

aging, which is closely linked to oxidative stress, is an independent risk factor for various cardiovascular diseases (1). The role of reactive oxygen species (ROS) such as superoxide (O2•−) in the aging process was initially proposed as the “free radical theory” by Harman in the 1950s (12), whereby ROS attack cellular molecules and cause the functional decline of organ systems, which eventually leads to death. Aging is dependent on mitochondrial dysfunction, cellular senescence, inflammation, and vascular dysfunction, which are associated with ROS (Fig. 1) (16). Underlying mechanisms remain unclear. In normal blood vessels, nitric oxide (NO) is released from the endothelium and diffuses into smooth muscle to produce vasorelaxation. In aged vessels, O2•− levels are increased by mitochondrial dysfunction or NADPH oxidase activation and negatively regulated (reduced) by antioxidant enzymes such as Mn-SOD (SOD-2) and cytosolic Cu,Zn-SOD (SOD-1) (1, 16). NO reacts with O2•− to form peroxynitrite (ONOO−), which can induce protein modifications and DNA damage (20). Reduction of endothelium-derived NO contributes to vasoconstriction and inflammation. Thus a decrease in NO bioavailability as a result of excess O2•− formation is a major cause of endothelial dysfunction in aging.
Fig. 1.
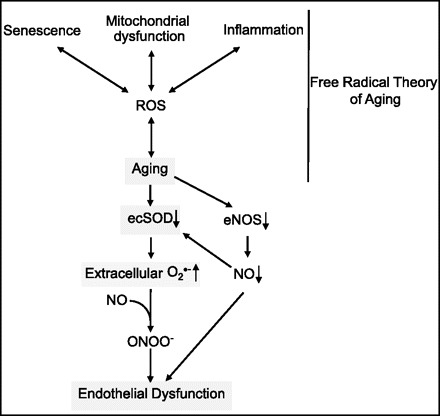
Role of extracellular superoxide dismutase (ecSOD) in endothelial function during aging. A causative role of reactive oxygen species (ROS) in the aging process has been referred to as the free radical theory of aging (12). In particular, senescence, mitochondrial dysfunction, and inflammation have been proposed to be linked to aging via ROS (16). In aged ecSOD−/− mice, endothelium-dependent vasorelaxation is markedly impaired. During aging, decreased expression of ecSOD leads to an increase in extracellular O2•−, resulting in oxidative inactivation of endothelial nitric oxide (NO) synthase (eNOS)-derived NO and peroxynitrite (ONOO−) formation, which in turn contribute to endothelial dysfunction. Given that eNOS-derived NO increases ecSOD transcription, the aging-induced decrease in eNOS expression may result in a decrease in ecSOD. This further increases extracellular O2•− levels and, thus, promoting impaired endothelial function.
The extracellular form of Cu,Zn-SOD (ecSOD, SOD-3) is the major SOD in the vascular extracellular space. It protects against inactivation of NO released from the endothelium by O2•− during diffusion to smooth muscle (19), thereby preserving endothelial function. In studies using ecSOD-deficient mice, ecSOD has been shown to play an important role in various oxidative stress-dependent pathophysiologies, including hypertension, ischemia-reperfusion injury, and lung injury (7, 8, 23). Several lines of evidence suggest a role for ecSOD in aging. The R213G polymorphism in the ecSOD gene has been linked to an increase in cardiovascular risk (15) that is not revealed until 70 yr of age in humans. Plasma levels of ecSOD decrease with aging (6), and gene transfer of ecSOD improves endothelial function in old rats (2). However, it remains unknown whether ecSOD expression or activity in blood vessels is regulated by aging and whether endogenous ecSOD is involved in regulation of vascular function during aging.
In this issue, using aged (old) and younger (adult) ecSOD−/− mice, Lund et al. (16a) provide the first evidence in aged (old) and old (adult) ecSOD−/− mice that endogenous ecSOD plays an essential role in protection against O2•−-mediated endothelial dysfunction during aging. They showed that acetylcholine-induced endothelium-dependent relaxation of the aorta from old (29 mo old) and adult (11 mo old) ecSOD−/− mice is significantly impaired compared with that from wild-type mice at each age, whereas sodium nitroprusside-induced relaxation is not altered. Moreover, the extent of endothelial dysfunction is much greater in aged ecSOD−/− than younger ecSOD−/− mice and is rescued by infusion of the SOD mimetic tempol. Impaired relaxation of arteries in response to acetylcholine with increasing age is consistent with previous studies in rats (2) and is presumably due to elevated levels of O2•− in aged arteries (2). Although Lund et al. did not measure O2•− levels in aged ecSOD−/− mice, tempol-induced restoration of relaxation to acetylcholine strongly supports the possibility that O2•− is increased in the aorta of these mice. Their data also suggest that impaired endothelial function in aged ecSOD−/− mice might be due to reduced bioavailability of NO, rather than impaired smooth muscle function. ecSOD expression is also significantly decreased in the aorta from old mice compared with young mice, and the correlation between endothelial function and ecSOD expression is much steeper in aged mice than younger mice. These findings suggest a greater protective effect of ecSOD against endothelial dysfunction in aged mice. This may be due to higher vascular O2•− levels in old mice than in younger animals (2). Consistent with this idea, Brown et al. (2) demonstrated that gene transfer of ecSOD in old rats reduces O2•− to the levels observed in young rats and restores vascular function without significant effects on young animals. These findings indicate that ecSOD more effectively protects against endothelial dysfunction in aged animals, which would have higher extracellular O2•− levels than younger animals.
Consistent with the results from ecSOD−/− mice, previous reports show that endothelium-dependent vasorelaxation is markedly impaired in old Cu,Zn-SOD+/− and Mn-SOD+/− mice because of enhanced O2•− production compared with young mice (3, 5). One of the underlying mechanisms has been explained by activation of poly(ADP-ribose) polymerase via oxidative DNA damage (21), because poly(ADP-ribose) polymerase inhibitor improves endothelial function in old Cu,Zn-SOD+/− mice (5). Importantly, this response is not observed in Mn-SOD+/− mice (3). Although Lund et al. (16a) show no effects on vessel contraction induced by PGF2α or serotonin in aged ecSOD−/− mice, vasoconstriction is enhanced in aged Cu,Zn-SOD+/− and Mn-SOD+/− mice. Thus each isoform of SOD, expressed in a different location of the vessel wall, seems to play an important role in protecting vascular function during aging through distinct mechanisms.
Lund et al. (16a) also reported that mRNA levels of ecSOD, but not Cu,Zn-SOD or Mn-SOD, are significantly reduced in aged (old) mice compared with younger (adult) mice. This finding is consistent with a previous report of no compensatory upregulation of Cu,Zn-SOD or Mn-SOD in ecSOD−/− mice (11, 14). Mechanisms for selective reduction of ecSOD with aging are not clear. Reduced levels of ecSOD have been reported in plasma of older patients (6). Because endothelial NO synthase (eNOS) is a positive regulator for ecSOD expression (9, 18) and the eNOS level tended to be decreased in aged mice in the study of Lund et al. (16a), reduced eNOS may also contribute to decreased ecSOD mRNA expression. Lu et al. (17) reported a decrease in cardiac Cu,Zn-SOD activity but no change in Cu,Zn-SOD protein levels in ecSOD−/− mice challenged with pressure overload. This might be caused by inactivation of Cu,Zn-SOD by ONOO−, which is formed by reaction of NO with O2•− due to ecSOD deficiency (10). It is tempting to speculate that a similar phenomenon may be observed in aged ecSOD−/− mice. Thus, not only mRNA, but also protein level and activity of Cu,Zn-SOD or Mn-SOD, in aged ecSOD−/− mice, as well as ecSOD protein and activity in aged mice, should be measured in a future study.
The findings of Lund et al. (16a) strongly support a critical role of ecSOD as a regulator of NO bioavailability and endothelial function during aging (Fig. 1). As they pointed out, their findings will have potential clinical implications in relation to a gene polymorphism (R213G) found in humans in the heparin-binding site of ecSOD (15). This gene variant impairs binding of ecSOD to the vessel wall and, thus, impairs protection of vessels from the increased levels of O2•− in the extracellular space (4). The findings of Lund et al. may explain why the risk of ischemic heart disease in subjects with ecSODR213G is confined to individuals >70 yr old (15). There are many unanswered questions. It has been shown that the level of ecSOD in cells is positively associated with the length of telomeres in human fibroblast lines (24). Is this relevant to the findings of Lund et al.? ecSOD activity and expression are regulated at transcriptional and posttranslational levels (25). For example, full activation of ecSOD requires copper carrier protein in order to obtain its catalytic copper (13, 22). Does this protein play a role in regulation of endothelial function during aging through control of ecSOD activity? Addressing these questions will be essential to an understanding of the mechanism of oxidative stress-dependent cardiovascular diseases and aging, in which extracellular O2•− may play an essential role.
GRANTS
This work was supported by National Heart, Lung, and Blood Institute Grant 5R01 HL-070187-08.
Acknowledgments
I thank Dr. Masuko Ushio-Fukai for critical review of the manuscript and helpful comments.
REFERENCES
- 1.BrandesRPBrandes RP, Fleming I, Busse R. Endothelial aging. Cardiovasc Res 66: 286–294, 2005. [DOI] [PubMed] [Google Scholar]
- 2.BrownKABrown KA, Chu Y, Lund DD, Heistad DD, Faraci FM. Gene transfer of extracellular superoxide dismutase protects against vascular dysfunction with aging. Am J Physiol Heart Circ Physiol 290: H2600–H2605, 2006. [DOI] [PubMed] [Google Scholar]
- 3.BrownKABrown KA, Didion SP, Andresen JJ, Faraci FM. Effect of aging, MnSOD deficiency, and genetic background on endothelial function: evidence for MnSOD haploinsufficiency. Arterioscler Thromb Vasc Biol 27: 1941–1946, 2007. [DOI] [PubMed] [Google Scholar]
- 4.ChuYChu Y, Alwahdani A, Iida S, Lund DD, Faraci FM, Heistad DD. Vascular effects of the human extracellular superoxide dismutase R213G variant. Circulation 112: 1047–1053, 2005. [DOI] [PubMed] [Google Scholar]
- 5.DidionSPDidion SP, Kinzenbaw DA, Schrader LI, Faraci FM. Heterozygous CuZn superoxide dismutase deficiency produces a vascular phenotype with aging. Hypertension 48: 1072–1079, 2006. [DOI] [PubMed] [Google Scholar]
- 6.Di MassimoCDi Massimo C, Scarpelli P, Di Lorenzo N, Caimi G, di Orio F, Ciancarelli MG. Impaired plasma nitric oxide availability and extracellular superoxide dismutase activity in healthy humans with advancing age. Life Sci 78: 1163–1167, 2006. [DOI] [PubMed] [Google Scholar]
- 7.FattmanCLFattman CL, Schaefer LM, Oury TD. Extracellular superoxide dismutase in biology and medicine. Free Radic Biol Med 35: 236–256, 2003. [DOI] [PubMed] [Google Scholar]
- 8.FukaiTFukai T, Folz RJ, Landmesser U, Harrison DG. Extracellular superoxide dismutase and cardiovascular disease. Cardiovasc Res 55: 239–249, 2002. [DOI] [PubMed] [Google Scholar]
- 9.FukaiTFukai T, Siegfried MR, Ushio-Fukai M, Cheng Y, Kojda G, Harrison DG. Regulation of the vascular extracellular superoxide dismutase by nitric oxide and exercise training. J Clin Invest 105: 1631–1639, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.GongoraMCGongora MC, Harrison DG. Sad heart from no SOD. Hypertension 51: 28–30, 2008. [DOI] [PubMed] [Google Scholar]
- 11.GongoraMCGongora MC, Qin Z, Laude K, Kim HW, McCann L, Folz JR, Dikalov S, Fukai T, Harrison DG. Role of extracellular superoxide dismutase in hypertension. Hypertension 48: 473–481, 2006. [DOI] [PubMed] [Google Scholar]
- 12.HarmanDHarman D. Aging: a theory based on free radical and radiation chemistry. J Gerontol 11: 298–300, 1956. [DOI] [PubMed] [Google Scholar]
- 13.ItohSItoh S, Ozumi K, Kim HW, Nakagawa O, McKinney RD, Folz RJ, Zelko IN, Ushio-Fukai M, Fukai T. Novel mechanism for regulation of extracellular SOD transcription and activity by copper: role of antioxidant-1. Free Radic Biol Med 46: 95–104, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.JungOJung O, Marklund SL, Geiger H, Pedrazzini T, Busse R, Brandes RP. Extracellular superoxide dismutase is a major determinant of nitric oxide bioavailability: in vivo and ex vivo evidence from ecSOD-deficient mice. Circ Res 93: 622–629, 2003. [DOI] [PubMed] [Google Scholar]
- 15.JuulKJuul K, Tybjaerg-Hansen A, Marklund S, Heegaard NH, Steffensen R, Sillesen H, Jensen G, Nordestgaard BG. Genetically reduced antioxidative protection and increased ischemic heart disease risk: the Copenhagen City Heart Study. Circulation 109: 59–65, 2004. [DOI] [PubMed] [Google Scholar]
- 16.KregelKCKregel KC, Zhang HJ. An integrated view of oxidative stress in aging: basic mechanisms, functional effects, and pathological considerations. Am J Physiol Regul Integr Comp Physiol 292: R18–R36, 2007. [DOI] [PubMed] [Google Scholar]
- 16a.LundDDLund DD, Chu Y, Miller JD, Heistad DD. Protective effect of extracellular superoxide dismutase on endothelial function during aging. Am J Physiol Heart Circ Physiol. 10.1152/ajpheart.01342.2008. [DOI] [PMC free article] [PubMed]
- 17.LuZLu Z, Xu X, Hu X, Zhu G, Zhang P, van Deel ED, French JP, Fassett JT, Oury TD, Bache RJ, Chen Y. Extracellular superoxide dismutase deficiency exacerbates pressure overload-induced left ventricular hypertrophy and dysfunction. Hypertension 51: 19–25, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.OppermannMOppermann M, Balz V, Adams V, Dao VT, Bas M, Suvorava T, Kojda G.Pharmacologic induction of vascular extracellular superoxide dismutase expression in-vivo. J Cell Mol Med. In press. [DOI] [PMC free article] [PubMed]
- 19.OuryTDOury TD, Day BJ, Crapo JD. Extracellular superoxide dismutase: a regulator of nitric oxide bioavailability. Lab Invest 75: 617–636, 1996. [PubMed] [Google Scholar]
- 20.PacherPPacher P, Beckman JS, Liaudet L. Nitric oxide and peroxynitrite in health and disease. Physiol Rev 87: 315–424, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.PacherPPacher P, Szabo C. Role of the peroxynitrite-poly(ADP-ribose) polymerase pathway in human disease. Am J Pathol 173: 2–13, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.QinZQin Z, Gongora MC, Ozumi K, Itoh S, Akram K, Ushio-Fukai M, Harrison DG, Fukai T. Role of Menkes ATPase in angiotensin II-induced hypertension: a key modulator for extracellular superoxide dismutase function. Hypertension 52: 945–951, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.QinZQin Z, Reszka KJ, Fukai T, Weintraub NL. Extracellular superoxide dismutase (ecSOD) in vascular biology: an update on exogenous gene transfer and endogenous regulators of ecSOD. Transl Res 151: 68–78, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.SerraVSerra V, von Zglinicki T, Lorenz M, Saretzki G. Extracellular superoxide dismutase is a major antioxidant in human fibroblasts and slows telomere shortening. J Biol Chem 278: 6824–6830, 2003. [DOI] [PubMed] [Google Scholar]
- 25.ZelkoINZelko IN, Mariani TJ, Folz RJ. Superoxide dismutase multigene family: a comparison of the CuZn-SOD (SOD1), Mn-SOD (SOD2), and EC-SOD (SOD3) gene structures, evolution, and expression. Free Radic Biol Med 33: 337–349, 2002. [DOI] [PubMed] [Google Scholar]