairments of endothelium-dependent dilation in rat soleus feed arteries. J Appl Physiol 110: 1171–1180, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ungvari Z, Kaley G, de Cabo R, Sonntag WE, Csiszar A. Mechanisms of vascular aging: new perspectives. J Gerontol A Biol Sci Med Sci 65: 1028–1041, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang M, Jiang L, Monticone RE, Lakatta EG. Proinflammation: the key to arterial aging. Trends Endocrinol Metab 25: 72–79, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang M, Khazan B, Lakatta EG. Central arterial aging and angiotensin II signaling. Curr Hypertens Rev 6: 266–281, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang M, Monticone RE, Lakatta EG. Arterial aging: a journey into subclinical arterial disease. Curr Opin Nephrol Hypertens 19: 201–207, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang M, Takagi G, Asai K, Resuello RG, Natividad FF, Vatner DE, Vatner SF, Lakatta EG. Aging increases aortic MMP-2 activity and angiotensin II in nonhuman primates. Hypertension 41: 1308–1316, 2003. [DOI] [PubMed] [Google Scholar]
- 37.Wang M, Zhang J, Jiang LQ, Spinetti G, Pintus G, Monticone R, Kolodgie FD, Virmani R, Lakatta EG. Proinflammatory profile within the grossly normal aged human aortic wall. Hypertension 50: 219–227, 2007. [DOI] [PubMed] [Google Scholar]
- 38.Wang M, Zhang J, Spinetti G, Jiang LQ, Monticone R, Zhao D, Cheng L, Krawczyk M, Talan M, Pintus G, Lakatta EG. Angiotensin II activates matrix metalloproteinase type II and mimics age-associated carotid arterial remodeling in young rats. Am J Pathol 167: 1429–1442, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yoon HE, Kim EN, Kim MY, Lim JH, Jang IA, Ban TH, Shin SJ, Park CW, Chang YS, Choi BS. Age-associated changes in the vascular renin-angiotensin system in mice. Oxid Med Cell Longev 2016: 6731093, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhao Y, Flavahan S, Leung SW, Xu A, Vanhoutte PM, Flavahan NA. Elevated pressure causes endothelial dysfunction in mouse carotid arteries by increasing local angiotensin signaling. Am J Physiol Heart Circ Physiol 308: H358–H363, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]