

Abstract
Epidemiological evidence links an individual's susceptibility to chronic disease in adult life to events during their intrauterine phase of development. Biologically this should not be unexpected, for organ systems are at their most plastic when progenitor cells are proliferating and differentiating. Influences operating at this time can permanently affect their structure and functional capacity, and the activity of enzyme systems and endocrine axes. It is now appreciated that such effects lay the foundations for a diverse array of diseases that become manifest many years later, often in response to secondary environmental stressors. Fetal development is underpinned by the placenta, the organ that forms the interface between the fetus and its mother. All nutrients and oxygen reaching the fetus must pass through this organ. The placenta also has major endocrine functions, orchestrating maternal adaptations to pregnancy and mobilizing resources for fetal use. In addition, it acts as a selective barrier, creating a protective milieu by minimizing exposure of the fetus to maternal hormones, such as glucocorticoids, xenobiotics, pathogens, and parasites. The placenta shows a remarkable capacity to adapt to adverse environmental cues and lessen their impact on the fetus. However, if placental function is impaired, or its capacity to adapt is exceeded, then fetal development may be compromised. Here, we explore the complex relationships between the placental phenotype and developmental programming of chronic disease in the offspring. Ensuring optimal placentation offers a new approach to the prevention of disorders such as cardiovascular disease, diabetes, and obesity, which are reaching epidemic proportions.
I. INTRODUCTION
The intrauterine phase of development is key to life-long health, for the foundations of the body plan and the major organ systems are laid down during this period. Perturbation of gene expression or cell proliferation and differentiation during vulnerable periods by nutritional and other environmental influences can alter the structure and functional capacity of major organ systems for life, a process known as developmental programming. These changes predispose the offspring to a variety of disorders that may become manifest in later life, often following exposure to a second precipitating challenge. This concept has profound implications for public health and our approach to the management of chronic diseases, some of which are now reaching epidemic proportions.
The programmed outcomes and the mechanisms by which they occur in the developing fetus, together with their significance for future health have been reviewed previously (37, 56, 215, 237, 374, 426, 528, 565). Here, we focus on the impact of the placenta, the organ that forms the interface between the mother and her offspring while in utero, on the causation of chronic disease. The placenta evolved to transfer nutrients to the fetus, and also to create a stable milieu in which the fetus can develop, isolated as far as possible from maternal and environmental stressors. To achieve these functions, it performs a remarkably diverse range of activities, including active and passive transport, endocrine secretion, immunological protection, and xenobiotic detoxification. As well as being multifunctional, the placenta is also a remarkably plastic organ, capable of considerable structural and functional adaptations that help to mitigate adverse maternal insults, such as nutrient deprivation, and exposure to drugs, toxins, or hypoxia. However, if normal placental function is impaired, or the organ's capacity for adaptation exceeded, then the fetal milieu may be perturbed with major consequences for the life-long health of the offspring (Figure 1). Ensuring women of childbearing age have access to sufficient and appropriate nutrition is essential, but so too is an understanding of maternal physiological adaptations during pregnancy, in particular the mechanisms by which resources are allocated such that her own needs, and those of her offspring, are suitably met. There is now compelling evidence that the placenta plays a central role in orchestrating this process.
FIGURE 1.

Diagrammatic illustration showing how the placenta may modulate and transduce environmental cues that lead to developmental programming of the fetus. The functional capacity of the placenta will depend on its development and its ability to adapt, as well as any reserve that exists.
To achieve our aim we will consider the following: 1) the various functions of the mammalian placenta; 2) how placental structure and development facilitate those functions in the human and in the two main experimental models, the mouse and the sheep; 3) the epidemiological evidence linking changes in human placental phenotype to adult disease; 4) the mechanisms by which placental cells may sense oxygen and nutrient availability; 5) how maternal nutrient supply and fetal demand may be integrated at the placental interface; 6) the mechanisms by which the placenta can impact on developmental programming of the offspring; and, finally, 7) areas for future research.
We start by briefly describing the general concept of developmental programming of chronic disease.
II. DEVELOPMENTAL PROGRAMMING OF CHRONIC DISEASE
It has long been known that the intrauterine environment has a major impact on development of the adult phenotype (558), but the significance of this phenomenon for adult health was first highlighted by David Barker and colleagues. In the late 1980s, they reported on ∼15,000 records from men and women in Hertfordshire, United Kingdom, and showed that rates of death from ischemic heart disease were ordered across the birth weight scale (40). Babies born at the lower end of the scale (5 lb. or 2.3 kg) had the highest mortality rates as adults, while those at the opposite end (9 lb. or 4.0 kg) were two-thirds lower. At the time, Barker and Osmond (31) had just concluded a study examining cardiac-related death rates across England. The finding that people in the industrial areas of the north of the country died more often of cardiovascular disease than those in the rural south was not surprising, since the impact of an adverse social environment on mortality was already known. The new insight gained was that their findings showed a similar geographic distribution as for the death rates of neonates some 60 years earlier.
The Barker team reasoned that both the neonates and adults died for the same reason, namely, that their development had been compromised before birth. Thus they suggested that an adverse intrauterine environment rendered them vulnerable to death as neonates, and more likely to acquire heart disease later if they survived childhood (33). This relationship between poor growth in the womb and the risk of adult disease has since been confirmed in many other countries, including Finland (26), Sweden (332), China (183), India (517), and the United States (464).
The conclusion that growth rates before birth predict later disease was initially received with skepticism, principally because a mechanistic explanation was not immediately apparent. Eventually, however, experimental evidence accumulated showing clear biological links between stresses that occurred during the first 1,000 days after conception and elevated risks for chronic conditions. These links revolve around permanent structural changes in organ systems, premature aging of tissues, and epigenetic changes. For example, a growth-restricted fetus has smaller coronary arteries (288), fewer but more immature cardiomyocytes (55, 348, 394), less elastin in the arteries (152, 362, 526), and fewer nephrons in the kidney (22, 352). In addition, the pancreas has fewer insulin-producing beta cells and reduced vascularization (159, 340, 473), and the structure and maturation of the brain (142), lungs (358, 359, 424, 460), and liver (209, 474) are compromised. All these outcomes have been linked experimentally to impaired placental function. These links go beyond an abnormal maternal nutrient supply and include, for example, intrauterine hypoxia (213), maternal social stress of the severity that leads to hypercortisolemia (128) and, increasingly, environmental toxins. Thus diverse stressors acting alone or in combination can lead to alterations in fetal development. Developmental plasticity is a well-described process in nature (43), but little research has addressed the phenomenon within the placenta. This is an area ripe for study.
The placenta does not function in isolation, however, and the mother's nutritional status has a powerful modifying influence on allocation of resources. Accumulating data show that maternal size, a marker of the mother's own growth history, and body composition, a marker of her current nutritional state, combine with placental size and shape to predict chronic disease outcomes. This is perhaps not surprising given that a proportion of the nutrients that support fetal growth, particularly in late pregnancy, come from turnover of maternal fat reserves that are built up in early pregnancy (414). More research is needed to understand how mothers and their offspring communicate through the placenta to regulate nutrient flow so that the needs of both parties are adequately met.
III. FUNCTIONS OF THE PLACENTA
When considering the potential impact that perturbation of placental function may have on developmental programming, it is essential to bear in mind the variety of activities that the organ performs. Different stressors, for example, undernutrition or hypoxia, may affect different placental functions, either in isolation or across the range. Here we consider those functions that have the greatest impact on the embryonic/fetal milieu, namely, the transport of nutrients and respiratory gases, the secretion of hormones, and its action as a selective barrier.
A. Transport of Nutrients and Mechanisms
Although a wide diversity of morphological types exists among mammals, a common feature is that the placenta provides for an extensive and intimate apposition of the maternal and fetal circulations. The tissue separating the two circulations is best referred to generically as the interhemal membrane, and it may vary in the number and nature of its cell layers (583). Transport across the membrane has recently been extensively reviewed (19, 61, 95), so this account is restricted to those aspects most pertinent to placental adaptations to environmental cues.
There are three main mechanisms by which exchange across the interhemal membrane can take place: diffusion, transporter-mediated mechanisms, and endocytosis/exocytosis (Figure 2).
FIGURE 2.
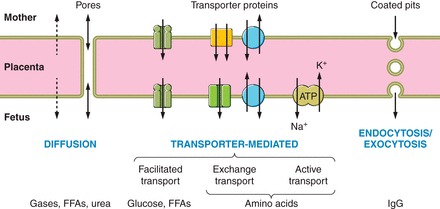
Diagrammatic representation of the three main processes by which materials can cross the interhemal placental membrane: diffusion, transporter-mediated, and endocytosis. The nature of the mechanism involved will determine how readily the placenta can adapt to facilitate transport under adverse conditions.
1. Diffusion
Simple diffusion is the passage of molecules through the lipid bilayers of the cell membranes and the intervening cytoplasm, and is a passive process that does not involve the expenditure of ATP. For small uncharged molecules, the rate of diffusion is governed by Fick's Law of diffusion, being proportional to the surface area for exchange and inversely proportional to the thickness of the interhemal membrane:
where Krogh's constant is a measure of the diffusivity of the molecule.
Small hydrophobic molecules cross cell membranes easily, so their transplacental flux depends principally on the concentration gradient driving exchange. The main factor maintaining that gradient is the rate of circulation of blood on either side of the membrane, refreshing and depleting the reservoir and recipient pools respectively. Hence, exchange of molecules such as the respiratory gases and lipophilic drugs is considered to be “flow-limited,” and changes in maternal or fetal blood flow have a profound impact on the net flux (573). However, under limiting conditions, such as pregnancy at high altitude, changes in surface area or membrane thickness may be considered adaptive responses to facilitate exchange (275, 369, 458). In contrast, more hydrophilic molecules traverse lipid bilayers slowly, so the transmembrane concentration gradient is generally more stable. Exchange of these molecules is said to be membrane- or “diffusion-limited,” and structural parameters such as surface area and membrane thickness will play a more major role in determining the flux.
Another mechanism influencing diffusion across the interhemal membrane is the presence of water-filled channels or pores. These are most relevant in species where the trophoblastic layer of the membrane is syncytial in nature and there are no paracellular pathways available, such as the human and mouse. The presence of such pores is evidenced by data showing that the human placenta is permeable to solutes of up to 5,200 Da (529, 575). Changing the number or diameter of these pores represents a potential mechanism by which the diffusion characteristics of the membrane could be altered in response to environmental cues. Identifying the morphological correlates of these pores has proven problematic in the human due to the complexity of the syncytiotrophoblast. Occasional membrane-lined clefts resembling intercellular spaces have been reported, but may represent areas of repair (339). However, the apical portions of such clefts are sealed by tight junctions and are impenetrable to the extracellular marker ruthenium red instilled at the time of postfixation. Another approach has been to perfuse the fetal vasculature of the placenta at elevated pressures. Pressures of 100 mmHg and above cause dilation of basal invaginations of the syncytiotrophoblast, and enlargement of vacuoles within the syncytioplasm (304), but connections with the apical surface are not found. Hence, the physiological significance of these morphological observations remains uncertain. An alternative explanation for the apparent existence of pores is that localized areas of damage to the syncytiotrophoblast represent paracellular routes of transport (68). These are discussed in more detail in section IIIC1.
2. Transporter-mediated mechanisms
Transporter-mediated processes are dependent on carrier proteins being inserted into cell membranes to facilitate the passage of highly hydrophilic molecules (Figure 2). Although contrasting in their functional characteristics, they are characterized by common features such as substrate specificity, saturation kinetics, and the ability to be competitively inhibited (19). Some transporter proteins are also capable of pumping against a concentration gradient, utilizing ATP. As will be discussed later, there is considerable evidence indicating that expression of the transporter proteins, and their insertion into the appropriate membrane, are responsive to nutritional and hormonal cues. This flexibility allows the placenta to adapt functionally, independent of structural changes. Transporter-mediated processes are responsible for the exchange of key nutrients such as glucose, amino acids, and fatty acids as outlined below. In addition, there are a variety of other transporter proteins localized to the apical surface of the syncytiotrophoblast in the human, including ones specific for micronutrients, such as copper, iron, and folate (49, 371).
a) glucose. Transport of glucose and related hexoses is dependent on the GLUT family of transporters that enable the sugar to pass down a concentration gradient at rates up to 10,000 times faster than possible by simple diffusion (169). Hence, it is commonly referred to as facilitated diffusion. The density of glucose transporter proteins is considerably greater on the apical surface of the syncytiotrophoblast of the human placenta than on the basal surface, which is thought to reflect the fact that much of the glucose taken up from the maternal circulation is used to meet the placenta's own considerable metabolic needs (141, 270). It is likely, therefore, that the density of the transporters on the basal surface represents the rate-limiting step for exchange. In the human, GLUT1 is the principal isoform involved in transport across the trophoblast, and protein levels in the apical membrane of the syncytiotrophoblast remain constant from 16 wk until term. In contrast, levels in the basal membrane double during the late second trimester (280), and this change may explain the increase in glucose transport seen towards term. GLUT1 in the placenta is insensitive to insulin.
GLUT3 is also present on the apical, but not the basal, membrane of the syncytiotrophoblast (67) and is the principal isoform on fetal capillary endothelial cells (245, 270). It has a higher affinity for glucose than GLUT1 and may be more important for transport during early pregnancy (67). In the murine placenta, GLUT1 has been immunolocalized at the ultrastructural level to the apical surface of layer II of the syncytiotrophoblast and the basal surface of layer III (409), suggesting these layers may operate in terms of glucose transport as one functional unit. Both GLUT1 and GLUT3 are expressed in the sheep placenta, but in different layers of the interhemal membrane (95, 582). GLUT1 is localized to the basal surfaces of the maternal-fetal synepithelium and the fetal trophoblast, while GLUT3 is present on the apical surface of the trophoblast. Therefore, a glucose molecule must interact with the two isoforms sequentially to transit between the circulations. Expression of GLUT1 and GLUT3 increases across gestation in the sheep, but the ratio alters with GLUT3 becoming more predominant towards term (165). The implications for glucose transport are not obvious, but clearly caution needs to be exercised when extrapolating data across species (270).
b) amino acids. Amino acid transport across the placenta is a key determinant of fetal growth as it provides the essentials for protein synthesis. Single amino acids diffuse slowly across cell membranes, and most uptake is mediated by a large family of transporter proteins. Amino acid transporters can be classified according to their properties, for example, whether they are sodium-coupled or not, and whether they convey neutral, cationic, or aromatic amino acids (19, 113). Alternatively, on a more functional basis, they fall into three broad categories: accumulative, exchange, and facilitative that interact to modulate net transfer across the placenta against a concentration gradient (335). Accumulative transporters are present on both the apical and basal cell membranes of the trophoblast and mediate the uptake of amino acids driven by the intra/extracellular electrochemical gradient previously described. These transporters generate a pool of amino acids within the trophoblast that drives the activity of other transporters. Efflux from the basal membrane is performed by facilitative transporters, and the rate is determined principally by the concentration gradient across the membrane. The gradient for specific amino acids is modulated by the action of exchange transporters, which, as their name suggests, exchange an amino acid of one type in the intracellular pool generated by the accumulative transporters for an amino acid of another type. Hence, interaction between the three groups of transporters is required to effect transfer, and the net flux per unit area will be dependent on the density of the transporter proteins in the apical and basal membranes, the metabolic and anabolic demands of the intervening trophoblastic cytoplasm, and the rate of blood flow in the two circulations.
c) lipids. Lipids are essential for the formation of cell membranes and may be an important fuel for fetal growth, especially among Asian Indians (319). Triglycerides cannot cross the placenta, but may be conveyed in lipoproteins. A number of binding sites for lipoproteins have been identified on the apical and basal membranes of the syncytiotrophoblast, including those for very-low-density (VLDL-R) (580), low-density (LDL-R) (179), and high-density lipoproteins (HDL-R) (9). The scavenger receptors SR-B1 and CLA-1 that bind LDL and HDL, respectively, are also present (179, 322). Expression of the mRNAs encoding the VLDL-R and LDL-R increases across gestation (402, 580), but is suppressed at term in pregnancies complicated by preeclampsia and severe growth restriction (402, 552). The impact of these changes is uncertain, as is, indeed, the contribution of lipoprotein uptake to overall lipid transport. However, placental lipoprotein uptake represents an important step in the maternal-fetal transfer of cholesterol (584).
Alternatively, triglycerides can be converted into free fatty acids (FFAs) by the actions of lipases. Endothelial lipase and lipoprotein lipase have been immunolocalized to the apical membrane of the syncytiotrophoblast during the first trimester, although only the former is seen at term (208). The mRNA encoding endothelial lipase is notably lower in growth-restricted placentas compared with normal counterparts (208), but the significance of this finding for transfer of FFAs is not known. The mechanisms underlying transport of FFAs across the placenta are not fully understood, but at least three membrane systems have been implicated that may act in concert, in addition to simple diffusion. A family of fatty acid transport proteins (FATP 1-6) has been identified in plasma membranes of the human placenta (162). These are particularly important for transfer of medium to long-chain fatty acids. Targeted deletion of FAT-4 results in embryonic lethality, but little is known regarding the specificity of the different transporters. There is also a fatty acid binding protein (FABPpm) located in the apical membrane of the syncytiotrophoblast that appears to preferentially bind and transport long-chain polyunsaturated fatty acids. Finally, fatty acid translocase (FAT/CD36) is present in both the apical and basal membranes of the syncytiotrophoblast. Expression of these transporters is responsive to nutrient availability through fatty acid activated transcription factors (PPARs, LXR, PXR, and SREBP-1) (162) and is also influenced by maternal obesity (156).
Computational modeling has suggested that transport of fatty acids across the placenta is modulated by the presence of an intracellular metabolic pool (438), which had been assumed to be within the syncytiotrophoblast. However, recent data derived from placental explants demonstrate that esterification of long-chain fatty acids and their incorporation into lipid droplets occurs within the cytotrophoblast cells, and not the syncytium (314). Further research into the role of the cytotrophoblast cells in lipid transfer to the fetus is therefore clearly required.
3. Endocytosis/exocytosis
Endocytosis/exocytosis is the final mechanism for transplacental transport (Figure 2). Immunoglubulin G (IgG), other large proteins, and cholesterol are considered to be transported by this route. Early studies suggested IgG binds to the apical membrane of the syncytiotrophoblast surface and then concentrates in clathrin-coated pits. However, further work has indicated that IgG is internalized initially through nonspecific endocytosis, and delivered, along with other proteins, to early endosomes (486). In the acidic microenvironment, IgG binds to the neonatal Fc receptor, FcRn, which routes it for transcytosis and exocytosis at the basal membrane. There, the more neutral pH of the interstitial fluid favors release of the IgG, promoting transport into the fetal circulation. In this way, a proportion of the IgG internalized is protected from lysosomal degradation, and specificity of transport of Ig subclasses is conferred.
Endocytosis of macro- and micronutrients is particularly important in the yolk sac of rodents during the period of early organogenesis (18, 45, 617). The multifunctional endocytic receptors megalin and cubilin have been immunolocalized to the visceral endoderm layer of the rodent yolk sac (13, 186), and potential ligands include folic acid, retinoic acid, vitamins B12 and D, cholesterol, insulin, and aminoglycosides (110). Targeted disruption of these receptors leads to failure of somite formation, indicating their key role in supporting early embryogenesis (506). Endocytic uptake of maternal proteins has been described in the human syncytiotrophoblast (325, 571) and is particularly prominent during the first trimester when maternal glycoproteins secreted by the endometrial glands, such as MUC-1 and glycodelin, are engulfed (83). A large proportion of the endosomes colocalize immunohistochemically with lysosomes (83), but some maternal glycodelin crosses the placenta intact and accumulates in the amniotic fluid (296). Megalin and cubilin are expressed in the syncytiotrophoblast and are also present in the yolk sac, raising the possibility that it too may play a role in nutrient exchange during the earliest stages of human pregnancy (72).
B. Endocrine Functions
The importance of the placenta's endocrine role is reflected in the fact that many of the large-placenta-specific gene families arising during evolution through gene duplication encode hormones (450). A wide array of hormones is secreted from the placenta with major impacts on maternal physiology, ranging from suppression of reproductive cycles to mobilization of nutrient resources. The evolution and function of the principal placental hormones was reviewed by Carter (95).
In the earliest stages of pregnancy, the most important function is to signal the presence of the conceptus to the mother, and prevent onset of the next ovarian cycle. In the human, chorionic gonadotropin (hCG) secreted by the syncytiotrophoblast acts via luteinizing hormone receptors to maintain progesterone output from the corpus luteum. In the sheep, secretion of interferon τ by the conceptus blocks endometrial production of the luteolytic prostaglandin F2α, and so establishes pregnancy. Continuing high levels of progesterone keep the myometrium in a quiescent state, and in the human prevent menstruation.
There is strong evidence in the sheep and other domestic species that interferon τ performs additional functions, combining with placental lactogens secreted by the trophoblast to upregulate the expression of genes encoding uterine milk proteins and growth factors in the endometrial glands (514). This signaling loop represents a mechanism by which the trophectoderm is able to enhance the nutrient supply to the conceptus, and stimulate early development of the placenta. Circumstantial evidence suggests that an equivalent mechanism may operate in the human based on hCG and placental lactogen from the trophoblast (76), but details of the pathways involved are not available as yet.
Progesterone also stimulates maternal appetite during early pregnancy, as does human placental lactogen (hPL), enabling the deposition of maternal adipose energy reserves that can be utilized later in pregnancy and during lactation (414). This build-up is facilitated by the development of leptin resistance (321), which prevents the negative feedback on appetite centers in the hypothalamus that would normally occur as leptin levels rise with fat accumulation. Evidence from rodent models suggests this central resistance may be mediated by placental lactogens. Deposition of fat reserves is also facilitated by increased levels of insulin secretion following stimulation of pancreatic β cell proliferation by placental lactogens in early pregnancy.
Later in pregnancy, a state of insulin resistance develops in the peripheral maternal tissues, mediated, in part, through the actions of placental growth hormone (23). There is also an accompanying rise in circulating triglycerides and FFAs. This may serve to enhance nutrient transfer to the fetus by elevating the concentration gradients across the villous membrane, particularly after meals. The placenta may further stimulate its own development by the action of placental growth hormone on the secretion of insulin-like growth factor I (IGF-I) by the maternal liver. IGF-I is a powerful mitogen that increases placental cell proliferation and increases maternal blood flow to the organ (196, 494).
Finally, it is important to note that the placenta secretes a number of hormones that are traditionally associated with the hypoxic kidney, including erythropoietin, angiotensin II, and adrenomedullin (123, 357). Erythropoietin, in particular, is synthesized at rates far higher than the fetal kidney and may mediate both classical hematopoietic responses to hypoxia and nonclassical changes, including increased placental vascularity and defense against oxidative stress (534).
C. Protective Functions of the Placenta
As well as facilitating the transport of nutrients to the fetus, the placenta plays an equally important role in minimizing xenobiotics, inorganic toxins, pathogens, and also maternal hormones from reaching the fetus. It therefore acts as a selective barrier to create an internal milieu in which the fetus, and in particular its endocrine systems, can develop independently. Nonetheless, perturbations of this function due to mechanical damage, polymorphisms (267), or environmental factors (287) may lead to increased fetal exposure. A range of drugs and toxins are well known to disrupt normal development and mediate teratogenesis, and one might speculate that lower doses, insufficient to cause malformations, may play a role in programming.
1. A physical barrier
The syncytiotrophoblast is often cited as a physical barrier, impeding the entry of pathogens and maternal immune cells into the fetal compartment. While this is true, defects in the surface are seen in all pregnancies and represent potential portals of entry. These defects, usually 10–20 μm in diameter, can arise through physical interactions between neighboring villi, or the rupture of syncytial bridges that form between terminal villi (73). Abnormal hemodynamics within the intervillous space as a result of deficient conversion of the spiral arteries may also cause damage to the syncytium (265). Defects in the villous surface stimulate activation of maternal platelets and deposition of fibrin (82). These deposits, which are seen in all pregnancies (367), have been demonstrated to be permeable to creatinine and so may represent sites of paracellular transport through the syncytiotrophoblast (68). They are also potential portals for infectious agents; indeed, incubation of placental villi with Listeria in vitro revealed that the bacteria are only able to penetrate at sites where the syncytiotrophoblast is damaged or absent (465). Despite these defects, the majority of pathogens and parasites do not cross the placenta, most likely due to the large number of marcophages within the villous stroma. These are actively phagocytic, and generally only those pathogens that can survive within the macrophages are associated with vertical transmission in utero (345, 346). Infection of the fetus can lead to growth restriction (3), and hence developmental programming.
2. Efflux transporters
Efflux transporters, such as members of the multidrug resistance protein family, the breast cancer resistance protein, P-glycoprotein, organic anion (OAT and OATP) and cation (OCTN) transporters, and the norepinephrine and serotonin transporters are present on the apical and basal surfaces of the syncytiotrophoblast and the fetal endothelial cells in the human placenta (20, 407, 500, 540). These transporters aid the efflux of a broad range of anionic and cationic organic compounds, and are thought to provide protection to the fetus from maternally administered drugs and exposure to environmental chemicals. The mRNA and protein levels of P-glycoprotein reduce across gestation, suggesting the fetus may be more exposed to toxic insults later in pregnancy (519).
Assessing the efficacy of these mechanisms in preventing placental transfer is difficult in the human, and in the clinical setting is limited to correlative studies. Thus, during the first trimester, the teratogenic effects of drugs that are targets of P-glycoprotein are greater if they are administered in combination with other P-glycoproteins substrates than by themselves, suggesting competitive interactions at the level of the transporter (140). Other studies have compared maternal and fetal blood levels at the time of delivery; for example, levels of dioxins in the fetal circulation were found to be approximately half those in the mother (536). Experimentation is obviously possible in animal models, but species differences in the expression of efflux transporters raise questions as to the applicability of the resultant data to the human (406).
Dual-perfusion of the delivered placenta provides an experimental system, albeit technically challenging, in which to explore transfer of drugs and toxins (266, 408). Comparison of the data with maternal-fetal in vivo measurements has validated transfer for ∼50 drugs (266). In addition, the system is manipulable; for example, inhibition of G-glycoprotein increases transfer of the antiretroviral drugs lopinavir and retinavir to the fetal perfusate, confirming its role as an efflux transporter (100).
3. Enzymatic defenses
A range of defensive enzymes capable of detoxifying xenobiotics and drugs is present within the syncytioplasm. This includes cytochrome P-450 enzymes, alcohol dehydrogenase, and glutathione transferase (407). These enzymes provide a measure of defense against agents such alcohol and components of cigarette smoke, but can be overwhelmed, as evidenced by the occurrence of fetal alcohol syndrome. Also present is the enzyme 11β-hydroxysteroid dehydrogenase 2 (11β-HSD2) that catalyzes the conversion of maternal cortisol to its inactive metabolite cortisone. Glucocorticoids are powerful inhibitors of cell proliferation for most fetal organs, except the heart and kidney, and the importance of this enzyme for normal development is demonstrated by the fact that there is a significant correlation between its activity in the human placenta and birth weight (497, 518). In addition, deletion of the 11β-HSD2 gene in mice is associated with fetal growth restriction (261). The amount of cortisol reaching the fetus will be dependent both on maternal circulating levels and the activity of placental 11β-HSD2. Maternal concentrations are elevated in response to stress, which may be emotional (300), induced by undernutrition (495), or the result of thermal (491) and other adverse stimuli. Equally, the expression and activity of placental 11β-HSD2 are influenced by a number of factors, including intrauterine growth restriction (221), the sex of the fetus (132), hypoxia (6), heavy metals such as cadmium that are present in tobacco smoke (591), and MAPK stress response pathways (498). Resultant exposure to elevated levels of cortisol may contribute to developmental programming of the fetal hypothalamic-pituitary-adrenal axis and other organ systems (127, 129, 306).
D. Sexual Dimorphism
The placenta is of the same genotype of the fetus, and there is increasing evidence that sexual dimorphism in terms of its gene expression may modulate its responses to environmental stimuli, and so influence the likelihood of fetal developmental programming. Placentas associated with female fetuses tend to have higher expression of genes involved in immune regulation, endocrine functions, and placental growth (71, 512), whilst those from males have more inflammatory profiles (136). These observations have led to the suggestion that females invest more resources in building the placenta, while males invest more in fetal growth and consequently may have less placental reserve capacity under adverse conditions (71). The situation is made more complex by the finding that sex-dependent expression patterns vary among the tissue types comprising the placenta, with differences being observed among purified isolates of syncytiotrophoblast, cytotrophoblast cells, and arterial and venous endothelial cells (136).
Nonetheless, sex-dependent differences in gene expression are likely to underlie the contrasting placental responses observed following exposure to high-fat/low-fat diets (202, 356), glucocorticoids (132), or hypoxia (133, 363) in mice. Similarly, lower levels of mRNAs encoding key enzymes regulating glucocorticoid transfer, including 11β-HSD2, were found in female placentas from women suffering anxiety or depression compared with male counterparts (381). Hence, female fetuses may be exposed to higher levels of maternal stress hormones in these cases, but as yet no data on protein levels or enzyme activity are available to confirm this suggestion.
At present, there are few details of the molecular mechanisms involved, but clearly the genetic sex plays an important role in determining the placenta's responses to environmental insults, and hence how it transduces these to the fetus. The impact of the sex of the placenta on its various functions is an important area for future research and may explain some of the sex-specific aspects of fetal developmental programming (63, 203, 472, 522).
IV. PLACENTAL STRUCTURE AND DEVELOPMENT
While the functions of the placenta are common across all species, its structure is the most varied of any organ. Although major differences exist among species in terms of the gross shape of the placenta, the most striking difference is in the degree of invasion by derivatives of the fetal chorion into the maternal tissues. This varies from no invasion in the epitheliochorial placenta of ruminants, equids, and suids, in which the trophoblast simply abuts the uterine epithelium, through the partially invasive endotheliochorial placenta of carnivores, to the fully invasive hemochorial placenta of the human and rodents where the trophoblast is bathed by maternal blood (583). The reduction in the number of tissue layers constituting the interhemal membrane as a result of increased invasion was considered for many years to represent an evolutionary progression. Molecular phylogenetic data have, however, overturned this view. It is now appreciated that the noninvasive epitheliochorial placenta is a derived form that arose by convergent evolution in different orders, and that the ancestral mammal was most likely a shrewlike creature with an invasive hemochorial placenta (96, 98, 171, 572). Epitheliochorial placentation avoids many of the immunological and hemodynamic problems associated with the invasive forms that underlie complications of human pregnancy, such as preeclampsia, and these may have operated as selective pressures over the millennia (131, 170, 231).
Placentas also vary in the degree of interdigitation at the maternal-fetal interface, which impacts on the surface area for exchange. Patterns vary from the folded type, characteristic of pigs where there are poorly branched ridgelike folds, through the more complex villous type, seen in the human and ruminants, to the labyrinthine type of rodents where intricate networks of maternal and fetal vascular channels permeate a block of trophoblast tissue (583). Comparative studies have demonstrated that species with a labyrinthine placenta have gestation lengths less than half those associated with a villous or folded placenta, although there are no relationships with birth weight or brain size (91, 92). Hence, the labyrinthine placenta is capable of delivering nutrients at a faster rate, which may be traded-off against gestational length to prevent maternal depletion. Short gestations are presumed to have a selective advantage in environments with marked seasonal changes in food availability.
Hence, the form of placentation needs to be considered in the context of the reproductive strategy of the species concerned and the environment and habitat that it lives within, for all forms are equally successful in supporting the development of live offspring. Nonetheless, extreme care needs to be taken when extrapolating data from one species to another. Extensive descriptions of different placental types are available elsewhere (395, 449, 583), and here we restrict our consideration to the human placenta and that of the two main animal species used in research into developmental programming, the mouse and the sheep. The mouse is favored because of the ease of genetic manipulations, which enable, for example, imbalances to be created among maternal supply, placental size, and fetal demands (482). Furthermore, placental transport capacity can be assayed in vivo (502), and assessed in relation to the maternal and fetal blood flows monitored using high-resolution ultrasound (399). While ultrasound permits longitudinal assessment of placental and fetal development, the small size of the mouse prohibits repeated blood sampling, which represents a significant limitation for metabolic studies. In contrast, the sheep offers opportunities for extended experimentation in conscious, ambulant animals through chronic catheterization of the maternal and fetal circulations. The neonate is also of approximately the same size as that of the human, and born at a similar degree of maturation.
Although research has been performed on other species, including the rat, rabbit, guinea pig, pig, horse, and non-human primates (94, 520), the data are limited in comparison. There is no perfect model of human placentation, except for the great apes in which experimentation is ethically unacceptable. Hence, one has to select the species most suitable for the question being addressed, giving consideration to factors such as the number of offspring, the histology of the interhemal membrane, the length of gestation, and the relative mass of the conceptus to that of the mother at term.
A. The Human Placenta
1. The mature placenta
The mature human placenta is usually a circular or oval disc ∼22 cm in diameter (435, 478). The disc is bounded on the fetal surface by the chorionic plate to which the umbilical cord is attached, and over which the branches and tributaries of the umbilical vessels radiate. The branching pattern of the chorionic arteries may be monopodial or dichotomous and varies depending on the site of insertion of the umbilical cord. The first two to three generations are always dichotomous, and thereafter are mostly monopodial if the cord insertion is marginal and dichotomous if it is central (223). Computational models indicate that energy losses are small in monopodial branching, and this may be beneficial when perfusing placental territory over a long distance (224). Conversely, dichotomous branching is more efficient in distributing blood over large areas near the bifurcation. On the maternal surface is the basal plate that abuts the decidua, and this is divided into a number of lobes by septa that are directed towards, but do not reach, the chorionic plate. Hence, the placenta is divided into a variable number of compartments, and this arrangement may assist in directing the flow of maternal blood (49). Lobes are alternatively known as cotyledons, but we prefer to use the former term to avoid confusion with the ovine placenta.
Internally, it comprises a series of highly branched villus trees that in total contribute a surface area for exchange of 12–14 m2 (75). Each tree arises via a stem villus from the chorionic plate and forms a lobule that is centered over the opening of a maternal spiral artery through the basal plate, so constituting an individual maternal-fetal exchange unit (Figure 3A). There may be one or more lobule(s) per lobe. While some villi, the anchoring villi, extend between the two plates, the majority are free-floating within the cavity of the placenta, the intervillous space. The finest branches of the villus tree, the terminal villi, are highly vascularized with fetal capillaries. Dilations of the capillaries, referred to as sinusoids, bring the endothelium into close apposition with the overlying syncytiotrophoblast, which is often locally thinned to form a vasculosyncytial membrane. Consequently, the diffusion distance between the two circulations may be reduced to 1–2 μm at these sites, aiding diffusional exchange (Figure 3B).
FIGURE 3.
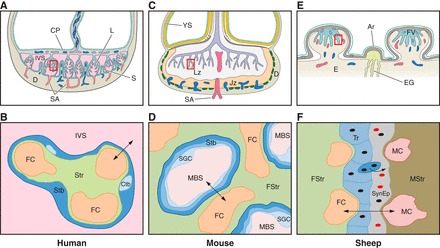
Diagrammatic representation of the gross morphology of the placenta and the histology of the interhemal membrane in the human, mouse, and sheep. In each case, the bottom panel represents detail of the area outlined by the square in the top panel. A: in the human, the fetal villi arise as a series of lobules (L) from the chorionic plate (CP). The basal plate abutting the maternal decidua (D) is thrown into a series of folds forming septae (S) that partially compartmentalize the placenta into lobes. Each lobe may contain one or more lobules. Maternal blood enters the intervillous space (IVS) from the spiral arteries (SA), passes between the villi, and drains into the openings of the uterine veins on the septae. B: a single layer of syncytiotrophoblast (Stb) covers each villus and is generated from underlying cytotrophoblast (Ctb) cells. It is bathed by maternal blood in the IVS from the start of the 2nd trimester onwards. Fetal capillaries (FC) within the stromal core (Str) invaginate to reduce the length of the diffusion pathway (arrowed). C: the mouse placenta is divided into an exchange labyrinth zone (Lz) and an endocrine junctional zone (Jz). The visceral endoderm layer of the inverted yolk sac (YS) is exposed to the decidua (D) after the outer parietal layer breaks down (dotted line). This represents an important route of nutrient exchange during early pregnancy and may continue until term. D: in the labyrinth, the syncytiotrophoblast (Stb) is two-layered, and an additional layer of sinusoidal giant cells (SGC) lines the maternal blood spaces (MBS). Little stromal tissue (Str) is interposed between the fetal capillaries (FC) and the trophoblast. E: in sheep, fetal villi (FV) interdigitate with maternal crypts within specialized areas of the endometrium (E), the caruncles, to form placentomes. In between placentomes, the trophoblast forms areolae (Ar) opposite the openings of the endometrial glands (EG). Histotroph from the glands is taken up by the trophoblast, representing another route for maternal-fetal transfer. F: within a placentome, there are six tissue layers interposed between the maternal (MC) and fetal (FC) capillaries: the maternal endothelium, maternal stromal tissue (MStr), the uterine epithelium which is converted into a synepithelium by the migration and fusion of fetal binucleate cells, the trophoblast (Tr), the fetal stroma (FStr), and the fetal endothelial cells. Differences in the nature of the interhemal interface mean that extrapolation of transport data from one species to another may not always be justified.
The syncytiotrophoblast forms the epithelial covering of the villus tree, and during the second and third trimesters and is bathed directly with the maternal blood circulating in the intervillous space (Figure 3B). Hence, the human placenta is described as being of the hemochorial type (49). The syncytiotrophoblast is a terminally differentiated, multinucleated syncytium. The apical surface bears numerous microvilli, amplifying the surface area for receptor-mediated transport by a factor of ∼7 (302). A wide variety of receptors have been localized to the microvillous surface, and their activity is responsive to maternal nutrition (204). Coated pits are observed at the base of the microvilli for endocytic transport (291, 420).
The syncytioplasm is dense with organelles, including rough endoplasmic reticulum and mitochondria, reflecting its high synthetic and metabolic activity. Hence, the tissue is vulnerable to oxidative and endoplasmic reticulum stress, which if not resolved leads to activation of the unfolded protein response. These stresses may impact severely on its endocrine and transport functions and are associated with growth restriction and other complications (85, 405, 596). In this respect, comparisons can be drawn between the syncytiotrophoblast and other endocrine-active cells, for example, pancreatic β cells (21).
The basal surface of the syncytiotrophoblast contacts either the progenitor unicellular cytotrophoblast cells, or the trophoblastic basement membrane. In early pregnancy, the cytotrophoblast cells form a complete layer, so nutrients must either pass through the cells or through the narrow intercellular clefts. Towards term these cells become more dispersed, with studies finding that they occupy 44% (290) or up to 90% (392) of the basement membrane, creating larger gaps for potential paracellular transport.
The fetal capillaries lie within the stromal core, often closely approximated to the trophoblastic basement membrane. They form the third layer of the interhemal membrane and potentially play an important role in regulating maternal-fetal transport (168, 185). The endothelial cells are of the nonfenestrated type and are connected by both tight and adherens junctions (339). The composition of these complexes differs with gestational age, and with their location in the villus tree. During the first trimester, the tight junctions lack occludin and claudin-1 and -2, suggesting that they are still plastic and highly permeable (329). This arrangement persists in later pregnancy within the terminal villi (330), pointing again to the importance of these villi for exchange. Numerous caveolae are present within the cytoplasm of the endothelial cells, which may play a role in transcellular endocytic transport. The GLUT3 transporter protein has also been immunolocalized to the endothelial cells (245), as has the multidrug resistance protein (540), tocopherol transfer protein (400), and phospholipid transfer protein (487). However, no data are yet available describing the importance of the different transcellular and paracellular pathways for the transfer of specific types of nutrients. In a coculture model of the interhemal membrane, the endothelial layer was found to offer greater resistance to the transport of glucose than the trophoblast layer (333). While a step forward, these results cannot necessarily be extrapolated to the in vivo condition as the unicellular HTR8 trophoblast cell line was used, which may not reflect the same transport properties as the syncytiotrophoblast. Insulin receptors are detectable on the cell surface from the start of the second trimester onwards (143) and may regulate villus angiogenesis in metabolic disorders (327).
2. Development
The human placenta undergoes major transformations in its structure during pregnancy, and it is important to be aware of these changes when considering the impact of environmental insults on fetal-placental development.
Placental development begins with differentiation of the trophoblast lineage at the morula stage, and there is evidence of plasticity at this early stage. For example, embryos derived from oocytes retrieved from women with a body mass index greater than 25 kg/m2 develop faster than those from lean women, and have fewer trophectoderm cells (331). The embryos also display differences in metabolism, with reduced glucose consumption and altered amino acid usage. Mammalian zygotes do not form functional gap junctions until around the 8-cell stage, so the individual cells behave metabolically in an autonomous fashion (62). It has been speculated that this lack of cell-cell communication heightens sensitivity to stressors. Thus the zygote may be affected by environmental cues transduced through the oviductal secretions during its passage into the uterus. Equally, it may be influenced by the culture conditions during assisted reproduction techniques (ART), which can have significant effects on birth weight (160). The impact of ART on pregnancy outcomes (508), and cardiovascular health (427), has recently been reviewed, but few data relating to placental development are available. A large study of over 500,000 births showed that placentas arising from ART are heavier than those from natural conceptions (233). The placental-fetal weight ratio is also increased in ART pregnancies, and this relationship is independent of the technique employed, the method of delivery and other potential confounders. However, the mechanism underpinning the effect, and the timing at which it operates, are still unknown.
The first morphological evidence of placental development is seen at implantation, which occurs around day 7 postfertilization. On attachment to the uterine epithelium, the trophectodermal cells differentiate and fuse to form the syncytiotrophoblast. Projections from the latter penetrate between the epithelial cells and into the underlying stroma so that the zygote is completely embedded within the superficial endometrium by day 11. The syncytiotrophoblast expands through the proliferation and fusion of underlying cytotrophoblast cells and surrounds the entire surface of the original blastocyst. As it expands, the syncytiotrophoblast erodes into dilated capillaries within the endometrium, and also the apical parts of the endometrial glands. As a result, maternal erythrocytes and gland secretions enter into spaces that form within the syncytiotrophoblastic mantle, the forerunners of the intervillous space (49). Development of the placenta is precocious, but the factors stimulating and regulating this rapid development are poorly understood, principally through the difficulty of obtaining suitable specimens. However, it is now accepted that during the first trimester the conceptus is supported by histotrophic secretions from the endometrial glands, the “uterine milk” (80, 83).
The full composition of the endometrial secretions during pregnancy is not yet known, but evidence from proteomic analysis during the secretory phase of the nonpregnant cycle indicate that they likely contain large glycoproteins, including MUC-1, glycodelin-A, and uteroglobin, as well as carbohydrates and lipids (46, 50, 238). These secretions are phagocytosed by the syncytiotrophoblast (83, 254), and maternal proteins and amino acids accumulate in the coelomic fluid inside the placental sac (282). From there, they may be transported to the embryo via the secondary yolk sac, which floats within the coelom. The yolk sac is the first of the extraembryonic membranes to be vascularized, and abnormalities in its development are associated with early pregnancy loss (416). Recent immunohistochemical studies have located transporter proteins, such as GLUT1, folate receptor-α, and tocopherol transfer protein, to the outer mesothelial surface (49, 281, 285), but no data are available regarding the yolk sac's functional capacity for uptake in vivo. However, deficiency in the transport of retinol and other key signaling molecules, potentially involving the yolk sac, has been implicated in the causation of major embryonic defects seen in chromosomally normal spontaneous miscarriages (440).
The glandular epithelial cells are also immunopositive during early pregnancy for an array of powerful mitogens, such as epithelial growth factor (EGF) and vascular endothelial growth factor (VEGF) (254), so the secretions may play an important role in stimulating early development of the placenta. Indeed, evidence from animal species indicates that the conceptus promotes its own development by signaling to the glands through placental lactogens and upregulating expression of uterine milk proteins and growth factors (514). It is suspected, but not yet proven, that the same happens in the human, possibly augmented by prolactin secreted by the decidual cells (76, 80). The fact that the morphology of the glandular epithelial cells changes to a characteristic hypersecretory type, the Arias-Stella reaction, suggests this may be the case (17).
Taken together, these data indicate that the endometrium plays a greater role in stimulating and supporting placental development during early pregnancy than previously anticipated. The first trimester is a critical period for placental development, for expression of markers of trophoblast stemness decline rapidly after 12 wk of gestation (252), suggesting loss of proliferative potential. Perturbation of endometrial and, in particular, gland function may therefore have a profound effect on the ultimate growth of the villus trees and the surface area for exchange. Whether the secretome is altered in response to maternal nutritional status, obesity, or other conditions during early pregnancy is not known. Further research is necessary to test this hypothesis, and also to determine whether the endometrial glands may themselves be subjected to developmental programming. Ultrasound data indicate that the size of the uterus is reduced in girls born with low birth weight (268), but whether the density or activity of the glands are also compromised is not known. If so, this could represent a mechanism mediating intergenerational effects on birth weight.
Placental metabolism is heavily glycolytic in early pregnancy due to the prevailing low oxygen concentration (286). The phylogenetically old polyol pathways are highly active, avoiding excessive fermentation of glucose to lactate (284). Whether these pathways are more robust to environmental stressors than oxidative phosphorylation is not known, but it is notable that the placental ATP/ADP ratio is the same as in later pregnancy and that there is no evidence of hypoxic stress in early placental tissues (112).
The maternal arterial circulation to the placenta is established towards the end of the first trimester and is associated with transformation of the early placenta to its definitive form. Establishing the circulation requires invasion into, and remodeling of, the endometrial spiral arteries. This is performed by a subpopulation of migratory trophoblast cells, the extravillous trophoblast, which in normal pregnancies penetrate the underlying decidua and reach as far as the inner third of the myometrium. Remodeling of the spiral arteries involves the loss of smooth muscle cells and elastic tissue from their walls, and their replacement by fibrinoid material (441, 570). As a result, the vessels lose their vasoreactivity, and their terminal portions dilate as they approach the basal plate of the placenta. Together, these changes ensure a constant flow of maternal blood into the placenta at a low velocity and pressure (84). Failure of trophoblast invasion and arterial remodeling is associated with the “Great Obstetrical Syndromes,” including growth restriction, preeclampsia, and late spontaneous abortion, due to impaired maternal perfusion (64). Early in pregnancy the invading trophoblast cells plug the maternal spiral arteries, preventing flow of maternal blood into the placenta (264). Towards the end of the first trimester these plugs dislocate, leading to onset of the maternal arterial placental circulation and the switch from predominantly histotrophic to hemotrophic nutrition.
Events at this stage appear to play a major role in determining the final size and shape of the organ. Villi initially form over the whole of the chorionic sac, but at around 7–8 wk of gestation, those over the superficial pole begin to regress, leaving the smooth membranes or chorion laeve. This regression is linked with locally high levels of oxidative stress and apoptosis, for blood flow starts in the periphery of the early placenta and gradually extends to the central region (283). This pattern reflects the extent of extravillous trophoblast invasion and plugging of the maternal spiral arteries across the placental bed (65). In normal pregnancies, this centripetal regression results in an approximately discoid placenta with the umbilical cord near the center. However, we have speculated that if onset of the circulation is more erratic, possibly due to uneven trophoblast invasion, excessive villous regression may lead to small, abnormally shaped placentas with eccentrically inserted umbilical cords (77). Unfortunately, this hypothesis cannot be tested experimentally. However, the site of cord insertion identified by ultrasound at the end of the first trimester correlates closely with that observed at delivery, confirming the location is determined early in pregnancy (479, 489). Equally, placentas that are growth restricted at term are smaller than normal at the end of the first trimester (122, 235), whereas the converse is the case for macrosomic placentas (490).
An important question that has not been fully addressed is whether compensatory lateral growth of the placenta is possible in later pregnancy. There are three aspects of human placentation that are critical when considering this possibility. First, the conceptus is completely embedded in the uterine wall, so it is not just a question of the placenta expanding over the uterine surface. Any enlargement with respect to the uterus must be associated with erosion into the maternal tissues. Second, there must be recruitment of additional spiral arteries to supply any significant increase in territory. Recruitment is possible during the first trimester when there is an alternative source of nutrients from the endometrial glands, and a prolific supply of extravillous trophoblast cells from the cytotrophoblast columns to initially plug the arteries while remodeling takes place. However, that supply wanes during the second trimester as the columns become short and sparse, and the villi at the margin of the disc regress. Thus it is probable that the final complement of arteries is essentially fixed at the end of the first trimester. Third, the uterus obviously expands and remodels as pregnancy advances, and consequently, the relative position of the placental attachment within the uterus changes with gestational age. This is not achieved through migration or trophotropism as suggested by early investigators (594), but is principally due to the drawing out of the lower uterine segment (257, 404). Hence, while in early pregnancy the placenta grows faster than the uterus and the syncytiotrophoblast mantle expands within the superficial endometrium (130), it is likely that the placental footprint is established around the end of the first trimester when formation of the chorion laeve is complete. Thereafter, it has been suggested that the placenta and uterine wall expand together (229). Rough estimates based on the density of the spiral arteries in the nonpregnant uterus and their final disposition in the placental bed at term indicates that this may be the case. The arteries are initially 2–3 mm apart (41), but at term must be 10–20 mm apart based on the diameter of the lobules that each supplies (253). Thus the placental bed has expanded approximately fivefold, whereas the diameter of the placenta increases similarly from 5 cm at 11 wk to 22 cm at term (49). Whether all areas of the uterus expand equally or whether some areas, such as the fundus, expand preferentially is not known. However, differential expansion could explain why some placentas are circular and others elliptical dependent on the implantation site. Equally, it is not known whether the density of the spiral arteries is uniform in the uterine wall. If not, then the site of implantation may affect the ultimate blood supply to the placenta. This linkage provides a potential mechanism by which the shape of the placenta may be associated with its functional capacity and the ensuing phenotype of the offspring.
If the first trimester sees the establishment of the framework of the placenta, the second and third trimesters see an increase in its functional capacity, principally owing to the exponential increase in villous surface area created by the formation of terminal villi and a reduction in the maternal-fetal diffusion distance (274). It is notable that the theoretical diffusing capacity for oxygen expressed per kilogram of fetal weight remains constant across gestational age (364, 368), suggesting placental development determines the rate of fetal growth or that the two are closely co-regulated. Formation of terminal villi is believed to be driven through angiogenesis causing capillary loops to obtrude from the side of the containing villus (303). Hence, it is likely to be heavily influenced by the prevailing oxygen tension (312). The vascular network appears to be particularly plastic during the first trimester due to its low coverage with stabilizing pericytes at that time (607). Pericyte coverage is also reduced in placentas from pregnancies at high altitude, which may facilitate vascular adaptations to increase gaseous exchange, as will be discussed later.
B. The Murine Placenta
1. The mature placenta
The mouse has a single, discoid hemochorial placenta that in terms of its gross morphology is similar to that of the human. Internally, however, there are significant differences (210), the most major being that the placenta is divided into two morphologically and functionally distinct zones: the labyrinth zone that is responsible primarily for exchange and the junctional zone that serves an endocrine function (Figure 3C). The proportion of these two zones displays considerable plasticity, varying within a normal litter depending on the overall placental size and also following dietary and other manipulations (114, 118, 119).
The labyrinth zone is closest to the chorionic plate and consists of a dense meshwork of interconnecting lamellae of trophoblast. Within the lamellae are the fetal capillaries, whereas between them lie the maternal blood spaces (Figure 3D). The labyrinthine trophoblast comprises three layers. The outer layer is formed of uninucleate cells that in the past were referred to as cytotrophoblast cells. However, it is now recognized that they do not equate in progenitor terms to the cells of the same name in the human placenta, and their expression of genes encoding placental lactogen suggests they have an endocrine function (504). They display a large nucleus with evidence of limited endoreduplication (117), so are now classified as sinusoidal giant cells (503). Beneath these cells are two layers of syncytiotrophoblast that are closely approximated to each other and linked by extensive gap junctions (378, 409). This arrangement is often referred to as hemotrichorial, although as gestation advances the sinusoidal giant cells become perforated, allowing maternal blood access to the outer layer of syncytiotrophoblast (117). The extent to which the two syncytiotrophoblast layers function as one is also debatable, for the presence of the gap junctions will allow small molecules to pass easily between them. This is evidenced by the fact that GLUT1 glucose transporter proteins are only immunolocalized to the apical surface of layer II and the basal surface of layer III, with none being located at the interface between the two layers (409). They are also not present on the layer I, the sinusoidal giant cells. Hence, the arrangement in the mouse may be more analogous to the single layer of trophoblast in the human than previously anticipated. These proteins, and a variety of amino acid transporters, appear responsive to maternal nutrition and genetic manipulations of the placental to fetal size ratio (14, 204, 566). The trophoblast layers rest on a basement membrane to which the fetal capillaries are closely apposed on the other side, with no intervening stromal cells. Unlike the human, the murine syncytiotrophoblast has no endocrine function (355).
The junctional zone, in contrast, does not contain fetal blood vessels and is only traversed by the maternal spiral artery delivering blood to the labyrinth and venous channels conveying maternal blood back to the uterine veins. It is composed of two principal cell types, spongiotrophoblast cells and glycogen cells, and the proportion of these changes with gestational age. Glycogen cells are sparse before E14.5, but numbers then expand before declining around E18.5 as they migrate into the decidua (115). As their name suggests, these cells accumulate large quantities of glycogen that may act as an energy reserve to be released when growth of the fetus is maximal. Spongiotrophoblast cells display large quantities of rough endoplasmic reticulum, suggesting a high secretory output. Many members of the placental lactogen family have been localized to these cells (355, 507), but the full range of their output is still unknown. These cells are more vulnerable to stress than the syncytiotrophoblast of the labyrinth, which may reflect a higher metabolic rate (599). The venous channels are lined by other types of polyploid trophoblast giant cells (4, 446, 503). These too have a potential endocrine function through the release of placental lactogens (504), raising the possibility that they may relay information to the mother concerning the composition of her blood following exchange with the fetus. Integration of signals from the sinusoidal giant cells and the giant cells lining the venous channels could thus provide an indicator of fetal demand.
In addition to the discoid chorioallantoic placenta, an inverted yolk sac placenta is functional in the mouse from early in pregnancy until term (583) (Figure 3C). The yolk sac is highly vascularized, and the visceral endodermal layer is exposed to the uterine lumen and any nutrients secreted by the endometrial glands. The apical surface of the cells resembles in many respects that of the syncytiotrophoblast in the human placenta. There is an abundance of microvilli and coated pits, and numerous absorptive droplets and vacuoles within the underlying cytoplasm (232). The absorptive function is reinforced by the presence of the multifunctional endocytic receptors megalin and cubilin that potentially transport a wide variety of vitamins and micronutrients (617). The large number of mitochondria and cisternae of rough endoplasmic reticulum suggest that the endodermal cells have a high metabolic rate. Experiments in the rat have revealed that more than 95% of amino acids transported during the period of organogenesis are derived from the uptake and subsequent breakdown of maternal proteins by the yolk sac (59, 344). Perturbation of yolk sac function can thus have profound effects on embryo development (455), so impact on yolk sac function is often targeted in screening of potential teratogens.
2. Development
Development of the placenta starts with differentiation of the trophectoderm lineage at around E3.5. This process shows considerable plasticity in response to environmental cues, such as maternal diet, that influence the ratio and number of trophectoderm and inner cell mass cells. A low-protein diet during the perimplantation period induces an increase in the total number of trophectoderm cells in the blastocyst, suggesting an early compensatory reaction (163). However, maintenance on such a diet throughout pregnancy results in reduced placental and pup weights, indicating that growth of the conceptus is ultimately constrained by the impoverished nutrient supply (120). Implantation commences at E4.5. At this time, the polar trophectoderm cells overlying the inner cell mass differentiate into two cell types: extraembryonic ectoderm and the ectoplacental cone. The remaining mural trophectoderm cells undergo limited proliferation before exiting the cell cycle and transforming into polyploid primary trophoblast giant cells (210, 503, 566). These mediate the initial invasion of the conceptus at the implantation site, and hence lie at the boundary between the mature placental disc and the decidua. They have an endocrine role, expressing several members of the prolactin/placental lactogen gene family, and are also thought to secrete angiogenic and vasodilatory factors (503).
It is likely that in rodents embryogenesis and early placental development take place in a low oxygen environment, as in the human, for at E6 the antimesometrial decidual cells form an avascular zone around the conceptus, separating it from the maternal blood (430). Nutrition at this time is histotrophic, absorbed principally through the visceral yolk sac. The yolk sac grows at a prolific rate and soon encapsulates the conceptus except for the region of the ectoplacental cone. Initially, the yolk sac comprises an outer avascular parietal layer formed from the primary trophoblast giant cells and endoderm, and an inner vascularized visceral layer. Nutrients must diffuse through the parietal layer to be absorbed by the visceral layer. Later in pregnancy the parietal layer breaks down with migration of the giant cells, exposing the visceral layer directly to the uterine epithelium and forming an “inverted” yolk sac (583).
The chorioallantoic placenta develops from both the ectoplacental cone and the extraembryonic ectoderm. The former gives rise to the spongiotrophoblast and glycogen cells of the junctional zone, and a second wave of giant cells (503). These secondary trophoblast giant cells invade along the lumens of the spiral arteries and are therefore analogous to the endovascular extravillous trophoblast of the human placenta. The extraembryonic ectoderm gives rise to the trophoblast forming the labyrinth. At E8.5 the allantois attaches to the expanding extraembryonic ectoderm, bringing in mesoderm from which the fetal vasculature differentiates. Allantoic attachment stimulates folding within the ectoderm layer, initiating the formation of the trabecular network of trophoblast and maternal blood spaces. The genes and transcriptional networks regulating placental development in the mouse have recently been extensively reviewed (511, 566).
The fetus becomes dependent on the chorioallantoic placenta from E10.5, and hence gene mutations that severely compromise placental function cause embryonic lethality at this time. The placenta undergoes rapid growth, with weight reaching a maximum around E16.5 and plateauing, or even declining, thereafter (116). In contrast, peak fetal growth is seen around E18.5. As in the human, it appears that trophoblast proliferative potential is limited to early pregnancy, for progenitor cells positive for EpCAM, a marker of stemness, are not detectable within the labyrinth after E14.5 (538). However, stereological analyses reveal that the labyrinth continues to expand in volume until E16.5 and more slowly thereafter (116). This enlargement is associated principally with an increase in the volume of the maternal blood spaces and fetal capillaries. While the surface area of the maternal blood spaces reaches a maximum at E16.5, that of the fetal capillaries continues to increase until term, allowing for the possibility of adaptations during late pregnancy. Continuing fetal placental angiogenesis is reflected in a progressive reduction in the thickness of the interhemal membrane, and consequently the theoretical diffusing capacity of the placenta rises until term (116). By the end of pregnancy, the conductance for oxygen in the murine placenta is approximately the same as in the mature human placenta (364).
In contrast, the volume of the junctional zone peaks at approximately E16.5 due to an increase in both the number and mean cell volume of the spongiotrophoblast and glycogen cells, and then declines (115). The decline in volume towards term reflects the migration of the glycogen cells into the decidua, but this cannot account for the whole change and there may be additional cell loss through apoptosis.
C. The Ovine Placenta
1. The mature placenta
Morphologically, the placenta of the sheep is very different from those of the human and the mouse, although there are many functional similarities. The ovine placenta is of the cotyledonary type, comprising ∼70 placentomes of 0.5–4.0 cm diameter in a singleton pregnancy (516). A placentome is formed when villous outgrowth creates a fetal cotyledon opposite a preexisting nonglandular specialization, a caruncle, in the wall of the uterus (Figure 3E). Thus placentomes are only formed at predetermined sites, and there is no villus regression as in the human. The fetal villi interdigitate with crypts in the maternal caruncle, and the complexity of branching increases with gestational age. Each cotyledon functions as an independent maternal-fetal exchange unit and is therefore analogous to a single lobule of the human placenta.
Histologically, the maternal-fetal interface is also different. The trophoblast covering the fetal villi remains unicellular, and the cells are linked at their apices by tight junctions to form a columnar epithelium. There is no invasion by the fetal tissues comparable to that seen in the human and murine placentas, and the interface is formed by a microvillar interdigitation with the maternal tissues (Figure 3F). The exception is the migration of binucleate cells that arise in the trophoblast layer just prior to implantation, and form 15–20% of the layer throughout gestation (583). These cells migrate across the interface and fuse with the uterine epithelial cells to form localized plaques of maternal-fetal syncytium that are interspersed amongst the otherwise unicellular uterine epithelium (583). The placental interface in the sheep is therefore referred to as synepitheliochorial. The binucleate cells contain large numbers of dense granules that are immunoreactive for ovine placental lactogen (581). Their migration appears to be a way of delivering this hormone, and possibly other effectors, into the maternal circulation, where it plays an important role in early pregnancy by stimulating activity of the endometrial glands and the secretion of uterine milk (415).
Dense capillary plexuses are present within both the fetal villi and the maternal crypts. The fetal capillaries display sinusoidal dilations, as in the human, which may serve to reduce the vascular resistance (234). Nutrients and respiratory gases thus have to pass through six tissue layers: the maternal endothelium, maternal stromal tissue, the maternal-fetal syncytium, the trophectoderm, fetal stromal tissue, and the fetal endothelium (Figure 3F). Diffusional exchange is facilitated by the invagination of the fetal capillaries into the trophectoderm, which along with the apposing syncytium is locally thinned, forming the equivalents of vasculosyncytial membranes in the human placenta. Exchange of glucose is aided by the presence of GLUT1 and GLUT3 that are expressed on different membranes (95, 582). Amino acid transporters have been characterized functionally in vivo, although not localized to individual cell layers (44).
In addition, there are two specialized accessory structures that contribute to nutrient transfer. First, in the center of each placentome is a hemophagous zone where maternal blood is released by limited degradation of the uterine tissues, sequestered, and then phagocytosed by the trophoblast cells (79). This is considered to be the principal pathway for the maternal-fetal transfer of iron. Since there is no evidence of circulation of maternal blood through these regions, they cannot be considered analogous to a hemochorial placenta. Second, openings of uterine glands are found clustered in the uterine wall between the placentomes. The trophoblast cells opposite are transformed from cuboidal to columnar and form small elevations known as areolae (Figure 3E). Histotrophic secretions from the glands are endocytosed by the trophoblast, representing a pathway for the transfer of large proteins. The areolae reach their maximum diameter of ∼3 mm in the last third of pregnancy, although this decreases considerably towards term, most likely due to diminishing activity of the glands (578).
2. Development
As in the human and mouse, maternal diet during the periconceptional period can influence both the number of cells in the blastocyst and also the ratio between the cell lineages (299). This sensitivity is further evidenced by the impact that embryo transfer or assisted reproductive technologies has on subsequent ovine placental development, particularly its vascularization and expression of sex steroid receptors (462). Implantation is relatively later in the sheep than in the other species, and there is no invasion of the maternal tissues. Consequently, the conceptus remains within the uterine lumen throughout the whole of pregnancy. After entering the uterus the blastocyst elongates rapidly, a response driven principally by the endometrial secretions. Their importance has been demonstrated by endocrine ablation of gland development in newborn lambs (227, 514, 515). Complete ablation results in a cessation of growth of the conceptus and loss of the pregnancy in the adult animal, whereas partial ablation leads to a small, nonexpanded conceptus that fails to attach. Following expansion, trophoblastic papillae project into the mouths of the endometrial glands and immobilize the conceptus. Villous development starts opposite caruncles around days 24–26 postfertilization, which corresponds to the timing of allantoic attachment with the chorion, as in the mouse (583). The number of cotyledons is fixed by ∼5–6 wk of pregnancy and does not increase thereafter. Each cotyledon does expand, however, reaching maximum weight at 80–90 days after which there is a decline due to loss of water and a reduction in the mesenchymal component of the fetal villi (516). Together, the caruncular and cotyledonary portions at each implantation site form 70–100 placentomes, which have been be classified into 4 different types on the basis of their gross morphology (545). The frequency distribution of the different placentome types changes with gestational age and suboptimal environmental conditions, although the functional significance of the different types and the mechanisms governing the shape changes remain unclear (198).
The surface area of the maternal-fetal interface increases in line with placental weight during gestation due to the increasing length and branching of the fetal villi. In addition, towards term the distal parts of the villi are thrown into folds that presumably generate further surface area for exchange, despite the decreasing placental weight (234). Vascularization of the villi appears to increase continuously throughout pregnancy, although it occurs at twice the rate on the fetal as on the maternal side (54). On the maternal side the changes are predominantly in capillary diameter rather than number, whereas on the fetal side there is more branching angiogenesis. Consequently, total fetal capillary surface area increases, favoring exchange, although mean diameter is reduced, as in the mouse (54, 116). These contrasting patterns reflect different expression of angiogenic factors in the maternal and fetal tissues (54). The increase in vascularization is matched by exponential rises in uterine and umbilical blood flows throughout pregnancy, which are of critical importance for fetal growth (461).
V. EPIDEMIOLOGICAL ASSOCIATIONS BETWEEN PLACENTAL PHENOTYPE AND ADULT DISEASE
Low birth weight usually implies inadequate placental function, either as a primary or secondary cause. In recent years, an increasing number of relationships have been discovered between placental phenotypic features, such as its weight, length, and width, and diseases in later life (Table 1). While correlation does not necessarily equate with causation, one possibility is that these gross morphological features are linked biologically to the functional capacity of the placenta. Alternatively, it may be that these features impose mechanical or other constraints on the developing embryo/fetus. Thus there is accumulating evidence that the vascular arrangement of the early embryo plays an important mechanical role in regulating gene expression in the developing heart. Cells in the common ventricle and the outflow tract of the embryonic heart are sensitive to wall and shear forces (342, 442), and increases in these forces can lead to heart defects (135, 260, 379). The heart of the human embryo begins beating around day 21 postfertilization. From then on, it is subject to the pulsatile pressures and flows generated by its own pumping action. At 6–8 wk, the heart has become four chambered, and the vitelline and allantoic circulations are increasingly perfused (Figure 4). These vascular beds offer mechanical resistance to blood flow, and there is increasing evidence that their inadequate growth promotes mechanical signals in the heart that result in structural defects (218, 260, 341, 379). We suspect that changes in the yolk sac and/or early placental vascular architecture underlie a broad spectrum of cardiovascular disorders, including a propensity for heart failure, but confirmatory data are not yet available.
Table 1.
Programmed conditions in the human associated with placental phenotype
| Condition | Phenotypic Association | Reference Nos. |
|---|---|---|
| Hypertension | Number of lobes/cotyledons | 24 |
| Low birth weight, large placenta | 25, 39, 541 | |
| Coronary heart disease | Very small or large placenta | 175, 361 |
| Heart failure | Small placental area | 175 |
| Sudden cardiac death | Placental thinness | 30 |
| Diabetes | Low birth weight, low placental weight | 236 |
| Large placenta-to-birth weight ratio | 28 | |
| Asthma | Placental length | 32 |
| Rheumatic heart disease | Long umbilical cord | 174, 222 |
| Lung cancer | Short mother, low ponderal index, large or small placenta | 38 |
| Hodgkin's lymphoma | Short placental length | 34 |
| Reduced life span | Short length, roundness, rapid childhood growth | 35 |
FIGURE 4.
The relationship between the vascular plexuses of the secondary yolk sac and the chorioallantoic placenta, and the developing heart. Because these two beds account for a substantial portion of the total vascular impedance to flow sensed by the embryonic heart, poor vascularity in these organs would offer an increased load to the heart, altering gene expression patterns and leading to congenital defects or a myocardium that is vulnerable for later disease. (Image is reprinted from www.netterimages.com, with permission.)
A. Placental Efficiency
While fetal weight generally correlates with placental weight, the efficiency of the placenta, defined as the amount of fetal body mass accumulated per gram of placenta, is a key indicator of the offspring's resilience and susceptibility to chronic disease in later life (576). This index is a simple one to calculate in epidemiological studies and encapsulates many different factors, such as placental exchange surface area, transporter density and activity, and blood flow rates, which would require more detailed individual stereological, molecular, or physiological analyses. However, while it provides an overview of placental function, changes in the index provide no insight into whether specific activities have been stimulated or compromised and, if so, the mechanisms involved. The physiological and endocrine regulation of placental efficiency has recently been reviewed (196). Efficiency, as estimated by this index, varies dramatically among species. The index is not related to the histological type of the placenta, but to the relative geometries of the maternal and fetal blood flows (137), emphasizing the importance of flow-limited transport. The human placenta is notably one of the least efficient on this basis, suggesting that other evolutionary selective pressures have been more important.
In the human, large placentas tend to be less efficient than smaller ones (384, 480), suggesting adaptations, such as changes in transporter expression, have been successful in the latter. The risk of cardiovascular disease in adult men in relation to the birth weight to placental weight ratio does not follow a linear fashion as one might expect, but rather a “U”-shaped pattern with heart-related death rates being lowest when placental weight is ∼19% of birth weight (Figure 5) (25, 217, 361). The explanation for this relationship is not intuitively obvious, for it might be assumed that the more weight achieved per gram of placenta, the better the fetal outcome. However, it is possible that very small placentas are symptomatic of a severely compromised and constraining maternal-fetal supply line and have a limited functional capacity (477). Large placentas, on the other hand, may imply that compensatory growth was stimulated by inadequate access to nutrients at key phases of development. This phenomenon is well known to farmers, who graze ewes on poor pasture after mating to stimulate placental growth, before placing them on good pasture during mid- to late-pregnancy. In doing so, they obtain larger lambs than ewes grazing on good pasture throughout (373). In the non-human primate, a reduction in placental mass can be compensated by expansion of the remaining placenta. However, this plasticity is lost as term approaches (466). When, and how, the human placenta is able to adapt to different nutritional conditions is poorly understood.
FIGURE 5.

Coronary heart disease mortality in 2,571 men born in Sheffield, UK, during 1907–1930 as a function of the placental-to-birth weight ratio expressed as a percentage. The lowest rates of death from heart disease were found among men where the placental weight was ∼19% of the newborn body weight (P = 0.03). [Adapted from Godfrey (217), with permission from Elsevier.]
There is considerable variation in placental efficiency across the birth weight range, as demonstrated by the relationship between 17,000 birth weight-placental weight pairs from deliveries in Saudi Arabia (11) (Figure 6). If the data are divided into quadrants based on the approximate median birth weight and placental weight then in the upper left quadrant, heavy babies were nourished by relatively light placentas, whereas in the lower right quadrant babies were light even though their placentas were at the heavier end of the scale. On the basis of the “U”-shaped pattern described above, one might speculate that people born in these quadrants may have higher than average risks for chronic conditions in later life (217). It would be interesting to know whether these extreme quadrants have different sex ratios. On average, boys have heavier birth weights per gram of placenta (173), and thus would be expected to be more numerous in the upper left quadrant. In contrast, girls tend to make larger placentas for any given birth weight and so may be more likely to populate the lower right quadrant. This remains to be tested.
FIGURE 6.

Birth and placental weights of 17,000 live births in Unizah, Saudi Arabia. The points in the top left box represent relatively low placental weights associated with relatively large babies, which have been defined as efficient placentas. The bottom right box shows low efficiency placentas where large placentas nourished low-birth-weight babies. These two extremes of efficiency may represent different kinds of programming. [Adapted from Alwasel et al. (11), with permission from Elsevier.]
There is increasing evidence that placental efficiency changes over time in any given population and is likely to be influenced by the nutritional environment. The data from Saudi Arabia mentioned above showed that placental efficiency decreased significantly over a decade (11). This change was due solely to an increase in placental weight, which rose by more than 100 g without a concomitant increase in birth weight. Studies in Mysore, India, suggest that maternal head circumference in conjunction with maternal fat mass predict placental efficiency (579). The former is related to the mother's early life nutrition, whereas the latter reflects levels of nutrition during adulthood. This finding suggests that the nutritional conditions across the life of a woman are highly influential in the establishment and growth of the placenta, and thus impact the lifelong health risks of her offspring. However, it has not been determined whether the optimal efficiency associated with low cardiovascular death rates originally found in the United Kingdom population (Figure 5) applies to populations in Saudi Arabia and elsewhere.
B. Placental Shape and Its Influence on Fetal Development
The human placenta is generally described as discoid, but in large populations it has often been found on average to be slightly elliptical (435). Among ∼6,000 placentas in the Helsinki Birth Cohort, the placentas were some 2.6 cm longer in one direction than in the other (172). Because the Helsinki data allow so many comparisons with a variety of diseases, it has become clear that the degree to which a placenta deviates from being perfectly round has a predictive value for specific diseases. Here, we define “length” of the delivered placenta as the longest dimension, and “width” as the longest distance measured perpendicular to the first. Assuming an elliptical surface, the average thickness can be estimated by the weight of the delivered placenta divided by the estimated surface area, the length × width × π/4 (172). A frequent finding is that although length and width correlate in any sample of placentas, the two measurements often have independent associations with fetal growth parameters as well as with postnatal disease. For example, simultaneous regression revealed that increasing ponderal index and the circumferences of the head, chest, abdomen, and thigh among newborns are all highly associated with placental width; however, none is related to placental length (12). For each centimeter increase in placental width, birth weight increased by 125 g (95% confidence interval, 88–162, P < 0.001) but only by 20 g for each centimeter increase in placental length (−13 to 53, P = 0.2). Mothers below the median height (157 cm) had the strongest associations between placental width and neonatal body size.
The biological links among placental shape, size, and function have not been defined. It has been proposed that different regions of the placenta have specific roles in nutrient transport and that the placenta has a polarity related to the rostral-caudal axis of the early embryo (12, 298). However, there are no experimental data to substantiate this speculation. A more likely explanation is that placental shape is a powerful proxy indicator of processes in placentation that are related to its transport and physiological functions. As described in section IVA2, placental shape may reflect the site of implantation, or events taking place during the transition of the early placenta to its definitive form. For the former, implantation at different sites within the uterus, such as on the anterior or lateral walls, close to the cervix or in the fundus, may lead to variable shapes depending on whether the uterus expands symmetrically during later pregnancy or preferentially in certain dimensions. If uterine vascularity is different at these implantation sites, in terms of the density of the arcuate and spiral arteries, then placental blood flows, and hence functional capacity, will correlate with placental shape. For the latter, excessive villous regression at the time of onset of the maternal circulation may be indicative of deficient extravillous trophoblast invasion (77), which may mean that the remaining spiral arteries are not fully remodeled, compromising placental blood flow.
The lack of certainty over the role of placental shape in driving biological function is based on a paucity of basic data detailing how the placenta actually grows, and how shape is determined. Placental growth patterns measured over intervals across gestation are needed, and this may be possible with the advent of high-resolution ultrasound from which volumetric and shape data can be obtained. Once the mechanisms are known, it should be possible to determine how maternal diet, body type, and lifestyle alter the placenta over its lifespan. We also need more detailed information on the regional distribution of spiral arteries, and how the uterus and placenta coexpand during pregnancy. The visualization of placental oxygenation, metabolism, and nutrient transport in real-time would further illuminate the relationships between shape and placental function, and may become possible through magnetic resonance imaging techniques. At present, data show that elliptical placentas tend to be less efficient than circular ones (590), but more research is needed to understand the underlying biology.
C. Placental Inflammation
A number of maternal conditions, both infectious and noninfectious, have been linked to placental inflammation (102, 453). Elevated levels of placental inflammation have been associated independently with slower postnatal growth in preterm infants, and so may have long-lasting effects for adult health (377).
It is becoming increasing clear that standard definitions of inflammation do not fit the findings in the placenta in noninfectious cases, such as obesity and diabetes, for it is possible for inflammatory pathways to be activated without the classic infiltration of granulocytes (418). Recently, new definitions have been applied to chronic inflammatory states in adult tissues (87). The basis for this reappraisal is the recognition that the same signaling cascades that are activated in classical inflammation in response to pathogens, including activator protein 1 (AP1), NF-κB, and interferon regulatory factors (IRFs), can be stimulated by cytokines arising from a variety of metabolically active tissues, such as adipose tissue, muscle, and liver, and their resident immune cells in response to excess nutrients (228, 567). The outcome has been termed metaflammation, or “cold, smoldering inflammation” as it is characterized by its chronicity. Since the tissues remain in an anabolic state, there is the capacity for tissue remodeling and gradual metabolic deterioration over time (87).
It is likely that the stressors known to lead to fetal programming, including insufficient or excess nutrition, social stress, and hypoxia, can lead to metaflammation in the placenta. Indeed, the same inflammatory pathways can be activated through the signaling cascades of the unfolded protein response (UPR) (228, 609), as will be discussed later. These cascades are activated in placentas from cases of growth restriction and early-onset preeclampsia (595, 596), but no data are yet available for pregnancies complicated by maternal obesity or other metabolic disorders. A sterile inflammatory state can also arise through senescence, when cells adopt the senescent-associated secretory phenotype (SASP) and release proinflammatory cytokines and proteases (425). Senescence has only recently been considered a potential feature of the syncytiotrophoblast in human placenta (220). While it may be part of the normal aging process, the fact that it can be induced by chronic stress, including oxidative and endoplasmic reticulum stress, suggests it may be more prevalent in complicated pregnancies.
Although the intermediate molecular mechanisms remain uncertain, we speculate that metaflammation is present in the human placenta in pregnancies complicated by malperfusion or maternal metabolic disorders (Figure 7). It may mediate programming of the fetus by either adversely affecting placental function, or by causing the release of proinflammatory cytokines into the fetal circulation.
FIGURE 7.
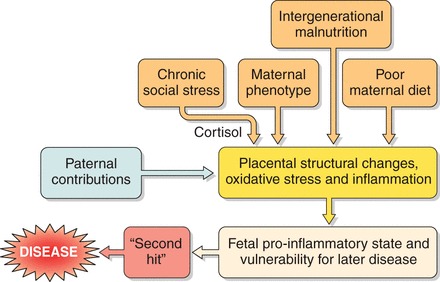
Schematic representation of how multiple environments may give rise to placental metaflammation or “cold, smoldering inflammation,” and how this may predispose the fetus to chronic disease.
D. Specific Examples Linking Placental Phenotype to Chronic Disease
Here, we explore associations between placental phenotypes and adult chronic diseases resulting from epidemiological studies. In each case the findings have been corrected for known confounders. Epidemiological studies are unable to separate cause from effect, but these associations provide the opportunity to investigate the underlying biology.
1. Hypertension
Most of the epidemiological data related to hypertension have arisen from studies of large cohorts followed from birth to the present day. One such is the Helsinki Birth Cohort that comprises 13,345 men and women born during 1934–1944. Among the 644 hypertensive subjects, treatment for hypertension was associated with low placental weight and surface area (39). Birth weight is linked to maternal body size and composition, as well as to growth of the placenta. When taking maternal characteristics into consideration, the associations were strongest among mothers whose stature was below average height (160 cm), or who were of low socioeconomic status (Figure 8). This suggests an interaction between the role played by the placenta and the nutritional state of the mother during her early development. Among these shorter women, the prevalence of hypertension fell from 38% if the placental area was 200 cm2 or less, to 21% if the area exceeded 320 cm2 (P = 0.0007). Poor maternal nutrition may exaggerate the adverse effects of small placental size on fetal development, possibly by restricting compensatory mechanisms.
FIGURE 8.

In the Helsinki Birth Cohort, hypertension is related to the surface area of the delivered placenta, in mothers of below median height (160 cm) (P = 0.002) but not for tall mothers (P = 0.72). [From Thornburg et al. (531), with permission from Elsevier, using data from Barker et al. (39).]
Among men who were exposed in utero to the starvation effects of the post-war famine in Holland, a reduced width of the placenta was associated with hypertension (471). The surface area of the placenta also predicted hypertension with an odds ratio of 1.34 (95% CI 0.99–1.80) for an increase in surface area of 40 cm2. However, hypertension was predicted by a short placental width and a more oval shape in men who were born after the war (471, 541).
There are also effects of placental development on blood pressure in children. A study of placentas of children in the longitudinal Alspac study of 13,971 births in Bristol, UK, showed the number of lobes on the maternal surface was related to the blood pressure of the children at age 9 yr (24). Increasing lobe number (range 4–40) was associated with higher blood pressures in both boys and girls. The larger the surface area of the placenta, the more lobes it contained and the heavier the birth weight of the offspring. However, among boys, the number of lobes was directly associated with higher systolic and diastolic pressure, but not with an increased pulse pressure. In that group, diastolic pressure rose by 2.2 mmHg (95% CI 0.6–3.7, P = 0.007) for every 10 additional lobes. A greater number of lobes was associated with higher systolic pressure and pulse pressure, but not with higher diastolic pressure, in girls. Pulse pressure rose by 2.7 mmHg (1.1–4.3, P < 0.001) for every 10 additional lobes. Adjustment for placental surface area, a powerful determinant of hypertension in adult men, did not change the relationships. Although we do not understand how lobation of the human placenta arises developmentally (49), these data are fascinating for two reasons. First, since septae are first observed projecting from the basal plate as early as 6 wk of gestation, they may represent an informative proxy marker of early events in placental development that are of physiological consequence. Second, they complicate the role of placental area as a predictor of adult hypertension, for the number of lobes is the more powerful index.
2. Heart failure
Returning to the Helsinki Birth Cohort, 187 patients were taking medications for chronic heart failure. Their disease was associated with a small surface area of the delivered placenta, and the odds ratio for chronic heart failure was 1.7 (1.1–2.5) in men and women born with a placental area <225 cm2 compared those with an area >295 cm2 (27). There was no relationship with placental weight. As with hypertension, the relationships were strongest among women of below median stature (Table 2), suggesting a link with the mother's own nutritional state during her fetal and childhood development.
Table 2.
Hazard ratios for chronic heart failure are linked to placental size and maternal weight
| Mother's Height, cm |
||
|---|---|---|
| Placental Area, cm2 | <160 | >160 |
| −225 | 2.3 (1.2–4.2) | 1.2 (0.6-2.4) |
| −255 | 1.1 (0.6–2.2) | 0.9 (0.5–1.9) |
| −295 | 1.8 (0.9–3.3) | 1.1 (0.6–2.1) |
| >295 | 1.0 (baseline) | 1.0 (baseline) |
| P value | 0.05 | 0.5 |
Confidence intervals are given in parentheses. Data from Barker et al. (27).
In a separate study of adult men, concentric enlargement of the left ventricle, a known predictor of coronary heart disease, was found to be associated with low weight at 1 yr (548). It was speculated that this was due to altered hemodynamics during the fetal period, or a persisting elevation of growth factors. Taken together, these data suggest that chronic heart failure in adult life may be initiated by impaired placental growth, which subsequently adversely affects cardiac development. In addition, people born with a vulnerable heart are more likely to develop chronic heart failure if they become insulin resistant (27).
3. Coronary heart disease
Numerous studies have shown a relationship between cardiovascular disease and restricted growth before birth (5, 182, 428). However, the finding that the placenta is a more powerful driver of coronary heart disease in men (182) than in women (175) is a recent discovery. A study of 6,975 men in the Helsinki Birth Cohort found that three different placental phenotypes were associated with disease of the coronary arteries (175). In the first, an increasing difference between the length and width of the placenta predicted the disease in the offspring of primiparous mothers who were of below median height. The hazard ratio for each centimeter in difference between length and width was 1.14 (95% CI 1.08–1.21, P = 0.0001). The second placental phenotype was a small surface area of the delivered placenta. In tall mothers whose body mass index was above the median, a 40 cm2 decrease in surface area was associated with a hazard ratio of 1.25 (1.10–1.42, P = 0.0007). The third phenotype was found also in tall mothers, but only those whose body mass index was below the median. In these mothers, the hazard ratio was 1.07 (1.02–1.13, P = 0.01) per 1% increase in the placental weight-to-birth weight ratio. These data suggest an interaction between a mother's body size and composition and placentation that leads to a particular capacity for nutrient exchange and efficiency. Coronary heart disease was associated with a low ponderal index (birth weight/length3) in all groups, suggesting that the fetuses were all undernourished because of poor placental growth.
Whereas placental size usually correlates with maternal body size, coronary heart disease in men was associated with a small placenta in tall mothers. This suggests poor placentation and poor placental growth throughout pregnancy. It was placental inefficiency that predicted the disease in the third group. In this group, thin tall women had large placentas in proportion to the size of the baby at term. In these women, placenta growth may have been stimulated by poor maternal nutrition at mid-gestation.
4. Sudden cardiac death
Because it is not possible to study the living hearts of people who have died suddenly from cardiac causes, it has been very difficult to pinpoint the electrical properties of the myocardium in hearts whose contractility suddenly becomes inadequate to sustain life. Nonetheless, aberrant functioning of the autonomic nervous system is considered the most common explanation for sudden cardiac death (201). While it is known that sympathetic tone may be exaggerated in people who had low birth weight, the links among maternal, placental, and fetal growth have only recently been explored through the Helsinki Birth Cohort. Sudden unexplained cardiac death outside hospital was associated with a thin placenta, and for each gram/cm2 decrease in thickness the hazard ratio was 1.47 (95% CI 1.11–1.93, P = 0.006) (30). A high placental-to-birth weight ratio also predicted sudden death among women, but not men. The determinants of placental thickness are not fully understood, but if the theory that abnormal autonomic function underlies sudden death holds then one might suspect a relationship between the nutritional function of a thin placenta and development of the autonomic nervous system during fetal life.
5. Lung and colorectal cancers
Susceptibility to developing cancer on exposure to carcinogens, such as those in tobacco smoke, differs among individuals. Through a combination of the Helsinki Birth Cohort with an older cohort born in 1924–1933, the smoking history was known for 6,822 men and women, of which 385 developed lung cancer by 2010. The cases were characterized by having a short mother and a high ponderal index (weight/length3) at birth, and the delivered placenta had either a small or a large surface area in three separate phenotypes (38). It was suggested that in each phenotype, low amino acid transport but normal glucose transfer was reflected in a newborn that was short in relation to its weight. These data indicate that both large and small placentas can limit the flow of nutrients, and that poor placentation occurs more often in women who are short in stature.
In the combined Helsinki cohorts, 275 had colorectal cancer (36). The risk for acquiring the disease increased as the placental surface became longer and more oval. Among people in whom the difference between the length and breadth of the surface exceeded 6 cm, the hazard ratio was 2.3 (95% CI 1.2–4.7) compared with those in whom there was no difference. Colorectal cancer was unrelated to other placental measurements or to body size at birth. Thus colorectal cancer had a graded association with placental elipticity.
VI. OXYGEN SENSING BY PLACENTAL CELLS
Placental cells, and in particular the trophoblast, are metabolically highly active due to their multiple functions. Thus it has been estimated that the placenta accounts for ∼40% of the oxygen consumption of the fetal-placental unit, and of that ∼33% is utilized in active transport and ∼33% in protein synthesis (97). It is therefore to be expected that placental cells are sensitive to oxygen availability. There are a number of potential pathways by which cells may sense the prevailing oxygen concentration (Figure 9), and there is evidence of considerable interplay between them (180, 324, 562). Given that oxygen is central to cell metabolism, some of these pathways also overlap with those sensing energy levels and nutrient supply. Here, the respective pathways will be considered according to their principal function. Equally, activation of the two sets of pathways causes, to a large extent, a common outcome, for under conditions of hypoxia or nutrient deprivation there is a need to conserve energy reserves by stimulating glycolysis and suppressing nonessential protein synthesis. Responses to hypoxia generally begin when the oxygen concentration falls below a cell's critical threshold that switches aerobic oxygen-regulated metabolism to anaerobic oxygen-conforming metabolism (225). That threshold is likely to be different for different cell types within the placenta dependent on their metabolic activity and other factors, but no data are available as to what the precise values might be. For most primary or transformed mammalian cells, it is within the range of ∼0.15–1.5% oxygen (225). Equally, although it is commonly asserted that the placenta is hypoxic in pathological states, such as preeclampsia, no measurements have been made in vivo to confirm whether or not this is the case. These claims must therefore be treated with caution.
FIGURE 9.
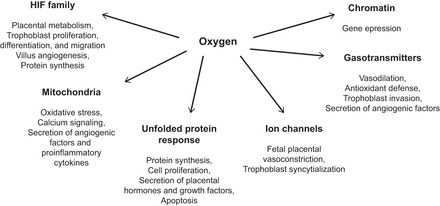
A summary of the principal mechanisms for oxygen sensing in cells and of the effects of modulating oxygen concentration on cell behavior that have been reported for the placenta.
There are a variety of signaling mechanisms that are activated in response to hypoxia, as follows.
A. Transcription Factors
Central to oxygen sensing in any cell is the family of hypoxia-inducible basic helix-loop-helix transcription factors, the HIFs, of which there are three members: HIF-1-3 (297). All members consist of an alpha and beta subunit, and it is the former that is oxygen dependent. In well-oxygenated conditions this subunit turns over rapidly and does not accumulate, whereas under hypoxia it is stabilized and the two subunits are able to combine to form an effective transcription factor. This binds to hypoxia response elements on a wide range of genes, the most important of which in the current context include those encoding VEGF, glucose transporters, and glycolytic enzymes. It can also inhibit mTOR signaling, and hence regulate protein synthesis. The actual oxygen sensors are the prolyl-4-hydroxylase (PHDs) enzymes that have an absolute requirement for molecular oxygen and hydroxylate conserved proline residues on the alpha subunit. This enables binding of the von Hippel-Lindau protein (pVHL), which targets the subunit for ubiquitination and subsequent degradation (493). HIF-1 thus provides a very rapid mechanism for responding within seconds or minutes to acute changes in oxygenation, whereas HIF-2 is thought to mediate longer-term adaptations to relatively modest changes (444).
Animal studies have confirmed that HIFs are expressed at the blastocyst stage of development (243) and that they are essential for normal placental development. Knockout in mice results in failure of allantoic attachment with the chorion, impaired vascularization in the labyrinth, and abnormal differentiation of the trophoblast subpopulations (161). Consistent with these effects, HIF-1 and HIF-2 have been immunolocalized in the human placenta to the trophoblast and endothelial cells throughout gestation (447). There has been considerable interest in the importance of HIF signaling during the first trimester when the intraplacental oxygen concentration is relatively low. Ontogenetic studies have reported contrasting results, with peaks of HIF-1α at 7–10 wk and then again at 14–18 wk (269), or a steady decline in HIF-1α from 5–8 wk to 18–21 wk and a rapid decline of HIF-2α between 5–8 wk and 9–11 wk (447). Interpretation of these data is difficult given that it is now realized that HIFs can be stabilized by factors other than hypoxia, including reactive oxygen and nitrogen species, angiotensin, growth factors, and cytokines (433, 444), many of which are changing during early pregnancy. Another potential confounder is stress induced on the tissues during collection, especially when this is performed by curettage when they are inevitably mixed with maternal blood (112). It is notable that HIF-1 and HIF-2 are undetectable in first trimester samples collected by a chorionic villus sampling (CVS) technique and processed immediately (112). Thus it is unlikely that HIF signaling plays a significant role under the steady-state, low oxygen conditions that prevail during the first trimester. This is supported by the observation that the ATP/ADP ratio is the same during the first and early second trimesters and at term (112). The tissues are therefore not energetically compromised, most likely due the high activity of glycolytic metabolism (284). In addition, the intraplacental oxygen concentration of ∼2.5% during the first trimester exceeds the evolutionary conserved range of maximal HIF activity of 0.5–2.0% oxygen (225, 286).
There is no doubt, however, that the HIF signaling machinery is competent in the human early placenta and can respond to acute changes in oxygenation. Experiments on first-trimester placental explants cultured at 3% oxygen compared with 21% indicate that HIF regulates trophoblast proliferation, migration, and invasion through its actions on TGF-β3 (89). Equally, HIF-1 and HIF-2 can be stabilized in CVS samples by stress and activate downstream targets, including VEGF (112).
In later pregnancy, natural selection acting on HIF-targeted or -regulatory genes has been implicated in mediating placental adaptations to the chronic hypobaric hypoxia experienced at high altitude (388). Increased levels of HIF-1α mRNA and protein have been reported in healthy placentas from pregnancies at 3,100 m, in association with elevated TGF-β3, suggestive of stimulation of HIF-mediated pathways (606). Another study of placentas from the same region found enhanced vascularization, consistent with a hypoxic response, but paradoxically HIF-DNA binding was less than in the low-altitude controls (533). Analysis of the placenta after delivery only provides a single snapshot, however, and this finding may reflect successful adaptations earlier in pregnancy, for these placentas showed no evidence of oxidative or glycolytic stress. An excessive hypoxic response may account for the increased incidence of chorangiomas in placentas from altitudes greater than 4,000 m (459, 510), but no data are available as to whether this is HIF-mediated.
Aberrant HIF signaling has been implicated in the pathophysiology of preeclampsia, in particular of the early-onset form when the PHDs do not appear to sense oxygen (90, 448, 467), and may be responsible for the abnormal placental secretion of angiogenic regulatory factors that is thought to precipitate the clinical syndrome (413).
A number of other transcription factors and coactivators have been identified that respond to changes in the redox potential of a cell rather than to oxygen directly. These include AP-1, CREB, Mash2, NFκB, p53, PCC-1α, SP-1, and STAT3 (16, 106, 155, 277). Often, activation involves conformational changes secondary to formation of disulfide bonds, so responses can be rapid. While some of these pathways have been implicated in stress responses, others are involved in trophoblast proliferation and differentiation, and secretion of extracellular matrix.
B. Epigenetics
1. Noncoding RNAs
A large number of miRNAs have been identified from the field of cancer biology as being regulated by hypoxia and mediate events such as cell proliferation, differentiation, invasion, and metastasis that are relevant to placental biology (109, 499). Many of these are miRNAS and are regulated by HIF, but others are HIF-independent. The human placenta expresses a wide variety of non-coding RNAs (398), but few data are available regarding their responsiveness to hypoxia. Most attention has focused on miR-210, which is HIF-dependent. It is increased in normal placentas from pregnancies at high altitude (121), and in placentas from pregnancies complicated by preeclampsia (272, 351, 401, 611). MiR-210 has a number of targets that are relevant to the placenta and developmental programming. Within mitochondria it targets regulatory proteins that assist in the assembly of the complexes of the electron transport chain, and suppresses respiration (103, 108). Consistent with this action, the complexes are reduced in these high-altitude placentas (121), as is the ATP/ADP ratio (534), suggesting energetic compromise that could adversely affect transport and synthetic activities of the organ. Furthermore, transfection of trophoblast cells with miR-210 reduces respiration and oxygen consumption (401). Besides mitochondria, other targets identified for miR-210 include the steroidogenic enzyme hydroxysteroid (17-β) dehydrogenase 1 (272), ephrin-A3, and homeobox-A9 which are involved in trophoblast cell migration and vascular remodeling (611), and thrombospondin type 1 domain containing 7A in the placental vasculature (351).
Other micro-RNAs identified as being differentially expressed under hypoxia include miR-93, miR-205, miR-224, miR-335, miR-424, miR-451 and miR-491 (396, 397), and mIR-34a (154), but little is known regarding their functional significance at present.
2. mRNA stability
Transcript levels are determined by both the rate of transcription and the rate of mRNA degradation. Stability of mRNAs is regulated by association with specific binding proteins or with micro-RNAs and can be influenced by hypoxia. A notable example is the mRNA encoding angiopoietin-1, which becomes less stable under low oxygen conditions, shifting the balance of angiopoietin-1:angiopoietin-2 in favor of angiopoietin-2 and vessel growth (608). In contrast, the half-life of the mRNA encoding VEGF is more than doubled under hypoxic conditions (334), again favoring angiogenesis. Such effects could contribute to the increased placental angiogenesis observed under hypoxic conditions.
3. DNA methylation and histone modifications
Hypoxia can potentially impact on DNA and histone methylation since the demethylase enzymes are dioxygenases, and so require oxygen and 2-oxoglutarate for their activity (501). Hence, there is the possibility of changes in chromatin structure in response to hypoxia. The significance for the placenta is still unknown, although it is well recognized that nuclei within the syncytiotrophoblast exhibit contrasting patterns of chromatin and different epigenetic states (187). Nuclei that display particularly condensed chromatin aggregate in syncytial knots and are transcriptionally inactive (188). Whether more subtle changes regulate gene expression under different environmental conditions awaits investigation.
Methylation of the DNA represents another level of control, affecting promoter availability. Methylation changes in response to intermittent hypoxia in experimental animals have been reported (66), and the realization that 5-hydroxymethylcytosine, an oxidation product of 5-methylcytosine, plays an important role in regulating transcription raises further possibilities for gene-environment interactions. The heavily condensed nuclei within syncytial knots stain particularly strongly for 5-hydroxymethylcytosine (187).
Epigenetics is a rapidly expanding field, and there are now many reports of changes in placental DNA methylation and histone proteins in response to environmental cues that have been associated with developmental programming of the offspring (203, 417, 522). Interpreting the significance of these findings is difficult at present, since they are based on analysis of placental homogenates. It is therefore impossible to determine in which tissue the changes have occurred, be it trophoblast, immune cell, or vascular endothelium. It is also impossible to assess whether the changes have functional significance for placental transport or hormone synthesis, or whether they are just epiphenomena. The situation may be resolved as the technology advances, enabling methylation studies to be performed on single cells or laser-capture microdissected tissues, allowing more specific analyses to be undertaken.
4. Mitochondrial pathways
As the principal site of oxygen consumption within cells, mitochondria likely play an important role in oxygen sensing. There are close interactions between mitochondria and HIF signaling that operate on a number of levels, not least because mitochondria and the PHDs compete for molecular oxygen (523). Therefore, one or the other may be favored depending on the precise concentration. In addition, the metabolic intermediate 2-oxoglutarate generated in the tricarboxylic acid cycle is a cofactor regulating PHD activity (48). Another means by which the pathways may interact is through reactive oxygen species (ROS). Mitochondria are the principal source of ROS, and production is stimulated under both hypoxic and hyperoxic conditions. Leakage of electrons from the complexes of the electron transport chain, in particular complexes I and III, generates the superoxide ion, which is then converted to hydrogen peroxide. Being nonpolar, hydrogen peroxide diffuses out of the organelle and can stabilize HIFs (48). In addition, it will influence the redox potential within the cytoplasm and contribute to activation of other redox-sensitive transcription factors. It is notable that complex I and III are downregulated at the protein level in the placenta at high altitude, which may be interpreted as an adaptation to reduce production of ROS and so limit HIF signaling (121). This may explain the reduced HIF-binding reported in these placentas (533).
5. Unfolded protein response
The unfolded protein response (UPR) is a set of three evolutionary conserved pathways whose primary function is to maintain homeostasis within the endoplasmic reticulum. However, they are now recognized as being a point of convergence of cell responses to a variety of stimuli, including hypoxia. Activation occurs generally at oxygen levels below those regulating HIF (225), and although the precise sensing mechanisms are not known, there are two main possibilities. First, the protein disulfide isomerase enzymes that facilitate formation of disulfide bonds during folding of nascent proteins have a requirement for molecular oxygen as an electron acceptor. Second, the protein folding machinery is dependent on a high concentration of Ca2+ ions within the lumen of the endoplasmic reticulum that is maintained by SERCA pumps in the membrane. Folding capacity may therefore be compromised if oxygen or ATP concentrations are limiting, but this is likely to be a secondary mechanism (315). The accumulation of misfolded proteins will activate the three pathways: PKR-like ER kinase (PERK), activating transcription factor 6 (ATF6), and inositol-requiring protein 1 (IRE1). PERK selectively suppresses formation of nonessential new proteins by phosphorylating eIF2α (eukaryotic initiation factor 2 subunit alpha) and blocking cap-dependent RNA translation. It also upregulates the transcription factor ATF4, which along with the ATF6 and IRE1 pathways selectively regulates gene expression to assist the cell to tolerate hypoxia (180, 343, 585). These adaptations include increased amino acid transporter activity and enzyme expression to boost the concentration of glutathione, the principal intracellular antioxidant, stimulation of hematopoiesis, and upregulation of the angiogenic growth factor VEGF. However, it is now appreciated that these transcription factors have broader targets (2), and, for example, the UPR has been implicated in regulating processes as diverse as the inflammatory response (609) and stemness within the intestinal epithelium (251).
Manipulations in mice have confirmed that the IRE1 pathway is essential for normal placental development, for knockout leads to downregulation of VEGF and abnormal angiogenesis within the labyrinth (273). Mild activation of the UPR, with phosphorylation of eIF2α alone, has been reported in the high-altitude human placenta, where it may mediate homeostatic adaptations to the hypobaric hypoxia experienced (598). More severe activation is seen in human placentas from growth-restricted pregnancies (596), and particularly in cases of early-onset preeclampsia (595). These findings can be replicated in trophoblast cell lines by exposure to hypoxia-reoxygenation, with activation of the three pathways being dependent on the severity of the stress (596).
The regulator eIF2α can be phosphorylated by at least three other kinases besides PERK. One of these is GCN2, which is activated by the presence of uncharged tRNAs (153). Hence, if either oxygen or amino acids are in short supply, protein synthesis is suppressed by a common mechanism.
6. Ion channels
Since ionic pumping is energy dependent, a number of ion channels are sensitive to the prevailing oxygen concentration. Most data have been derived from the carotid body, but may also be applicable to the placenta. The proximal sensor is still uncertain, but two main theories have been proposed: the mitochondrial and membrane models (324). In the former, it is proposed that under hypoxia the accumulation of ROS leads to opening of the mitochondrial permeability transition pore and the efflux of Ca2+ from the mitochondrial endoplasmic reticulum complex. In the latter, there are various K+ channels that are influenced by hypoxia, including voltage-gated K+ channels, Ca2+-activated K+ channels, and ATP-sensitive K+ channels (308, 347). Suppression of these channels under hypoxia is thought to lead to membrane depolarization and influx of extracellular calcium through voltage-gated calcium channels. In addition, transient receptor potential (TRP) channels that are responsive to ROS have been identified in a number of cell types and provide another route of entry for calcium (589). The end result of all these pathways is a rise in intracellular calcium, which at physiological levels can regulate transcription of a number of genes that assist in adaptations to hypoxia, including those encoding the ion channels themselves (347). Excessive calcium influx can lead to activation of apoptotic and necrotic cell death, dependent on the severity.
A variety of K+ channel subtypes, many of them oxygen sensitive, are present in the vasculature of the human placenta (307, 563, 564). Hypoxia-induced fetoplacental vasoconstriction, equivalent to that seen in the pulmonary circulation, has often been proposed but never proven (563), but could assist in matching flow in the two placental circulations. Voltage-dependent K+ channels have also been immunolocalized to the syncytiotrophoblast, cytotrophoblast, and some stromal cells in the first trimester and term placenta (382, 383), so they may play a broader role in placental biology, including regulating the secretion of hCG (574). In addition, Ca2+-activated K+ channels have recently been demonstrated to be important for trophoblast syncytialization and for syncytial volume homeostasis (145). The significance of these findings for the expansion and functional well-being of the syncytiotrophoblast under different environmental conditions awaits confirmation.
7. Gasotransmitters
Allied to the functions of these ion channels are the gasotransmitters, and in particular hydrogen sulfide. Increasing evidence suggests that this evolutionary ancient gas can act as an oxygen sensor (423, 436). Hydrogen sulfide is generated from cysteine by two enzymes cystathionine-γ-lyase (CSE) and cystathionine-β-synthase (CBS). It is notable that expression of CSE is induced by the UPR through the PERK pathway and ATF4 (146), illustrating crosstalk between these pathways. Hydrogen sulfide is metabolized principally in the mitochondria by oxidation to thiosulfate, but if oxygen availability is limited, then the concentration in the cytosol will increase (423). There, it can activate ATP-sensitive K+ channels with all the downstream consequences of hyperpolarization. In addition, it can scavenge ROS and so act as a defense against ischemia-reperfusion injury (310), affect the redox balance, and also inhibit a number of related enzymes, including NADPH oxidase and nitric oxide synthase.
CBS and CSE have been immunolocalized to the syncytiotrophoblast and also to the smooth muscle cells surrounding the stem villus arteries in the human placenta (111, 262). Perfusion experiments ex vivo have demonstrated that hydrogen sulfide is a powerful vasodilator of the fetal placental vasculature and that its effects are mediated through actions on both ATP-sensitive K+ channels and nitric oxide (111). The functional significance of the gas is reflected in the observation that downregulation of CSE is associated with evidence of increased resistance within the umbilical circulation as assessed by Doppler waveforms. Its role in trophoblast biology has not yet been explored, although it is likely to impact on the redox potential within the cytosol. Inhibition of CSE activity also inhibits trophoblast invasion from first trimester explants in vitro, and increases the release on anti-angiogenic factors from umbilical vein endothelial cells (559). It has been proposed that dysregulation of placental CSE activity may contribute to the syndrome of preeclampsia (559).
VII. NUTRIENT AND ENERGY SENSING BY PLACENTAL CELLS
In the same way that placental cells are sensitive to oxygen due to their high proliferative and metabolic rates, they are also sensitive to variations in the nutrient supply. The available evidence suggests that they utilize pathways common to most mammalian cells, in particular the mTOR/AKT and the AMPK pathways (Figure 10). These networks combine with the UPR to regulate protein synthesis over the short- and long-term periods to promote cell survival.
FIGURE 10.
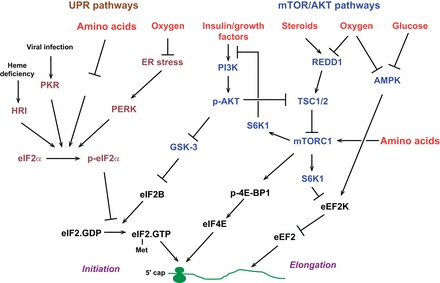
Diagrammatic summary of the principal ways by which the Unfolded Protein Response pathway may interact with the mTOR/AKT pathway to modulate protein synthesis within the placenta. Both pathways receive input at various levels regarding oxygen and nutrient availability and will influence cell proliferation and growth. See text for details.
A. mTOR/AKT Pathway
The mTOR (mechanistic/mammalian target of rapamycin) complex integrates signals from a diverse array of pathways, including oxygen- and nutrient-sensitive sensors. As it is central to the control of cell proliferation, the complex also has strong input from the AKT (protein kinase B) pathway that transduces growth factor stimulation. These pathways will therefore be considered together (Figure 10).
mTOR comprises two major complexes, mTORC1 and mTORC2, which share the same core kinase but have the different adaptor proteins, raptor and rictor, respectively. mTORC1 has the most direct effect on cell proliferation due to its actions on cap-dependent RNA translation mediated through phosphorylation of the binding protein 4E-BP1, and the ribosomal protein S6. In its nonphosphorylated state, 4E-BP1 modulates formation of the ribosomal complex (246), while activation of S6K1 (p70 ribosomal protein S6 kinase 1) has multiple actions. These include phosphorylation of S6, a component of the 40S subunit, of regulators of translation elongation (560, 561), and of the insulin receptor substrate (IRS) 1, creating a negative-feedback loop that limits signaling through the insulin/PI3K/AKT pathway (Figure 10). Conversely, mTOR inhibits autophagy and other catabolic processes by promoting the ubiquitination of ULK1 (410). Hence, stimulation of mTORC1 promotes protein synthesis, increases cell mass, and leads to cell proliferation and growth.
mTORC1 is regulated by amino acid availability, although for many years the mechanism has been uncertain. Early experiments revealed that withdrawal of amino acids, in particular leucine, led to inhibition of mTOR, growth restriction, and the stimulation of autophagy. It was also clear that the amino acids acted independently of insulin, and so were not sensed through the insulin/PI3K pathway. More recent research has identified a Ragulator-RAG GTPases multiprotein complex associated with the lysosomal surface that regulates mTOR activity by controlling its subcellular location, recruiting it to the lysosome (164). The membrane-resident amino acid transporter SLC38A9 is a key component of this sensing machinery (452).
Glucose availability also regulates mTORC1, with accumulation of ADP and AMP under conditions of energy shortage stimulating AMPK (AMP-activated protein kinase). AMPK has a direct inhibitory action on raptor, the adaptor protein for mTORC1, and can also stimulate the tuberous sclerosis complex TSC1/2 (164). TSC1/2 is a major upstream regulator of mTORC1, suppressing activity under stress conditions. This complex plays a major role in integrating insulin and growth factor signaling through the PI3K and AKT pathway (Figure 10), although recent data suggest growth factors may also stimulate mTORC1 by enhancing the delivery of amino acid-laden macropinosomes to the Ragulator complex (593). TSC1/2 is also responsive to hypoxia through the actions of REDD (70). The latter involves phosphorylation of HIF-1α (88), illustrating again the overlap between these pathways. TSC1/2 also acts on mTORC2, which in turn regulates the activity and substrate specificity of AKT through phosphorylation (597).
AKT is a serine/threonine protein kinase that has a wide range of targets, but the most important for cell growth are TSC1/2 and glycogen synthase kinase 3β (GSK-3β). The latter plays a major role in glucose homeostasis and is likely to be key to the deposition of glycogen in the human extravillous trophoblast cell and the murine glycogen cells. It is also a major regulator of protein synthesis through its actions on eIF2B (568).
The involvement of mTOR/AKT signaling in the regulation of placental growth has only recently been addressed, but there is evidence that it is of key importance from the earliest stages. mTOR/AKT signaling is essential for maintenance of embryonic and hematopoietic stem cells (419), and the same is likely to be true for trophoblast. Indeed, treatment of mouse blastocysts with rapamycin or knockout of mTOR causes lethality at E5.5 associated with a failure of trophoblast outgrowth and maintenance of stem cells in the inner cell mass (206, 360). Disruption of just Akt1 causes placental and fetal growth restriction in the mouse, with a particularly severe effect on the development of the glycogen cells (592). Equally, a reduction of AKT and mTOR at the protein level, and reduced phosphorylation of mTOR, TSC1/2, 4E-BP1, and GSK-3β have been reported in growth-restricted human placentas associated with maternal vascular compromise, but there was no effect on S6Ks and eEF2K (596). Reduced p-S6K1, but no change in p-4E-BP1, was observed in placentas with growth restriction of unknown origin (468). In contrast, increased placental mTOR signaling is associated with large for gestational age babies delivered by obese women (279). mTOR expression is also inversely correlated with levels of maternal exercise, and the total sugar content in her diet (60).
The effects of diet on placental mTOR signaling have been more fully explored in animal models. Evidence from downstream signaling indicates that nutrient restriction leads to reduced mTOR activity, along with reduced insulin and AKT signaling, in rats fed a low-protein diet (470), and mice fed 80% of the control ad libitum diet (495), as might be expected. However, no effect was observed at mid- to late-gestation in sheep fed 50% of the control diet (353). Data arising from overnutrition models have been more conflicting. Thus, while an obesogenic diet has been shown to cause activation of mTOR in rat placentas (205), the opposite was found following overnutrition in sheep (150% of control diet) and mice fed an obesogenic diet (323, 615).
Nonetheless, it seems reasonable to conclude that the mTOR/AKT pathway plays an important role in matching placental growth to the available nutrient supply, an idea originally proposed a decade ago (569). Indeed, a linear relationship between placental mTOR activity and birth weight has been found across a wide range of maternal body mass index (279). In addition, the mTOR pathway regulates the activity of system A, system L, and taurine amino acid transporters in the placenta at the posttranslational level, either through modifications or by influencing translocation to the apical membrane (469). Thus activity of system A, but not system L, transporters in the apical membrane of the syncytiotrophoblast correlates positively with birth weight and may contribute to fetal overgrowth in cases of maternal obesity (279).
The mTOR/AKT pathway thus plays a central role in modulating anabolic and catabolic pathways in response to fluctuations in the nutrient and oxygen supply reaching the placenta. Such fluctuations may arise from either variations in maternal diet or compromise of utero-placental blood flow secondary to deficient trophoblast invasion. By regulating the activity of amino acid transporters, the pathway will also be pivotal in integrating the maternal supply and fetal demand signals that underpin resource allocation between the mother and her fetus. Genes encoding components of the mTOR and protein translation pathways are among the most sexually dimorphically expressed genes in the placenta (71). This could account for the different growth rates displayed by male and female placentas, and the variations in their adaptations observed in response to stress. Other functions, such as the regulation of extravillous trophoblast invasion (86), have also been proposed.
B. AMP-Activated Protein Kinase
AMPK is an evolutionarily conserved and ubiquitously expressed regulator of cell metabolism that is activated by depletion of ATP. Hence, it acts as a key metabolic sensor to match energy demand with supply (240). It comprises three subunits, each of which have multiple isoforms and confer tissue specificity (549). Classically, it acts to promote glucose uptake and mitochondrial biogenesis, while reducing energy demands by inhibiting mTORC1 (239). It may also regulate more physiological functions as it has been implicated in stimulating endothelial nitric oxide production and regulating vascular smooth muscle tone (219).
Within the placental field there are limited data concerning the involvement of AMPK in placental development and function. It has recently been implicated in controlling uterine blood flow, and in adaptations to pregnancy at high altitude (505). Activation has been demonstrated to be a key step in the differentiation of mouse trophoblast stem cells under stress conditions (613), confirming that the pathway is functional in this cell type. Knockdown of the isoforms AMPKα1 and AMPKα2 in murine SM10 trophoblast progenitor cells has been shown to affect cell nutrient transport, inhibiting expression of Glut3 and blocking translocation of the protein to the cell surface, but increasing the activity of system A transporters (93). In addition, knockdown inhibits cell proliferation and cytokine-induced differentiation. Dietary restriction (50% of controls) during early to mid-gestation in ewes resulted in increased activation of AMPK in the fetal cotyledonary tissues at d78 but not later on d135 (353), whereas the reverse was the case with overnutrition (150% of controls) (615). In the latter situation there was reduced vascularity within the placentomes, suggesting perturbation of VEGF signaling. Similar inhibition of AMPK signaling has been reported in the placenta of rats fed an obesogenic diet of high-saturated fats (205).
C. Protein Synthesis Inhibition
A common, immediate response of cells to oxygen or nutrient deprivation is to suppress nonessential energy-demanding processes to harbor what resources are remaining and maximize the chance of survival. Protein synthesis represents one of these processes, for incorporation of a single amino acid into a polypeptide chain involves four high-energy bonds, two ATP and two GTP. Most of the data relating to regulation of protein synthesis in response to hypoxia come from the cancer field (542, 585, 586), but it appears that the same pathways are operative in the placenta under physiological and pathological conditions (596, 598). There are two principal mechanisms that operate, a rapid response involving phosphorylation of eIF2α through PERK and the UPR, and a longer term response dependent on suppression of the mTOR pathway (181, 585). Both of these mechanisms regulate cap-dependent mRNA translation.
It should be appreciated that these blocks lead to a selective rather than a global inhibition of protein synthesis. Indeed, one of the actions of the UPR is to upregulate cellular antioxidant defenses and ER chaperone proteins, and to stimulate ER biogenesis to increase the folding capacity and promote cell survival. Selected mRNAs must therefore be able to bypass this translational arrest, and it has been suggested that those containing small upstream open reading frames (uORFs) within their 5'-UTR regions or internal ribosome entry site (IRES) sequences are able to do so (278, 349). More recently, it has been proposed that HIF-2α is able to combine with RBM4 at hypoxia response elements within the 3'UTR of a selective subset of mRNAs to initiate translation under hypoxic conditions (539). It is to be expected, therefore, that the secretory output of the trophoblast might change both quantitatively and qualitatively under conditions of stress.
Protein synthesis is essential for normal development, and blocking dephosphorylation of eIF2α in mice results in severe growth restriction and early embryonic lethality (241). Equally, evidence of translational arrest has been reported in human placentas from normal healthy pregnancies at high altitude (3,100 m) where growth of the villus tree is impaired (598). These placentas display increased phosphorylation of eIF2α, reduced phosphorylation of AKT and 4E-BP1, and an increase in total 4E-BP1 that will favor sequestration of eIF4E. These changes can be recapitulated by exposing trophoblast cell lines to hypoxia (1% O2), when they are associated with reduced cell proliferation. Such mechanisms may provide the homeostatic means for matching placental and fetal growth to the reduced ambient oxygen concentration, as previously described. The same placentas also show a decrease in the complexes of the mitochondrial electron transport chain at the protein, but not mRNA, level, consistent with translational arrest (121). Indeed, treating trophoblast cell lines with salubrinal, an eIF2α-phosphatase inhibitor, is sufficient to lower the complexes under normoxic conditions (121). There is thus a danger of a feed-forward vicious circle developing if glycolysis is insufficient to maintain energy levels under stress conditions.
Evidence of more severe translational arrest has been reported in placentas from growth-restricted pregnancies of maternal vascular origin when placental weight is significantly reduced (596). Marked increased phosphorylation of eIF2α and decreased phosphorylation of 4E-BP1 were observed, and more significantly, all three isoforms of AKT were reduced at the protein, but not at the mRNA, level. Activation of the UPR has also been reported in placentas from cases of early-onset, but not late-onset, preeclampsia (595), when there is often accompanying growth restriction. It is notable that certain placental proteins, such as leptin, VEGF, and its receptor-soluble fms-like tyrosine kinase (sFlt-1), are markedly increased in early-onset preeclampsia. Genomic sequence analysis revealed that the encoding genes contain either uORFs or IRES sequences or both. These factors may be responsible for the maternal endothelial cell activation that typifies preeclampsia.
VIII. INTEGRATION OF SUPPLY AND DEMAND AT THE PLACENTAL INTERFACE
The placenta is not just a passive conduit for nutrient transfer. It has a dynamic role in optimising resource allocation between the fetus and mother during pregnancy (74). This is particularly apparent in late gestation when the fetus is growing rapidly in absolute terms or when resources are scarce due to poor maternal nutrition or nutrient reserves. The importance of this balance to the successful outcome of pregnancy also depends on the total uterine mass relative to maternal body size both within and between species, and on the particular mix of nutrients required by the fetus(es) relative to the metabolic ability of the mother to supply these nutrients in the correct proportions and absolute amounts (193).
The fetus demands nutrients from the mother via the placenta to improve its fitness since a large neonate with significant fuel reserves is more likely to survive at birth and onto reproductive age (372). This drive is mediated partly through the expression of imprinted genes that are expressed from paternal alleles and promote growth of the placental tissues (42). In evolutionary terms, the role of imprinting has been explained as a means by which the male optimizes the spread of his genome through the population (391). While the mother also benefits from this resource investment as her genes too are transmitted to the next generation, a balance must be struck as maternal investment in the current fetus leaves less reserves for future pregnancies (193). There is, therefore, both cooperation and conflict between the mother and her fetus, and between siblings in litter-bearing species, in resource allocation at the placental interface (193). This leads to adaptations in placental phenotype designed to optimize maternal-fetal fitness with respect to the conditions prevailing during the current pregnancy. Changes in placental phenotype in response to environmental cues can, therefore, be seen as a coadaptive response mutually beneficial to the mother and fetus in the successful outcome of pregnancy (305). However, in the polyandrous mating systems used by many mammals, competition between mother and fetus at the placental level is potentially more intense because of the differing contributions to the fetal genomes of half-siblings in demanding resources from the mother to the detriment of other half-siblings in future pregnancies (390). This leads to differential selection pressures on maternally and paternally inherited alleles in the conceptus with respect to resource allocation and is manifested as genomic imprinting, a mechanism for monoallelically regulating gene dosage in a parent-of-origin fashion (305, 391). Imprinted genes, therefore, have an important role in the developmental plasticity of the placenta, particularly with respect to resource allocation (189, 445).
Inter- and intraspecies crosses between breeds of several species have shown that the mother can constrain the fetal genetic drive for growth while, conversely, the fetal genome can influence the mother to provide more resources with consequences for fetal growth and pregnancy outcome (8, 457, 551). Similarly, direct manipulation of the fetal genome by gene deletion or disruption is known to alter resource allocation to fetal growth via alterations in placental phenotype (198). Thus the placenta acts as an environmental sensor, integrating signals of the current availability of oxygen and nutrients, maternal stores of nutrient reserves, and the fetal nutrient demands for growth driven by the genes and actual mass of the fetus (74, 204). The adaptive responses of the placenta to these signals depend on the species, stage of gestation, total uterine mass, evolutionary history, and the specific nature of the environmental cues (546). However, the integration of nutrient-response systems in the placenta has not been fully determined.
A. Hypoxia
Hypoxia is one of the most common complications of human pregnancy, occurring in 9–10% of pregnancies at sea level and all pregnancies at high altitude (484, 600). It affects both the placenta and fetus and is associated with impaired trophoblast invasion, poor villous development, altered vascularity, reduced spiral artery remodeling, and low blood flow on both sides of the placenta (387, 543, 600). The placental effects of hypoxia have been studied in a number of species using both in vivo and in vitro approaches. In humans, the studies have concentrated on the chronic hypoxia of high altitude and pathologies such as preeclampsia, whereas, in experimental animals like sheep and mice, they have focussed more on short term, acute hypoxia at different stages of pregnancy (255, 271, 363, 543, 600). At the placental and fetal levels, maternal hypoxia, the diminished availability of oxygen, manifests most frequently as hypoxemia, a low level of oxygen in the blood.
1. Placental size, morphology, and blood flow
Development of the human placenta at high altitude has been studied in populations in Saudia Arabia, Kirghizstan, the Himalayas, and both South and North America (7, 276, 458, 532, 607, 612). These show no consistent effect of chronic hypoxia on placental weight or size with increases, decreases, and no change depending on the study and/or population. Among studies at high altitude, the most consistent findings are alterations in the uterine vasculature and blood flow and an increase in fetal capillary density in the placenta in association with fetal growth restriction. On average, there is a 100 g decrease in birth weight per 1,000 m elevation in altitude, although the exact figure varies with the ethnicity of the population (387). Lower birth weight at higher altitudes is seen with both long and short residency at high altitude regardless of nutrition or socioeconomic class (214, 386, 389). However, the high-altitude decline in birth weight is less in populations with longer residency at high altitude, such as the Tibetans and Andeans, than in populations like the Han and Europeans who have settled at altitude more recently (214, 387, 443). These ethnic differences in birth weight have been related to better placental adaptation to low Po2 in populations with a longer evolutionary history at high altitude.
Although alterations in the uterine vasculature and blood flow are a common feature of pregnancy at high altitude, both increases and decreases in these parameters have been reported during late gestation relative to lowland populations (387). This is likely to relate to differences in the ethnicity, altitude, and obstetric history of the populations studied and in the methods used to measure and calculate blood flow in the different studies (69). However, in the majority of studies, the normal pregnancy-induced rise in uterine blood flow is blunted at high altitude in association with a reduced diameter of the uterine arteries and/or lower nitric oxide synthesis, regardless of ancestry (294, 295, 577, 603, 604). This suggests that remodeling of the uterine arteries during human pregnancy is impaired at high altitude (532), in line with the greater incidence of preeclampsia in these populations (429). An increase in placental weight was recently reported in mice exposed to 13% oxygen throughout gestation, but only in those associated with male fetuses (363) (Table 3). Male placentas also showed more resistance to oxidative stress, and fetal growth restriction was notably significantly less than in their female littermates, suggesting the placentas had been able to compensate. In pregnant sheep at high altitude, the luminal cross-sectional area, but not the number, of maternal vessels in the placentomes is increased, which results in a greater percentage area of maternal blood vessels and normal fetal growth (317, 437). Sheep evolved at higher altitudes than human populations and, hence, may be better adapted to pregnancy in hypoxic conditions (600).
Table 3.
Effect of maternal hypoxia during pregnancy on the morphological and transport phenotype of the mouse placenta
| Weight, % Control |
Placental Characteristics |
||||||
|---|---|---|---|---|---|---|---|
| Hypoxic Challenge | Duration, days (D) | Age at Study, days (D) | Placenta | Fetus | Morphology | Transport | Reference Nos. |
| 16% O2 | D1 onwards | D19 | No change | No change | No data | No data | 363 |
| 13% O2 | D1 onwards | D19 | ↑10% | ↓13% | ↑Maternal blood spaces | 363 | |
| D11–D16 | D16 | No change | No change | ↑Maternal blood spaces | No change in glucose or MeAIB transport | 256 | |
| ↑Lz volume and surface area | |||||||
| D14–D19 | D19 | No change | ↓5% | ↑Fetal capillary volume | ↑Glucose transport | ||
| ↓Barrier thickness | ↑Slc38a1 | ||||||
| 12% O2 | D15–D19 | D19 | No change | ↓6–7% | ↓ Lz blood vessel volume | ↓Slc2a1 and GLUT1 (female only) | 133 |
| ↑Slc38a1, ↓Slc38a2 | |||||||
| D16–D19 | D19 | ↓6% | ↓28% | ↑Lz specific genes | ↓Passive permeability | 535 | |
| 10.5% O2 | D11–D19 | D19 | No change | ↓38% | ↓Umbilical artery flow | 475 | |
| D16–D18 | D18 | No data | No data | ↑Lz vascular density | ↓Slc7a2 | 212 | |
| 10% O2 | D14–D19 | D19 | No change | ↓21% | ↓Maternal blood spaces | No change in glucose transport | 256 |
| ↓Fetal capillary % volume | |||||||
| ↑Barrier thickness | ↓MeAIB transport | ||||||
| 7% O2 | D10–D11 | D11 | No data | No data | No gross morphological changes | 483 | |
In human populations, the adverse consequences of high altitude on uterine artery diameter and uterine blood flow are less pronounced with long than short ancestry at high altitude (295, 387). Pregnant Tibetan women have higher uterine artery diameters and blood velocity than Han women at high altitude (107, 389), while Andean women have twice the increment in uterine artery diameter during pregnancy than European women at high altitude (295, 577, 604). This results in differences in uterine blood flow between Andean and European women, which are detectable at 20 wk of gestation before fetal growth slows (295). In some studies, the protective effect of Andean ancestry on uterine artery hemodynamics is only seen at high altitude while in others, the ethnic differences are also evident at sea level (295, 604). Indeed, in Andean women, the increase in uterine blood flow during pregnancy at high altitude can exceed that seen at low altitude (69, 295). Thus, in some studies of Andeans, absolute oxygen delivery to the gravid uterus at high altitude is maintained or even increased above lowland values, despite the low maternal Po2, while in others the uterine oxygen supply is less at high than low altitude irrespective of ancestry (69, 295, 443, 604). However, for any given altitude or ancestry group, fetal size is related to the absolute rate of uterine oxygen delivery, so weight-specific rates of uterine oxygen delivery vary less with altitude and ancestry than the absolute values (295, 604). The fetus is, therefore, growing in relation to its overall oxygen availability.
At high altitude, there are also changes in the vasculature and blood flow on the fetal side of the placenta in both humans and sheep (317, 431, 443). In human infants near term, the diameters of the umbilical vein and artery are both smaller at high than low altitude, which results in a lower absolute blood flow in the umbilical circulation, irrespective of ethnicity (295, 443). However, babies of European ancestry are more adversely affected than those of Andean descent (443). In all high-altitude populations studied to date, there is increased villous vascularization as a result of increased vasculogenesis and angiogenesis (7, 78, 176, 509, 532). Depending on the study, the increase in fetal capillary density in the human placenta may be due to an increased capillary number, diameter, or length. At high altitude, there is increased branching and reduced coiling of the fetal capillaries in the placenta with more densely packed capillary loops in the terminal villi responsible for gas exchange (7, 532). The fetal capillaries are also longer and thinner in Andean than European/Mestizo placentas (275). This increase in villous vascularization at high altitude appears to occur without any consistent increase in villous surface area or volume (176, 365, 370, 387, 458). In addition, there is thinning of the interhemal membrane in the human placenta at high altitude (276, 366, 458). This is achieved through selective dilation of the capillary sinusoids at the vasculosyncytial membranes (78), a mechanism that increases placental diffusing capacity but has minimal effects on extracorporeal blood volume and the load on the fetal heart.
Similar increases in fetal vascularity are seen in ovine placentomes at high altitude, but, in contrast to the findings in the human placenta, these are accompanied by an increase in the total surface area of fetal-maternal contact for gas exchange (317, 431). In the mouse placenta, vascularity has been shown to be increased in late gestation by 48 h of hypoxia, but decreased by longer exposures (133, 212) (Table 3). Earlier in gestation, there appears to be little, if any, change in placental morphology during severe hypoxia of the mouse dam (483). In neither of these species is there evidence for thinning of the interhemal membrane in response to hypoxic conditions (133, 317). Overall among species, the morphological adaptations of the placenta will increase its oxygen diffusion capacity and aid oxygen delivery to the fetus at low maternal Po2 (370, 387, 431). Indeed, per kilogram of fetus, placental oxygen delivery to the human fetus and its rate of oxygen consumption near term are normal at high altitude, although the fetuses are smaller (443, 605). Similarly, in sheep, fetal oxygen consumption is maintained when fetal-placental hypoxia is induced by restricting uterine blood flow and, hence, uterine oxygen delivery for 24 h (53, 263).
2. Placental metabolism and nutrient transport
In addition to the morphological adaptations in the high-altitude placenta, there are changes in placental metabolism that may spare oxygen for onward passage to the fetus (271). In Bolivian women, oxygen consumption by the uteroplacental tissues near term appears to be ∼20% less at high than low altitude in the absence of any change in placental weight (605). This is greater than the 13–15% reduction in absolute uterine oxygen delivery observed between these high- and low-altitude populations of pregnant women (604, 605). Measurements of absolute rates of uterine and umbilical glucose uptake suggest that the placenta may be using up to 60% more glucose at high altitude (605). The placental content of glucose and lactate also tend to be lower and higher, respectively, at high relative to low altitude (534). In addition, in the high-altitude human placenta, there is reduced abundance of all four complexes of the mitochondrial electron transport system (ETS) responsible for oxidative phosphorylation (121). The lower ATP/ADP ratio and the trend towards higher levels of the energy store, phosphocreatine, in these high-altitude placentas also suggest that there is a greater coupling of ATP demand to production and alternative sources of energy other than oxidative phosphorylation at high altitude. Similarly, during in vitro studies of cultured mouse trophoblast cells, hypoxia decreases abundance of cytochrome oxidase c, a component of the ETS involved in generating the proton gradient used for mitochondrial ATP synthesis (588). Collectively, these observations suggest that the chronically hypoxic placenta at high altitude may switch from oxidative phosphorylation to a greater dependence on anaerobic glycolysis to meet its ATP requirements, which may increase fetal oxygen availability albeit at the expense of the fetal glucose supply (271, 403). The finding that placental mitochondrial oxygen consumption under state III conditions (ADP stimulated) is higher in Tibetan than Han women also suggests that these metabolic adaptions may be dependent on ethnicity and/or duration of high-altitude residency (612).
The proposed metabolic switch in the high-altitude placenta will have consequences for fetal metabolism because, although fetal oxygen consumption is maintained on a weight-specific basis, the fetus has lower than normal glucose concentrations and uses less glucose per kilogram body weight measured as umbilical glucose uptake (605). Fetal oxygen consumption must, therefore, be maintained by oxidation of substrates other than glucose, such as amino acids and/or fats, which will then reduce their availability for other purposes (403). Since the percentage decrease in fetal glucose delivery appears to be greater than the percentage reduction in net fetal glucose consumption measured as umbilical glucose uptake at high altitude (605), there may also be activation of gluconeogenesis from lactate and amino acids by the high-altitude fetus near term. There is an increase in the fetal arterial concentration of lactate in these fetuses, but little is known about their rate of lactate consumption or about the rates of placental production and delivery of lactate at high altitude (534, 605). The reduced availability of both glucose and amino acids for tissue accretion will, therefore, decrease fetal growth in line with the oxygen supply.
With shorter episodes of severe hypoxia (<48 h) in pregnant sheep, there is evidence for activation of fetal glucogenesis and increased delivery of glucose and lactate to the placenta from the fetal circulation (230, 263, 292). This short-term type of hypoxic challenge also induces changes in umbilical blood flow, with increases or decreases in flow immediately after the onset of maternal hypoxia depending on its severity (380, 524). Similarly, umbilical flow increases transiently 1–4 h after inducing placental-fetal hypoxemia by uterine artery constriction in ewes, but then normalizes as the period of restricted uterine flow is extended to 24 h or more (52, 230, 263, 380, 524). In line with the alterations in umbilical flow, there are changes in the placental delivery and fetal consumption of oxygen. Initially, fetal oxygen consumption decreases in line with placental delivery but then recovers to normal values despite the sustained low delivery by increasing oxygen extraction (230, 263, 380). Placental oxygen consumption is maintained at normal values for up to 48 h of hypoxemia (230, 263). Consequently, there is little evidence for placental oxygen sparing in response to acute normobaric hypoxia in the sheep (230, 263, 380), as may occur in the human placenta in response to the chronic hypoxia of high altitude (271).
In contrast to oxygen, the ovine placenta appears to spare glucose for onward passage to the fetus during acute hypoxemia as fetal glucose delivery and consumption are maintained at the expense of utero-placental glucose consumption for periods of up to 24 h (53, 230, 263, 380, 525). In these circumstances, the normal rate of placental oxygen consumption may be maintained by oxidation of lactate derived from the lactacidemic, hypoxemic fetus (230, 263). Certainly, lactate production by ovine utero-placental tissues is reduced by 24 h of placental-fetal hypoxemia induced by restricting uterine blood flow or placental growth by hyperthermia (263, 456). Little is known about the changes in abundance of the glucose or lactate transporters in the ovine placenta during hypoxic conditions (610). In the human placenta in vitro, acute hypoxia leads to upregulated expression of the GLUT1 and GLUT3 (178, 249). In contrast, in the high-altitude human placenta, GLUT1 is reduced at the basal but not the microvillous membranes, consistent with the decreased placental transfer of glucose to the fetus in conditions of chronic maternal hypoxia (601, 605). There is also a sex-linked decrease in placental Slc2a1 (GLUT1) gene expression in the mouse placenta after 4 days of maternal hypoxia in late gestation (Table 3).
Much less is known about fetal-placental amino acid metabolism during either acute or chronic hypoxemia. In the human placenta, concentrations of several key essential amino acids were unaffected by altitude, with the exception of glutamine and the antioxidant taurine which were higher in concentration at high altitude than at sea level (534). However, there is evidence for a decrease in protein synthesis in the placenta of nonnative women at high altitude (598). In human placentas in vitro, exposure to acute episodes of hypoxia reduces expression and/or activity of accumulative system A amino acid transporters but increases activity of system L amino acid transport (313, 412, 547). In pregnant sheep, acute hypoxia for 4 h decreases the placental supply and fetal use of leucine in association with a general increase in amino-nitrogen availability in the fetal circulation and reduced rates of fetal protein synthesis and proteolysis (380). The net effect of these changes is a reduction in protein accretion by the fetus. In the mouse placenta, maternal hypoxia for 2–4 days in late pregnancy leads to reduced expression of an isoform of the y+ system of cationic amino acid transporters and increased expression of the Slc38a1 isoform of the System A amino acid transporters (Table 3).
Collectively, the studies show that the placenta tolerates hypoxemia well, but adapts its metabolism and transport characteristics to cope with the reduced oxygen availability (485). However, its strategy appears to differ with species, duration, severity, and timing of the hypoxic insult, and/or the presence of fetal hypoxemia (393). At high altitude with chronic maternal hypoxemia but mild fetal hypoxemia, the human placenta reduces consumption of oxygen but increases use of glucose (271). At low altitude in response to acute hypoxia and fetal hypoxemia, the sheep placenta maintains its rate of oxygen consumption but reduces its use of glucose, while increasing uptake of lactate from the fetal circulation. In both scenarios, fetal metabolism is altered. In the high-altitude human fetus, there is reduced glucose consumption but normal oxygen consumption, while in fetal sheep exposed to 4 h or more of acute hypoxia, normal rates of glucose and oxygen consumption are maintained coupled with fetal glucogenesis and altered amino acid turnover. In both species, the changes in placental and fetal metabolism will have adverse consequences for fetal growth.
3. Placental endocrine function
Both in vivo and in vitro studies have shown that hypoxia affects placental production of a wide variety of hormones including protein, glycoprotein, eicosanoid, and steroid hormones (Table 4). Altered placental endocrine function is seen in response to both chronic and acute hypoxia, and reflects changes in gene expression, protein synthesis, and metabolism and secretion of hormones (Table 4). These endocrine changes do not appear to be a strategy to reduce placental energy expenditure, as there are both increases and decreases in placental hormone production (Table 4). In addition to the endocrine outcomes of poor oxygen availability, there are also paracrine changes within the placenta itself, which will contribute to the adaptations in its morphological and transport phenotype (191). Furthermore, hypoxia induces changes in the placental barrier influencing transfer of maternal hormones to the fetal circulation (Table 4). For instance, in human and mouse placentas hypoxia reduces expression of 11β-HSD2, potentially compromising the inactivation of maternal cortisol and exposing the fetus to hypercortisolemia (133, 242). Conversely, hypoxia increases expression of the thyroid hormone binding protein involved in transferring maternal thyroid hormones to the human fetus (434).
Table 4.
Effects of hypoxia on placental secretion and metabolism of hormones determined either in culture or by secretion into the uterine or umbilical circulations
| Species | Experimental Approach | Age Studied | Endocrine Change | Reference Nos. |
|---|---|---|---|---|
| Human | ||||
| In vivo | High altitude 3,100 m (≈14% O2) | Term | ↑P4 ↓E2 | 602 |
| In vitro | Cytotrophoblast culture 2% O2 | Mid term | ↓CYP19 mRNA (aromatase) | 289 |
| Cytotrophoblast culture 1% O2 24 h | Term | ↓11βHSD2 mRNA | 242 | |
| Trophoblast cell line 1% O2 24–48 h | ↑Leptin production | 375 | ||
| Trophoblast culture 1–3% O2 24 h | Term | ↑Thyroid hormone binding protein | 434 | |
| Trophoblast culture 72 h | Term | ↓ hCG, ↓hPL, ↓P4, ↓E2 secretion | 177 | |
| Trophoblast culture 1–2% O2 24–72 h | Term | ↓ hCG &↑PGE2 secretion, ↑PGH synthase2 | 411 | |
| Villous explant 3% O2 24–48 h | Term | ↑ABCB1(P-glycoprotein) | 287 | |
| Villous explant 2% O2 24 h | Term | ↓PGE2 production | 51 | |
| Perfused whole placenta 11% O2 | Term | Altered rations of PGE2/PGF2α | 167 | |
| Sheep | ||||
| In vivo | High altitude 3,589 m (≈13.5% O2) | 100 day term | ↑P4 | 432 |
| High altitude 3,820 m (≈13% O2) | 130 days | ↑Leptin mRNA | 158 | |
| Mouse | ||||
| In vivo | Maternal inhalation hypoxia 12% O2 | 14.5–18.5 days | ↓Igf2 mRNA, ↓11βHsd2 mRNA, ↓Ace-2 mRNA | 133, 134 |
| Maternal inhalation hypoxia 10.5% O2 | 15.5–17.5 days | ↑Angiotensin, ↑ACE-1, ↑ACE-2 proteins | 226 | |
| Maternal inhalation hypoxia 10% O2 | 5.5–11.5 days | ↑Prolactin-like protein-E mRNA | 259 |
The changes in placental hormone synthesis and metabolism in response to hypoxia are likely to have consequences for both the mother and her fetus. They may contribute to the observed changes in uterine and umbilical blood flows, and influence the maternal metabolic adaptation to pregnancy during hypoxic conditions. Certainly, the normal pregnancy-induced increase in maternal insulin resistance associated with increased placental production of somatotrophic and steroid hormones is absent in women chronically hypoxic at high altitude (316). Studies in women and sheep at high altitude have also shown that placental steroid production depends on the length of residency at altitude (105, 432). In turn, this may explain some of the ethnic differences in birth weight seen between populations with long and short ancestry at altitude (69).
B. Nutrition
The ability of the mother to provide nutrients to the fetus is determined, in part, by her nutritional state. This involves her diet, body composition, and fuel reserves both before and during pregnancy, as well as her metabolic and physiological adaptations to the pregnancy per se. In addition, maternal nutrition is a major determinant of placental development and alters the morphological, transport, and endocrine characteristics of the placenta with consequences for the fetal nutrient supply in a wide range of species, including laboratory and farm species as well as human and non-human primates (47, 144, 204, 522, 550). Both under- and overnutrition during pregnancy are effective at altering placental development in human and other species. There are also interactions between the current nutritional environment of the mother and her past nutritional history, as indicated by her body mass index (BMI) and body composition, in determining placental phenotype (29, 521, 527, 556). In experimental animals, the role of nutrition in regulating placental phenotype has been studied by varying dietary composition and maternal intake of calories, macro- and micronutrients and of other substances with metabolic actions such as alcohol, antioxidants and hormones (546). These dietary manipulations have been applied to induce obesity, for example, prior to conception and/or after establishment of pregnancy, or to investigate the effects of more acute nutritional changes later in gestation. In addition, changes in nutritional state and food intake during pregnancy often accompany, and are confounding factors in studying, other environmental challenges such as hypoxia, heat stress, exercise, and alterations in housing, lighting, and noise levels (482).
1. Placental size, morphology, and blood flow
Relative to the hypoxia of high altitude, much less is known about the effects of nutrition on development of the human placenta. The size of the human placenta at term is known to be affected by the calorie intake and dietary composition during the pregnancy (216, 350, 471). It is also positively related to maternal BMI across the normal spectrum, from the underweight to the morbidly obese (556). Variations in the balance between protein and carbohydrate intake at different stages of human pregnancy are related to placental and infant size at birth, with reduced protein intake in late gestation associated with a smaller placenta (216). Women who gain weight between pregnancies are more likely to have a large placenta subsequently, whereas those with significant inter-pregnancy weight loss are more prone to placental growth restriction in their second pregnancy (553). Formal fasting in late pregnancy during Ramadan is also associated with reduced placental weight at term in Saudi Arabian and Tunisian women (10, 12). In the Dutch hunger winter populations, placental weight at term was increased when the famine occurred in the first trimester but was reduced, along with infant birth weight, in women who were in their third trimester at the time of the famine (350, 471). However, despite this, placental efficiency measured as the fetal to placental weight ratio was greater in response to the undernutrition in late pregnancy than seen in control pregnancies before the famine (350, 471). Similar increases in placental efficiency are seen in the human populations fasting for Ramadan and in underweight women delivering small infants with a small placenta (10, 556). Taken together, these observations suggest that the human placenta either has a significant reserve capacity or can adapt its nutrient transport capacity during nutritional compromise to support fetal growth. There is also some evidence of changes in angiogenesis in the placenta of obese women (157). In non-human primates, there are increases in placental infarction and reductions in uteroplacental blood flow in response to feeding a high-fat diet, which are more pronounced in mothers that become obese than in those who remained lean on the diet (200).
Similar changes in placental growth and efficiency are seen in murine and ovine placentas in response to maternal undernutrition (198, 546, 550). In pregnant mice, decreases in placental vascularity are seen in response to maternal obesity, calorie restriction, and feeding diets high in fat or low in protein, particularly in the labyrinthine zone (Table 5). There are also changes in the relative proportions of the placental zones and reductions in surface area of the labyrinth and thickness of the interhemal membrane in response to nutritional manipulations before, and during, mouse pregnancy, irrespective of the level of calorie intake or maternal adiposity (Table 5). Even relatively modest changes in dietary composition within the range recommended for rodent pregnancy can alter placental weight and zonal proportions near term (119, 301).
Table 5.
Effect of nutritional manipulation during pregnancy on the morphological and transport phenotype of the mouse placenta
| Weight, % Control |
Placental Characteristics |
||||||
|---|---|---|---|---|---|---|---|
| Nutritional Challenge | Duration, days (D) | Age at Study, days (D) | Placenta | Fetus | Morphology | Transport | Reference Nos. |
| Undernutrition | |||||||
| Low calories | |||||||
| ↓50% | D2 onwards | D12 | ↓12% | No change | ↓Placental area | 488 | |
| ↓Jz area | |||||||
| ↓Lz fetal capillary spaces | |||||||
| D10 onwards | D19 | ↓38% | ↓48% | ↓Transport of glucose and leucine | 207 | ||
| ↑System A uptake | |||||||
| ↓Slc2a3, ↑Slc2a1 | |||||||
| ↑Slc38a1 and Slc38a2 | |||||||
| ↓LAT2 protein | |||||||
| ↓20% | D3 onwards | D16 | ↓6% | No change | ↓Jz volume | ↓Slc2a1 | 120 |
| D19 | ↓9% | ↓13% | ↓Lz fetal capillary volumes | ↑MeAIB transport | |||
| ↓Lz surface area | No change glucose transport | ||||||
| ↑Slc38a1& a2, ↓Slc38a4 | |||||||
| D3 onwards | D19 | ↓9% | ↓13% | ↓Lz fetal capillary volumes | ↑MeAIB transport | 495 | |
| ↓Lz surface area | ↑Slc38a2 | ||||||
| Low protein | |||||||
| 10 vs. 20% | D11 onwards | D18 | No change | ↓16% | ↑Apoptotic gene expression | ↓ y+ system transporter mRNA | 211 |
| 9 vs. 23% | D16 | No change | No change | ↓Jz % volume | ↑Glucose transport | 119 | |
| No change MeAIB transport | |||||||
| ↑Slc2a1, ↑Slc38a2 | |||||||
| 9 vs. 18% | D1 onwards | D19 | ↑5% | ↓9% | No change zone volumes | No change glucose or MeAIB transport | |
| ↓Slc38a1 and a4, No change Slc2a1 and 3 | |||||||
| D1 onwards | D19 | ↓27% | ↓13% | ↓Vessel lengths | 476 | ||
| Overnutrition | |||||||
| High fat | |||||||
| 58 vs. 10% | −17 wk onwards | D15 | No change | No change | ↑Slc38a2 and a4 mRNA | 311 | |
| 45 vs. 10% | −8 wk onwards | D18 | No change | ↑14% | ↓%Lz, ↓ cell proliferation | 309 | |
| 15 vs. 5% | −4 wk onwards | D19 | No change | ↑20% | ↑Lipid uptake and transport | 451 | |
| No change GLUT1, no change FATP1 and 4 proteins | |||||||
| 32 vs. 11% | −4 wk onwards | D19 | No change | ↑30% | ↑Glucose transport | 293 | |
| ↑MeAIB transport | |||||||
| ↑GLUT1 and SNAT2 proteins | |||||||
| 60 vs. 5% | -D30 onwards | D19 | ↓30% | No data | Fibrosis, endothelial damage | 337 | |
| Lz vascular pathology | |||||||
| High fat and sugar | |||||||
| Fat 30 vs. 11% Sugar 36 vs. 7% | D1 onwards | D16 | ↓11% | ↓9% | ↓Fetal capillary volume | ↑Glucose transport | 496 |
| ↑MeAIB transport | |||||||
| ↑Slc2a3, ↑Slc38a2 | |||||||
| D19 | ↓8% | No change | ↓Maternal blood spaces | No change glucose or MeAIB transport | |||
| ↓Lz surface area | No change Slc2a or Slc38a isoforms | ||||||
| ↓Barrier thickness | ↑FATP1 protein | ||||||
In sheep, undernutrition during the early stages of pregnancy when the placenta is growing most rapidly leads to an increase in term placental weight, whereas nutrient restriction later in pregnancy once the placenta is fully formed leads to reduced placental weight at term (198). These changes in weight are accompanied by alterations in the gross morphology of the ovine placentomes and in their vascularity (454, 463). Similar changes in placental morphology are also seen in response to overnutrition in juvenile and adult sheep (250, 354, 454). In particular, there are decreases in capillary vascular density and/or volume in the caruncular part of the ovine placentomes, irrespective of whether the mothers were over- or undernourished during pregnancy (454, 463). In contrast, vascularity of the fetal cotyledonary part of the placentomes is increased in response to undernutrition during mid-gestation (614). When overnutrition begins before ovine pregnancy, there are increases in placentome capillary diameter at mid-gestation that are not sustained until term (354). In some, but not all, studies these changes in placental vascularity are accompanied by decreases in cell proliferation, angiogenesis, and uterine and/or umbilical blood flow (166, 550, 557). At present, the mechanisms that regulate the responses to under- or overnutrition are unknown, but likely involve the mTOR pathway and/or the generation of ROS.
2. Placental metabolism and nutrient transport
There have been relatively few studies of placental metabolism and nutrient transport with respect to nutritional state in women. The majority have concentrated on maternal obesity rather than on undernutrition or dietary composition. A recent study has shown that placental expression of GLUT1 is positively related to sugar intake in normal pregnant women (60). System A amino acid transport activity is lower in the placenta of women with smaller upper arm muscle areas, which suggests that reduced protein accretion in the mother, a proxy measure of nutritional state, may limit fetal amino acid availability (336). Reduced taurine and system A transport activity, and lower SNAT4 expression, are also seen in placental villous fragments from obese women delivering infants of normal birth weight (151, 184). In contrast, in obese women delivering larger infants, placental GLUT1 expression and system A activity are higher in the basal and microvillous membranes, respectively, than seen in women with a lean BMI (1, 279). There are also changes in placental fatty acid transport and transporter expression in obese relative to lean women irrespective of the weight of their infants (101, 156). Furthermore, mitochondrial density and expression of the electron transport chain ETS complexes decrease in the human placenta as maternal BMI increases, with the result that placental respiration is lower in obese than lean women (244, 376). Taken together, these observations suggest that nutritional state and obesity, in particular, alter the energetics and nutrient transport capacity of the human placenta, which will have consequences for fetal development. However, the heterogeneity of the placental responses to maternal obesity in relation to infant birth weight suggests that there may be additional metabolic or other factors involved in regulating placental transport phenotype in these circumstances. Certainly, the specific changes in placental ETS function in obese women appear to depend, in part, on the degree of maternal glucose intolerance (244).
More is known about the effects of maternal nutrition on placental transport and consumption of nutrients in experimental animals. In pregnant mice, both under- and overnutrition influence the transport phenotype of the placenta in late gestation (Table 5). Even relatively minor changes in dietary composition are known to alter placental clearance of glucose and amino acids with consequences for growth of the mouse pups near term (119). In part, the responses of the mouse placenta depend on the severity and duration of the altered dietary regime, and on the degree of placental and/or fetal growth restriction (Table 5). For example, a 50% reduction in food intake for the second half of mouse pregnancy leads to reduced placental glucose and amino acid delivery and severe fetal-placental growth restriction, whereas a 20% reduction in food intake for most of pregnancy upregulates amino acid transport per gram of placenta in association with relatively small reductions in fetal-placental weight close to term (120, 207). With several of the dietary manipulations including those inducing maternal obesity, the compromised growth and morphology of the mouse placenta is associated with upregulation of nutrient transport or transporter expression (Table 5). Collectively, these studies suggest that a smaller, morphologically compromised placenta adapts its transport characteristics to help maintain fetal growth in late gestation (Table 5). For instance, feeding a diet high in fat and sugar reduces fetal-placental growth at day 16 of mouse pregnancy yet upregulates placental glucose and amino acid clearance with the result that fetal weight is restored to normal by day 19, despite persisting placental growth restriction (496). However, the extent to which this strategy is successful in altering maternal-fetal resource allocation in favor of the mouse fetus depends on the actual nutritional and endocrine environment of the mother, and on the mass, gestational age, and genetic background of her litter (74, 193, 546).
In sheep, changes in maternal glucose levels induced by fasting, overnutrition, or direct experimental manipulation by maternal glucose or insulin infusion alter uterine glucose uptake and, hence, placental consumption and transfer of glucose in relation to the transplacental glucose concentration gradient driving glucose flux (247, 557). Even when hyperglycemia or hypoglycemia is prolonged in the ewe, placental glucose consumption still varies directly with the maternal glucose concentration (99, 147). Similarly, in overnourished obese adolescent ewes, there is no evidence for a change in the placental glucose transfer capacity, despite fetal-placental growth restriction, as placental glucose consumption and transfer vary normally with maternal glucose concentrations on a weight-specific basis (554). However, in late gestation once the sheep fetus has developed the capacity for glucogenesis, the placenta can consume glucose from the fetal circulation if the maternal supply is limited or concentration gradients are manipulated experimentally (190, 194, 248). With prolonged maternal hypoglycemia, distribution of uterine glucose uptake between the ovine utero-placental and fetal tissues shifts to favour the utero-placental tissues, although absolute rates of glucose consumption remain lower than normal (99). This too may reflect the ability of the sheep fetus to supplement its own glucose supply by activating gluconeogenesis, thereby reducing its demand for maternal glucose and sparing glucose for placental functions essential to maintaining pregnancy (148, 194). Ovine placental GLUT expression is altered in response to longer term variations in maternal glycemia with decreases in GLUT1 abundance in hypoglycemia conditions, and in both GLUT1 and GLUT3 abundance in response to hyperglycemia (138, 139, 247, 353). However, these changes may not have significant effects on glucose transport as the maximal capacity for placental glucose transport is much greater than the actual transport rate in the ewe (247).
Variations in maternal nutritional state have little effect on oxygen consumption by the ovine utero-placental tissues (99, 148, 190, 554). Consequently, when glucose consumption is reduced, for example, by fasting, the utero-placental tissues must be oxidizing other substrates. There are increases in the uterine uptake and utero-placental utilization of some branched chain amino acids in response to fasting ewes, which may provide an alternative source of energy (338). In addition, undernutrition of ewes from early to mid-gestation increases expression of several molecules involved in transplacental fatty acid transport at mid-gestation, although few of these changes persist until late gestation (353). Certainly, there is no measureable net uptake of fatty acids by ovine utero-placental tissues in late gestation after prolonged hypoglycemia (99). In contrast, diet-induced obesity of ewes increases expression of several fatty acid transporters in the placenta at both mid and late gestation (616).
3. Placental endocrine function
Changes in placental size due to variations in maternal nutrition or obesity are likely to affect placental hormone synthesis, and hence circulating hormone concentrations. There are reductions in maternal progesterone and estrogen concentrations in obese women and overnourished adolescent ewes (326, 328, 555). In the latter animals, the decrease in maternal progesterone levels was associated with reductions in both placental weight and expression of CYP11A1 mRNA at term compared with moderately fed adolescent ewes (328). In adult ewes, there are changes in the utero-placental synthesis and metabolism of prostaglandins in response to acute nutritional manipulations during late gestation (195). In particular, there is increased utero-placental production of prostaglandins E and F2α when maternal glucose levels fall due to fasting or insulin infusion in the ewe (192, 197). This may reflect the switch of hypoglycemic placental tissues from glucose to fat metabolism, and the concomitant increase in availability of arachidonic acid, the prostaglandin precursor (195). Certainly, infusion of glucose into the fasted ewes to restore normoglycemia also normalizes prostaglandin output by the utero-placental tissues (192). In addition, production of ovine placental lactogen (oPL) is nutritionally responsive and is increased in late gestation by short-term fasting and periconceptional undernutrition (421, 422). It is also increased by short-term glucose infusion (421). In contrast, maternal oPL concentrations are low for most of gestation in overnourished adolescent ewes in association with placental growth restriction (328). In the mouse, manipulation of dietary fat content during pregnancy also alters expression of both the growth hormone gene (Gh1) and the extensive family of prolactin and prolactin-like genes in the placenta during late gestation (356). However, the extent to which any of these alterations in hormone synthesis and metabolism are due directly to the changes in nutrient availability remains unclear, as other factors known to affect production of these hormones, such as glucocorticoid bioavailability, are also influenced by nutritional state (128, 191).
C. Genetic Manipulation of Nutrient Supply and Demand
The responses of the placenta to environmental cues, such as hypoxia and nutrition, indicate that there is significant interaction between maternal nutrient availability and fetal demands for these resources in determining maternal-fetal nutrient allocation at the level of the placenta (74, 144). The specific nature of these interactions is difficult to establish in vivo when the maternal, fetal, and placental contributions to the dynamics of resource allocation all change simultaneously, for example, in response to undernutrition. Consequently, gene manipulations in mice have been used to induce more discrete changes in fetal demand relative to the placental supply of nutrients. These studies have tended to concentrate on the imprinted genes, which are known to have a disproportionately important role in fetal-placental development and are involved in resource allocation more widely (126, 189, 385, 544). However, even with the imprinted genes, relatively few studies have examined the functional consequences for the mouse placenta of altering its growth and morphological development in relation to the fetal genetic demands for growth (537, 544).
Measurements of placental nutrient transfer or transporters have been made in a number of the genetic mutants with deletions in imprinted and other genes involved in resource allocation (Table 6). There is often an increased fetal-to-placental weight ratio that is accompanied by upregulation of glucose and amino acid transfer per gram of placenta, particularly when placental growth is restricted early in mouse development (Table 6). This helps to support fetal growth despite the reduced passive permeability of the small, morphologically compromised mutant placenta (Table 6). Indeed, for nutrients actively transported to the fetus, the reduced placental permeability may enhance net transfer by preventing backflux of nutrients into the placenta (150). Comparison of the placental-specific Igf2P0 with the complete Igf2 null mutant demonstrates clearly that the small placenta can become more efficient by increasing its nutrient transfer and transporter abundance when there is a maintained drive for growth by fetal-placental tissues still expressing Igf2 (124, 125). Even in the large H19−/+ mutant placenta, restriction of its transport capacity by simultaneous deletion of the Igf2P0 transcript leads to upregulation of MeAIB transport and Slc38a4 expression to meet the larger demand of the overgrown mutant fetus, with increased Igf2 expression in all its other tissues (14). Since changes in placental Igf2P0 expression occur in response to maternal undernutrition and feeding an obesogenic diet (120, 496), this gene transcript may have an important role in adapting placental phenotype to environmental cues. Certainly, the changes in the placental capacity for nutrient transfer induced by maternal undernutrition do not occur in the Igf2P0 null mutant (495).
Table 6.
Effects of deleting genes likely to affect intrauterine development on the morphological and transport phenotype of the mouse placenta in late gestation
| Weight, % Wild Type |
Placental Phenotype |
|||||
|---|---|---|---|---|---|---|
| Gene Deletion | Age at Study, days (D) | Placenta | Fetus | Morphology | Transport | Reference Nos. |
| Imprinted genes | ||||||
| Igf2−/− | D16 | ↓38% | ↓34% | No change glucose or MeAIB transport | 124 | |
| No change Slc38a isoforms | ||||||
| D19 | ↓40% | ↓51% | ↓Placental volume, ↓Lz% | ↓Passive permeability | 118, 124 | |
| ↓Lz surface area | ↓MeAIB transport | |||||
| ↑Barrier thickness | ||||||
| Igf2P0+/− | D16 | ↓21% | ↓4% | ↑Glucose and MeAIB transport | 124 | |
| ↑Slc2a3, ↑Slc38a4 | ||||||
| D19 | ↓33% | ↓23% | ↓Placental volume | ↓Passive permeability | 118, 125, 149, 502 | |
| ↓Lz surface area | ↑Glucose and MeAIB transport | |||||
| ↑Barrier thickness | ↑Slc38a2 | |||||
| ↑Ca2+ transport | ||||||
| Rtl1+/− | D16 | No data | No change | Abnormal vasculature | ↓Passive permeability | 492 |
| No change MeAIB transport | ||||||
| D19 | ↓18% | ↓20% | Abnormal vasculature | |||
| H19−/+ | D16 | ↑30% | ↑12% | ↑Lz surface area, no change zone % | ↓Passive permeability | 14 |
| ↑M and F vessel volumes | ↓Glucose transport, ↓Slc2a3 | |||||
| No change MeAIB transport | ||||||
| D19 | ↑45% | ↑30% | ↑Lz surface area, no change zone % | ↓Passive permeability | ||
| ↑M vessel volumes | ↓Glucose and MeAIB transport | |||||
| ↓Slc38a4 | ||||||
| Phlda2−/+ | D19 | ↑30% | ↓4% | Lz surface area, Jz% | ↓MeAIB transport | 15, 199 |
| Grb10−/+ | D19 | ↑30% | ↑45% | ↑ Lz% | No change Slc38a4 | 104 |
| Other genes | ||||||
| Glut3+/− | D19 | No change | No change | ↓Glucose uptake and transport | 207 | |
| ↑Leucine transport | ||||||
| ↑MeAIB transport | ||||||
| ↑SNAT1 and SNAT2 | ||||||
| 11βHsd−/− | D15 | ↓11% | No change | No change zone % or blood spaces | ↑MeAIB transport, ↑Slc38a2 and a4 | 587 |
| No change glucose transport | ||||||
| No change Slc2a isoforms | ||||||
| D18 | ↓6% | ↓12% | ↓Fetal capillary volume, length and density | No change MeAIB transport | ||
| ↓Lz% | ↓Glucose transport | |||||
| No change Slc38a isoforms | ||||||
| ↓Slc2a3, no change Slc2a1 | ||||||
| eNOS−/− | D18 | No change | No change | ↓Fetal capillary length and density | ↓MeAIB transport | 318, 320 |
| No change Slc38a isoforms | ||||||
| DnmtΔ1o | D18 | ↑30% | ↑20% | ↑Jz%, ↓Lz% | ↑Slc2a3 | 258 |
| ↓Lz surface area | ||||||
| ↓Fetal vasculature | ||||||
When the fetal demand and supply are mismatched in the naturally small placenta, there are changes in expression of several imprinted genes, including Igf2 in association with sparing of the labyrinth zone, increased MeAIB and glucose transport, and upregulated expression of the Slc38a2 amino acid transporters (114). As a result, the naturally small placenta also supports more fetal growth per gram and maintains a normal fetal growth rate in late gestation (114). Similarly, when the placental supply of glucose to the fetus is disrupted by deletion of the Slc2a3 gene, the placenta compensates by upregulating placental amino acid transport and transporter expression sufficiently to maintain normal fetal growth and metabolism until term (207). Furthermore, when placental growth is restricted by glucocorticoid overexposure in the 11βHsd2-null mutant, there is upregulation of placental amino acid transfer and transporter expression in association with maintained fetal growth for the first 15 days of gestation (587). However, this upregulation is not maintained into late gestation, probably due to the decreased fetal demand associated with the direct growth inhibitory effects of excess glucocorticoids on fetal tissues late in gestation (191, 587). Thus a mismatch between nutrient supply and demand appears to drive an upregulated capacity for transplacental nutrient transport, particularly when the placenta is small in relation to the fetal genetic drive for growth and the mother has the ability to provide the additional nutrients.
Conflict between the fetal demands for nutrients and the maternal capacity to supply them may also underlie the altered transport characteristics of those mouse mutants with placentomegaly (Table 6). The reduced passive permeability, nutrient transport, and/or transporter expression of these mutant placentas may reflect the dominance of maternal signals that constrain maternal-fetal resource allocation in the face of the increased drain posed by the overgrown conceptuses in late gestation (14, 15). There is also evidence for intersibling competition for nutrients between mutant and wild-type fetuses within mixed litters, which affects the size of the wild-type pups (14, 104, 124). Taken together, these observations suggest that maternal constraint is an important factor in regulating placental phenotype, not only when maternal nutritional resources are limited but also when fetal demands increase rapidly in late gestation due either to large litter sizes or genetically induced conceptus overgrowth. Certainly, upregulation of placental amino acid transport is maintained until day 19 in mice with complete Igf2P0-null litters but not in Igf2P0 mutants of dams with mixed litters of mutants and wild types, which have a greater total conceptus mass and, thus, demand for nutrients in late gestation (124, 495). In addition, variations in fetal-placental growth induced genetically are known to alter the metabolic and endocrine environment of the dam (439, 495). However, whether these maternal changes are a consequence of altered placental endocrine function or alterations in fetal-placental nutrient demand remains unknown. The placenta is, therefore, integrating maternal and fetal signals of resource needs along with its own growth and metabolic requirements in controlling maternal nutrient allocation to the gravid uterus. This dynamic adaptation in placental phenotype optimizes fetal fitness in the prevailing conditions while maintaining sufficient maternal resource for lactation and subsequent pregnancies (144, 193).
IX. POSSIBLE MECHANISMS LINKING THE PLACENTA AND DEVELOPMENTAL PROGRAMING
The placenta clearly plays a critical role in the maternal-fetal supply line, but its potential influence on programming must be set in the context of other nonplacental candidates, including gametogenesis in both parents, fertilization, transport of the conceptus in the oviduct, lactation, and postnatal nutrition. Attributing causation, or even a proportion of it, to placental changes is therefore problematic, as the same environmental insult may affect several different systems. In addition, the placental changes may be secondary to programming within the embryo/fetus, or simply a parallel response to the same insult independent of that of the offspring (58). Nonetheless, the placenta is in a key position to modulate signals coming from the mother before they are transduced to the embryo/fetus and can influence programming in at least four ways (Figure 11).
FIGURE 11.
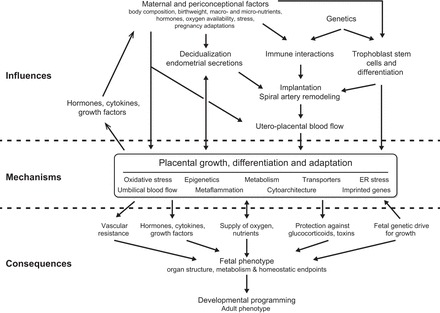
Schematic summary showing how various environmental influences may interact with, and be modulated by, the placenta and the consequences for developmental programming of the fetus.
First, its capacity to deliver sufficient oxygen and macro- and micronutrients to sustain normal fetal growth may be impaired. This may be due to a number of causes, but establishment of an adequate maternal blood flow to the placenta represents the final common pathway for many in the human. The maternal circulation to the placenta is dependent on remodeling of the spiral arteries, which in turn is reliant on invasion of the endometrium by extravillous trophoblast cells during the first and early second trimesters. The remodeling process is still far from understood, but brings together genetic, endocrinological, and local endometrial factors. Deficient remodeling leads to malperfusion of the placenta, causing loss of function through oxidative and ER stress, diminished surface area through reduced growth and increased infarction, mechanical damage to the syncytiotrophoblast, and hypoxemia. The end result will be altered development of the fetal organs, as seen in cases of severe maternal undernutrition (215, 374, 565).
Second, the same stresses may compromise the protective, barrier functions of the placenta, allowing exposure of the embryo/fetus to abnormally high levels of maternal glucocorticoids, drugs (both therapeutic and recreational), xenobiotics, and pathogens (129).
Third, the placenta secretes a variety of factors into both the maternal and fetal circulations. Perturbation of that secretion may impact on fetal development, either directly or indirectly via alterations in maternal metabolism. For example, placental prostaglandins or the release of pro-inflammatory cytokines, such as TNF-α in response to oxidative or ER stress, may influence fetal cardiovascular development through their effects on the ductus venosus and endothelial cells respectively. Equally, placental hormones, such as the IGFs, stimulate fetal organ growth, and impact on maternal nutrient supply through their actions on appetite, the endometrial glands, pancreatic β cells, and peripheral insulin resistance as discussed in section IIIB. The impact of placental release of microRNAs in exosomes is only just beginning to be explored, but this represents another potentially powerful signaling mechanism relaying information bidirectionally from the organ.
Fourth, there may be mechanical influences imposed by the placental and vitelline vascular beds on the developing cardiovascular system (Figure 4). The resistance offered by the extracorporeal circulations exerts a powerful influence on the development of the entire fetal arterial tree, in addition to its effects on the heart (530). The heart appears to be particularly vulnerable during the early embryonic and late fetal periods of development (528), so both the vitelline and placenta must be considered. With regards to the latter, placental surface area was found to be inversely related to ultrasound measurements of umbilical arterial resistance in a prospective cohort of nulliparous pregnancies (481). The effect on the developing heart will be exaggerated in pathological pregnancies, where poor placental development is associated with absent or reversed end-diastolic umbilical arterial flow (513).
It is also possible that the placenta may modify fetal growth and development by mechanisms as yet unknown. For example, it may provide stem cells to the mother and fetus, or may alter the maternal vascular tree so that nutrient flow is reduced. Equally, it may provide molecules that influence epigenetic alterations in the offspring, to suggest but a few possibilities.
All these factors will interact with the fetal drive for growth determined by its genotype, and the outcome will depend on the timing and severity of the insult (Figure 11). Their impact will also be influenced by the placenta's ability to adapt and compensate, for example, by increasing vascularity, enzyme, or transporter expression. There is also the question as to whether the placenta has a functional reserve capacity, or whether it is operating to its maximum potential in normal pregnancies. They are clearly differences in placental efficiency within healthy pregnancies that deliver babies within the normal birth weight range, but placental weight is an uninformative proxy measure for placental function. At the crudest levels it does not distinguish between maternal blood and placental tissue and is heavily influenced by the mode of delivery and processing of the organ (57). Quantifying physical parameters that determine the theoretical diffusing capacity of the placenta provides a more objective assessment of placental function. The fact that the value expressed per kilogram of fetus remains constant across gestation suggests that development of the placenta and fetus are closely interlinked (368). But such analyses cannot take into account changes in, for example, placental blood flows, maternal-fetal concentration gradients, hemoglobin binding affinities, transporter activity, and placental metabolism. The speed with which these and many other physiological adaptations on both the maternal and fetal sides can occur makes it difficult to determine if any reserve capacity exists. There might also be detrimental effects for the fetus of building too large a placenta for its immediate needs, as this will consume extra resources and place an additional burden on the extracorporeal circulation.
X. FUTURE RESEARCH
The placenta remains the most poorly understood and underresearched organ. One of the biggest gaps in our knowledge is how the human trophoblast interacts with the endometrium to establish the placenta during the first few weeks of pregnancy. The events taking place then are of critical importance to generating the framework of the placenta, and remodeling of the maternal circulation to perfuse it. We now know this phase of development is stimulated and supported by the endometrial glands, but what are the contents of those secretions, how are they regulated, and are they affected by maternal diet? Emerging evidence suggests that the yolk sac plays a key role in the transport of nutrients from the glands to the embryo, but what is its functional capacity, and how does its vascularization impact on the developing heart?
The maternal circulation becomes fully established at the start of the send trimester, and the extent of villus regression at this time appears to be a major determinant of final placental size and shape. But how does unplugging of the spiral arteries occur? Is it purely a mechanical event, or is it related in some way to decline of gland function, coordinating the switch from histotrophic to hemotrophic nutrition? Is onset of the circulation abnormal in pregnancies complicated by early-onset preeclampsia or growth restriction? Equally, little is known about the spiral arteries and growth of the uterus during pregnancy. Are the arteries evenly distributed within the nonpregnant endometrium, or are there regional differences that might affect placental efficiency? Does the uterus expand symmetrically during gestation, and if so, does the shape of the delivered placenta reflect the implantation site and its possible arterial supply? What determines placental thickness? How do genetic interactions between the invading extravillous trophoblast cells and the uterine natural killer cells influence birth weight and obstetric outcome mechanistically?
The placenta clearly receives signals from the mother regarding her nutritional resources and reserves, and from the fetus relating to its demands, but what is the nature of those signals and how are they integrated? Imprinted genes are important, but what is the role of epigenetic factors, and in what tissues are these mediated? The placenta is also sending signals in the form of growth factors, hormones, and potentially exosomes into the maternal and fetal circulations to modify the maternal ability to support the pregnancy and fetal growth. The precise nature of these signals, their regulation in response to environmental cues, and their effects remain to be determined.
Answers to these, and other questions, will come in part through advances in imaging technologies, and the ability to monitor placental development, oxygenation, and metabolism in real-time. Magnetic resonance imaging and associated techniques, such as BOLD, promise much, but they must be capable of being applied during early pregnancy to capture the most fundamental events in placentation. In part, the answers will come through better phenotyping of the neonates and of the delivered placenta, at both the clinical and molecular levels. Longitudinal assessments of fetal growth trajectories in utero are needed to identify which neonates are potentially subject to programming. For the placenta, we need more information than just weight and shape at delivery. Greater attention needs to be paid to the mode of delivery, and the collection of samples to avoid possible artifacts (81), as well as to the sex of the placenta. Ultimately, we require more comprehensive phenotyping of the placenta, including quantification of structural parameters such as surface area and interhemal distance, characterization of maternal and fetal circulations, expression and activity levels of the different types of transporters, measurement of endocrine function and enzyme activities, assessment of placental metabolism and regulatory signaling pathways, and, ideally, single-cell transcriptomics and epigenetics. Such a comprehensive approach will require multidisciplinary research groups and/or collaborations over samples, but only then might we be able to tease apart the multiple interactions occurring during pregnancy and attribute causation with some degree of certainty.
XI. CONCLUSIONS
Over the last decade, a mass of epidemiological evidence associating the gross placental phenotype with predisposition to chronic disease has been accumulated. The statistical associations are so strong and have been confirmed in so many different cohorts across the globe that they are incontrovertible. However, the associations are complex, for they integrate the long-term nutritional status of the mother, environmental cues and stressors, the demands of the fetus, and development of the placenta. The challenge now is to elucidate the developmental mechanisms that link the placental phenotype to chronic disease in the offspring. These may operate at different levels for different diseases and at different times during gestation.
The pioneering epidemiological studies of David Barker inspired a new approach to our understanding of chronic disease, highlighting the importance of prenatal growth as the foundation for a healthy body. As life expectancy continues to increase, we need to ensure that the next generations are built optimally from the outset so that their organ systems endure. The placenta plays a pivotal role in this process as it represents the platform on which the individual is constructed. Its transience should not belittle its importance.
GRANTS
We thank the various funding agencies that have generously supported our research over the years: G. J. Burton, Medical Research Council, Wellcome Trust, and Action Medical Research; A. L. Fowden, Biotechnology and Biological Sciences Council, Medical Research Council, and Wellcome Trust; K. L. Thornburg, National Institutes of Child Health and Human Development, National Heart, Lung, and Blood Institute, National Institute of Diabetes and Digestive and Kidney Diseases, National Institute of Aging, American Heart Association, and M. Lowell Edwards Endowment.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
acknowledgments
This review is in honor of the memory of David Barker, FRS, FMedSci, a friend and colleague.
We gratefully acknowledge the invaluable contributions from our many research colleagues who have helped to develop and refine the ideas presented here.
Address for reprint requests and other correspondence: G. J. Burton, Physiological Laboratory, Downing Street, Cambridge CB2 3EG, UK (e-mail: gjb2@cam.ac.uk).
REFERENCES
- 1.Acosta O, Ramirez VI, Lager S, Gaccioli F, Dudley DJ, Powell TL, Jansson T. Increased glucose and placental GLUT-1 in large infants of obese nondiabetic mothers. Am J Obstet Gynecol 212: e221–227, 2015. [DOI] [PubMed] [Google Scholar]
- 2.Acosta-Alvear D, Zhou Y, Blais A, Tsikitis M, Lents NH, Arias C, Lennon CJ, Kluger Y, Dynlacht BD. XBP1 controls diverse cell type- and condition-specific transcriptional regulatory networks. Mol Cell 27: 53–66, 2007. [DOI] [PubMed] [Google Scholar]
- 3.Adams Waldorf KM, McAdams RM. Influence of infection during pregnancy on fetal development. Reproduction 146: R151–162, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Adamson SL, Lu Y, Whiteley KJ, Holmyard D, Hemberger M, Pfarrer C, Cross JC. Interactions between trophoblast cells and the maternal and fetal circulation in the mouse placenta. Dev Biol 250: 358–373, 2002. [DOI] [PubMed] [Google Scholar]
- 5.Alastalo H, Raikkonen K, Pesonen AK, Osmond C, Barker DJ, Kajantie E, Heinonen K, Forsen TJ, Eriksson JG. Cardiovascular health of Finnish war evacuees 60 years later. Ann Med 41: 66–72, 2009. [DOI] [PubMed] [Google Scholar]
- 6.Alfaidy N, Gupta S, DeMarco C, Caniggia I, Challis JRG. Oxygen regulation of placental 11ß-hydroxysteroid dehydrogenase 2: physiological and pathological implications. J Clin Endocrinol Metab 87: 4797–4805, 2002. [DOI] [PubMed] [Google Scholar]
- 7.Ali KZM, Burton GJ, Morad N, Ali ME. Does hypercapillarization influence the branching pattern of terminal villi in the human placenta at high altitude? Placenta 17: 677–682, 1996. [DOI] [PubMed] [Google Scholar]
- 8.Allen WR, Wilsher S, Turnbull C, Stewart F, Ousey J, Rossdale PD, Fowden AL. Influence of maternal size on placental, fetal and postnatal growth in the horse. I. Development in utero. Reproduction 123: 445–453, 2002. [PubMed] [Google Scholar]
- 9.Alsat E, Malassine A. High density lipoprotein interaction with human placenta: biochemical and ultrastructural characterization of binding to microvillous receptor and lack of internalization. Mol Cell Endocrinol 77: 97–108, 1991. [DOI] [PubMed] [Google Scholar]
- 10.Alwasel SH, Abotalib Z, Aljarallah JS, Osmond C, Alkharaz SM, Alhazza IM, Badr G, Barker DJ. Changes in placental size during Ramadan. Placenta 31: 607–610, 2010. [DOI] [PubMed] [Google Scholar]
- 11.Alwasel SH, Abotalib Z, Aljarallah JS, Osmond C, Alkharaz SM, Alhazza IM, Harrath A, Thornburg K, Barker DJ. Secular increase in placental weight in Saudi Arabia. Placenta 32: 391–394, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Alwasel SH, Harrath A, Aljarallah JS, Abotalib Z, Osmond C, Al Omar SY, Khaled I, Barker DJ. Intergenerational effects of in utero exposure to Ramadan in Tunisia. Am J Hum Biol 25: 341–343, 2013. [DOI] [PubMed] [Google Scholar]
- 13.Ambroso JL, Larsen SV, Brabec RK, Harris C. Fluorometric analysis of endocytosis and lysosomal proteolysis in the rat visceral yolk sac during whole embryo culture. Teratology 56: 201–209, 1997. [DOI] [PubMed] [Google Scholar]
- 14.Angiolini E, Coan PM, Sandovici I, Iwajomo OH, Peck G, Burton GJ, Sibley CP, Reik W, Fowden AL, Constancia M. Developmental adaptations to increased fetal nutrient demand in mouse genetic models of Igf2-mediated overgrowth. FASEB J 25: 1737–1745, 2011. [DOI] [PubMed] [Google Scholar]
- 15.Angiolini E, Fowden A, Coan P, Sandovici I, Smith P, Dean W, Burton G, Tycko B, Reik W, Sibley C, Constancia M. Regulation of placental efficiency for nutrient transport by imprinted genes. Placenta 27 Suppl A: S98–102, 2006. [DOI] [PubMed] [Google Scholar]
- 16.Arany Z, Foo SY, Ma Y, Ruas JL, Bommi-Reddy A, Girnun G, Cooper M, Laznik D, Chinsomboon J, Rangwala SM, Baek KH, Rosenzweig A, Spiegelman BM. HIF-independent regulation of VEGF and angiogenesis by the transcriptional coactivator PGC-1alpha. Nature 451: 1008–1012, 2008. [DOI] [PubMed] [Google Scholar]
- 17.Arias-Stella J. The Arias-Stella reaction: facts and fancies four decades after. Adv Anat Pathol 9: 12–23, 2002. [DOI] [PubMed] [Google Scholar]
- 18.Assemat E, Vinot S, Gofflot F, Linsel-Nitschke P, Illien F, Chatelet F, Verroust P, Louvet-Vallee S, Rinninger F, Kozyraki R. Expression and role of cubilin in the internalization of nutrients during the peri-implantation development of the rodent embryo. Biol Reprod 72: 1079–1086, 2005. [DOI] [PubMed] [Google Scholar]
- 19.Atkinson DE, Boyd RDH, Sibley CP. Placental transfer. In: Knobil & Neill's Physiology of Reproduction, edited by Neill JD. Amsterdam: Elsevier, 2006, p. 2787–2846. [Google Scholar]
- 20.Aye IL, Keelan JA. Placental ABC transporters, cellular toxicity and stress in pregnancy. Chem Biol Interact 203: 456–466, 2013. [DOI] [PubMed] [Google Scholar]
- 21.Back SH, Kaufman RJ. Endoplasmic reticulum stress and type 2 diabetes. Annu Rev Biochem 81: 767–793, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bagby SP. Maternal nutrition, low nephron number, and hypertension in later life: pathways of nutritional programming. J Nutr 137: 1066–1072, 2007. [DOI] [PubMed] [Google Scholar]
- 23.Barbour LA, Shao J, Qiao L, Leitner W, Anderson M, Friedman JE, Draznin B. Human placental growth hormone increases expression of the p85 regulatory unit of phosphatidylinositol 3-kinase and triggers severe insulin resistance in skeletal muscle. Endocrinology 145: 1144–1150, 2004. [DOI] [PubMed] [Google Scholar]
- 24.Barker D, Osmond C, Grant S, Thornburg K, Cooper C, Ring S, Davey-Smith G. Maternal cotyledons at birth predict blood pressure in childhood. Placenta 34: 672–675, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Barker DJ, Bull AR, Osmond C, Simmonds SJ. Fetal and placental size and risk of hypertension in adult life. BMJ 301: 259–262, 1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Barker DJ, Eriksson JG, Forsen T, Osmond C. Fetal origins of adult disease: strength of effects and biological basis. Int J Epidemiol 31: 1235–1239, 2002. [DOI] [PubMed] [Google Scholar]
- 27.Barker DJ, Gelow J, Thornburg K, Osmond C, Kajantie E, Eriksson JG. The early origins of chronic heart failure: impaired placental growth and initiation of insulin resistance in childhood. Eur J Heart Fail 12: 819–825, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Barker DJ, Hales CN, Fall CH, Osmond C, Phipps K, Clark PM. Type 2 (non-insulin-dependent) diabetes mellitus, hypertension and hyperlipidaemia (syndrome X): relation to reduced fetal growth. Diabetologia 36: 62–67, 1993. [DOI] [PubMed] [Google Scholar]
- 29.Barker DJ, Lampl M, Roseboom T, Winder N. Resource allocation in utero and health in later life. Placenta 33 Suppl 2: e30–34, 2012. [DOI] [PubMed] [Google Scholar]
- 30.Barker DJ, Larsen G, Osmond C, Thornburg KL, Kajantie E, Eriksson JG. The placental origins of sudden cardiac death. Int J Epidemiol 41: 1394–1399, 2012. [DOI] [PubMed] [Google Scholar]
- 31.Barker DJ, Osmond C. Infant mortality, childhood nutrition, and ischaemic heart disease in England and Wales. Lancet 1: 1077–1081, 1986. [DOI] [PubMed] [Google Scholar]
- 32.Barker DJ, Osmond C, Forsen TJ, Thornburg KL, Kajantie E, Eriksson JG. Foetal and childhood growth and asthma in adult life. Acta Paediatr 102: 732–738, 2013. [DOI] [PubMed] [Google Scholar]
- 33.Barker DJ, Osmond C, Golding J, Kuh D, Wadsworth ME. Growth in utero, blood pressure in childhood and adult life, and mortality from cardiovascular disease. BMJ 298: 564–567, 1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Barker DJ, Osmond C, Thornburg KL, Kajantie E, Eriksson JG. The intrauterine origins of Hodgkin's lymphoma. Cancer Epidemiol 37: 321–323, 2013. [DOI] [PubMed] [Google Scholar]
- 35.Barker DJ, Osmond C, Thornburg KL, Kajantie E, Eriksson JG. The lifespan of men and the shape of their placental surface at birth. Placenta 32: 783–787, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Barker DJ, Osmond C, Thornburg KL, Kajantie E, Eriksson JG. The shape of the placental surface at birth and colorectal cancer in later life. Am J Hum Biol 25: 566–568, 2013. [DOI] [PubMed] [Google Scholar]
- 37.Barker DJ, Thornburg KL. The obstetric origins of health for a lifetime. Clin Obstet Gynecol 56: 511–519, 2013. [DOI] [PubMed] [Google Scholar]
- 38.Barker DJ, Thornburg KL, Osmond C, Kajantie E, Eriksson JG. The prenatal origins of lung cancer. II. The placenta. Am J Hum Biol 22: 512–516, 2010. [DOI] [PubMed] [Google Scholar]
- 39.Barker DJ, Thornburg KL, Osmond C, Kajantie E, Eriksson JG. The surface area of the placenta and hypertension in the offspring in later life. Int J Dev Biol 54: 525–530, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Barker DJ, Winter PD, Osmond C, Margetts B, Simmonds SJ. Weight in infancy and death from ischaemic heart disease. Lancet 2: 577–580, 1989. [DOI] [PubMed] [Google Scholar]
- 41.Bartelmez GW. Histological studies on the menstruating mucous membrane of the human uterus. Contrib Embryol 24: 141–186, 1933. [Google Scholar]
- 42.Barton SC, Surani MA, Norris ML. Role of paternal and maternal genomes in mouse development. Nature 311: 374–376, 1984. [DOI] [PubMed] [Google Scholar]
- 43.Bateson P, Barker D, Clutton-Brock T, Deb D, D'Udine B, Foley RA, Gluckman P, Godfrey K, Kirkwood T, Lahr MM, McNamara J, Metcalfe NB, Monaghan P, Spencer HG, Sultan SE. Developmental plasticity and human health. Nature 430: 419–421, 2004. [DOI] [PubMed] [Google Scholar]
- 44.Battaglia FC. In vivo characteristics of placental amino acid transport and metabolism in ovine pregnancy–a review. Placenta 23 Suppl A: S3–8, 2002. [DOI] [PubMed] [Google Scholar]
- 45.Beckman DA, Lloyd JB, Brent RL. Quantitative studies on the mechanisms of amino acid supply to rat embryos during organogenesis. Reprod Toxicol 12: 197–200, 1998. [DOI] [PubMed] [Google Scholar]
- 46.Beier-Hellwig K, Sterzik K, Bonn B, Beir HM. Contribution to the physiology and pathology of endometrial receptivity: the determination of protein patterns in human uterine secretions. Hum Reprod 4 Suppl: 115–120, 1989. [DOI] [PubMed] [Google Scholar]
- 47.Belkacemi L, Nelson DM, Desai M, Ross MG. Maternal undernutrition influences placental-fetal development. Biol Reprod 83: 325–331, 2010. [DOI] [PubMed] [Google Scholar]
- 48.Bell EL, Emerling BM, Chandel NS. Mitochondrial regulation of oxygen sensing. Mitochondrion 5: 322–332, 2005. [DOI] [PubMed] [Google Scholar]
- 49.Benirschke K, Burton GJ, Baergen RN. Pathology of the Human Placenta. Heidelberg: Springer, 2012, p. 941. [Google Scholar]
- 50.Berlanga O, Bradshaw HB, Vilella-Mitjana F, Garrido-Gomez T, Simon C. How endometrial secretomics can help in predicting implantation. Placenta 32 Suppl 3: S271–275, 2011. [DOI] [PubMed] [Google Scholar]
- 51.Blumenstein M, Keelan JA, Mitchell MD. Hypoxia attenuates PGE2 but increases prostacyclin and thromboxane production in human term villous trophoblast. Placenta 22: 519–525, 2001. [DOI] [PubMed] [Google Scholar]
- 52.Bocking AD, Gagnon R, White SE, Homan J, Milne KM, Richardson BS. Circulatory responses to prolonged hypoxemia in fetal sheep. Am J Obstet Gynecol 159: 1418–1424, 1988. [DOI] [PubMed] [Google Scholar]
- 53.Bocking AD, White SE, Homan J, Richardson BS. Oxygen consumption is maintained in fetal sheep during prolonged hypoxaemia. J Dev Physiol 17: 169–174, 1992. [PubMed] [Google Scholar]
- 54.Borowicz PP, Arnold DR, Johnson ML, Grazul-Bilska AT, Redmer DA, Reynolds LP. Placental growth throughout the last two thirds of pregnancy in sheep: vascular development and angiogenic factor expression. Biol Reprod 76: 259–267, 2007. [DOI] [PubMed] [Google Scholar]
- 55.Botting KJ, McMillen IC, Forbes H, Nyengaard JR, Morrison JL. Chronic hypoxemia in late gestation decreases cardiomyocyte number but does not change expression of hypoxia-responsive genes. J Am Heart Assoc 3: e000531, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bouret S, Levin BE, Ozanne SE. Gene-environment interactions controlling energy and glucose homeostasis and the developmental origins of obesity. Physiol Rev 95: 47–82, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bouw GM, Stolte LAM, Baak JPA, Oort J. Quantitative morphology of the placenta. I. Standardization of sampling. Eur J Obstet Gynecol Reprod Biol 6: 325–331, 1976. [Google Scholar]
- 58.Boyd R, Boyd R. The placenta and developmental programming. Some reflections. In: The Placenta and Human Developmental Programming, edited by Burton GJ, Barker DJ, Moffett A, and Thornburg K. Cambridge, UK: Cambridge Univ. Press, 2011, p. 233–235. [Google Scholar]
- 59.Brent RL, Fawcett LB. Nutritional studies of the embryo during early organogenesis with normal embryos and embryos exhibiting yolk sac dysfunction. J Pediatr 132: S6–16, 1998. [DOI] [PubMed] [Google Scholar]
- 60.Brett KE, Ferraro ZM, Holcik M, Adamo KB. Prenatal physical activity and diet composition affect the expression of nutrient transporters and mTOR signaling molecules in the human placenta. Placenta 36: 204–212, 2015. [DOI] [PubMed] [Google Scholar]
- 61.Brett KE, Ferraro ZM, Yockell-Lelievre J, Gruslin A, Adamo KB. Maternal-fetal nutrient transport in pregnancy pathologies: the role of the placenta. Int J Mol Sci 15: 16153–16185, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Brison DR, Sturmey RG, Leese HJ. Metabolic heterogeneity during preimplantation development: the missing link? Hum Reprod Update 20: 632–640, 2014. [DOI] [PubMed] [Google Scholar]
- 63.Bronson SL, Bale TL. The placenta as a mediator of stress effects on neurodevelopmental reprogramming. Neuropsychopharmacology 41: 207–218, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Brosens I, Pijnenborg R, Vercruysse L, Romero R. The “Great Obstetrical Syndromes” are associated with disorders of deep placentation. Am J Obstet Gynecol 204: 193–201, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Brosens IA. The utero-placental vessels at term - the distribution and extent of physiological changes. Trophoblast Res 3: 61–67, 1988. [Google Scholar]
- 66.Brown CJ, Rupert JL. Hypoxia and environmental epigenetics. High Altitude Med Biol 15: 323–330, 2014. [DOI] [PubMed] [Google Scholar]
- 67.Brown K, Heller DS, Zamudio S, Illsley NP. Glucose transporter 3 (GLUT3) protein expression in human placenta across gestation. Placenta 32: 1041–1049, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Brownbill P, Mahendran D, Owen D, Swanson P, Thornburg KL, Nelson DM, Sibley CP. Denudations as paracellular routes for alphafetoprotein and creatinine across the human syncytiotrophoblast. Am J Physiol Regul Integr Comp Physiol 278: R677–R683, 2000. [DOI] [PubMed] [Google Scholar]
- 69.Browne VA, Julian CG, Toledo-Jaldin L, Cioffi-Ragan D, Vargas E, Moore LG. Uterine artery blood flow, fetal hypoxia and fetal growth. Philos Trans R Soc Lond B Biol Sci 370: 20140068, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Brugarolas J, Lei K, Hurley RL, Manning BD, Reiling JH, Hafen E, Witters LA, Ellisen LW, Kaelin WG Jr. Regulation of mTOR function in response to hypoxia by REDD1 and the TSC1/TSC2 tumor suppressor complex. Genes Dev 18: 2893–2904, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Buckberry S, Bianco-Miotto T, Bent SJ, Dekker GA, Roberts CT. Integrative transcriptome meta-analysis reveals widespread sex-biased gene expression at the human fetal-maternal interface. Mol Hum Reprod 20: 810–819, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Burke KA, Jauniaux E, Burton GJ, Cindrova-Davies T. Expression and immunolocalisation of the endocytic receptors megalin and cubilin in the human yolk sac and placenta across gestation. Placenta 34: 1105–1109, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Burton GJ. The fine structure of the human placenta as revealed by scanning electron microscopy. Scanning Microsc 1: 1811–1828, 1987. [PubMed] [Google Scholar]
- 74.Burton GJ, Fowden AL. Review: the placenta and developmental programming: balancing fetal nutrient demands with maternal resource allocation. Placenta 33 Suppl: S23–27, 2012. [DOI] [PubMed] [Google Scholar]
- 75.Burton GJ, Jauniaux E. Sonographic, stereological and Doppler flow velocimetric assessments of placental maturity. Br J Obstet Gynaecol 102: 818–825, 1995. [DOI] [PubMed] [Google Scholar]
- 76.Burton GJ, Jauniaux E, Charnock-Jones DS. Human early placental development: potential roles of the endometrial glands. Placenta 28 Suppl A: S64–69, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Burton GJ, Jauniaux E, Charnock-Jones DS. The influence of the intrauterine environment on human placental development. Int J Dev Biol 54: 303–312, 2010. [DOI] [PubMed] [Google Scholar]
- 78.Burton GJ, Reshetnikova OS, Milovanov AP, Teleshova OV. Stereological evaluation of vascular adaptations of human placental villi to differing forms of hypoxic stress. Placenta 17: 49–55, 1996. [DOI] [PubMed] [Google Scholar]
- 79.Burton GJ, Samuel CA, Steven DH. Ultrastructural studies of the placenta of the ewe: phagocytosis of erythrocytes by the chorionic epithelium at the central depression of the cotyledon. Q J Exp Physiol Cogn Med Sci 61: 275–286, 1976. [DOI] [PubMed] [Google Scholar]
- 80.Burton GJ, Scioscia M, Rademacher TW. Endometrial secretions: creating a stimulatory microenvironment within the human early placenta. Implications for the etiopathogenesis of pre-eclampsia. J Reprod Immunol 89: 118–125, 2011. [DOI] [PubMed] [Google Scholar]
- 81.Burton GJ, Sebire NJ, Myatt L, Tannetta D, Wang YL, Sadovsky Y, Staff AC, Redman CW. Optimising sample collection for placental research. Placenta 35: 9–22, 2014. [DOI] [PubMed] [Google Scholar]
- 82.Burton GJ, Watson AL. The structure of the human placenta: implications for initiating and defending against viral infections. Rev Med Virol 7: 219–228, 1997. [DOI] [PubMed] [Google Scholar]
- 83.Burton GJ, Watson AL, Hempstock J, Skepper JN, Jauniaux E. Uterine glands provide histiotrophic nutrition for the human fetus during the first trimester of pregnancy. J Clin Endocrinol Metab 87: 2954–2959, 2002. [DOI] [PubMed] [Google Scholar]
- 84.Burton GJ, Woods AW, Jauniaux E, Kingdom JC. Rheological and physiological consequences of conversion of the maternal spiral arteries for uteroplacental blood flow during human pregnancy. Placenta 30: 473–482, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Burton GJ, Yung HW, Cindrova-Davies T, Charnock-Jones DS. Placental endoplasmic reticulum stress and oxidative stress in the pathophysiology of unexplained intrauterine growth restriction and early onset preeclampsia. Placenta 30 Suppl A: S43–48, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Busch S, Renaud SJ, Schleussner E, Graham CH, Markert UR. mTOR mediates human trophoblast invasion through regulation of matrix-remodeling enzymes and is associated with serine phosphorylation of STAT3. Exp Cell Res 315: 1724–1733, 2009. [DOI] [PubMed] [Google Scholar]
- 87.Calay ES, Hotamisligil GS. Turning off the inflammatory, but not the metabolic, flames. Nat Med 19: 265–267, 2013. [DOI] [PubMed] [Google Scholar]
- 88.Cam H, Easton JB, High A, Houghton PJ. mTORC1 signaling under hypoxic conditions is controlled by ATM-dependent phosphorylation of HIF-1alpha. Mol Cell 40: 509–520, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Caniggia I, Mostachfi H, Winter J, Gassmann M, Lye SJ, Kuliszewski M, Post M. Hypoxia-inducible factor-1 mediates the biological effects of oxygen on human trophoblast differentiation through TGFbeta(3). J Clin Invest 105: 577–587, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Caniggia I, Winter J, Lye SJ, Post M. Oxygen and placental development during the first trimester: implications for the pathophysiology of pre-eclampsia. Placenta 21 Suppl A: S25–30, 2000. [DOI] [PubMed] [Google Scholar]
- 91.Capellini I. The evolutionary significance of placental interdigitation in mammalian reproduction: contributions from comparative studies. Placenta 33: 763–768, 2012. [DOI] [PubMed] [Google Scholar]
- 92.Capellini I, Venditti C, Barton RA. Placentation and maternal investment in mammals. Am Nat 177: 86–98, 2011. [DOI] [PubMed] [Google Scholar]
- 93.Carey EA, Albers RE, Doliboa SR, Hughes M, Wyatt CN, Natale DR, Brown TL. AMPK knockdown in placental trophoblast cells results in altered morphology and function. Stem Cells Dev 23: 2921–2930, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Carter AM. Animal models of human placentation–a review. Placenta 28 Suppl A: S41–47, 2007. [DOI] [PubMed] [Google Scholar]
- 95.Carter AM. Evolution of placental function in mammals: the molecular basis of gas and nutrient transfer, hormone secretion, and immune responses. Physiol Rev 92: 1543–1576, 2012. [DOI] [PubMed] [Google Scholar]
- 96.Carter AM. Evolution of the placenta and fetal membranes seen in the light of molecular phylogenetics. Placenta 22: 800–807, 2001. [DOI] [PubMed] [Google Scholar]
- 97.Carter AM. Placental oxygen consumption. Part I: in vivo studies–a review. Placenta 21 Suppl A: S31–37, 2000. [DOI] [PubMed] [Google Scholar]
- 98.Carter AM, Mess A. Evolution of the placenta in eutherian mammals. Placenta 28: 259–262, 2007. [DOI] [PubMed] [Google Scholar]
- 99.Carver TD, Hay WW Jr. Uteroplacental carbon substrate metabolism and O2 consumption after long-term hypoglycemia in pregnant sheep. Am J Physiol Endocrinol Metab 269: E299–E308, 1995. [DOI] [PubMed] [Google Scholar]
- 100.Ceccaldi PF, Gavard L, Mandelbrot L, Rey E, Farinotti R, Treluyer JM, Gil S. Functional role of P-glycoprotein and binding protein effect on the placental transfer of lopinavir/ritonavir in the ex vivo human perfusion model. Obstet Gynecol Int 2009: 726593, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Cetin I, Parisi F, Berti C, Mando C, Desoye G. Placental fatty acid transport in maternal obesity. J Dev Origins Health Dis 3: 409–414, 2012. [DOI] [PubMed] [Google Scholar]
- 102.Challier JC, Basu S, Bintein T, Minium J, Hotmire K, Catalano PM, Hauguel-de Mouzon S. Obesity in pregnancy stimulates macrophage accumulation and inflammation in the placenta. Placenta 29: 274–281, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Chan SY, Zhang YY, Hemann C, Mahoney CE, Zweier JL, Loscalzo J. MicroRNA-210 controls mitochondrial metabolism during hypoxia by repressing the iron-sulfur cluster assembly proteins ISCU1/2. Cell Metab 10: 273–284, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Charalambous M, Cowley M, Geoghegan F, Smith FM, Radford EJ, Marlow BP, Graham CF, Hurst LD, Ward A. Maternally-inherited Grb10 reduces placental size and efficiency. Dev Biol 337: 1–8, 2010. [DOI] [PubMed] [Google Scholar]
- 105.Charles SM, Julian CG, Vargas E, Moore LG. Higher estrogen levels during pregnancy in Andean than European residents of high altitude suggest differences in aromatase activity. J Clin Endocrinol Metab 99: 2908–2916, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Chen CP, Aplin JD. Placental extracellular matrix: gene expression, deposition by placental fibroblasts and the effect of oxygen. Placenta 24: 316–325, 2003. [DOI] [PubMed] [Google Scholar]
- 107.Chen D, Zhou X, Zhu Y, Zhu T, Wang J. [Comparison study on uterine and umbilical artery blood flow during pregnancy at high altitude and at low altitude]. Zhonghua fu chan ke za zhi 37: 69–71, 2002. [PubMed] [Google Scholar]
- 108.Chen Z, Li Y, Zhang H, Huang P, Luthra R. Hypoxia-regulated microRNA-210 modulates mitochondrial function and decreases ISCU and COX10 expression. Oncogene 29: 4362–4368, 2010. [DOI] [PubMed] [Google Scholar]
- 109.Choudhry H, Harris AL, McIntyre A. The tumour hypoxia induced non-coding transcriptome. Mol Aspects Med 47–48: 35–53, 2016. [DOI] [PubMed] [Google Scholar]
- 110.Christensen EI, Birn H. Megalin and cubilin: multifunctional endocytic receptors. Nat Rev Mol Cell Biol 3: 256–266, 2002. [DOI] [PubMed] [Google Scholar]
- 111.Cindrova-Davies T, Herrera EA, Niu Y, Kingdom J, Giussani DA, Burton GJ. Reduced cystathionine gamma-lyase and increased miR-21 expression are associated with increased vascular resistance in growth-restricted pregnancies: hydrogen sulfide as a placental vasodilator. Am J Pathol 182: 1448–1458, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Cindrova-Davies T, van Patot MT, Gardner L, Jauniaux E, Burton GJ, Charnock-Jones DS. Energy status and HIF signalling in chorionic villi show no evidence of hypoxic stress during human early placental development. Mol Hum Reprod 21: 296–308, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Cleal JK, Lewis RM. The mechanisms and regulation of placental amino acid transport to the human foetus. J Neuroendocrinol 20: 419–426, 2008. [DOI] [PubMed] [Google Scholar]
- 114.Coan PM, Angiolini E, Sandovici I, Burton GJ, Constancia M, Fowden AL. Adaptations in placental nutrient transfer capacity to meet fetal growth demands depend on placental size in mice. J Physiol 586: 4567–4576, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Coan PM, Conroy N, Burton GJ, Ferguson-Smith AC. Origin and characteristics of glycogen cells in the developing murine placenta. Dev Dynamics 235: 3280–3294, 2006. [DOI] [PubMed] [Google Scholar]
- 116.Coan PM, Ferguson-Smith AC, Burton GJ. Developmental dynamics of the definitive mouse placenta assessed by stereology. Biol Reprod 70: 1806–1813, 2004. [DOI] [PubMed] [Google Scholar]
- 117.Coan PM, Ferguson-Smith AC, Burton GJ. Ultrastructural changes in the interhaemal membrane and junctional zone of the murine chorioallantoic placenta across gestation. J Anat 207: 783–796, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Coan PM, Fowden AL, Constancia M, Ferguson-Smith AC, Burton GJ, Sibley CP. Disproportional effects of Igf2 knockout on placental morphology and diffusional exchange characteristics in the mouse. J Physiol 586: 5023–5032, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Coan PM, Vaughan OR, McCarthy J, Mactier C, Burton GJ, Constancia M, Fowden AL. Dietary composition programmes placental phenotype in mice. J Physiol 589: 3659–3670, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Coan PM, Vaughan OR, Sekita Y, Finn SL, Burton GJ, Constancia M, Fowden AL. Adaptations in placental phenotype support fetal growth during undernutrition of pregnant mice. J Physiol 588: 527–538, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Colleoni F, Padmanabhan N, Yung HW, Watson ED, Cetin I, Tissot van Patot M, Burton GJ, Murray AJ. Suppression of mitochondrial electron transport chain function in the hypoxic human placenta: a role for miR-210 and protein synthesis inhibition. PLoS One 8: e55194, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Collins SL, Stevenson GN, Noble JA, Impey L. Rapid calculation of standardized placental volume at 11 to 13 weeks and the prediction of small for gestational age babies. Ultrasound Med Biol 39: 253–260, 2013. [DOI] [PubMed] [Google Scholar]
- 123.Conrad KP, Benyo DF, Westerhausen-Larsen A, Miles TM. Expression of erythropoietin by the human placenta. FASEB J 10: 760–768, 1996. [DOI] [PubMed] [Google Scholar]
- 124.Constancia M, Angiolini E, Sandovici I, Smith P, Smith R, Kelsey G, Dean W, Ferguson-Smith A, Sibley CP, Reik W, Fowden A. Adaptation of nutrient supply to fetal demand in the mouse involves interaction between the Igf2 gene and placental transporter systems. Proc Natl Acad Sci USA 102: 19219–19224, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Constancia M, Hemberger M, Hughes J, Dean W, Ferguson-Smith A, Fundele R, Stewart F, Kelsey G, Fowden A, Sibley C, Reik W. Placental-specific IGF-II is a major modulator of placental and fetal growth. Nature 417: 945–948, 2002. [DOI] [PubMed] [Google Scholar]
- 126.Constancia M, Kelsey G, Reik W. Resourceful imprinting. Nature 432: 53–57, 2004. [DOI] [PubMed] [Google Scholar]
- 127.Correia-Branco A, Keating E, Martel F. Maternal undernutrition and fetal developmental programming of obesity: the glucocorticoid connection. Reprod Sci 22: 138–145, 2015. [DOI] [PubMed] [Google Scholar]
- 128.Cottrell EC, Holmes MC, Livingstone DE, Kenyon CJ, Seckl JR. Reconciling the nutritional and glucocorticoid hypotheses of fetal programming. FASEB J 26: 1866–1874, 2012. [DOI] [PubMed] [Google Scholar]
- 129.Cottrell EC, Seckl JR, Holmes MC, Wyrwoll CS. Foetal and placental 11beta-HSD2: a hub for developmental programming. Acta Physiol 210: 288–295, 2014. [DOI] [PubMed] [Google Scholar]
- 130.Craven CM, Zhao L, Ward K. Lateral placental growth occurs by trophoblast cell invasion of decidual veins. Placenta 21: 160–169, 2000. [DOI] [PubMed] [Google Scholar]
- 131.Crosley EJ, Elliot MG, Christians JK, Crespi BJ. Placental invasion, preeclampsia risk and adaptive molecular evolution at the origin of the great apes: evidence from genome-wide analyses. Placenta 34: 127–132, 2013. [DOI] [PubMed] [Google Scholar]
- 132.Cuffe JS, Dickinson H, Simmons DG, Moritz KM. Sex specific changes in placental growth and MAPK following short term maternal dexamethasone exposure in the mouse. Placenta 32: 981–989, 2011. [DOI] [PubMed] [Google Scholar]
- 133.Cuffe JS, Walton SL, Singh RR, Spiers JG, Bielefeldt-Ohmann H, Wilkinson L, Little MH, Moritz KM. Mid- to late term hypoxia in the mouse alters placental morphology, glucocorticoid regulatory pathways and nutrient transporters in a sex-specific manner. J Physiol 592: 3127–3141, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Cuffe JS, Walton SL, Steane SE, Singh RR, Simmons DG, Moritz KM. The effects of gestational age and maternal hypoxia on the placental renin angiotensin system in the mouse. Placenta 35: 953–961, 2014. [DOI] [PubMed] [Google Scholar]
- 135.Culver JC, Dickinson ME. The effects of hemodynamic force on embryonic development. Microcirculation 17: 164–178, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Cvitic S, Longtine MS, Hackl H, Wagner K, Nelson MD, Desoye G, Hiden U. The human placental sexome differs between trophoblast epithelium and villous vessel endothelium. PLoS One 8: e79233, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Dantzer V, Leiser R, Kaufmann P, Luckhardt M. Comparative morphological aspects of placental vascularisation. Trophoblast Res 3: 235–260, 1988. [Google Scholar]
- 138.Das UG, He J, Ehrhardt RA, Hay WW Jr, Devaskar SU. Time-dependent physiological regulation of ovine placental GLUT-3 glucose transporter protein. Am J Physiol Regul Integr Comp Physiol 279: R2252–R2261, 2000. [DOI] [PubMed] [Google Scholar]
- 139.Das UG, Sadiq HF, Soares MJ, Hay WW Jr, Devaskar SU. Time-dependent physiological regulation of rodent and ovine placental glucose transporter (GLUT-1) protein. Am J Physiol Regul Integr Comp Physiol 274: R339–R347, 1998. [DOI] [PubMed] [Google Scholar]
- 140.Daud AN, Bergman JE, Bakker MK, Wang H, Kerstjens-Frederikse WS, de Walle HE, Groen H, Bos JH, Hak E, Wilffert B. P-glycoprotein-mediated drug interactions in pregnancy and changes in the risk of congenital anomalies: a case-reference study. Drug Saf 38: 651–659, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Day PE, Cleal JK, Lofthouse EM, Hanson MA, Lewis RM. What factors determine placental glucose transfer kinetics? Placenta 34: 953–958, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.De Rooij SR, Wouters H, Yonker JE, Painter RC, Roseboom TJ. Prenatal undernutrition and cognitive function in late adulthood. Proc Natl Acad Sci USA 107: 16881–16886, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Desoye G, Hartmann M, Jones CJ, Wolf HJ, Kohnen G, Kosanke G, Kaufmann P. Location of insulin receptors in the placenta and its progenitor tissues. Microsc Res Tech 38: 63–75, 1997. [DOI] [PubMed] [Google Scholar]
- 144.Diaz P, Powell TL, Jansson T. The role of placental nutrient sensing in maternal-fetal resource allocation. Biol Reprod 91: 82, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Diaz P, Wood AM, Sibley CP, Greenwood SL. Intermediate conductance Ca2+-activated K+ channels modulate human placental trophoblast syncytialization. PLoS One 9: e90961, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Dickhout JG, Carlisle RE, Jerome DE, Mohammed-Ali Z, Jiang H, Yang G, Mani S, Garg SK, Banerjee R, Kaufman RJ, Maclean KN, Wang R, Austin RC. Integrated stress response modulates cellular redox state via induction of cystathionine gamma-lyase: cross-talk between integrated stress response and thiol metabolism. J Biol Chem 287: 7603–7614, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.DiGiacomo JE, Hay WW Jr. Placental-fetal glucose exchange and placental glucose consumption in pregnant sheep. Am J Physiol Endocrinol Metab 258: E360–E367, 1990. [DOI] [PubMed] [Google Scholar]
- 148.DiGiacomo JE, Hay WW Jr. Regulation of placental glucose transfer and consumption by fetal glucose production. Pediatr Res 25: 429–434, 1989. [DOI] [PubMed] [Google Scholar]
- 149.Dilworth MR, Kusinski LC, Cowley E, Ward BS, Husain SM, Constancia M, Sibley CP, Glazier JD. Placental-specific Igf2 knockout mice exhibit hypocalcemia and adaptive changes in placental calcium transport. Proc Natl Acad Sci USA 107: 3894–3899, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Dilworth MR, Sibley CP. Review: transport across the placenta of mice and women. Placenta 34 Suppl: S34–39, 2013. [DOI] [PubMed] [Google Scholar]
- 151.Ditchfield AM, Desforges M, Mills TA, Glazier JD, Wareing M, Mynett K, Sibley CP, Greenwood SL. Maternal obesity is associated with a reduction in placental taurine transporter activity. Int J Obesity 39: 557–564, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Dodson RB, Rozance PJ, Fleenor BS, Petrash CC, Shoemaker LG, Hunter KS, Ferguson VL. Increased arterial stiffness and extracellular matrix reorganization in intrauterine growth-restricted fetal sheep. Pediatr Res 73: 147–154, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Dong J, Qiu H, Garcia-Barrio M, Anderson J, Hinnebusch AG. Uncharged tRNA activates GCN2 by displacing the protein kinase moiety from a bipartite tRNA-binding domain. Mol Cell 6: 269–279, 2000. [DOI] [PubMed] [Google Scholar]
- 154.Doridot L, Houry D, Gaillard H, Chelbi ST, Barbaux S, Vaiman D. miR-34a expression, epigenetic regulation, and function in human placental diseases. Epigenetics 9: 142–151, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Droge W. Free radicals in the physiological control of cell function. Physiol Rev 82: 47–95, 2002. [DOI] [PubMed] [Google Scholar]
- 156.Dube E, Gravel A, Martin C, Desparois G, Moussa I, Ethier-Chiasson M, Forest JC, Giguere Y, Masse A, Lafond J. Modulation of fatty acid transport and metabolism by maternal obesity in the human full-term placenta. Biol Reprod 87: 14, 2012. [DOI] [PubMed] [Google Scholar]
- 157.Dubova EA, Pavlov KA, Borovkova EI, Bayramova MA, Makarov IO, Shchegolev AI. Vascular endothelial growth factor and its receptors in the placenta of pregnant women with obesity. Bull Exp Biol Med 151: 253–258, 2011. [DOI] [PubMed] [Google Scholar]
- 158.Ducsay CA, Hyatt K, Mlynarczyk M, Kaushal KM, Myers DA. Long-term hypoxia increases leptin receptors and plasma leptin concentrations in the late-gestation ovine fetus. Am J Physiol Regul Integr Comp Physiol 291: R1406–R1413, 2006. [DOI] [PubMed] [Google Scholar]
- 159.Dumortier O, Blondeau B, Duvillie B, Reusens B, Breant B, Remacle C. Different mechanisms operating during different critical time-windows reduce rat fetal beta cell mass due to a maternal low-protein or low-energy diet. Diabetologia 50: 2495–2503, 2007. [DOI] [PubMed] [Google Scholar]
- 160.Dumoulin JC, Land JA, Van Montfoort AP, Nelissen EC, Coonen E, Derhaag JG, Schreurs IL, Dunselman GA, Kester AD, Geraedts JP, Evers JL. Effect of in vitro culture of human embryos on birthweight of newborns. Hum Reprod 25: 605–612, 2010. [DOI] [PubMed] [Google Scholar]
- 161.Dunwoodie SL. The role of hypoxia in development of the mammalian embryo. Dev Cell 17: 755–773, 2009. [DOI] [PubMed] [Google Scholar]
- 162.Duttaroy AK. Transport of fatty acids across the human placenta: a review. Prog Lipid Res 48: 52–61, 2009. [DOI] [PubMed] [Google Scholar]
- 163.Eckert JJ, Porter R, Watkins AJ, Burt E, Brooks S, Leese HJ, Humpherson PG, Cameron IT, Fleming TP. Metabolic induction and early responses of mouse blastocyst developmental programming following maternal low protein diet affecting life-long health. PLoS One 7: e52791, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Efeyan A, Zoncu R, Sabatini DM. Amino acids and mTORC1: from lysosomes to disease. Trends Mol Med 18: 524–533, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Ehrhardt RA, Bell AW. Developmental increases in glucose transporter concentration in the sheep placenta. Am J Physiol Regul Integr Comp Physiol 273: R1132–R1141, 1997. [DOI] [PubMed] [Google Scholar]
- 166.Eifert AW, Wilson ME, Vonnahme KA, Camacho LE, Borowicz PP, Redmer DA, Romero S, Dorsam S, Haring J, Lemley CO. Effect of melatonin or maternal nutrient restriction on vascularity and cell proliferation in the ovine placenta. Anim Reprod Sci 153: 13–21, 2015. [DOI] [PubMed] [Google Scholar]
- 167.Ekblad U, Erkkola R, Uotila P. The effect of acute hypoxia on prostaglandin release in perfused human fetal placenta. Prostaglandins 33: 553–560, 1987. [DOI] [PubMed] [Google Scholar]
- 168.Elad D, Levkovitz R, Jaffa AJ, Desoye G, Hod M. Have we neglected the role of fetal endothelium in transplacental transport? Traffic 15: 122–126, 2014. [DOI] [PubMed] [Google Scholar]
- 169.Elbrink J, Bihler I. Membrane transport: its relation to cellular metabolic rates. Science 188: 1177–1184, 1975. [DOI] [PubMed] [Google Scholar]
- 170.Elliot MG, Crespi BJ. Genetic recapitulation of human pre-eclampsia risk during convergent evolution of reduced placental invasiveness in eutherian mammals. Philos Trans R Soc Lond B Biol Sci 370: 20140069, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Elliot MG, Crespi BJ. Phylogenetic evidence for early hemochorial placentation in eutheria. Placenta 30: 949–967, 2009. [DOI] [PubMed] [Google Scholar]
- 172.Eriksson JG, Gelow J, Thornburg KL, Osmond C, Laakso M, Uusitupa M, Lindi V, Kajantie E, Barker DJ. Long-term effects of placental growth on overweight and body composition. Int J Pediatr 2012: 324185, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Eriksson JG, Kajantie E, Osmond C, Thornburg K, Barker DJ. Boys live dangerously in the womb. Am J Hum Biol 22: 330–335, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Eriksson JG, Kajantie E, Phillips DI, Osmond C, Thornburg KL, Barker DJ. The developmental origins of chronic rheumatic heart disease. Am J Hum Biol 25: 655–658, 2013. [DOI] [PubMed] [Google Scholar]
- 175.Eriksson JG, Kajantie E, Thornburg KL, Osmond C, Barker DJ. Mother's body size and placental size predict coronary heart disease in men. Eur Heart J 32: 2297–2303, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Espinoza J, Sebire NJ, McAuliffe F, Krampl E, Nicolaides KH. Placental villus morphology in relation to maternal hypoxia at high altitude. Placenta 22: 606–608, 2001. [DOI] [PubMed] [Google Scholar]
- 177.Esterman A, Finlay TH, Dancis J. The effect of hypoxia on term trophoblast: hormone synthesis and release. Placenta 17: 217–222, 1996. [DOI] [PubMed] [Google Scholar]
- 178.Esterman A, Greco MA, Mitani Y, Finlay TH, Ismail-Beigi F, Dancis J. The effect of hypoxia on human trophoblast in culture: morphology, glucose transport and metabolism. Placenta 18: 129–136, 1997. [DOI] [PubMed] [Google Scholar]
- 179.Ethier-Chiasson M, Duchesne A, Forest JC, Giguere Y, Masse A, Mounier C, Lafond J. Influence of maternal lipid profile on placental protein expression of LDLr and SR-BI. Biochem Biophys Res Commun 359: 8–14, 2007. [DOI] [PubMed] [Google Scholar]
- 180.Fahling M. Cellular oxygen sensing, signalling and how to survive translational arrest in hypoxia. Acta Physiol 195: 205–230, 2009. [DOI] [PubMed] [Google Scholar]
- 181.Fahling M. Surviving hypoxia by modulation of mRNA translation rate. J Cell Mol Med 13: 2770–2779, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Fall CH, Barker DJ, Osmond C, Winter PD, Clark PM, Hales CN. Relation of infant feeding to adult serum cholesterol concentration and death from ischaemic heart disease. BMJ 304: 801–805, 1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Fan Z, Zhang ZX, Li Y, Wang Z, Xu T, Gong X, Zhou X, Wen H, Zeng Y. Relationship between birth size and coronary heart disease in China. Ann Med 42: 596–602, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Farley DM, Choi J, Dudley DJ, Li C, Jenkins SL, Myatt L, Nathanielsz PW. Placental amino acid transport and placental leptin resistance in pregnancies complicated by maternal obesity. Placenta 31: 718–724, 2010. [DOI] [PubMed] [Google Scholar]
- 185.Firth JA, Leach L. Not trophoblast alone: a review of the contribution of the fetal microvasculature to transplacental exchange. Placenta 17: 89–96, 1996. [DOI] [PubMed] [Google Scholar]
- 186.Fisher CE, Howie SE. The role of megalin (LRP-2/Gp330) during development. Dev Biol 296: 279–297, 2006. [DOI] [PubMed] [Google Scholar]
- 187.Fogarty NM, Burton GJ, Ferguson-Smith AC. Different epigenetic states define syncytiotrophoblast and cytotrophoblast nuclei in the trophoblast of the human placenta. Placenta 36: 796–802, 2015. [DOI] [PubMed] [Google Scholar]
- 188.Fogarty NM, Ferguson-Smith AC, Burton GJ. Syncytial knots (Tenney-Parker Changes) in the human placenta: evidence of loss of transcriptional activity and oxidative damage. Am J Pathol 183: 144–152, 2013. [DOI] [PubMed] [Google Scholar]
- 189.Fowden AL, Coan PM, Angiolini E, Burton GJ, Constancia M. Imprinted genes and the epigenetic regulation of placental phenotype. Prog Biophys Mol Biol 106: 281–288, 2011. [DOI] [PubMed] [Google Scholar]
- 190.Fowden AL, Forhead AJ. Adrenal glands are essential for activation of glucogenesis during undernutrition in fetal sheep near term. Am J Physiol Endocrinol Metab 300: E94–E102, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Fowden AL, Forhead AJ, Sferruzzi-Perri AN, Burton GJ, Vaughan OR. Endocrine regulation of placental phenotype. Placenta 10.1016/jplacenta.2014.11.018: 2015. [DOI] [PubMed]
- 192.Fowden AL, Harding R, Ralph MM, Thorburn GD. The nutritional regulation of plasma prostaglandin E concentrations in the fetus and pregnant ewe during late gestation. J Physiol 394: 1–12, 1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Fowden AL, Moore T. Maternal-fetal resource allocation: co-operation and conflict. Placenta 33 Suppl 2: e11–15, 2012. [DOI] [PubMed] [Google Scholar]
- 194.Fowden AL, Mundy L, Silver M. Developmental regulation of glucogenesis in the sheep fetus during late gestation. J Physiol 508: 937–947, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Fowden AL, Ralph MM, Silver M. Nutritional regulation of uteroplacental prostaglandin production and metabolism in pregnant ewes and mares during late gestation. Exp Clin Endocrinol 102: 212–221, 1994. [DOI] [PubMed] [Google Scholar]
- 196.Fowden AL, Sferruzzi-Perri AN, Coan PM, Constancia M, Burton GJ. Placental efficiency and adaptation: Endocrine regulation. J Physiol 587: 3459–3472, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Fowden AL, Silver M. The effect of the nutritional state on uterine prostaglandin F metabolite concentrations in the pregnant ewe during late gestation. Q J Exp Physiol 68: 337–349, 1983. [DOI] [PubMed] [Google Scholar]
- 198.Fowden AL, Ward JW, Wooding FP, Forhead AJ, Constancia M. Programming placental nutrient transport capacity. J Physiol 572: 5–15, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Frank D, Fortino W, Clark L, Musalo R, Wang W, Saxena A, Li CM, Reik W, Ludwig T, Tycko B. Placental overgrowth in mice lacking the imprinted gene Ipl. Proc Natl Acad Sci USA 99: 7490–7495, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Frias AE, Morgan TK, Evans AE, Rasanen J, Oh KY, Thornburg KL, Grove KL. Maternal high-fat diet disturbs uteroplacental hemodynamics and increases the frequency of stillbirth in a nonhuman primate model of excess nutrition. Endocrinology 152: 2456–2464, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Fukuda K, Kanazawa H, Aizawa Y, Ardell JL, Shivkumar K. Cardiac innervation and sudden cardiac death. Circ Res 116: 2005–2019, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Gabory A, Ferry L, Fajardy I, Jouneau L, Gothie JD, Vige A, Fleur C, Mayeur S, Gallou-Kabani C, Gross MS, Attig L, Vambergue A, Lesage J, Reusens B, Vieau D, Remacle C, Jais JP, Junien C. Maternal diets trigger sex-specific divergent trajectories of gene expression and epigenetic systems in mouse placenta. PLoS One 7: e47986, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Gabory A, Roseboom TJ, Moore T, Moore LG, Junien C. Placental contribution to the origins of sexual dimorphism in health and diseases: sex chromosomes and epigenetics. Biol Sex Differ 4: 5, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Gaccioli F, Lager S, Powell TL, Jansson T. Placental transport in response to altered maternal nutrition. J Dev Origins Health Dis 4: 101–115, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Gaccioli F, White V, Capobianco E, Powell TL, Jawerbaum A, Jansson T. Maternal overweight induced by a diet with high content of saturated fat activates placental mTOR and eIF2alpha signaling and increases fetal growth in rats. Biol Reprod 89: 96, 2013. [DOI] [PubMed] [Google Scholar]
- 206.Gangloff YG, Mueller M, Dann SG, Svoboda P, Sticker M, Spetz JF, Um SH, Brown EJ, Cereghini S, Thomas G, Kozma SC. Disruption of the mouse mTOR gene leads to early postimplantation lethality and prohibits embryonic stem cell development. Mol Cell Biol 24: 9508–9516, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Ganguly A, Collis L, Devaskar SU. Placental glucose and amino acid transport in calorie-restricted wild-type and Glut3 null heterozygous mice. Endocrinology 153: 3995–4007, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Gauster M, Hiden U, Blaschitz A, Frank S, Lang U, Alvino G, Cetin I, Desoye G, Wadsack C. Dysregulation of placental endothelial lipase and lipoprotein lipase in intrauterine growth-restricted pregnancies. J Clin Endocrinol Metab 92: 2256–2263, 2007. [DOI] [PubMed] [Google Scholar]
- 209.Gentili S, Morrison JL, McMillen IC. Intrauterine growth restriction and differential patterns of hepatic growth and expression of IGF1, PCK2, and HSDL1 mRNA in the sheep fetus in late gestation. Biol Reprod 80: 1121–1127, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Georgiades P, Ferguson-Smith AC, Burton GJ. Comparative developmental anatomy of the murine and human definitive placenta. Placenta 23: 3–19, 2002. [DOI] [PubMed] [Google Scholar]
- 211.Gheorghe CP, Goyal R, Holweger JD, Longo LD. Placental gene expression responses to maternal protein restriction in the mouse. Placenta 30: 411–417, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Gheorghe CP, Mohan S, Oberg KC, Longo LD. Gene expression patterns in the hypoxic murine placenta: a role in epigenesis? Reprod Sci 14: 223–233, 2007. [DOI] [PubMed] [Google Scholar]
- 213.Giussani DA, Niu Y, Herrera EA, Richter HG, Camm EJ, Thakor AS, Kane AD, Hansell JA, Brain KL, Skeffington KL, Itani N, Wooding FB, Cross CM, Allison BJ. Heart disease link to fetal hypoxia and oxidative stress. Adv Exp Med Biol 814: 77–87, 2014. [DOI] [PubMed] [Google Scholar]
- 214.Giussani DA, Phillips PS, Anstee S, Barker DJ. Effects of altitude versus economic status on birth weight and body shape at birth. Pediatr Res 49: 490–494, 2001. [DOI] [PubMed] [Google Scholar]
- 215.Gluckman PD, Hanson MA, Cooper C, Thornburg KL. Effect of in utero and early-life conditions on adult health and disease. N Engl J Med 359: 61–73, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Godfrey K, Robinson S, Barker DJ, Osmond C, Cox V. Maternal nutrition in early and late pregnancy in relation to placental and fetal growth. BMJ 312: 410–414, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Godfrey KM. The role of the placenta in fetal programming—a review. Placenta 23 Suppl A: S20–27, 2002. [DOI] [PubMed] [Google Scholar]
- 218.Goenezen S, Rennie MY, Rugonyi S. Biomechanics of early cardiac development. Biomech Model Mechanobiol 11: 1187–1204, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Goirand F, Solar M, Athea Y, Viollet B, Mateo P, Fortin D, Leclerc J, Hoerter J, Ventura-Clapier R, Garnier A. Activation of AMP kinase alpha1 subunit induces aortic vasorelaxation in mice. J Physiol 581: 1163–1171, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Goldman-Wohl D, Yagel S. United we stand not dividing: the syncytiotrophoblast and cell senescence. Placenta 35: 341–344, 2014. [DOI] [PubMed] [Google Scholar]
- 221.Gomez-Roig MD, Mazarico E, Cardenas D, Fernandez MT, Diaz M, Ruiz de Gauna B, Vela A, Gratacos E, Figueras F. Placental 11B-hydroxysteroid dehydrogenase type 2 mRNA levels in intrauterine growth restriction versus small-for-gestational-age fetuses. Fetal Diagn Ther 39: 147–151, 2016. [DOI] [PubMed] [Google Scholar]
- 222.Goodman A, Kajantie E, Osmond C, Eriksson J, Koupil I, Thornburg K, Phillips DI. The relationship between umbilical cord length and chronic rheumatic heart disease: a prospective cohort study. Eur J Prev Cardiol 22: 1154–1160, 2015. [DOI] [PubMed] [Google Scholar]
- 223.Gordon Z, Elad D, Almog R, Hazan Y, Jaffa AJ, Eytan O. Anthropometry of fetal vasculature in the chorionic plate. J Anat 211: 698–706, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Gordon Z, Eytan O, Jaffa AJ, Elad D. Fetal blood flow in branching models of the chorionic arterial vasculature. Ann NY Acad Sci 1101: 250–265, 2007. [DOI] [PubMed] [Google Scholar]
- 225.Gorr TA, Wichmann D, Hu J, Hermes-Lima M, Welker AF, Terwilliger N, Wren JF, Viney M, Morris S, Nilsson GE, Deten A, Soliz J, Gassmann M. Hypoxia tolerance in animals: biology and application. Physiol Biochem Zool 83: 733–752, 2010. [DOI] [PubMed] [Google Scholar]
- 226.Goyal R, Papamatheakis DG, Loftin M, Vrancken K, Dawson AS, Osman NJ, Blood AB, Pearce WJ, Longo LD, Wilson SM. Long-term maternal hypoxia: the role of extracellular Ca2+ entry during serotonin-mediated contractility in fetal ovine pulmonary arteries. Reprod Sci 18: 948–962, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Gray CA, Taylor KM, Ramsey WS, Hill JR, Bazer FW, Bartol FF, Spencer TE. Endometrial glands are required for preimplantation conceptus elongation and survival. Biol Reprod 64: 1608–1613, 2001. [DOI] [PubMed] [Google Scholar]
- 228.Gregor MF, Hotamisligil GS. Inflammatory mechanisms in obesity. Annu Rev Immunol 29: 415–445, 2011. [DOI] [PubMed] [Google Scholar]
- 229.Gruenwald P. Expansion of placental site and maternal blood supply of primate placentas. Anat Rec 173: 189–203, 1972. [DOI] [PubMed] [Google Scholar]
- 230.Gu W, Jones CT, Parer JT. Metabolic and cardiovascular effects on fetal sheep of sustained reduction of uterine blood flow. J Physiol 368: 109–129, 1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Gundling WE Jr, Wildman DE. A review of inter- and intraspecific variation in the eutherian placenta. Philos Trans R Soc Lond B Biol Sci 370: 20140072, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Haar JL, Ackerman GA. Ultrastructural changes in mouse yolk sac associated with the initiation of vitelline circulation. Anat Rec 170: 437–455, 1971. [DOI] [PubMed] [Google Scholar]
- 233.Haavaldsen C, Tanbo T, Eskild A. Placental weight in singleton pregnancies with and without assisted reproductive technology: a population study of 536,567 pregnancies. Hum Reprod 27: 576–582, 2012. [DOI] [PubMed] [Google Scholar]
- 234.Hafez SA, Borowicz P, Reynolds LP, Redmer DA. Maternal and fetal microvasculature in sheep placenta at several stages of gestation. J Anat 216: 292–300, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Hafner E, Metzenbauer M, Hofinger D, Munkel M, Gassner R, Schuchter K, Dillinger-Paller B, Philipp K. Placental growth from the first to the second trimester of pregnancy in SGA-foetuses and pre-eclamptic pregnancies compared to normal foetuses. Placenta 24: 336–342, 2003. [DOI] [PubMed] [Google Scholar]
- 236.Hales CN, Barker DJ. The thrifty phenotype hypothesis. Br Med Bull 60: 5–20, 2001. [DOI] [PubMed] [Google Scholar]
- 237.Hanson MA, Gluckman PD. Early developmental conditioning of later health and disease: physiology or pathophysiology? Physiol Rev 94: 1027–1076, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Haouzi D, Dechaud H, Assou S, De Vos J, Hamamah S. Insights into human endometrial receptivity from transcriptomic and proteomic data. Reprod Biomed Online 24: 23–34, 2012. [DOI] [PubMed] [Google Scholar]
- 239.Hardie DG. Sensing of energy and nutrients by AMP-activated protein kinase. Am J Clin Nutr 93: 891S–896, 2011. [DOI] [PubMed] [Google Scholar]
- 240.Hardie DG, Ross FA, Hawley SA. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol 13: 251–262, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Harding HP, Zhang Y, Scheuner D, Chen JJ, Kaufman RJ, Ron D. Ppp1r15 gene knockout reveals an essential role for translation initiation factor 2 alpha (eIF2alpha) dephosphorylation in mammalian development. Proc Natl Acad Sci USA 106: 1832–1837, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Hardy DB, Yang K. The expression of 11 beta-hydroxysteroid dehydrogenase type 2 is induced during trophoblast differentiation: effects of hypoxia. J Clin Endocrinol Metab 87: 3696–3701, 2002. [DOI] [PubMed] [Google Scholar]
- 243.Harvey AJ, Kind KL, Pantaleon M, Armstrong DT, Thompson JG. Oxygen-regulated gene expression in bovine blastocysts. Biol Reprod 71: 1108–1119, 2004. [DOI] [PubMed] [Google Scholar]
- 244.Hastie R, Lappas M. The effect of pre-existing maternal obesity and diabetes on placental mitochondrial content and electron transport chain activity. Placenta 35: 673–683, 2014. [DOI] [PubMed] [Google Scholar]
- 245.Hauguel-de Mouzon S, Challier JC, Kacemi A, Cauzac M, Malek A, Girard J. The GLUT3 glucose transporter isoform is differentially expressed within human placental cell types. J Clin Endocrinol Metab 82: 2689–2694, 1997. [DOI] [PubMed] [Google Scholar]
- 246.Hay N, Sonenberg N. Upstream and downstream of mTOR. Genes Dev 18: 1926–1945, 2004. [DOI] [PubMed] [Google Scholar]
- 247.Hay WW., Jr Placental-fetal glucose exchange and fetal glucose metabolism. Trans Am Clin Climatol Assoc 117: 321–340, 2006. [PMC free article] [PubMed] [Google Scholar]
- 248.Hay WW Jr, Molina RA, DiGiacomo JE, Meschia G. Model of placental glucose consumption and glucose transfer. Am J Physiol Regul Integr Comp Physiol 258: R569–R577, 1990. [DOI] [PubMed] [Google Scholar]
- 249.Hayashi M, Sakata M, Takeda T, Yamamoto T, Okamoto Y, Sawada K, Kimura A, Minekawa R, Tahara M, Tasaka K, Murata Y. Induction of glucose transporter 1 expression through hypoxia-inducible factor 1alpha under hypoxic conditions in trophoblast-derived cells. J Endocrinol 183: 145–154, 2004. [DOI] [PubMed] [Google Scholar]
- 250.Heasman L, Clarke L, Stephenson TJ, Symonds ME. The influence of maternal nutrient restriction in early to mid-pregnancy on placental and fetal development in sheep. Proc Nutr Soc 58: 283–288, 1999. [DOI] [PubMed] [Google Scholar]
- 251.Heijmans J, van Lidth de Jeude JF, Koo BK, Rosekrans SL, Wielenga MC, van de Wetering M, Ferrante M, Lee AS, Onderwater JJ, Paton JC, Paton AW, Mommaas AM, Kodach LL, Hardwick JC, Hommes DW, Clevers H, Muncan V, van den Brink GR. ER stress causes rapid loss of intestinal epithelial stemness through activation of the unfolded protein response. Cell Reports 3: 1128–1139, 2013. [DOI] [PubMed] [Google Scholar]
- 252.Hemberger M, Udayashankar R, Tesar P, Moore H, Burton GJ. ELF5-enforced transcriptonal networks define an epigentically regulated trophoblast stem cell compartment in the human placenta. Mol Hum Genet 19: 2456–2467, 2010. [DOI] [PubMed] [Google Scholar]
- 253.Hempstock J, Bao YP, Bar-Issac M, Segaren N, Watson AL, Charnock Jones DS, Jauniaux E, Burton GJ. Intralobular differences in antioxidant enzyme expression and activity reflect oxygen gradients within the human placenta. Placenta 24: 517–523, 2003. [DOI] [PubMed] [Google Scholar]
- 254.Hempstock J, Cindrova-Davies T, Jauniaux E, Burton GJ. Endometrial glands as a source of nutrients, growth factors and cytokines during the first trimester of human pregnancy: a morphological and immunohistochemical study. Reprod Biol Endocrinol 2: 58, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Herrera EA, Krause B, Ebensperger G, Reyes RV, Casanello P, Parra-Cordero M, Llanos AJ. The placental pursuit for an adequate oxidant balance between the mother and the fetus. Front Pharmacol 5: 149, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Higgins JS, Vaughan OR, Fernandez de Liger E, Fowden AL, Sferruzzi-Perri AN. Placental phenotype and resource allocation to fetal growth are modified by the timing and degree of hypoxia during mouse pregnancy. J Physiol 594: 1341–1356, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Hill LM, DiNofrio DM, Chenevey P. Transvaginal sonographic evaluation of first-trimester placenta previa. Ultrasound Obstet Gynecol 5: 301–303, 1995. [DOI] [PubMed] [Google Scholar]
- 258.Himes KP, Koppes E, Chaillet JR. Generalized disruption of inherited genomic imprints leads to wide-ranging placental defects and dysregulated fetal growth. Dev Biol 373: 72–82, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Ho-Chen JK, Bustamante JJ, Soares MJ. Prolactin-like protein-f subfamily of placental hormones/cytokines: responsiveness to maternal hypoxia. Endocrinology 148: 559–565, 2007. [DOI] [PubMed] [Google Scholar]
- 260.Hogers B, DeRuiter MC, Gittenberger-de Groot AC, Poelmann RE. Unilateral vitelline vein ligation alters intracardiac blood flow patterns and morphogenesis in the chick embryo. Circ Res 80: 473–481, 1997. [DOI] [PubMed] [Google Scholar]
- 261.Holmes MC, Abrahamsen CT, French KL, Paterson JM, Mullins JJ, Seckl JR. The mother or the fetus? 11Beta-hydroxysteroid dehydrogenase type 2 null mice provide evidence for direct fetal programming of behavior by endogenous glucocorticoids. J Neurosci 26: 3840–3844, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Holwerda KM, Bos EM, Rajakumar A, Ris-Stalpers C, van Pampus MG, Timmer A, Erwich JJ, Faas MM, van Goor H, Lely AT. Hydrogen sulfide producing enzymes in pregnancy and preeclampsia. Placenta 33: 518–521, 2012. [DOI] [PubMed] [Google Scholar]
- 263.Hooper SB, Walker DW, Harding R. Oxygen, glucose, and lactate uptake by fetus and placenta during prolonged hypoxemia. Am J Physiol Regul Integr Comp Physiol 268: R303–R309, 1995. [DOI] [PubMed] [Google Scholar]
- 264.Hustin J, Schaaps JP. Echographic and anatomic studies of the maternotrophoblastic border during the first trimester of pregnancy. Am J Obstet Gynecol 157: 162–168, 1987. [DOI] [PubMed] [Google Scholar]
- 265.Hutchinson ES, Brownbill P, Jones NW, Abrahams VM, Baker PN, Sibley CP, Crocker IP. Utero-placental haemodynamics in the pathogenesis of pre-eclampsia. Placenta 30: 634–641, 2009. [DOI] [PubMed] [Google Scholar]
- 266.Hutson JR, Garcia-Bournissen F, Davis A, Koren G. The human placental perfusion model: a systematic review and development of a model to predict in vivo transfer of therapeutic drugs. Clin Pharmacol Ther 90: 67–76, 2011. [DOI] [PubMed] [Google Scholar]
- 267.Hutson JR, Koren G, Matthews SG. Placental P-glycoprotein and breast cancer resistance protein: influence of polymorphisms on fetal drug exposure and physiology. Placenta 31: 351–357, 2010. [DOI] [PubMed] [Google Scholar]
- 268.Ibanez L, Potau N, Enriquez G, Marcos MV, de Zegher F. Hypergonadotrophinaemia with reduced uterine and ovarian size in women born small-for-gestational-age. Hum Reprod 18: 1565–1569, 2003. [DOI] [PubMed] [Google Scholar]
- 269.Ietta F, Wu Y, Winter J, Xu J, Wang J, Post M, Caniggia I. Dynamic HIF1A regulation during human placental development. Biol Reprod 75: 112–121, 2006. [DOI] [PubMed] [Google Scholar]
- 270.Illsley NP. Glucose transporters in the human placenta. Placenta 21: 14–22, 2000. [DOI] [PubMed] [Google Scholar]
- 271.Illsley NP, Caniggia I, Zamudio S. Placental metabolic reprogramming: do changes in the mix of energy-generating substrates modulate fetal growth? Int J Dev Biol 54: 409–419, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Ishibashi O, Ohkuchi A, Ali MM, Kurashina R, Luo SS, Ishikawa T, Takizawa T, Hirashima C, Takahashi K, Migita M, Ishikawa G, Yoneyama K, Asakura H, Izumi A, Matsubara S, Takeshita T, Takizawa T. Hydroxysteroid (17-beta) dehydrogenase 1 is dysregulated by miR-210 and miR-518c that are aberrantly expressed in preeclamptic placentas: a novel marker for predicting preeclampsia. Hypertension 59: 265–273, 2012. [DOI] [PubMed] [Google Scholar]
- 273.Iwawaki T, Akai R, Yamanaka S, Kohno K. Function of IRE1 alpha in the placenta is essential for placental development and embryonic viability. Proc Natl Acad Sci USA 106: 16657–16662, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Jackson MR, Mayhew TM, Boyd PA. Quantitative description of the elaboration and maturation of villi from 10 weeks of gestation to term. Placenta 13: 357–370, 1992. [DOI] [PubMed] [Google Scholar]
- 275.Jackson MR, Mayhew TM, Haas JD. Morphometric studies on villi in human term placentae and the effects of altitude, ethnic grouping and sex of newborn. Placenta 8: 487–495, 1987. [DOI] [PubMed] [Google Scholar]
- 276.Jackson MR, Mayhew TM, Haas JD. On the factors which contribute to thinning of the villous membrane at high altitude. II. An increase in the degree of peripheralization of fetal capillaries. Placenta 9: 9–18, 1988. [DOI] [PubMed] [Google Scholar]
- 277.James JL, Stone PR, Chamley LW. The regulation of trophoblast differentiation by oxygen in the first trimester of pregnancy. Hum Reprod Update 12: 137–144, 2006. [DOI] [PubMed] [Google Scholar]
- 278.Jan E, Thompson SR, Wilson JE, Pestova TV, Hellen CU, Sarnow P. Initiator Met-tRNA-independent translation mediated by an internal ribosome entry site element in cricket paralysis virus-like insect viruses. Cold Spring Harb Symp Quant Biol 66: 285–292, 2001. [DOI] [PubMed] [Google Scholar]
- 279.Jansson N, Rosario FJ, Gaccioli F, Lager S, Jones HN, Roos S, Jansson T, Powell TL. Activation of placental mTOR signaling and amino acid transporters in obese women giving birth to large babies. J Clin Endocrinol Metab 98: 105–113, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Jansson T, Wennergren M, Illsley NP. Glucose transporter protein expression in human placenta throughout gestation and in intrauterine growth retardation. J Clin Endocrinol Metab 77: 1554–1562, 1993. [DOI] [PubMed] [Google Scholar]
- 281.Jauniaux E, Cindrova-Davies T, Johns J, Dunster C, Hempstock J, Kelly FJ, Burton GJ. Distribution and transfer pathways of antioxidant molecules inside the first trimester human gestational sac. J Clin Endocrinol Metab 89: 1452–1459, 2004. [DOI] [PubMed] [Google Scholar]
- 282.Jauniaux E, Gulbis B. Fluid compartments of the embryonic environment. Hum Reprod Update 6: 268–278, 2000. [DOI] [PubMed] [Google Scholar]
- 283.Jauniaux E, Hempstock J, Greenwold N, Burton GJ. Trophoblastic oxidative stress in relation to temporal and regional differences in maternal placental blood flow in normal and abnormal early pregnancies. Am J Pathol 162: 115–125, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Jauniaux E, Hempstock J, Teng C, Battaglia F, Burton GJ. Polyol concentrations in the fluid compartments of the human conceptus during the first trimester of pregnancy: maintenance of redox potential in a low oxygen environment. J Clin Endocrinol Metab 90: 1171–1175, 2005. [DOI] [PubMed] [Google Scholar]
- 285.Jauniaux E, Johns J, Gulbis B, Spasic-Boskovic O, Burton GJ. Transfer of folic acid inside the first-trimester gestational sac and the effect of maternal smoking. Am J Obstet Gynecol 197: 58 e51–56, 2007. [DOI] [PubMed] [Google Scholar]
- 286.Jauniaux E, Watson AL, Hempstock J, Bao YP, Skepper JN, Burton GJ. Onset of maternal arterial bloodflow and placental oxidative stress: a possible factor in human early pregnancy failure. Am J Pathol 157: 2111–2122, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Javam M, Audette MC, Iqbal M, Bloise E, Gibb W, Matthews SG. Effect of oxygen on multidrug resistance in term human placenta. Placenta 35: 324–330, 2014. [DOI] [PubMed] [Google Scholar]
- 288.Jiang B, Godfrey KM, Martyn CN, Gale CR. Birth weight and cardiac structure in children. Pediatrics 117: e257–261, 2006. [DOI] [PubMed] [Google Scholar]
- 289.Jiang B, Kamat A, Mendelson CR. Hypoxia prevents induction of aromatase expression in human trophoblast cells in culture: potential inhibitory role of the hypoxia-inducible transcription factor Mash-2 (mammalian achaete-scute homologous protein-2). Mol Endocrinol 14: 1661–1673, 2000. [DOI] [PubMed] [Google Scholar]
- 290.Jones CJ, Harris LK, Whittingham J, Aplin JD, Mayhew TM. A re-appraisal of the morphophenotype and basal lamina coverage of cytotrophoblasts in human term placenta. Placenta 29: 215–219, 2008. [DOI] [PubMed] [Google Scholar]
- 291.Jones CJP, Fox H. Ultrastructure of the normal human placenta. Electron Microsc Rev 4: 129–178, 1991. [DOI] [PubMed] [Google Scholar]
- 292.Jones CT, Ritchie JW, Walker D. The effects of hypoxia on glucose turnover in the fetal sheep. J Dev Physiol 5: 223–235, 1983. [PubMed] [Google Scholar]
- 293.Jones HN, Woollett LA, Barbour N, Prasad PD, Powell TL, Jansson T. High-fat diet before and during pregnancy causes marked up-regulation of placental nutrient transport and fetal overgrowth in C57/BL6 mice. FASEB J 23: 271–278, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Julian CG, Galan HL, Wilson MJ, Desilva W, Cioffi-Ragan D, Schwartz J, Moore LG. Lower uterine artery blood flow and higher endothelin relative to nitric oxide metabolite levels are associated with reductions in birth weight at high altitude. Am J Physiol Regul Integr Comp Physiol 295: R906–R915, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Julian CG, Wilson MJ, Lopez M, Yamashiro H, Tellez W, Rodriguez A, Bigham AW, Shriver MD, Rodriguez C, Vargas E, Moore LG. Augmented uterine artery blood flow and oxygen delivery protect Andeans from altitude-associated reductions in fetal growth. Am J Physiol Regul Integr Comp Physiol 296: R1564–R1575, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Julkunen M, Rutanen EM, Koskimies A, Ranta T, Bohn H, Seppala M. Distribution of placental protein 14 in tissues and body fluids during pregnancy. Br J Obstet Gynaecol 92: 1145–1151, 1985. [DOI] [PubMed] [Google Scholar]
- 297.Kaelin WG Jr, Ratcliffe PJ. Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. Mol Cell 30: 393–402, 2008. [DOI] [PubMed] [Google Scholar]
- 298.Kajantie E, Thornburg KL, Eriksson JG, Osmond C, Barker DJ. In preeclampsia, the placenta grows slowly along its minor axis. Int J Dev Biol 54: 469–473, 2010. [DOI] [PubMed] [Google Scholar]
- 299.Kakar MA, Maddocks S, Lorimer MF, Kleemann DO, Rudiger SR, Hartwich KM, Walker SK. The effect of peri-conception nutrition on embryo quality in the superovulated ewe. Theriogenology 64: 1090–1103, 2005. [DOI] [PubMed] [Google Scholar]
- 300.Kane HS, Dunkel Schetter C, Glynn LM, Hobel CJ, Sandman CA. Pregnancy anxiety and prenatal cortisol trajectories. Biol Psychol 100: 13–19, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.Kappen C, Kruger C, MacGowan J, Salbaum JM. Maternal diet modulates placenta growth and gene expression in a mouse model of diabetic pregnancy. PLoS One 7: e38445, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.Karimu AL, Burton GJ. The distribution of microvilli over the villous surface of the normal human term placenta is homogenous. Reprod Fertil Dev 7: 1269–1273, 1995. [DOI] [PubMed] [Google Scholar]
- 303.Kaufmann P, Bruns U, Leiser R, Luckhardt M, Winterhager E. The fetal vascularisation of term placental villi. II. Intermediate and terminal villi. Anat Embryol 173: 203–214, 1985. [DOI] [PubMed] [Google Scholar]
- 304.Kertschanska S, Kosanke G, Kaufmann P. Pressure dependence of so-called transtrophoblastic channels during fetal perfusion of human placental villi. Microsc Res Tech 38: 52–62, 1997. [DOI] [PubMed] [Google Scholar]
- 305.Keverne EB. Mammalian viviparity: a complex niche in the evolution of genomic imprinting. Heredity 113: 138–144, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.Khorram O, Ghazi R, Chuang TD, Han G, Naghi J, Ni Y, Pearce WJ. Excess maternal glucocorticoids in response to in utero undernutrition inhibit offspring angiogenesis. Reprod Sci 21: 601–611, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307.Kiernan MF, Barrie A, Szkolar J, Mills TA, Wareing M. Functional evidence for oxygen-sensitive voltage-gated potassium channels in human placental vasculature. Placenta 31: 553–555, 2010. [DOI] [PubMed] [Google Scholar]
- 308.Kim D. K+ channels in O2 sensing and postnatal development of carotid body glomus cell response to hypoxia. Respir Physiol Neurobiol 185: 44–56, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309.Kim DW, Young SL, Grattan DR, Jasoni CL. Obesity during pregnancy disrupts placental morphology, cell proliferation, and inflammation in a sex-specific manner across gestation in the mouse. Biol Reprod 90: 130, 2014. [DOI] [PubMed] [Google Scholar]
- 310.King AL, Lefer DJ. Cytoprotective actions of hydrogen sulfide in ischaemia-reperfusion injury. Exp Physiol 96: 840–846, 2011. [DOI] [PubMed] [Google Scholar]
- 311.King V, Hibbert N, Seckl JR, Norman JE, Drake AJ. The effects of an obesogenic diet during pregnancy on fetal growth and placental gene expression are gestation dependent. Placenta 34: 1087–1090, 2013. [DOI] [PubMed] [Google Scholar]
- 312.Kingdom JCP, Kaufmann P. Oxygen and placental villous development: origins of fetal hypoxia. Placenta 18: 613–621, 1997. [DOI] [PubMed] [Google Scholar]
- 313.Kleppa MJ, Erlenwein SV, Darashchonak N, von Kaisenberg CS, von Versen-Hoynck F. Hypoxia and the anticoagulants dalteparin and acetylsalicylic acid affect human placental amino acid transport. PLoS One 9: e99217, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314.Kolahi K, Louey S, Varlamov O, Thornburg K. Real-time tracking of BODIPY-C12 long-chain fatty acid in human term placenta reveals unique lipid dynamics in cytotrophoblast cells. PLoS One 11: e0153522, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 315.Koumenis C. ER stress, hypoxia tolerance and tumor progression. Curr Mol Med 6: 55–69, 2006. [DOI] [PubMed] [Google Scholar]
- 316.Krampl E, Kametas NA, Nowotny P, Roden M, Nicolaides KH. Glucose metabolism in pregnancy at high altitude. Diabetes Care 24: 817–822, 2001. [DOI] [PubMed] [Google Scholar]
- 317.Krebs C, Longo LD, Leiser R. Term ovine placental vasculature: comparison of sea level and high altitude conditions by corrosion cast and histomorphometry. Placenta 18: 43–51, 1997. [DOI] [PubMed] [Google Scholar]
- 318.Kulandavelu S, Whiteley KJ, Bainbridge SA, Qu D, Adamson SL. Endothelial NO synthase augments fetoplacental blood flow, placental vascularization, and fetal growth in mice. Hypertension 61: 259–266, 2013. [DOI] [PubMed] [Google Scholar]
- 319.Kulkarni SR, Kumaran K, Rao SR, Chougule SD, Deokar TM, Bhalerao AJ, Solat VA, Bhat DS, Fall CH, Yajnik CS. Maternal lipids are as important as glucose for fetal growth: findings from the Pune Maternal Nutrition Study. Diabetes Care 36: 2706–2713, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320.Kusinski LC, Stanley JL, Dilworth MR, Hirt CJ, Andersson IJ, Renshall LJ, Baker BC, Baker PN, Sibley CP, Wareing M, Glazier JD. eNOS knockout mouse as a model of fetal growth restriction with an impaired uterine artery function and placental transport phenotype. Am J Physiol Regul Integr Comp Physiol 303: R86–R93, 2012. [DOI] [PubMed] [Google Scholar]
- 321.Ladyman SR, Augustine RA, Grattan DR. Hormone interactions regulating energy balance during pregnancy. J Neuroendocrinol 22: 805–817, 2010. [DOI] [PubMed] [Google Scholar]
- 322.Lafond J, Charest MC, Alain JF, Brissette L, Masse A, Robidoux J, Simoneau L. Presence of CLA-1 and HDL binding sites on syncytiotrophoblast brush border and basal plasma membranes of human placenta. Placenta 20: 583–590, 1999. [DOI] [PubMed] [Google Scholar]
- 323.Lager S, Samulesson AM, Taylor PD, Poston L, Powell TL, Jansson T. Diet-induced obesity in mice reduces placental efficiency and inhibits placental mTOR signaling. Physiol Reports 2: e00242, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 324.Lahiri S, Roy A, Baby SM, Hoshi T, Semenza GL, Prabhakar NR. Oxygen sensing in the body. Prog Biophys Mol Biol 91: 249–286, 2006. [DOI] [PubMed] [Google Scholar]
- 325.Lambot N, Lybaert P, Boom A, Delogne-Desnoeck J, Vanbellinghen AM, Graff G, Lebrun P, Meuris S. Evidence for a clathrin-mediated recycling of albumin in human term placenta. Biol Reprod 75: 90–97, 2006. [DOI] [PubMed] [Google Scholar]
- 326.Lassance L, Haghiac M, Minium J, Catalano P, Hauguel-de Mouzon S. Obesity-induced down-regulation of the mitochondrial translocator protein (TSPO) impairs placental steroid production. J Clin Endocrinol Metab 100: E11–18, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 327.Lassance L, Miedl H, Absenger M, Diaz-Perez F, Lang U, Desoye G, Hiden U. Hyperinsulinemia stimulates angiogenesis of human fetoplacental endothelial cells: a possible role of insulin in placental hypervascularization in diabetes mellitus. J Clin Endocrinol Metab 98: E1438–1447, 2013. [DOI] [PubMed] [Google Scholar]
- 328.Lea RG, Wooding P, Stewart I, Hannah LT, Morton S, Wallace K, Aitken RP, Milne JS, Regnault TR, Anthony RV, Wallace JM. The expression of ovine placental lactogen, StAR and progesterone-associated steroidogenic enzymes in placentae of overnourished growing adolescent ewes. Reproduction 133: 785–796, 2007. [DOI] [PubMed] [Google Scholar]
- 329.Leach L. The phenotype of the human materno-fetal endothelial barrier: molecular occupancy of paracellular junctions dictate permeability and angiogenic plasticity. J Anat 200: 599–606, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 330.Leach L, Lammiman MJ, Babawale MO, Hobson SA, Bromilou B, Lovat S, Simmonds MJ. Molecular organization of tight and adherens junctions in the human placental vascular tree. Placenta 21: 547–557, 2000. [DOI] [PubMed] [Google Scholar]
- 331.Leary C, Leese HJ, Sturmey RG. Human embryos from overweight and obese women display phenotypic and metabolic abnormalities. Hum Reprod 30: 122–132, 2015. [DOI] [PubMed] [Google Scholar]
- 332.Leon DA, Lithell HO, Vagero D, Koupilova I, Mohsen R, Berglund L, Lithell UB, McKeigue PM. Reduced fetal growth rate and increased risk of death from ischaemic heart disease: cohort study of 15 000 Swedish men and women born 1915-29. BMJ 317: 241–245, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 333.Levkovitz R, Zaretsky U, Jaffa AJ, Hod M, Elad D. In vitro simulation of placental transport. Part II. Glucose transfer across the placental barrier model. Placenta 34: 708–715, 2013. [DOI] [PubMed] [Google Scholar]
- 334.Levy AP, Levy NS, Goldberg MA. Post-transcriptional regulation of vascular endothelial growth factor by hypoxia. J Biol Chem 271: 2746–2753, 1996. [DOI] [PubMed] [Google Scholar]
- 335.Lewis RM, Brooks S, Crocker IP, Glazier J, Hanson MA, Johnstone ED, Panitchob N, Please CP, Sibley CP, Widdows KL, Sengers BG. Review: modelling placental amino acid transfer–from transporters to placental function. Placenta 34 Suppl: S46–51, 2013. [DOI] [PubMed] [Google Scholar]
- 336.Lewis RM, Greenwood SL, Cleal JK, Crozier SR, Verrall L, Inskip HM, Cameron IT, Cooper C, Sibley CP, Hanson MA, Godfrey KM. Maternal muscle mass may influence system A activity in human placenta. Placenta 31: 418–422, 2010. [DOI] [PubMed] [Google Scholar]
- 337.Liang C, DeCourcy K, Prater MR. High-saturated-fat diet induces gestational diabetes and placental vasculopathy in C57BL/6 mice. Metabolism Clin Exp 59: 943–950, 2010. [DOI] [PubMed] [Google Scholar]
- 338.Liechty EA, Kelley J, Lemons JA. Effect of fasting on uteroplacental amino acid metabolism in the pregnant sheep. Biol Neonate 60: 207–214, 1991. [DOI] [PubMed] [Google Scholar]
- 339.Lievano S, Alarcon L, Chavez-Munguia B, Gonzalez-Mariscal L. Endothelia of term human placentae display diminished expression of tight junction proteins during preeclampsia. Cell Tissue Res 324: 433–448, 2006. [DOI] [PubMed] [Google Scholar]
- 340.Limesand SW, Jensen J, Hutton JC, Hay WW Jr. Diminished beta-cell replication contributes to reduced beta-cell mass in fetal sheep with intrauterine growth restriction. Am J Physiol Regul Integr Comp Physiol 288: R1297–R1305, 2005. [DOI] [PubMed] [Google Scholar]
- 341.Lindsey SE, Butcher JT, Yalcin HC. Mechanical regulation of cardiac development. Front Physiol 5: 318, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 342.Liu A, Nickerson A, Troyer A, Yin X, Cary R, Thornburg K, Wang R, Rugonyi S. Quantifying blood flow and wall shear stresses in the outflow tract of chick embryonic hearts. Comput Struct 89: 855–867, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 343.Liu L, Wise DR, Diehl JA, Simon MC. Hypoxic reactive oxygen species regulate the integrated stress response and cell survival. J Biol Chem 283: 31153–31162, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 344.Lloyd JB, Beckman DA, Brent RL. Nutritional role of the visceral yolk sac in organogenesis-stage rat embryos. Reprod Toxicol 12: 193–195, 1998. [DOI] [PubMed] [Google Scholar]
- 345.Loke YW. Transmission of parasites across the placenta. Adv Parasitol 21: 155–228, 1982. [DOI] [PubMed] [Google Scholar]
- 346.Loke YW, Eremin O, Ashby J, Day S. Characterization of the phagocytic cells isolated from the human placenta. J Reticuloendothel Soc 31: 317–324, 1982. [PubMed] [Google Scholar]
- 347.Lopez-Barneo J, del Toro R, Levitsky KL, Chiara MD, Ortega-Saenz P. Regulation of oxygen sensing by ion channels. J Appl Physiol 96: 1187–1195, 2004. [DOI] [PubMed] [Google Scholar]
- 348.Louey S, Jonker SS, Giraud GD, Thornburg KL. Placental insufficiency decreases cell cycle activity and terminal maturation in fetal sheep cardiomyocytes. J Physiol 580: 639–648, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 349.Lu PD, Harding HP, Ron D. Translation reinitiation at alternative open reading frames regulates gene expression in an integrated stress response. J Cell Biol 167: 27–33, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 350.Lumey LH. Compensatory placental growth after restricted maternal nutrition in early pregnancy. Placenta 19: 105–111, 1998. [DOI] [PubMed] [Google Scholar]
- 351.Luo R, Wang Y, Xu P, Cao G, Zhao Y, Shao X, Li YX, Chang C, Peng C, Wang YL. Hypoxia-inducible miR-210 contributes to preeclampsia via targeting thrombospondin type I domain containing 7A. Sci Rep 6: 19588, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 352.Luyckx VA, Brenner BM. Birth weight, malnutrition and kidney-associated outcomes–a global concern. Nat Rev Nephrol 11: 135–149, 2015. [DOI] [PubMed] [Google Scholar]
- 353.Ma Y, Zhu MJ, Uthlaut AB, Nijland MJ, Nathanielsz PW, Hess BW, Ford SP. Upregulation of growth signaling and nutrient transporters in cotyledons of early to mid-gestational nutrient restricted ewes. Placenta 32: 255–263, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 354.Ma Y, Zhu MJ, Zhang L, Hein SM, Nathanielsz PW, Ford SP. Maternal obesity and overnutrition alter fetal growth rate and cotyledonary vascularity and angiogenic factor expression in the ewe. Am J Physiol Regul Integr Comp Physiol 299: R249–R258, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 355.Malassiné A, Frendo JL, Evain-Brion D. A comparison of placental development and endocrine functions between the human and mouse model. Hum Reprod Update 9: 531–539, 2003. [DOI] [PubMed] [Google Scholar]
- 356.Mao J, Zhang X, Sieli PT, Falduto MT, Torres KE, Rosenfeld CS. Contrasting effects of different maternal diets on sexually dimorphic gene expression in the murine placenta. Proc Natl Acad Sci USA 107: 5557–5562, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 357.Marinoni E, Casciani V, Marianetti V, Di Rocco A, Moscarini M, Di Iorio R. Localization and distribution of adrenomedullin receptor in the human placenta: changes with gestational age. J Reprod Med 52: 831–838, 2007. [PubMed] [Google Scholar]
- 358.Maritz GS, Cock ML, Louey S, Joyce BJ, Albuquerque CA, Harding R. Effects of fetal growth restriction on lung development before and after birth: a morphometric analysis. Pediatr Pulmonol 32: 201–210, 2001. [DOI] [PubMed] [Google Scholar]
- 359.Maritz GS, Cock ML, Louey S, Suzuki K, Harding R. Fetal growth restriction has long-term effects on postnatal lung structure in sheep. Pediatr Res 55: 287–295, 2004. [DOI] [PubMed] [Google Scholar]
- 360.Martin PM, Sutherland AE. Exogenous amino acids regulate trophectoderm differentiation in the mouse blastocyst through an mTOR-dependent pathway. Dev Biol 240: 182–193, 2001. [DOI] [PubMed] [Google Scholar]
- 361.Martyn CN, Barker DJ, Osmond C. Mothers' pelvic size, fetal growth, and death from stroke and coronary heart disease in men in the UK. Lancet 348: 1264–1268, 1996. [DOI] [PubMed] [Google Scholar]
- 362.Martyn CN, Greenwald SE. A hypothesis about a mechanism for the programming of blood pressure and vascular disease in early life. Clin Exp Pharmacol Physiol 28: 948–951, 2001. [DOI] [PubMed] [Google Scholar]
- 363.Matheson H, Veerbeek JH, Charnock-Jones DS, Burton GJ, Yung HW. Morphological and molecular changes in the murine placenta exposed to normobaric hypoxia throughout pregnancy. J Physiol 594: 1371–1388, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 364.Mayhew TM. Allometric studies on growth and development of the human placenta: growth of tissue compartments and diffusive conductances in relation to placental volume and fetal mass. J Anat 208: 785–794, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 365.Mayhew TM. Scaling placental oxygen diffusion to birthweight: studies on placentae from low- and high-altitude pregnancies. J Anat 175: 187–194, 1991. [PMC free article] [PubMed] [Google Scholar]
- 366.Mayhew TM. Thinning of the intervascular tissue layers of the human placenta is an adaptive response to passive diffusion in vivo and may help to predict the origins of fetal hypoxia. Eur J Obstet Gynecol Reprod Biol 81: 101–109, 1998. [DOI] [PubMed] [Google Scholar]
- 367.Mayhew TM, Bowles C, Orme G. A stereological method for testing whether or not there is random deposition of perivillous fibrin-type fibrinoid at the villous surface: description and pilot applications to term placentae. Placenta 21: 684–692, 2000. [DOI] [PubMed] [Google Scholar]
- 368.Mayhew TM, Jackson MR, Boyd PA. Changes in oxygen diffusive conductances of human placental during gestation (10–41 weeks) are commensurate with the gain in fetal weight. Placenta 14: 51–61, 1993. [DOI] [PubMed] [Google Scholar]
- 369.Mayhew TM, Jackson MR, Haas JD. Microscopical morphology of the human placenta and its effects on oxygen diffusion: a morphometric study. Placenta 7: 121–131, 1986. [DOI] [PubMed] [Google Scholar]
- 370.Mayhew TM, Jackson MR, Haas JD. Oxygen diffusive conductances of human placentae from pregnancies at low and high altitudes. Placenta 11: 493–503, 1990. [DOI] [PubMed] [Google Scholar]
- 371.McArdle HJ, Andersen HS, Jones H, Gambling L. Copper and iron transport across the placenta: regulation and interactions. J Neuroendocrinol 20: 427–431, 2008. [DOI] [PubMed] [Google Scholar]
- 372.McCormick MC. The contribution of low birth weight to infant mortality and childhood morbidity. N Engl J Med 312: 82–90, 1985. [DOI] [PubMed] [Google Scholar]
- 373.McCrabb GJ, Egan AR, Hasking BJ. Maternal undernutrition during mid-pregnancy in sheep: variable effects on placental growth. J Agricult Sci 118: 127–132, 1992. [Google Scholar]
- 374.McMillen IC, Robinson JS. Developmental origins of the metabolic syndrome: prediction, plasticity, and programming. Physiol Rev 85: 571–633, 2005. [DOI] [PubMed] [Google Scholar]
- 375.Meissner U, Spranger R, Lehner M, Allabauer I, Rascher W, Dotsch J. Hypoxia-induced leptin production in human trophoblasts does not protect from apoptosis. Eur J Endocrinol 153: 455–461, 2005. [DOI] [PubMed] [Google Scholar]
- 376.Mele J, Muralimanoharan S, Maloyan A, Myatt L. Impaired mitochondrial function in human placenta with increased maternal adiposity. Am J Physiol Endocrinol Metab 307: E419–E425, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 377.Mestan K, Yu Y, Matoba N, Cerda S, Demmin B, Pearson C, Ortiz K, Wang X. Placental inflammatory response is associated with poor neonatal growth: preterm birth cohort study. Pediatrics 125: e891–898, 2010. [DOI] [PubMed] [Google Scholar]
- 378.Metz J, Heinrich D, Forssmann WG. Gap junctions in hemodichorial and hemotrichorial placentae. Cell Tissue Res 171: 305–315, 1976. [DOI] [PubMed] [Google Scholar]
- 379.Midgett M, Rugonyi S. Congenital heart malformations induced by hemodynamic altering surgical interventions. Front Physiol 5: 287, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 380.Milley JR. Ovine fetal leucine kinetics and protein metabolism during decreased oxygen availability. Am J Physiol Endocrinol Metab 274: E618–E626, 1998. [DOI] [PubMed] [Google Scholar]
- 381.Mina TH, Raikkonen K, Riley SC, Norman JE, Reynolds RM. Maternal distress associates with placental genes regulating fetal glucocorticoid exposure and IGF2: role of obesity and sex. Psychoneuroendocrinology 59: 112–122, 2015. [DOI] [PubMed] [Google Scholar]
- 382.Mistry HD, Kurlak LO, Whitley GS, Cartwright JE, Broughton Pipkin F, Tribe RM. Expression of voltage-dependent potassium channels in first trimester human placentae. Placenta 35: 337–340, 2014. [DOI] [PubMed] [Google Scholar]
- 383.Mistry HD, McCallum LA, Kurlak LO, Greenwood IA, Broughton Pipkin F, Tribe RM. Novel expression and regulation of voltage-dependent potassium channels in placentas from women with preeclampsia. Hypertension 58: 497–504, 2011. [DOI] [PubMed] [Google Scholar]
- 384.Molteni RA, Stys SJ, Battaglia FC. Relationship of fetal and placental weight in human beings: fetal/placental weight ratios at various gestational ages and birth weight distributions. J Reprod Med 21: 327–334, 1978. [PubMed] [Google Scholar]
- 385.Moore GE, Ishida M, Demetriou C, Al-Olabi L, Leon LJ, Thomas AC, Abu-Amero S, Frost JM, Stafford JL, Chaoqun Y, Duncan AJ, Baigel R, Brimioulle M, Iglesias-Platas I, Apostolidou S, Aggarwal R, Whittaker JC, Syngelaki A, Nicolaides KH, Regan L, Monk D, Stanier P. The role and interaction of imprinted genes in human fetal growth. Philos Trans R Soc Lond B Biol Sci 370: 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 386.Moore LG. Uterine blood flow as a determinant of fetoplacental development. In: The Placenta and Human Developmental Programming, edited by Burton GJ, Barker DJP, Moffett A, and Thornburg T. Cambridge, UK: Cambridge Univ. Press, 2010, p. 126–144. [Google Scholar]
- 387.Moore LG, Charles SM, Julian CG. Humans at high altitude: hypoxia and fetal growth. Respir Physiol Neurobiol 178: 181–190, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 388.Moore LG, Shriver M, Bemis L, Hickler B, Wilson M, Brutsaert T, Parra E, Vargas E. Maternal adaptation to high-altitude pregnancy: an experiment of nature–a review. Placenta 25 Suppl A: S60–71, 2004. [DOI] [PubMed] [Google Scholar]
- 389.Moore LG, Young D, McCullough RE, Droma T, Zamudio S. Tibetan protection from intrauterine growth restriction (IUGR) and reproductive loss at high altitude. Am J Hum Biol 13: 635–644, 2001. [DOI] [PubMed] [Google Scholar]
- 390.Moore T. Review: parent-offspring conflict and the control of placental function. Placenta 33 Suppl: S33–36, 2012. [DOI] [PubMed] [Google Scholar]
- 391.Moore T, Haig D. Genomic imprinting in mammalian development: a parental tug-of-war. Trends Genet 7: 45–49, 1991. [DOI] [PubMed] [Google Scholar]
- 392.Mori M, Ishikawa G, Luo SS, Mishima T, Goto T, Robinson JM, Matsubara S, Takeshita T, Kataoka H, Takizawa T. The cytotrophoblast layer of human chorionic villi becomes thinner but maintains its structural integrity during gestation. Biol Reprod 76: 164–172, 2007. [DOI] [PubMed] [Google Scholar]
- 393.Morrison JL. Sheep models of intrauterine growth restriction: fetal adaptations and consequences. Clin Exp Pharmacol Physiol 35: 730–743, 2008. [DOI] [PubMed] [Google Scholar]
- 394.Morrison JL, Botting KJ, Dyer JL, Williams SJ, Thornburg KL, McMillen IC. Restriction of placental function alters heart development in the sheep fetus. Am J Physiol Regul Integr Comp Physiol 293: R306–R313, 2007. [DOI] [PubMed] [Google Scholar]
- 395.Mossman HW. Vertebrate Fetal Membranes:Comparative Ontogeny and Morphology; Evolution; Phylogenetic Significance; Basic Functions; Research Opportunities. London: Macmillan, 1987, p. 383. [Google Scholar]
- 396.Mouillet JF, Chu T, Nelson DM, Mishima T, Sadovsky Y. MiR-205 silences MED1 in hypoxic primary human trophoblasts. FASEB J 24: 2030–2039, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 397.Mouillet JF, Donker RB, Mishima T, Cronqvist T, Chu T, Sadovsky Y. The unique expression and function of miR-424 in human placental trophoblasts. Biol Reprod 89: 25, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 398.Mouillet JF, Ouyang Y, Coyne CB, Sadovsky Y. MicroRNAs in placental health and disease. Am J Obstet Gynecol 213: S163–172, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 399.Mu J, Adamson SL. Developmental changes in hemodynamics of uterine artery, utero- and umbilicoplacental, and vitelline circulations in mouse throughout gestation. Am J Physiol Heart Circ Physiol 291: H1421–H1428, 2006. [DOI] [PubMed] [Google Scholar]
- 400.Muller-Schmehl K, Beninde J, Finckh B, Florian S, Dudenhausen JW, Brigelius-Flohe R, Schuelke M. Localization of alpha-tocopherol transfer protein in trophoblast, fetal capillaries' endothelium and amnion epithelium of human term placenta. Free Radic Res 38: 413–420, 2004. [DOI] [PubMed] [Google Scholar]
- 401.Muralimanoharan S, Maloyan A, Mele J, Guo C, Myatt LG, Myatt L. MIR-210 modulates mitochondrial respiration in placenta with preeclampsia. Placenta 33: 816–823, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 402.Murata M, Kodama H, Goto K, Hirano H, Tanaka T. Decreased very-low-density lipoprotein and low-density lipoprotein receptor messenger ribonucleic acid expression in placentas from preeclamptic pregnancies. Am J Obstet Gynecol 175: 1551–1556, 1996. [DOI] [PubMed] [Google Scholar]
- 403.Murray AJ. Oxygen delivery and fetal-placental growth: beyond a question of supply and demand? Placenta 33 Suppl 2: e16–22, 2012. [DOI] [PubMed] [Google Scholar]
- 404.Mustafa SA, Brizot ML, Carvalho MH, Watanabe L, Kahhale S, Zugaib M. Transvaginal ultrasonography in predicting placenta previa at delivery: a longitudinal study. Ultrasound Obstet Gynecol 20: 356–359, 2002. [DOI] [PubMed] [Google Scholar]
- 405.Myatt L, Cui X. Oxidative stress in the placenta. Histochem Cell Biol 122: 369–382, 2004. [DOI] [PubMed] [Google Scholar]
- 406.Myllynen P, Kummu M, Sieppi E. ABCB1 and ABCG2 expression in the placenta and fetus: an interspecies comparison. Expert Opin Drug Metab Toxicol 6: 1385–1398, 2010. [DOI] [PubMed] [Google Scholar]
- 407.Myllynen P, Pasanen M, Pelkonen O. Human placenta: a human organ for developmental toxicology research and biomonitoring. Placenta 26: 361–371, 2005. [DOI] [PubMed] [Google Scholar]
- 408.Myllynen P, Vahakangas K. Placental transfer and metabolism: an overview of the experimental models utilizing human placental tissue. Toxicol In Vitro 27: 507–512, 2013. [DOI] [PubMed] [Google Scholar]
- 409.Nagai A, Takebe K, Nio-Kobayashi J, Takahashi-Iwanaga H, Iwanaga T. Cellular expression of the monocarboxylate transporter (MCT) family in the placenta of mice. Placenta 31: 126–133, 2010. [DOI] [PubMed] [Google Scholar]
- 410.Nazio F, Strappazzon F, Antonioli M, Bielli P, Cianfanelli V, Bordi M, Gretzmeier C, Dengjel J, Piacentini M, Fimia GM, Cecconi F. mTOR inhibits autophagy by controlling ULK1 ubiquitylation, self-association and function through AMBRA1 and TRAF6. Nat Cell Biol 15: 406–416, 2013. [DOI] [PubMed] [Google Scholar]
- 411.Nelson DM, Johnson RD, Smith SD, Anteby EY, Sadovsky Y. Hypoxia limits differentiation and up-regulates expression and activity of prostaglandin H synthase 2 in cultured trophoblast from term human placenta. Am J Obstet Gynecol 180: 896–902, 1999. [DOI] [PubMed] [Google Scholar]
- 412.Nelson DM, Smith SD, Furesz TC, Sadovsky Y, Ganapathy V, Parvin CA, Smith CH. Hypoxia reduces expression and function of system A amino acid transporters in cultured term human trophoblasts. Am J Physiol Cell Physiol 284: C310–C315, 2003. [DOI] [PubMed] [Google Scholar]
- 413.Nevo O, Soleymanlou N, Wu Y, Xu J, Kingdom J, Many A, Zamudio S, Caniggia I. Increased expression of sFlt-1 in in vivo and in vitro models of human placental hypoxia is mediated by HIF-1. Am J Physiol Regul Integr Comp Physiol 291: R1085–R1093, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 414.Newbern D, Freemark M. Placental hormones and the control of maternal metabolism and fetal growth. Curr Opin Endocrinol Diabetes Obes 18: 409–416, 2011. [DOI] [PubMed] [Google Scholar]
- 415.Noel S, Herman A, Johnson GA, Gray CA, Stewart MD, Bazer FW, Gertler A, Spencer TE. Ovine placental lactogen specifically binds to endometrial glands of the ovine uterus. Biol Reprod 68: 772–780, 2003. [DOI] [PubMed] [Google Scholar]
- 416.Nogales FF, Beltran E, Gonzalez F. Morphological changes of the secondary human yolk sac in early pregnancy wastage. In: The Human Yolk Sac and Yolk Sac Tumours, edited by Nogales FF. Berlin: Springer-Verlag, 1993, p. 174–194. [Google Scholar]
- 417.Nugent BM, Bale TL. The omniscient placenta: Metabolic and epigenetic regulation of fetal programming. Front Neuroendocrinol 39: 28–37, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 418.O'Tierney PF, Lewis RM, McWeeney SK, Hanson MA, Inskip HM, Morgan TK, Barker DJ, Bagby G, Cooper C, Godfrey KM, Thornburg KL. Immune response gene profiles in the term placenta depend upon maternal muscle mass. Reprod Sci 19: 1041–1056, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 419.Ochocki JD, Simon MC. Nutrient-sensing pathways and metabolic regulation in stem cells. J Cell Biol 203: 23–33, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 420.Ockleford CD, Whyte A. Differeniated regions of human placental cell surface associated with exchange of materials between maternal and foetal blood: coated vesicles. J Cell Sci 25: 293–312, 1977. [DOI] [PubMed] [Google Scholar]
- 421.Oliver MH, Harding JE, Breier BH, Evans PC, Gluckman PD. The nutritional regulation of circulating placental lactogen in fetal sheep. Pediatr Res 31: 520–523, 1992. [DOI] [PubMed] [Google Scholar]
- 422.Oliver MH, Hawkins P, Harding JE. Periconceptional undernutrition alters growth trajectory and metabolic and endocrine responses to fasting in late-gestation fetal sheep. Pediatr Res 57: 591–598, 2005. [DOI] [PubMed] [Google Scholar]
- 423.Olson KR. Hydrogen sulfide as an oxygen sensor. Antioxid Redox Signal 22: 377–397, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 424.Orgeig S, Crittenden TA, Marchant C, McMillen IC, Morrison JL. Intrauterine growth restriction delays surfactant protein maturation in the sheep fetus. Am J Physiol Lung Cell Mol Physiol 298: L575–L583, 2010. [DOI] [PubMed] [Google Scholar]
- 425.Ovadya Y, Krizhanovsky V. Senescent cells: SASPected drivers of age-related pathologies. Biogerontology 15: 627–642, 2014. [DOI] [PubMed] [Google Scholar]
- 426.Ozanne SE, Constancia M. Mechanisms of disease: the developmental origins of disease and the role of the epigenotype. Nat Clin Pract Endocrinol Metab 3: 539–546, 2007. [DOI] [PubMed] [Google Scholar]
- 427.Padhee M, Zhang S, Lie S, Wang KC, Botting KJ, McMillen IC, MacLaughlin SM, Morrison JL. The periconceptional environment and cardiovascular disease: does in vitro embryo culture and transfer influence cardiovascular development and health? Nutrients 7: 1378–1425, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 428.Painter RC, de Rooij SR, Bossuyt PM, Simmers TA, Osmond C, Barker DJ, Bleker OP, Roseboom TJ. Early onset of coronary artery disease after prenatal exposure to the Dutch famine. Am J Clin Nutr 84: 322–327, 2006. [DOI] [PubMed] [Google Scholar]
- 429.Palmer SK, Moore LG, Young D, Cregger B, Berman JC, Zamudio S. Altered blood pressure course during normal pregnancy and increased preeclampsia at high altitude (3100 meters) in Colorado. Am J Obstet Gynecol 180: 1161–1168, 1999. [DOI] [PubMed] [Google Scholar]
- 430.Parr MB, Tung HN, Parr EL. The ultrastructure of the rat primary decidual zone. Am J Anat 176: 423–436, 1986. [DOI] [PubMed] [Google Scholar]
- 431.Parraguez VH, Atlagich MA, Urquieta B, Galleguillos M, De Los Reyes M, Kooyman DL, Araneda S, Raggi LA. Expression of vascular endothelial growth factor and endothelial nitric oxide synthase is increased in the placenta of sheep at high altitude in the Andes. Can J Vet Res 74: 193–199, 2010. [PMC free article] [PubMed] [Google Scholar]
- 432.Parraguez VH, Urquieta B, De los Reyes M, Gonzalez-Bulnes A, Astiz S, Munoz A. Steroidogenesis in sheep pregnancy with intrauterine growth retardation by high-altitude hypoxia: effects of maternal altitudinal status and antioxidant treatment. Reprod Fertil Dev 25: 639–645, 2013. [DOI] [PubMed] [Google Scholar]
- 433.Patel J, Landers K, Mortimer RH, Richard K. Regulation of hypoxia inducible factors (HIF) in hypoxia and normoxia during placental development. Placenta 31: 951–957, 2010. [DOI] [PubMed] [Google Scholar]
- 434.Patel J, Landers KA, Mortimer RH, Richard K. Expression and uptake of the thyroxine-binding protein transthyretin is regulated by oxygen in primary trophoblast placental cells. J Endocrinol 212: 159–167, 2012. [DOI] [PubMed] [Google Scholar]
- 435.Pathak S, Hook E, Hackett G, Murdoch E, Sebire NJ, Jessop F, Lees C. Cord coiling, umbilical cord insertion and placental shape in an unselected cohort delivering at term: relationship with common obstetric outcomes. Placenta 31: 963–968, 2010. [DOI] [PubMed] [Google Scholar]
- 436.Peng YJ, Nanduri J, Raghuraman G, Souvannakitti D, Gadalla MM, Kumar GK, Snyder SH, Prabhakar NR. H2S mediates O2 sensing in the carotid body. Proc Natl Acad Sci USA 107: 10719–10724, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 437.Penninga L, Longo LD. Ovine placentome morphology: effect of high altitude, long-term hypoxia. Placenta 19: 187–193, 1998. [DOI] [PubMed] [Google Scholar]
- 438.Perazzolo S, Hirschmugl B, Wadsack C, Desoye G, Lewis RM, Sengers BG. Computational modelling of fatty acid transport in the human placenta. Conf Proc IEEE Eng Med Biol Soc 2015: 8054–8057, 2015. [DOI] [PubMed] [Google Scholar]
- 439.Petry CJ, Evans ML, Wingate DL, Ong KK, Reik W, Constancia M, Dunger DB. Raised late pregnancy glucose concentrations in mice carrying pups with targeted disruption of H19delta13. Diabetes 59: 282–286, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 440.Philipp T, Philipp K, Reiner A, Beer F, Kalousek DK. Embryoscopic and cytogenetic analysis of 233 missed abortions: factors involved in the pathogenesis of developmental defects of early failed pregnancies. Hum Reprod 18: 1724–1732, 2003. [DOI] [PubMed] [Google Scholar]
- 441.Pijnenborg R, Vercruysse L, Hanssens M. The uterine spiral arteries in human pregnancy: facts and controversies. Placenta 27: 939–958, 2006. [DOI] [PubMed] [Google Scholar]
- 442.Poelmann RE, Gittenberger-de Groot AC, Hierck BP. The development of the heart and microcirculation: role of shear stress. Med Biol Eng Comput 46: 479–484, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 443.Postigo L, Heredia G, Illsley NP, Torricos T, Dolan C, Echalar L, Tellez W, Maldonado I, Brimacombe M, Balanza E, Vargas E, Zamudio S. Where the O2 goes to: preservation of human fetal oxygen delivery and consumption at high altitude. J Physiol 587: 693–708, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 444.Pringle KG, Kind KL, Sferruzzi-Perri AN, Thompson JG, Roberts CT. Beyond oxygen: complex regulation and activity of hypoxia inducible factors in pregnancy. Hum Reprod Update 16: 415–431, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 445.Radford EJ, Ferron SR, Ferguson-Smith AC. Genomic imprinting as an adaptative model of developmental plasticity. FEBS Lett 585: 2059–2066, 2011. [DOI] [PubMed] [Google Scholar]
- 446.Rai A, Cross JC. Development of the hemochorial maternal vascular spaces in the placenta through endothelial and vasculogenic mimicry. Dev Biol 387: 131–141, 2014. [DOI] [PubMed] [Google Scholar]
- 447.Rajakumar A, Conrad KP. Expression, ontogeny, and regulation of hypoxia-inducible transcription factors in the human placenta. Biol Reprod 63: 559–569, 2000. [DOI] [PubMed] [Google Scholar]
- 448.Rajakumar A, Michael HM, Daftary A, Jeyabalan A, Gilmour C, Conrad KP. Proteasomal activity in placentas from women with preeclampsia and intrauterine growth restriction: implications for expression of HIF-alpha proteins. Placenta 29: 290–299, 2008. [DOI] [PubMed] [Google Scholar]
- 449.Ramsey EM. The Placenta: Human and Animal. New York: Praeger, 1982, p. 187. [Google Scholar]
- 450.Rawn SM, Cross JC. The evolution, regulation, and function of placenta-specific genes. Annu Rev Cell Dev Biol 24: 159–181, 2008. [DOI] [PubMed] [Google Scholar]
- 451.Rebholz SL, Burke KT, Yang Q, Tso P, Woollett LA. Dietary fat impacts fetal growth and metabolism: uptake of chylomicron remnant core lipids by the placenta. Am J Physiol Endocrinol Metab 301: E416–E425, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 452.Rebsamen M, Pochini L, Stasyk T, de Araujo ME, Galluccio M, Kandasamy RK, Snijder B, Fauster A, Rudashevskaya EL, Bruckner M, Scorzoni S, Filipek PA, Huber KV, Bigenzahn JW, Heinz LX, Kraft C, Bennett KL, Indiveri C, Huber LA, Superti-Furga G. SLC38A9 is a component of the lysosomal amino acid sensing machinery that controls mTORC1. Nature 519: 477–481, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 453.Redline RW. Placental inflammation. Semin Neonatol 9: 265–274, 2004. [DOI] [PubMed] [Google Scholar]
- 454.Redmer DA, Wallace JM, Reynolds LP. Effect of nutrient intake during pregnancy on fetal and placental growth and vascular development. Domestic Anim Endocrinol 27: 199–217, 2004. [DOI] [PubMed] [Google Scholar]
- 455.Reece EA, Pinter E, Naftolin F. Experimental models of injury in the mammalian yolk sac. In: The Human Yolk Sac and Yolk Sac Tumours, edited by Nogales FF. Berlin: Springer-Verlag, 1993, p. 135–160. [Google Scholar]
- 456.Regnault TR, de Vrijer B, Galan HL, Wilkening RB, Battaglia FC, Meschia G. Development and mechanisms of fetal hypoxia in severe fetal growth restriction. Placenta 28: 714–723, 2007. [DOI] [PubMed] [Google Scholar]
- 457.Rennie MY, Detmar J, Whiteley KJ, Jurisicova A, Adamson SL, Sled JG. Expansion of the fetoplacental vasculature in late gestation is strain dependent in mice. Am J Physiol Heart Circ Physiol 302: H1261–H1273, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 458.Reshetnikova OS, Burton GJ, Milovanov AP. Effects of hypobaric hypoxia on the feto-placental unit; the morphometric diffusing capacity of the villous membrane at high altitude. Am J Obstet Gynecol 1 71: 1560–1565, 1994. [DOI] [PubMed] [Google Scholar]
- 459.Reshetnikova OS, Burton GJ, Milovanov AP, Fokin EI. Increased incidence of placental chorangioma in high altitude pregnancies; hypobaric hypoxia as a possible aetiological factor. Am J Obstet Gynecol 174: 557–561, 1996. [DOI] [PubMed] [Google Scholar]
- 460.Rexhaj E, Bloch J, Jayet PY, Rimoldi SF, Dessen P, Mathieu C, Tolsa JF, Nicod P, Scherrer U, Sartori C. Fetal programming of pulmonary vascular dysfunction in mice: role of epigenetic mechanisms. Am J Physiol Heart Circ Physiol 301: H247–H252, 2011. [DOI] [PubMed] [Google Scholar]
- 461.Reynolds LP, Borowicz PP, Caton JS, Vonnahme KA, Luther JS, Hammer CJ, Maddock Carlin KR, Grazul-Bilska AT, Redmer DA. Developmental programming: the concept, large animal models, and the key role of uteroplacental vascular development. J Anim Sci 88: E61–72, 2010. [DOI] [PubMed] [Google Scholar]
- 462.Reynolds LP, Borowicz PP, Palmieri C, Grazul-Bilska AT. Placental vascular defects in compromised pregnancies: effects of assisted reproductive technologies and other maternal stressors. Adv Exp Med Biol 814: 193–204, 2014. [DOI] [PubMed] [Google Scholar]
- 463.Reynolds LP, Caton JS, Redmer DA, Grazul-Bilska AT, Vonnahme KA, Borowicz PP, Luther JS, Wallace JM, Wu G, Spencer TE. Evidence for altered placental blood flow and vascularity in compromised pregnancies. J Physiol 572: 51–58, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 464.Rich-Edwards JW, Stampfer MJ, Manson JE, Rosner B, Hankinson SE, Colditz GA, Willett WC, Hennekens CH. Birth weight and risk of cardiovascular disease in a cohort of women followed up since 1976. BMJ 315: 396–400, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 465.Robbins JR, Skrzypczynska KM, Zeldovich VB, Kapidzic M, Bakardjiev AI. Placental syncytiotrophoblast constitutes a major barrier to vertical transmission of Listeria monocytogenes. PLoS Pathog 6: e1000732, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 466.Roberts VH, Rasanen JP, Novy MJ, Frias A, Louey S, Morgan TK, Thornburg KL, Spindel ER, Grigsby PL. Restriction of placental vasculature in a non-human primate: a unique model to study placental plasticity. Placenta 33: 73–76, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 467.Rolfo A, Many A, Racano A, Tal R, Tagliaferro A, Ietta F, Wang J, Post M, Caniggia I. Abnormalities in oxygen sensing define early and late onset preeclampsia as distinct pathologies. PLoS One 5: e13288, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 468.Roos S, Jansson N, Palmberg I, Saljo K, Powell TL, Jansson T. Mammalian target of rapamycin in the human placenta regulates leucine transport and is down-regulated in restricted fetal growth. J Physiol 582: 449–459, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 469.Roos S, Kanai Y, Prasad PD, Powell TL, Jansson T. Regulation of placental amino acid transporter activity by mammalian target of rapamycin. Am J Physiol Cell Physiol 296: C142–C150, 2009. [DOI] [PubMed] [Google Scholar]
- 470.Rosario FJ, Jansson N, Kanai Y, Prasad PD, Powell TL, Jansson T. Maternal protein restriction in the rat inhibits placental insulin, mTOR, and STAT3 signaling and down-regulates placental amino acid transporters. Endocrinology 152: 1119–1129, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 471.Roseboom TJ, Painter RC, de Rooij SR, van Abeelen AF, Veenendaal MV, Osmond C, Barker DJ. Effects of famine on placental size and efficiency. Placenta 32: 395–399, 2011. [DOI] [PubMed] [Google Scholar]
- 472.Rosenfeld CS. Sex-specific placental responses in fetal development. Endocrinology 156: 3422–3434, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 473.Rozance PJ, Anderson M, Martinez M, Fahy A, Macko AR, Kailey J, Seedorf GJ, Abman SH, Hay WW Jr, Limesand SW. Placental insufficiency decreases pancreatic vascularity and disrupts hepatocyte growth factor signaling in the pancreatic islet endothelial cell in fetal sheep. Diabetes 64: 555–564, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 474.Rueda-Clausen CF, Dolinsky VW, Morton JS, Proctor SD, Dyck JR, Davidge ST. Hypoxia-induced intrauterine growth restriction increases the susceptibility of rats to high-fat diet-induced metabolic syndrome. Diabetes 60: 507–516, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 475.Rueda-Clausen CF, Stanley JL, Thambiraj DF, Poudel R, Davidge ST, Baker PN. Effect of prenatal hypoxia in transgenic mouse models of preeclampsia and fetal growth restriction. Reprod Sci 21: 492–502, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 476.Rutland CS, Latunde-Dada AO, Thorpe A, Plant R, Langley-Evans S, Leach L. Effect of gestational nutrition on vascular integrity in the murine placenta. Placenta 28: 734–742, 2007. [DOI] [PubMed] [Google Scholar]
- 477.Salafia CM, Charles AK, Maas EM. Placenta and fetal growth restriction. Clin Obstet Gynecol 49: 236–256, 2006. [DOI] [PubMed] [Google Scholar]
- 478.Salafia CM, Yampolsky M, Misra DP, Shlakhter O, Haas D, Eucker B, Thorp J. Placental surface shape, function, and effects of maternal and fetal vascular pathology. Placenta 31: 958–962, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 479.Salafia CM, Yampolsky M, Shlakhter A, Mandel DH, Schwartz N. Variety in placental shape: when does it originate? Placenta 33: 164–170, 2012. [DOI] [PubMed] [Google Scholar]
- 480.Salafia CM, Zhang J, Miller RK, Charles AK, Shrout P, Sun W. Placental growth patterns affect birth weight for given placental weight. Birth Defects Res A Clin Mol Teratol 79: 281–288, 2007. [DOI] [PubMed] [Google Scholar]
- 481.Salavati N, Sovio U, Mayo RP, Charnock-Jones DS, Smith GC. The relationship between human placental morphometry and ultrasonic measurements of utero-placental blood flow and fetal growth. Placenta 38: 41–48, 2016. [DOI] [PubMed] [Google Scholar]
- 482.Sandovici I, Hoelle K, Angiolini E, Constancia M. Placental adaptations to the maternal-fetal environment: implications for fetal growth and developmental programming. Reprod Biomed Online 25: 68–89, 2012. [DOI] [PubMed] [Google Scholar]
- 483.Schaffer L, Vogel J, Breymann C, Gassmann M, Marti HH. Preserved placental oxygenation and development during severe systemic hypoxia. Am J Physiol Regul Integr Comp Physiol 290: R844–R851, 2006. [DOI] [PubMed] [Google Scholar]
- 484.Schneider H. Oxygenation of the placental-fetal unit in humans. Respir Physiol Neurobiol 178: 51–58, 2011. [DOI] [PubMed] [Google Scholar]
- 485.Schneider H. Tolerance of human placental tissue to severe hypoxia and its relevance for dual ex vivo perfusion. Placenta 30 Suppl A: S71–76, 2009. [DOI] [PubMed] [Google Scholar]
- 486.Schneider H, Miller RK. Receptor-mediated uptake and transport of macromolecules in the human placenta. Int J Dev Biol 54: 367–375, 2010. [DOI] [PubMed] [Google Scholar]
- 487.Scholler M, Wadsack C, Metso J, Chirackal Manavalan AP, Sreckovic I, Schweinzer C, Hiden U, Jauhiainen M, Desoye G, Panzenboeck U. Phospholipid transfer protein is differentially expressed in human arterial and venous placental endothelial cells and enhances cholesterol efflux to fetal HDL. J Clin Endocrinol Metab 97: 2466–2474, 2012. [DOI] [PubMed] [Google Scholar]
- 488.Schulz LC, Schlitt JM, Caesar G, Pennington KA. Leptin and the placental response to maternal food restriction during early pregnancy in mice. Biol Reprod 87: 120, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 489.Schwartz N, Mandel D, Shlakhter O, Coletta J, Pessel C, Timor-Tritsch IE, Salafia CM. Placental morphologic features and chorionic surface vasculature at term are highly correlated with 3-dimensional sonographic measurements at 11 to 14 weeks. J Ultrasound Med 30: 1171–1178, 2011. [DOI] [PubMed] [Google Scholar]
- 490.Schwartz N, Quant HS, Sammel MD, Parry S. Macrosomia has its roots in early placental development. Placenta 35: 684–690, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 491.Sejian V, Maurya VP, Naqvi SM. Adaptive capability as indicated by endocrine and biochemical responses of Malpura ewes subjected to combined stresses (thermal and nutritional) in a semi-arid tropical environment. Int J Biometeorol 54: 653–661, 2010. [DOI] [PubMed] [Google Scholar]
- 492.Sekita Y, Wagatsuma H, Nakamura K, Ono R, Kagami M, Wakisaka N, Hino T, Suzuki-Migishima R, Kohda T, Ogura A, Ogata T, Yokoyama M, Kaneko-Ishino T, Ishino F. Role of retrotransposon-derived imprinted gene, Rtl1, in the feto-maternal interface of mouse placenta. Nat Genet 40: 243–248, 2008. [DOI] [PubMed] [Google Scholar]
- 493.Semenza GL. Hypoxia-inducible factor 1: master regulator of O2 homeostasis. Curr Opin Genet Dev 8: 588–594, 1998. [DOI] [PubMed] [Google Scholar]
- 494.Sferruzzi-Perri AN, Owens JA, Pringle KG, Roberts CT. The neglected role of insulin-like growth factors in the maternal circulation regulating fetal growth. J Physiol 589: 7–20, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 495.Sferruzzi-Perri AN, Vaughan OR, Coan PM, Suciu MC, Darbyshire R, Constancia M, Burton GJ, Fowden AL. Placental-specific Igf2 deficiency alters developmental adaptations to undernutrition in mice. Endocrinology 152: 3202–3212, 2011. [DOI] [PubMed] [Google Scholar]
- 496.Sferruzzi-Perri AN, Vaughan OR, Haro M, Cooper WN, Musial B, Charalambous M, Pestana D, Ayyar S, Ferguson-Smith AC, Burton GJ, Constancia M, Fowden AL. An obesogenic diet during mouse pregnancy modifies maternal nutrient partitioning and the fetal growth trajectory. FASEB J 27: 3928–3937, 2013. [DOI] [PubMed] [Google Scholar]
- 497.Shams M, Kilby MD, Somerset DA, Howie AJ, Gupta A, Wood PJ, Afnan M, Stewart PM. 11Beta-hydroxysteroid dehydrogenase type 2 in human pregnancy and reduced expression in intrauterine growth restriction. Hum Reprod 13: 799–804, 1998. [DOI] [PubMed] [Google Scholar]
- 498.Sharma A, Guan H, Yang K. The p38 mitogen-activated protein kinase regulates 11beta-hydroxysteroid dehydrogenase type 2 (11beta-HSD2) expression in human trophoblast cells through modulation of 11beta-HSD2 messenger ribonucleic acid stability. Endocrinology 150: 4278–4286, 2009. [DOI] [PubMed] [Google Scholar]
- 499.Shen G, Li X, Jia YF, Piazza GA, Xi Y. Hypoxia-regulated microRNAs in human cancer. Acta Pharmacol Sin 34: 336–341, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 500.Shiverick K, Ino K, Harada T, Keelan J, Kikkawa F. Placental enzymes and transporters: new functions and genetic polymorphisms–a workshop report. Placenta 28 Suppl A: S125–128, 2007. [DOI] [PubMed] [Google Scholar]
- 501.Shmakova A, Batie M, Druker J, Rocha S. Chromatin and oxygen sensing in the context of JmjC histone demethylases. Biochem J 462: 385–395, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 502.Sibley CP, Coan PM, Ferguson-Smith AC, Dean W, Hughes J, Smith P, Reik W, Burton GJ, Fowden AL, Constancia M. Placental-specific insulin-like growth factor 2 (Igf2) regulates the diffusional exchange characteristics of the mouse placenta. Proc Natl Acad Sci USA 101: 8204–8208, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 503.Simmons DG, Fortier AL, Cross JC. Diverse subtypes and developmental origins of trophoblast giant cells in the mouse placenta. Dev Biol 304: 567–578, 2007. [DOI] [PubMed] [Google Scholar]
- 504.Simmons DG, Rawn S, Davies A, Hughes M, Cross JC. Spatial and temporal expression of the 23 murine Prolactin/Placental Lactogen-related genes is not associated with their position in the locus. BMC Genomics 9: 352, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 505.Skeffington KL, Higgins JS, Mahmoud AD, Evans AM, Sferruzzi-Perri AN, Fowden AL, Yung HW, Burton GJ, Giussani DA, Moore LG. Hypoxia, AMPK activation and uterine artery vasoreactivity. J Physiol 594: 1357–1369, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 506.Smith BT, Mussell JC, Fleming PA, Barth JL, Spyropoulos DD, Cooley MA, Drake CJ, Argraves WS. Targeted disruption of cubilin reveals essential developmental roles in the structure and function of endoderm and in somite formation. BMC Dev Biol 6: 30, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 507.Soares MJ. The prolactin and growth hormone families: pregnancy-specific hormones/cytokines at the maternal-fetal interface. Reprod Biol Endocrinol 2: 51, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 508.Society of Obstetricians annd Gynaecologists of Canada, Okun N, Sierra S. Pregnancy outcomes after assisted human reproduction. J Obstet Gynaecol Can 36: 64–83, 2014. [DOI] [PubMed] [Google Scholar]
- 509.Soma H, Hata T, Oguro T, Fujita K, Kudo M, Vaidya U. Characteristics of histopathological and ultrastructural features of placental villi in pregnant Nepalese women. Med Mol Morphol 38: 92–103, 2005. [DOI] [PubMed] [Google Scholar]
- 510.Soma H, Watanabe Y, Hata T. Chorangiosis and chorangioma in three cohorts of placentas from Nepal, Tibet and Japan. Reprod Fertil Dev 7: 1533–1538, 1995. [DOI] [PubMed] [Google Scholar]
- 511.Soncin F, Natale D, Parast MM. Signaling pathways in mouse and human trophoblast differentiation: a comparative review. Cell Mol Life Sci 72: 1291–1302, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 512.Sood R, Zehnder JL, Druzin ML, Brown PO. Gene expression patterns in human placenta. Proc Natl Acad Sci USA 103: 5478–5483, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 513.Soregaroli M, Bonera R, Danti L, Dinolfo D, Taddei F, Valcamonico A, Frusca T. Prognostic role of umbilical artery Doppler velocimetry in growth-restricted fetuses. J Matern Fetal Neonatal Med 11: 199–203, 2002. [DOI] [PubMed] [Google Scholar]
- 514.Spencer TE. Biological roles of uterine glands in pregnancy. Semin Reprod Med 32: 346–357, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 515.Spencer TE, Bazer FW. Uterine and placental factors regulating conceptus growth in domestic animals. J Anim Sci 82 E Suppl: E4–13, 2004. [DOI] [PubMed] [Google Scholar]
- 516.Stegeman J. Placental development in the sheep and its relation to fetal development. A qualitative and quantitative anatomic and histologic study. Bijdragen tot de Dierkunde 44: 1–72, 1974. [Google Scholar]
- 517.Stein CE, Fall CH, Kumaran K, Osmond C, Cox V, Barker DJ. Fetal growth and coronary heart disease in south India. Lancet 348: 1269–1273, 1996. [DOI] [PubMed] [Google Scholar]
- 518.Stewart PM, Rogerson FM, Mason JI. Type 2 11 beta-hydroxysteroid dehydrogenase messenger ribonucleic acid and activity in human placenta and fetal membranes: its relationship to birth weight and putative role in fetal adrenal steroidogenesis. J Clin Endocrinol Metab 80: 885–890, 1995. [DOI] [PubMed] [Google Scholar]
- 519.Sun M, Kingdom J, Baczyk D, Lye SJ, Matthews SG, Gibb W. Expression of the multidrug resistance P-glycoprotein, (ABCB1 glycoprotein) in the human placenta decreases with advancing gestation. Placenta 27: 602–609, 2006. [DOI] [PubMed] [Google Scholar]
- 520.Swanson AM, David AL. Animal models of fetal growth restriction: considerations for translational medicine. Placenta 36: 623–630, 2015. [DOI] [PubMed] [Google Scholar]
- 521.Swanson LD, Bewtra C. Increase in normal placental weights related to increase in maternal body mass index. J Matern Fetal Neonatal Med 21: 111–113, 2008. [DOI] [PubMed] [Google Scholar]
- 522.Tarrade A, Panchenko P, Junien C, Gabory A. Placental contribution to nutritional programming of health and diseases: epigenetics and sexual dimorphism. J Exp Biol 218: 50–58, 2015. [DOI] [PubMed] [Google Scholar]
- 523.Taylor CT. Mitochondria and cellular oxygen sensing in the HIF pathway. Biochem J 409: 19–26, 2008. [DOI] [PubMed] [Google Scholar]
- 524.Tchirikov M, Strohner M, Scholz A. Cardiac output and blood flow volume redistribution during acute maternal hypoxia in fetal sheep. J Perinat Med 38: 387–392, 2010. [DOI] [PubMed] [Google Scholar]
- 525.Tchirikov M, Tchirikov M, Buchert R, Wilke F, Brenner W. Glucose uptake in the placenta, fetal brain, heart and liver related to blood flow redistribution during acute hypoxia. J Obstet Gynaecol Res 37: 979–985, 2011. [DOI] [PubMed] [Google Scholar]
- 526.Thompson JA, Gimbel SA, Richardson BS, Gagnon R, Regnault TR. The effect of intermittent umbilical cord occlusion on elastin composition in the ovine fetus. Reprod Sci 18: 990–997, 2011. [DOI] [PubMed] [Google Scholar]
- 527.Thornburg K, O'Tierney PF, Morgan T, Louey S. The placental roots of cardiovascular disease. In: The Placenta and Human Developmental Programming, edited by Burton GJ, Barker DJP, Moffett A, and Thornburg K. Cambridge, UK: Cambridge Univ. Press, 2011, p. 201–215. [Google Scholar]
- 528.Thornburg KL. The programming of cardiovascular disease. J Dev Origins Health Dis 1–11, 2015. [DOI] [PMC free article] [PubMed]
- 529.Thornburg KL, Burry KJ, Adams AK, Kirk EP, Faber JJ. Permeability of placenta to inulin. Am J Obstet Gynecol 158: 1165–1169, 1988. [DOI] [PubMed] [Google Scholar]
- 530.Thornburg KL, Challis JR. How to build a healthy heart from scratch. Adv Exp Med Biol 814: 205–216, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 531.Thornburg KL, O'Tierney PF, Louey S. Review: the placenta is a programming agent for cardiovascular disease. Placenta 31 Suppl: S54–59, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 532.Tissot van Patot M, Grilli A, Chapman P, Broad E, Tyson W, Heller DS, Zwerdlinger L, Zamudio S. Remodelling of uteroplacental arteries is decreased in high altitude placentae. Placenta 24: 326–335, 2003. [DOI] [PubMed] [Google Scholar]
- 533.Tissot van Patot MC, Bendrick-Peart J, Beckey VE, Serkova N, Zwerdlinger L. Greater vascularity, lowered HIF-1/DNA binding, and elevated GSH as markers of adaptation to in vivo chronic hypoxia. Am J Physiol Lung Cell Mol Physiol 287: L525–L532, 2004. [DOI] [PubMed] [Google Scholar]
- 534.Tissot van Patot MC, Murray AJ, Beckey V, Cindrova-Davies T, Johns J, Zwerdlinger L, Jauniaux E, Burton GJ, Serkova NJ. Human placental metabolic adaptation to chronic hypoxia, high altitude: hypoxic preconditioning. Am J Physiol Regul Integr Comp Physiol 298: R166–R172, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 535.Tomlinson TM, Garbow JR, Anderson JR, Engelbach JA, Nelson DM, Sadovsky Y. Magnetic resonance imaging of hypoxic injury to the murine placenta. Am J Physiol Regul Integr Comp Physiol 298: R312–R319, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 536.Tsukimori K, Morokuma S, Hori T, Takahashi K, Hirata T, Otera Y, Fukushima K, Kawamoto T, Wake N. Characterization of placental transfer of polychlorinated dibenzo-p-dioxins, dibenzofurans and polychlorinated biphenyls in normal pregnancy. J Obstet Gynaecol Res 39: 83–90, 2013. [DOI] [PubMed] [Google Scholar]
- 537.Tunster SJ, Jensen AB, John RM. Imprinted genes in mouse placental development and the regulation of fetal energy stores. Reproduction 145: R117–137, 2013. [DOI] [PubMed] [Google Scholar]
- 538.Ueno M, Lee LK, Chhabra A, Kim YJ, Sasidharan R, Van Handel B, Wang Y, Kamata M, Kamran P, Sereti KI, Ardehali R, Jiang M, Mikkola HK. c-Met-dependent multipotent labyrinth trophoblast progenitors establish placental exchange interface. Dev Cell 27: 373–386, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 539.Uniacke J, Holterman CE, Lachance G, Franovic A, Jacob MD, Fabian MR, Payette J, Holcik M, Pause A, Lee S. An oxygen-regulated switch in the protein synthesis machinery. Nature 486: 126–129, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 540.Vahakangas K, Myllynen P. Drug transporters in the human blood-placental barrier. Br J Pharmacol 158: 665–678, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 541.Van Abeelen AF, de Rooij SR, Osmond C, Painter RC, Veenendaal MV, Bossuyt PM, Elias SG, Grobbee DE, van der Schouw YT, Barker DJ, Roseboom TJ. The sex-specific effects of famine on the association between placental size and later hypertension. Placenta 32: 694–698, 2011. [DOI] [PubMed] [Google Scholar]
- 542.Van den Beucken T, Koritzinsky M, Wouters BG. Translational control of gene expression during hypoxia. Cancer Biol Ther 5: 749–755, 2006. [DOI] [PubMed] [Google Scholar]
- 543.Van Patot MC, Ebensperger G, Gassmann M, Llanos AJ. The hypoxic placenta. High Altitude Med Biol 13: 176–184, 2012. [DOI] [PubMed] [Google Scholar]
- 544.Varmuza S, Miri K. What does genetics tell us about imprinting and the placenta connection? Cell Mol Life Sci 72: 51–72, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 545.Vatnick I, Schoknecht PA, Darrigrand R, Bell AW. Growth and metabolism of the placenta after unilateral fetectomy in twin pregnant ewes. J Dev Physiol 15: 351–356, 1991. [PubMed] [Google Scholar]
- 546.Vaughan OR, Sferruzzi-Perri AN, Fowden AL. Maternal corticosterone regulates nutrient allocation to fetal growth in mice. J Physiol 590: 5529–5540, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 547.Versen-Hoynck FMV, Rajakumar A, Roberts JM, Rath W, Powers RW. Placental Amino Acid transport is decreased after exposure to hypoxia. Geburtshilfe Frauenheilkd 68: PO. Geb.02.08, 2008. [Google Scholar]
- 548.Vijayakumar M, Fall CH, Osmond C, Barker DJ. Birth weight, weight at one year, and left ventricular mass in adult life. Br Heart J 73: 363–367, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 549.Viollet B, Horman S, Leclerc J, Lantier L, Foretz M, Billaud M, Giri S, Andreelli F. AMPK inhibition in health and disease. Crit Rev Biochem Mol Biol 45: 276–295, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 550.Vonnahme KA, Lemley CO, Shukla P, O'Rourke ST. 2011 and 2012 Early Careers Achievement Awards: Placental programming: how the maternal environment can impact placental function. J Anim Sci 91: 2467–2480, 2013. [DOI] [PubMed] [Google Scholar]
- 551.Vonnahme KA, Wilson ME, Ford SP. Conceptus competition for uterine space: different strategies exhibited by the Meishan and Yorkshire pig. J Anim Sci 80: 1311–1316, 2002. [DOI] [PubMed] [Google Scholar]
- 552.Wadsack C, Tabano S, Maier A, Hiden U, Alvino G, Cozzi V, Huttinger M, Schneider WJ, Lang U, Cetin I, Desoye G. Intrauterine growth restriction is associated with alterations in placental lipoprotein receptors and maternal lipoprotein composition. Am J Physiol Endocrinol Metab 292: E476–E484, 2007. [DOI] [PubMed] [Google Scholar]
- 553.Wallace JM, Bhattacharya S, Campbell DM, Horgan GW. Inter-pregnancy weight change impacts placental weight and is associated with the risk of adverse pregnancy outcomes in the second pregnancy. BMC Pregnancy Childbirth 14: 40, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 554.Wallace JM, Bourke DA, Aitken RP, Milne JS, Hay WW Jr. Placental glucose transport in growth-restricted pregnancies induced by overnourishing adolescent sheep. J Physiol 547: 85–94, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 555.Wallace JM, Da Silva P, Aitken RP, Cruickshank MA. Maternal endocrine status in relation to pregnancy outcome in rapidly growing adolescent sheep. J Endocrinol 155: 359–368, 1997. [DOI] [PubMed] [Google Scholar]
- 556.Wallace JM, Horgan GW, Bhattacharya S. Placental weight and efficiency in relation to maternal body mass index and the risk of pregnancy complications in women delivering singleton babies. Placenta 33: 611–618, 2012. [DOI] [PubMed] [Google Scholar]
- 557.Wallace JM, Luther JS, Milne JS, Aitken RP, Redmer DA, Reynolds LP, Hay WW Jr. Nutritional modulation of adolescent pregnancy outcome—a review. Placenta 27 Suppl A: S61–68, 2006. [DOI] [PubMed] [Google Scholar]
- 558.Walton A, Hammond J. The maternal effects on growth and conformation in Shire horse-Shetland pony crosses. Proc R Soc Lond Ser B Biol Sci 125: 311–335, 1938. [Google Scholar]
- 559.Wang K, Ahmad S, Cai M, Rennie J, Fujisawa T, Crispi F, Baily J, Miller MR, Cudmore M, Hadoke PW, Wang R, Gratacos E, Buhimschi IA, Buhimschi CS, Ahmed A. Dysregulation of hydrogen sulfide producing enzyme cystathionine gamma-lyase contributes to maternal hypertension and placental abnormalities in preeclampsia. Circulation 127: 2514–2522, 2013. [DOI] [PubMed] [Google Scholar]
- 560.Wang X, Li W, Williams M, Terada N, Alessi DR, Proud CG. Regulation of elongation factor 2 kinase by p90(RSK1) and p70 S6 kinase. EMBO J 20: 4370–4379, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 561.Wang X, Proud CG. The mTOR pathway in the control of protein synthesis. Physiology 21: 362–369, 2006. [DOI] [PubMed] [Google Scholar]
- 562.Ward JP. Oxygen sensors in context. Biochim Biophys Acta 1777: 1–14, 2008. [DOI] [PubMed] [Google Scholar]
- 563.Wareing M. Oxygen sensitivity, potassium channels, and regulation of placental vascular tone. Microcirculation 21: 58–66, 2014. [DOI] [PubMed] [Google Scholar]
- 564.Wareing M, Greenwood SL. Review: potassium channels in the human fetoplacental vasculature. Placenta 32 Suppl 2: S203–206, 2011. [DOI] [PubMed] [Google Scholar]
- 565.Warner MJ, Ozanne SE. Mechanisms involved in the developmental programming of adulthood disease. Biochem J 427: 333–347, 2010. [DOI] [PubMed] [Google Scholar]
- 566.Watson ED, Cross JC. Development of structures and transport functions in the mouse placenta. Physiology 20: 180–193, 2005. [DOI] [PubMed] [Google Scholar]
- 567.Wellen KE, Thompson CB. Cellular metabolic stress: considering how cells respond to nutrient excess. Mol Cell 40: 323–332, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 568.Welsh GI, Miller CM, Loughlin AJ, Price NT, Proud CG. Regulation of eukaryotic initiation factor eIF2B: glycogen synthase kinase-3 phosphorylates a conserved serine which undergoes dephosphorylation in response to insulin. FEBS Lett 421: 125–130, 1998. [DOI] [PubMed] [Google Scholar]
- 569.Wen HY, Abbasi S, Kellems RE, Xia Y. mTOR: a placental growth signaling sensor. Placenta 26 Suppl A: S63–69, 2005. [DOI] [PubMed] [Google Scholar]
- 570.Whitley GS, Cartwright JE. Cellular and molecular regulation of spiral artery remodelling: lessons from the cardiovascular field. Placenta 31: 465–474, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 571.Wild AE. Endocytic mechanisms in protein transfer across the placenta. Placenta Suppl 1: 165–186, 1981. [Google Scholar]
- 572.Wildman DE, Chen C, Erez O, Grossman LI, Goodman M, Romero R. Evolution of the mammalian placenta revealed by phylogenetic analysis. Proc Natl Acad Sci USA 103: 3203–3208, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 573.Wilkening RB, Meschia G. Current topic: comparative physiology of placental oxygen transport. Placenta 13: 1–15, 1992. [DOI] [PubMed] [Google Scholar]
- 574.Williams JL, Fyfe GK, Sibley CP, Baker PN, Greenwood SL. K+ channel inhibition modulates the biochemical and morphological differentiation of human placental cytotrophoblast cells in vitro. Am J Physiol Regul Integr Comp Physiol 295: R1204–R1213, 2008. [DOI] [PubMed] [Google Scholar]
- 575.Willis DM, O'Grady JP, Faber JJ, Thornburg KL. Diffusion permeability of cyanocobalamin in human placenta. Am J Physiol Regul Integr Comp Physiol 250: R459–R464, 1986. [DOI] [PubMed] [Google Scholar]
- 576.Wilson ME, Ford SP. Comparative aspects of placental efficiency. Reprod Suppl 58: 223–232, 2001. [PubMed] [Google Scholar]
- 577.Wilson MJ, Lopez M, Vargas M, Julian C, Tellez W, Rodriguez A, Bigham A, Armaza JF, Niermeyer S, Shriver M, Vargas E, Moore LG. Greater uterine artery blood flow during pregnancy in multigenerational (Andean) than shorter-term (European) high-altitude residents. Am J Physiol Regul Integr Comp Physiol 293: R1313–R1324, 2007. [DOI] [PubMed] [Google Scholar]
- 578.Wimsatt WA. New histological observations on the placenta of the sheep. Am J Anat 87: 391–457, 1950. [DOI] [PubMed] [Google Scholar]
- 579.Winder NR, Krishnaveni GV, Veena SR, Hill JC, Karat CL, Thornburg KL, Fall CH, Barker DJ. Mother's lifetime nutrition and the size, shape and efficiency of the placenta. Placenta 32: 806–810, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 580.Wittmaack FM, Gafvels ME, Bronner M, Matsuo H, McCrae KR, Tomaszewski JE, Robinson SL, Strickland DK, Strauss JF 3rd. Localization and regulation of the human very low density lipoprotein/apolipoprotein-E receptor: trophoblast expression predicts a role for the receptor in placental lipid transport. Endocrinology 136: 340–348, 1995. [DOI] [PubMed] [Google Scholar]
- 581.Wooding FB. Localization of ovine placental lactogen in sheep placentomes by electron microscope immunocytochemistry. J Reprod Fertil 62: 15–19, 1981. [DOI] [PubMed] [Google Scholar]
- 582.Wooding FB, Fowden AL, Bell AW, Ehrhardt RA, Limesand SW, Hay WW. Localisation of glucose transport in the ruminant placenta: implications for sequential use of transporter isoforms. Placenta 26: 626–640, 2005. [DOI] [PubMed] [Google Scholar]
- 583.Wooding FP, Burton GJ. Comparative Placentation. Structures, Functions and Evolution. Berlin: Springer, 2008, p. 301. [Google Scholar]
- 584.Woollett LA. Review: transport of maternal cholesterol to the fetal circulation. Placenta 32 Suppl 2: S218–221, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 585.Wouters BG, Koritzinsky M. Hypoxia signalling through mTOR and the unfolded protein response in cancer. Nat Rev Cancer 8: 851–864, 2008. [DOI] [PubMed] [Google Scholar]
- 586.Wouters BG, van den Beucken T, Magagnin MG, Lambin P, Koumenis C. Targeting hypoxia tolerance in cancer. Drug Resist Update 7: 25–40, 2004. [DOI] [PubMed] [Google Scholar]
- 587.Wyrwoll CS, Seckl JR, Holmes MC. Altered placental function of 11beta-hydroxysteroid dehydrogenase 2 knockout mice. Endocrinology 150: 1287–1293, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 588.Xie Y, Zhou S, Jiang Z, Dai J, Puscheck EE, Lee I, Parker G, Huttemann M, Rappolee DA. Hypoxic stress induces, but cannot sustain trophoblast stem cell differentiation to labyrinthine placenta due to mitochondrial insufficiency. Stem Cell Res 13: 478–491, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 589.Yamamoto S, Takahashi N, Mori Y. Chemical physiology of oxidative stress-activated TRPM2 and TRPC5 channels. Prog Biophys Mol Biol 103: 18–27, 2010. [DOI] [PubMed] [Google Scholar]
- 590.Yampolsky M, Salafia CM, Shlakhter O, Haas D, Eucker B, Thorp J. Modeling the variability of shapes of a human placenta. Placenta 29: 790–797, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 591.Yang K, Julan L, Rubio F, Sharma A, Guan H. Cadmium reduces 11 beta-hydroxysteroid dehydrogenase type 2 activity and expression in human placental trophoblast cells. Am J Physiol Endocrinol Metab 290: E135–E142, 2006. [DOI] [PubMed] [Google Scholar]
- 592.Yang ZZ, Tschopp O, Hemmings-Mieszczak M, Feng J, Brodbeck D, Perentes E, Hemmings BA. Protein kinase B alpha/Akt1 regulates placental development and fetal growth. J Biol Chem 278: 32124–32131, 2003. [DOI] [PubMed] [Google Scholar]
- 593.Yoshida S, Pacitto R, Yao Y, Inoki K, Swanson JA. Growth factor signaling to mTORC1 by amino acid-laden macropinosomes. J Cell Biol 211: 159–172, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 594.Young GB. The peripatetic placenta. Radiology 128: 183–188, 1978. [DOI] [PubMed] [Google Scholar]
- 595.Yung HW, Atkinson D, Campion-Smith T, Olovsson M, Charnock-Jones DS, Burton GJ. Differential activation of placental unfolded protein response pathways implies heterogeneity in causation of early- and late-onset pre-eclampsia. J Pathol 234: 262–276, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 596.Yung HW, Calabrese S, Hynx D, Hemmings BA, Cetin I, Charnock-Jones DS, Burton GJ. Evidence of placental translation inhibition and endoplasmic reticulum stress in the etiology of human intrauterine growth restriction. Am J Pathol 173: 451–462, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 597.Yung HW, Charnock-Jones DS, Burton GJ. Regulation of AKT phosphorylation at Ser473 and Thr308 by endoplasmic reticulum stress modulates substrate specificity in a severity dependent manner. PLoS One 6: e17894, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 598.Yung HW, Cox M, Tissot van Patot M, Burton GJ. Evidence of endoplasmic reticulum stress and protein synthesis inhibition in the placenta of non-native women at high altitude. FASEB J 26: 1970–1981, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 599.Yung HW, Hemberger M, Watson ED, Senner CE, Jones CP, Kaufman RJ, Charnock-Jones DS, Burton GJ. Endoplasmic reticulum stress disrupts placental morphogenesis: implications for human intrauterine growth restriction. J Pathol 228: 554–564, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 600.Zamudio S. The placenta at high altitude. High Altitude Med Biol 4: 171–191, 2003. [DOI] [PubMed] [Google Scholar]
- 601.Zamudio S, Baumann MU, Illsley NP. Effects of chronic hypoxia in vivo on the expression of human placental glucose transporters. Placenta 27: 49–55, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 602.Zamudio S, Leslie KK, White M, Hagerman DD, Moore LG. Low serum estradiol and high serum progesterone concentrations characterize hypertensive pregnancies at high altitude. J Soc Gynecol Invest 1: 197–205, 1994. [DOI] [PubMed] [Google Scholar]
- 603.Zamudio S, Palmer SK, Droma T, Stamm E, Coffin C, Moore LG. Effect of altitude on uterine artery blood flow during normal pregnancy. J Appl Physiol 79: 7–14, 1995. [DOI] [PubMed] [Google Scholar]
- 604.Zamudio S, Postigo L, Illsley NP, Rodriguez C, Heredia G, Brimacombe M, Echalar L, Torricos T, Tellez W, Maldonado I, Balanza E, Alvarez T, Ameller J, Vargas E. Maternal oxygen delivery is not related to altitude- and ancestry-associated differences in human fetal growth. J Physiol 582: 883–895, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 605.Zamudio S, Torricos T, Fik E, Oyala M, Echalar L, Pullockaran J, Tutino E, Martin B, Belliappa S, Balanza E, Illsley NP. Hypoglycemia and the origin of hypoxia-induced reduction in human fetal growth. PLoS One 5: e8551, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 606.Zamudio S, Wu Y, Ietta F, Rolfo A, Cross A, Wheeler T, Post M, Illsley NP, Caniggia I. Human placental hypoxia-inducible factor-1alpha expression correlates with clinical outcomes in chronic hypoxia in vivo. Am J Pathol 170: 2171–2179, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 607.Zhang EC, Burton GJ, Smith SK, Charnock-Jones DS. Placental vessel adaptation during gestation and to high altitude: changes in diameter and perivascular cell coverage. Placenta 23: 751–762, 2002. [DOI] [PubMed] [Google Scholar]
- 608.Zhang EG, Smith SK, Baker PN, Charnock-Jones DS. The regulation and localization of angiopoietin-1, -2, and their receptor Tie2 in normal and pathologic human placentae. Mol Med 7: 624–635, 2001. [PMC free article] [PubMed] [Google Scholar]
- 609.Zhang K, Kaufman RJ. From endoplasmic-reticulum stress to the inflammatory response. Nature 454: 455–462, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 610.Zhang S, Regnault TR, Barker PL, Botting KJ, McMillen IC, McMillan CM, Roberts CT, Morrison JL. Placental adaptations in growth restriction. Nutrients 7: 360–389, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 611.Zhang Y, Fei M, Xue G, Zhou Q, Jia Y, Li L, Xin H, Sun S. Elevated levels of hypoxia-inducible microRNA-210 in pre-eclampsia: new insights into molecular mechanisms for the disease. J Cell Mol Med 16: 249–259, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 612.Zhao XX, Gao WX, Gao YQ, Suo L, Chen J. [Placental mitochondrial respiratory function of native Tibetan at high altitude]. Zhonghua Yi Xue Za Zhi 87: 894–897, 2007. [PubMed] [Google Scholar]
- 613.Zhong W, Xie Y, Abdallah M, Awonuga AO, Slater JA, Sipahi L, Puscheck EE, Rappolee DA. Cellular stress causes reversible, PRKAA1/2-, and proteasome-dependent ID2 protein loss in trophoblast stem cells. Reproduction 140: 921–930, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 614.Zhu MJ, Du M, Hess BW, Nathanielsz PW, Ford SP. Periconceptional nutrient restriction in the ewe alters MAPK/ERK1/2 and PI3K/Akt growth signaling pathways and vascularity in the placentome. Placenta 28: 1192–1199, 2007. [DOI] [PubMed] [Google Scholar]
- 615.Zhu MJ, Du M, Nijland MJ, Nathanielsz PW, Hess BW, Moss GE, Ford SP. Down-regulation of growth signaling pathways linked to a reduced cotyledonary vascularity in placentomes of over-nourished, obese pregnant ewes. Placenta 30: 405–410, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 616.Zhu MJ, Ma Y, Long NM, Du M, Ford SP. Maternal obesity markedly increases placental fatty acid transporter expression and fetal blood triglycerides at midgestation in the ewe. Am J Physiol Regul Integr Comp Physiol 299: R1224–R1231, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 617.Zohn IE, Sarkar AA. The visceral yolk sac endoderm provides for absorption of nutrients to the embryo during neurulation. Birth Defects Res A Clin Mol Teratol 88: 593–600, 2010. [DOI] [PubMed] [Google Scholar]