

Abstract
Fluid shear stress is an important environmental cue that governs vascular physiology and pathology, but the molecular mechanisms that mediate endothelial responses to flow are only partially understood. Gating of ion channels by flow is one mechanism that may underlie many of the known responses. Here, we review the literature on endothelial ion channels whose activity is modulated by flow with an eye toward identifying important questions for future research.
Fluid shear stress, the frictional drag force caused by blood flow over the endothelial luminal surface, is one of the environmental cues sensed by the endothelial cells that line the vasculature. Endothelial cell (ECs) experience a range of shear stress profiles in vivo that differ in magnitude, direction, and temporal characteristics, depending on location within the vasculature. These shear profiles are sensed by the endothelial cells and modulate cell behavior. Long-term unidirectional flow within the physiological range (typically 10-40 dynes/cm2 for human arteries) promotes EC quiescence, with low proliferation and low inflammatory gene expression. Conversely, low flow or flow with changes in direction (multidirectional or oscillatory flow) promotes EC proliferation and turnover, and activates inflammatory pathways (22). In vivo, these flow patterns confer sensitivity to other risk factors such as hyperlipidemia, smoking, and diabetes, resulting in development of atherosclerotic plaques. On short time scales, decreases or increases of shear stress in resistance arteries trigger vasoconstriction or dilation, respectively. In the longer term, sustained changes in shear initiate inward or outward vessel remodeling (40, 67, 96). Fluid shear stress is also an important factor in controlling vascular development and the maturation of blood vessels (69).
Although our understanding of mechanotransduction is still fragmentary, several primary receptors of shear stress have been proposed, including G-protein-coupled receptors (GPCRs), junctional proteins, primary cilia, membrane lipids, and the apical glycocalyx (5). Mechanosensitive channels have also been proposed as primary detectors of shear stress, with many research groups reporting effects of shear stress on plasma membrane permeability and ionic flow. ECs are not excitable, so they do not express high levels of voltage-gated channels and do not show a propagated response to depolarization or hyperpolarization like neurons or muscle cells. However, the ionic conductances regulated by shear stress could influence a broad range of EC and vascular functions. The electrical coupling between ECs and smooth muscle cells (SMCs) also means that EC channels could directly control vascular tone (33). This review will discuss the changes in ion permeability observed in endothelial cells exposed to fluid shear stress and the roles various ionic fluxes may play in shear stress responses (Table 1).
Table 1.
Channels implicated in shear stress responses in endothelial cells
| Channel Subtype | Candidate Genes* | Permeability | Mechanisms of Modulation by Flow | Other Pathways Modulated by Channel Activity |
|---|---|---|---|---|
| Purinergic | P2RX4 | Cation | Activation by ATP release (122) Reduced inactivation (63) | Neuropathic pain (115) Endolysosomal membrane fusion (16) |
| Transient receptor potential | TRPV4 | Cation | Membrane tension? (73) Lipid second messengers? (119) Channel translocation to membrane? (7) | Cellular and systemic osmoregulation (39) |
| PKD1 and PKD2 | Cation | Cilial bending? (45) | Renal function (87) | |
| Mechanosensitive cation | PIEZO1 | Cation | Membrane tension? (26) | Mechanotransduction and mechanosensation (26, 95) |
| Inwardly rectifying potassium | KCNJ2 | Potassium | Membrane tension or cellular deformation? (53) | Craniofacial patterning (27) Cardiac excitability (72) |
| Calcium-activated potassium | KCNN3 | Potassium | Shear-induced calcium influx (15) | Cardiac hyperpolarization (130) |
| ATP-sensitive potassium | KCNJ11 | Potassium | ND | Insulin homeostasis Smooth muscle ischemic response (81) |
| Calcium-activated chloride | ND | Chloride | Shear-induced calcium influx (15) | Cellular volume regulation Fluid secretion Rhythmic smooth muscle contraction (9)† |
| Voltage-gated sodium | ND | Sodium | Endothelial cell depolarization? | Action potential propagation† |
| Epithelial sodium | ND | Sodium | Hydrostatic pressure? (47) | Sodium reabsorption Taste (42)† |
ND, not determined.
Human gene name.
General functions ascribed to the channel family; individual members may have different functions.
Calcium
Calcium is a critically important cellular signal. It acts locally at the cell membrane, affecting ion channel and receptor activity, and globally throughout the cell, controlling transcription factors and functioning as a cofactor for enzymes (24). It is thus unsurprising that calcium is postulated to play a role in endothelial signaling in general and flow sensing in particular. The best-defined role for shear stress-activated calcium signaling in ECs is flow-induced vasodilation (FIV). Increased shear stress in arteries results in elevated EC cytosolic calcium, leading to activation of endothelial nitric oxide synthase (eNOS) and production of nitric oxide (NO). NO diffuses from the endothelium and triggers relaxation of smooth muscle cells, causing dilation of the vessels and thereby reducing wall shear stress to maintain homeostasis (35). In the FIV response, increased cytosolic calcium also triggers opening of calcium-activated potassium channels, which hyperpolarize the endothelium (82). The adjacent smooth muscle cells also hyperpolarize, likely through direct electrical coupling via gap junctions between the two cell types, increasing smooth muscle cell relaxation (46). Calcium signaling can also modulate several processes that are affected by shear stress. In addition to controlling NO production, calcium regulates endothelial prostacyclin synthesis, release of endothelin-1, and junctional permeability and contractility through myosin light chain phosphorylation (17, 41, 57). However, whether changes in calcium are required for the effects of shear stress on these processes is unknown.
There have been many reports that show that onset of shear stress increases intracellular calcium in a magnitude-dependent manner (3, 4, 31, 44, 52, 66, 80, 84, 88, 105–107, 109, 123). Yet there is little agreement among different studies about the characteristics of this response, with large differences reported in threshold and half-maximal activation, latency, persistence of the calcium rise during application of shear stress, decay after stopping shear stress, and synchrony of the calcium response within a population of cells. This variability, which can presumably be attributed to differences in cell type, culture conditions, shear apparatus, and measurement techniques, suggests that calcium is regulated by multiple shear-sensitive components and multiple mechanisms.
Calcium Sources, Stores vs. Plasma Membrane Channels
Cytosolic calcium levels can be increased due to influx of extracellular calcium through the plasma membrane and through release from intracellular stores. Influx in response to shear stress could be controlled by either direct mechanical gating of calcium permeable membrane channels or indirect activation of these channels through other pathways (3, 106). Release from stores is generally controlled through activation of phospholipase C (PLC), which hydrolyzes phosphatidylinositol 4,5-bisphosphate (PIP2) to produce diacylglycerol (DAG) and inositol 1,4,5-trisphosphate (IP3). The latter gates calcium-permeable IP3R channels on the endoplasmic reticulum (ER), releasing calcium into the cytosol. Both RTKs and G proteins can stimulate PLC and are activated by shear stress (8, 18, 28, 59, 70). Activation of PLC and increased IP3 and DAG have also been observed within minutes after the application of shear (12, 89, 98). Thus elevation of cytosolic calcium via release from intracellular stores is expected.
Removing calcium from the extracellular solution blocks influx through the plasma membrane but does not affect release from stores; stores can be specifically depleted using thapsagargin. A number of studies have used these techniques to show that shear-stimulated increases in cytosolic calcium were wholly or partially dependent on release from stores, consistent with activation of the PLC pathway (3, 31, 44, 56, 76, 109). However, in some conditions, calcium increases were reported to be entirely due to influx from the medium (66, 86, 106, 107). There is little information about how calcium stores affect longer-term behavior of cells in vitro and virtually nothing about the role for this type of calcium signaling in endothelial cells in vivo.
ATP and Its Receptors
Shear stress can activate membrane receptors via both ligand-dependent and -independent mechanisms. There is evidence for ligand-independent activation of both VEGF receptors and GPCRs by flow; however, calcium signaling downstream of these receptors has not been reported (18, 59, 61). The best studied ligand-dependent shear-sensitive pathway involves adenosine triphosphate (ATP) receptors whose response to ATP is potentiated by shear stress. Several reports have demonstrated a rise in cytosolic calcium in response to onset of shear stress that requires extracellular ATP (31, 58, 118, 123–125). At low extracellular ATP concentrations, this rise is due to an influx of calcium through P2X4, an ATP-sensitive cation channel (123, 124). However, purinergic GPCRs like P2Y2 also contribute to calcium responses at higher ATP concentrations, or higher shear stresses, through activation of PLC and release of calcium from stores (118, 123). Strikingly, loss of P2X4 in ECs in vitro also decreases shear-induced transcription of Krüppel-like factor 2 (Klf2), an important flow-dependent transcription factor (102). In animal models, loss of P2X4 leads to a defect in both FIV and nitric oxide production, and defects in high shear stress-induced outward remodeling (125). However, since this is a global knockout, non-endothelial responses or developmental defects in the vasculature could contribute to this phenotype. When floxed P2Y2 was inducibly deleted in endothelial cells in adult mice, FIV decreased and mean arterial blood pressure increased, strongly suggesting that this receptor regulates shear stress responses (118). Whether the effect of P2Y2 loss is dependent on loss of calcium signaling was not investigated.
The mechanism by which shear stress potentiates purinergic receptor activity is unclear. Both P2Y2- and P2X4-dependent shear responses are blocked by apyrase, an enzyme that degrades extracellular ATP, indicating that these responses are ATP-dependent (118, 126). Consistent with this result, P2X4 is not directly gated by shear when expressed in heterologous systems (63). ATP is released by endothelial cells in response to shear stress and can act locally on time scales of a few seconds (122, 126). Coupled with local hydrolysis of ATP at the membrane and increased agonist delivery due to an increase in fluid flow rate, this release can partially explain the ATP-dependent calcium responses (31). However, because the mechanism of ATP release by shear stress is unknown, it has not been possible to determine whether the responses of P2X4 and P2Y2 are wholly due to increased concentration of extracellular ATP or whether there are additional signaling components that alter receptor sensitivity or change calcium dynamics. P2X4 does appear to display ATP release-independent responses to shear. In heterologous systems, P2X4 shows reduced inactivation and higher current density in response to ATP under shear stress compared with static conditions, which suggests that shear (directly or indirectly) stabilizes the open conformation of the channel (63). There are several possible mechanisms by which shear stress could alter P2X4 activity. For example, phosphoinositides inhibit inactivation and internalization of P2X4; thus shear stress-induced changes in phosphoinositide metabolism could influence channel activity (10).
Force-Gated Channels, TRPV4, and Piezo1
The simplest mechanism by which shear stress could induce calcium influx would be through direct activation of mechanically gated calcium permeable channels. Such channels could either be sensitive to membrane tension and/or curvature, or can respond to cellular deformation through tethering to the cytoskeleton and/or the extracellular matrix. Putative mechanosensitive calcium permeable channels have been reported in endothelial cells, and activities with similar pharmacological and/or physiological properties have been observed in response to shear stress (60, 68, 86, 106, 107, 110). It has therefore been hypothesized that such a channel may be partly or wholly responsible for calcium transients observed at the onset of shear in ECs. The identity of this channel or channels was not known at the time of these experiments; however, further studies have identified two mechanosensitive calcium permeable channels that may contribute to shear-evoked calcium responses in ECs. The first is transient receptor potential cation channel V4 (TRPV4), a member of the polymodal TRP channel family. TRPV4 is activated by cell swelling in several cell types, suggesting activation by membrane tension; it can also be activated by shear stress in ECs when expressed heterologously, suggesting direct mechanical activation (73, 80, 111). However, osmotic swelling-induced activation of TRPV4 has been reported to be indirect, mediated by accumulation of arachidonic acid, a lipid mediator known to activate TRPV4 (117, 119). Additionally, rapid translocation of TRPV4 to the cell surface in response to shear stress has been reported in heterologous systems, which could contribute to shear stress-activated TRPV4 currents (7). Interestingly, TRPV4 is one of the few TRP channels that has been shown to form heteromers with other TRP channel family members. It has been suggested that TRPV4 forms heterodimers with TRPC1 as well as a heterotrimeric complex of TRPV4, TRPC1, and Polycystin-2 (Pkd2), and that these heteromers conduct the majority of the TRPV4-dependent shear-sensitive calcium response (30, 77). Although it is unclear whether TRPC1 or Pkd2 alter the shear sensitivity of TRPV4 channels, TRPC1 appears to alter inactivation of the heteromer and may play a role in the adaptation of the channel to prolonged shear stimuli (77).
Significant work has been done to address the role of TRPV4 in vivo in the vasculature. Genetic deletion of TRPV4 reduced both FIV in mice and calcium responses to onset of flow in ECs in vitro (48, 80). However, interpretation of this result is complicated by the effects of TRPV4 in other cell types, such as smooth muscle cells, and other systemic effects that might influence vascular function, such as osmoregulation, which could cause differences in blood pressure and serum ion concentrations (32, 74, 83). Pharmacological tools have been used to address the issue of systemic effects. Activation of TRPV4 with the phorbol ester 4α-phorbol-12,13-didecanoate (4αPDD) in isolated arteries induces vasodilation, whereas blockade of TRPV4 with the non-specific TRP channel blocker ruthenium red (RR) inhibited FIV (65). Pharmacological activation of TRPV4 with 4αPDD also increased, whereas inhibition with RR reduced flow-induced cerebral arteriogenesis (104), providing some suggestive evidence for its involvement in flow-dependent remodeling in vivo. However, these pharmacological tools come with many caveats, especially RR, which has many off-target effects.
The other candidate is Piezo1 (Fam38a), which with Piezo2 forms a family of atypical channels directly activated by mechanical force and implicated in several mechanosensory processes (26, 95, 101, 120, 128). Like TRPV4, Piezo1 can be activated by shear stress when expressed heterologously in HEK cells, although the threshold for shear-induced currents seems to be above the threshold for calcium entry in ECs (100). Deletion of Piezo1 in ECs reduced calcium influx in response to the onset of shear stress and blocked their alignment in flow, a physiologically important flow response that has not been reported for other channels. Unlike loss of TRPV4 and P2X4, genetic deletion of Piezo1 in mice causes severe defects in vascular development, leading to embryonic lethality even with endothelial specific knockout (71, 100). Loss of Piezo1 also reduced the activation of the key flow-dependent transcription factor Klf2. The severity of the Piezo1 knockout phenotype suggests that this channel has a role beyond contributing to calcium entry in the short-term FIV response. Interestingly, loss or inhibition of Piezo1 in red blood cells reduced shear-stimulated release of ATP (23). If applicable to ECs, loss of ATP release could affect shear-induced activation of P2Y2 and P2X4, which could contribute to the Piezo1−/− phenotype. However, other downstream effects are likely, and the role of Piezo1 in sensing cellular stretch, independent of shear stress, may also contribute.
Primary Cilia and Calcium Regulation
In several cell types, flow-induced calcium signaling is controlled by a mechanosensory complex localized to primary cilia, a signaling complex that includes the TRP channel Pkd1 and its regulator Pkd2 (87). Bending of the cilia is an attractive mechanism for sensing very low flows (97). Embryonic endothelial cells exhibit primary cilia and show flow-induced calcium responses that are blocked by knockdown or genetic deletion of proteins involved in cilia formation, or of Pkd1 or Pkd2 (1, 88). Cilia appear to be required for flow-induced calcium responses in early zebrafish embryos, and loss of key cilia genes causes defects in embryonic angiogenesis (45, 62). In mice, loss of Pkd1 also affects vascular function, and knockout mice die in utero with pronounced hemorrhaging and moderate edema, whereas Pkd2-null mice show pronounced edema and focal hemorrhages (64, 121). However, global knockout mice also show profound defects in both vascular smooth muscle behavior and lymphatic development, which complicates the interpretation of the vascular phenotype (49, 93, 99). In adults, cilia are confined to regions of the vasculature that are under low flow, which correlates well with disruption of EC primary cilia by higher shear (54) and suggest that ciliary signaling is likely most important in early embryogenesis or in specific low-flow regions of adult vessels. However, recent work blocking cilia formation in endothelial and hematopoietic lineage cells through mutation of Ift88 produced no defect in retinal angiogenesis, suggesting that cilia per se may not be important for vascular development in mice (29). The difference between this phenotype and loss of Pkd2 or Pkd1 could be due to cilia-independent mechanosensory defects, since Pkd1 and 2 are not restricted to cilia (95, 108). Pkd2 in particular may interact with both TRPV4 and Piezo1 to affect mechanosensitivity through both of these channels (30, 95). Interestingly, mutation of Ift88 exacerbates atherosclerosis in apolipoprotein E-null mice, suggesting that ciliary signaling may mitigate inflammatory signaling in areas of low flow (29). However, Ift88 also has cilia-independent functions, so this phenotype may also involve other mechanisms (13, 14).
Potassium
In addition to calcium influx, onset of shear stress triggers rapid EC hyperpolarization. The characteristics of this hyperpolarization are consistent with the activation of potassium channels and efflux of cytoplasmic potassium (92). Under static conditions, control of potassium flux is important for establishing resting membrane potential. In cells that express voltage-gated channels, this affects excitability and action potential dynamics; in ECs, which are nonexcitable, the role of potassium is less well understood. Inhibition of potassium channel activity in endothelial cells has been linked to lower shear stress-induced transforming growth factor β1 (TGFβ1) transcription and activity; it is also associated with production of cyclic guanosine monophosphate (cGMP) and, in isolated arteries ex vivo, with flow-induced vasoconstriction (25, 90, 91). However, these studies rely on broad spectrum potassium channel blockers and do not distinguish between subtypes that may have very different roles. Blocking potassium channels can also modulate other ionic fluxes, including calcium. Hyperpolarization due to potassium efflux increases the driving force for calcium influx, increasing calcium entry through calcium channels. Conversely, calcium-sensitive potassium channels can be activated in response to an influx of calcium. This cross talk between the two ionic fluxes can make it difficult to separate their roles. Although flow-induced changes in potassium flux were first reported in the 1980s (92), the effects of this response on endothelial cell behavior are still not well understood.
Kir2.1 and Kca2.3
The potassium current activated by the onset of shear stress is inwardly rectifying and blocked by barium, both characteristics of the potassium voltage-gated channel subfamily J (KCNJ) channels (55, 85, 92). This family contains 18 members, with KCNJ2 (Kir2.1) being the predominant member in ECs (37). Kir2.1 can be activated by shear in heterologous systems, making it a good candidate for the fast hyperpolarizing, flow-activated channel (53). Kir2.1 is also linked to a number of other flow-sensitive pathways. Kir2.1 activity and cell surface expression are regulated by GPCR signaling, 5′ AMP-activated protein kinase (AMPK), protein kinase A (PKA), and protein kinase C (PKC), all of which are affected by shear stress (2, 34, 129). Surprisingly, although the role of potassium efflux has been examined in ECs using the potassium channel blocker barium, Kir2.1 has not been targeted specifically, in part due to a lack of pharmacological tools. It is therefore unclear whether Kir2.1 is required for flow-induced activation of potassium efflux in ECs and whether activation of this channel interacts with other shear stress-sensitive kinases such as Akt or extracellular signal-regulated kinases (ERK1 and ERK2). Interestingly, Kir-like currents are also reduced by VLDL, which blocks flow-induced activation of Kir. This is consistent with reduced FIV in animal models of hypercholesterolemia, suggesting that these channels may play a role in pathological responses to shear in athero-prone model systems (36).
Genetic ablation of Kir2.1 inhibits extracellular potassium-evoked dilation in cerebral arteries, although this has been primarily attributed to the effect of Kir2.1 in smooth muscle cells (127). Loss of Kir2.1 also causes perinatal lethality, due at least in part to a cleft pallet in these mice, making this mouse non-ideal for specific analysis of the role of Kir2.1 in endothelial cells (127). However, recent work using Kir2.1 heterozygotes, which show greatly reduced Kir currents in isolated endothelial cells, suggests that this channel directly affects FIV (Levitan I, personal communication). Previous work linked potassium channel activity, specifically, the activation of small conductance calcium-activated potassium channels (Kca2.3, KCNN3), to NO-independent FIV (15). Unlike Kca2.3, the effects of Kir2.1 are NO-dependent (Levitan I, personal communication). This result suggests that Kir2.1 activation potentiates eNOS activity; increased calcium signaling is a likely mechanism, but this remains to be demonstrated. By contrast, Kca2.3 activation appears to be downstream of flow-induced calcium influx and contributes to FIV by increasing release of endothelial hyperpolarization factor or through direct electrically coupling with smooth muscle cells (15).
Potassium and Cessation of Flow
Cessation of flow has been studied as an aspect of ischemia, although fast responses to flow cessation occur without hypoxia or other metabolic sequelae. Several studies have shown that flow-adapted endothelial cells depolarize upon cessation of flow in vitro (19–21). This response is blocked by cromakalim, an activator of ATP-sensitive potassium channels, suggesting that depolarization is due to closing of these potassium channels (19). This would suggest that ATP-sensitive potassium channels in endothelial cells, like Kir channels, are activated in response to shear stress. Long-term shear upregulates a number of potassium channels, including the ATP-sensitive channel Kir6.2 (KCNJ11) and the calcium-sensitive channel Kca3.1 (KCNN4), which may explain why this depolarization is not observed in cells under shear for short times (19, 113). Kir6.2-dependent depolarization of ECs after cessation of flow in mouse lungs ex vivo has also been reported (20). Although this response likely contributes to the effect of ischemia in vivo, it is unclear whether depolarization is a general response to decreasing shear stress and whether it plays a role in vasoconstriction or in long-term adaption to decreased shear stress.
Chloride
After the fast, potassium-based hyperpolarization, shear stress induces a subsequent large depolarization in ECs in vitro. Surprisingly, this depolarization is reduced by chloride channel blockers, suggesting a role of chloride efflux in shear stress signaling (6, 43, 75). Although opening of chloride channels is most frequently associated with influx of chloride and therefore hyperpolarization, some cell types show high levels of cytosolic chloride, flipping the driving force and causing chloride efflux in response to channel opening. Since this current has a fairly slow latency, it is unlikely that chloride efflux is due to mechanically gated chloride channels. Rather, it could be downstream of flow-induced calcium influx, since inhibitors of the chloride current primarily target calcium-activated chloride channels (CaCCs), which are activated by cytosolic calcium. Interestingly, blocking CaCCs reduces shear-induced activation of Akt (43), which is downstream of the PECAM-mediated junctional signaling and upstream of eNOS activation (among other pathways) (116). Membrane depolarization also seems to affect Akt signaling in the flow cessation/ischemia model, suggesting that ECs may respond to all depolarizing signals by increasing phosphoinositide 3-kinase (PI3K) and Akt activity (20). Chloride channel activation shows a low shear threshold, similar to potassium channel activation, but saturates well below 10 dynes and is suppressed by higher shear stresses; by contrast, the potassium signal saturates ∼12–15 dynes and does not appear to be inhibited by high shear stress (43, 92). These findings suggest that chloride signaling may play a role in the response to low shear stress and thus may be part of an inflammatory response. However, without knowing the identity of the channel(s) involved, the importance of this flow-activated current is difficult to ascertain due to the paucity of specific pharmacological tools for chloride channels.
Sodium
Sodium, along with potassium, is a major regulator of membrane potential. Dietary intake of high salt is correlated with hypertension in humans and reduces FIV in animal models (78). However, since sodium influx is important for smooth muscle contraction, it is difficult to determine to what extent endothelial cells mediate these effects. In lieu of a method to directly alter endothelial sodium influx without altering smooth muscle cells, a few groups have examined the role of sodium channels in endothelial shear stress responses in vitro. Although all non-specific cation channels conduct sodium ions to some degree, there are only two families of sodium-specific cation channels: the voltage-gated sodium channels (Nav) and epithelial sodium channels (ENaC). Although there is debate over whether either of these types of channels are expressed in ECs, and, if they are expressed, what their function might be, the role of both types of channels in shear stress signaling has been examined. One group reported that both loss of extracellular sodium and blocking Nav channels with tetrodotoxin increased ERK phosphorylation in response to shear stress (114). Although some Nav channels are directly sensitive to mechanical forces, the shear stress-induced depolarization of endothelial cells would likely be sufficient to activate these channels, potentiating depolarization (6, 11). This could suggest that the shear activation of the ERK pathway is in some way sensitive to membrane potential, much like the PI3K-Akt pathway. Another group observed ENaC-like channel activation in endothelial cells by flow and hydrostatic pressure (47). Since ENaC channels are activated by flow in heterologuous systems, it is possible that these channels contribute directly to the mechanical response to flow (103). However, significant work remains to prove that sodium channels are important for endothelial responses to shear in vitro and in vivo. Information on different channels is summarized in FIGURE 1.
FIGURE 1.
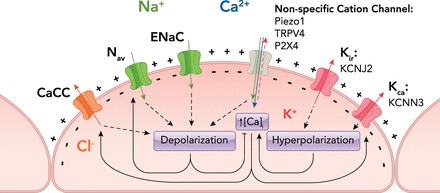
Diagram of putative shear stress-activated channel subtypes in endothelial cells and their interactions
Specific candidate genes are listed below their subtype. Flow activates non-specific cation channels, which depolarize the cell and increase cytoplasmic calcium. Flow also activates voltage-gated and epithelial sodium channels (Nav and ENaC), which increase sodium influx, and calcium-sensitive chloride channels (CaCC), which increase chloride efflux. Activation of both of these channels leads to depolarization. Conversely, flow activates calcium-activated and inwardly rectifying potassium channels (Kca and Kir), leading to hyperpolarization. Calcium influx directly activates both CaCCs and Kca channels. In contrast, hyperpolarization potentiates calcium influx through open channels by increasing the driving force, which propels calcium into the cell, whereas depolarization decreases this driving force. Additionally, depolarization directly activates voltage-gated channels like Nav,whereas hyperpolarization inhibits these channels.
Channel Activity and Shear Profiles
Flow magnitude, pulsatility, direction, and temporal variations have major effects on EC phenotype. However, few studies have addressed how differing shear profiles affect channel activity. Early work suggested that calcium influx in ECs can be stimulated by steady or pulsatile flow but not oscillatory flow, suggesting that calcium influx is related to the anti-inflammatory adaptive response to laminar shear (51). This group also reported that pulsatile flow increased the frequency of EC calcium oscillations more strongly than laminar flow of the same average magnitude (52). Pulsatile flow, typical of arteries, activates some anti-inflammatory, vessel-stabilization pathways more strongly than steady flow; it will be interesting to test whether calcium oscillations contribute to these effects. However, work in animal models have suggested that TRPV4, which contributes to calcium responses to flow, is important for responses to oscillatory flow in zebrafish endocardium during valve development (50). Additionally, ciliary calcium signaling in adult mice, which theoretically contributes to shear sensing in regions of disturbed flow, modulates development of atherosclerosis (29). These studies suggest that calcium may also respond to oscillatory flow and be functionally important.
In terms of other ionic fluxes, laminar but not oscillatory shear was found to induce CaCC-mediated depolarization; by contrast, fast hyperpolarization, presumably due to Kir activation, was seen under all flow conditions (75). This result suggests that the slow depolarization of endothelial cells may be specific to laminar shear stress and could play a role in the anti-inflammatory adaptive response to shear stress, whereas the fast hyperpolarization may play a role in the inflammatory long-term response to disturbed shear. However, without more detailed information about the mechanisms of channel activation and their downstream consequences, it is difficult to determine how or whether channel activity is related to endothelial cell behavior in vivo.
Concluding Remarks
Given the amount of evidence for shear stress-activated (or -inhibited) conductances in endothelial cells, it is somewhat surprising that there is so little consensus about their function in shear stress signaling. In some cases, the specific channels responsible for changes in endothelial cell permeability have yet to be identified, but even where candidate channels are known, their role in shear stress signaling remains poorly understood. Although it is clear that FIV involves shear stress-induced activation of endothelial calcium and potassium channels, only a handful of reports address the effects of EC channels on other long-term physiological responses to shear stress, from development to remodeling to atherosclerosis. Similarly, few papers have addressed the role of channel activity in modulation of established shear stress EC responses, such as activation of kinases like Akt or ERK, changes in gene expression, and changes in cytoskeletal organization and EC alignment. Overall, although channels are clearly activated directly and indirectly by shear stress, functional roles remain to be explored in depth.
As targets for pharmaceutical intervention, channels are a mixed bag. Opening or closing a channel through pharmacological intervention can have profound effects on cell function or even viability, independent of more fine-tuned signaling responces. For example, chronic reduction of intracellular potassium activates inflammasomes, whereas chronically elevated calcium can trigger apoptosis (79, 94). However, if mechanosensitivity could be targeted independently of basal activity (e.g., shifting the threshold of activation by force), pharmacological tools could alter shear responses in a potentially useful way. This would be of particular relevance to channels like Kir2.1, which show altered flow responses in pathological conditions (36). Although there are currently few drugs that target mechanosensitive channels, recent work identifying the Piezo1 agonist Yoda1 offers a proof of principal for a drug that modulates the mechanosensitivity of a channel (112). However, without better understanding the role of specific channels in the short- and long-term responses to shear stress, it is difficult to propose specific targets for drug development.
Despite these limitations, available evidence supports the importance of regulated channel activity in shear stress signaling. One aspect of potential interest concerns the ability of ECs to distinguish fine temporal features of shear stress profiles, such that subsecond dynamics influence signaling outputs and EC phenotype (38). Sensing changes in flow on this time scale would require pathways that are both activated and inactivated with suitably fast kinetics. Although G-protein signaling can be activated in seconds, rates of inactivation are slower and thus seem incompatible with sensing flow pulsatility. Channels, however, often have on and off rates on these time scales, particularly Piezo channels, which show rapid inactivation as well as activation (26). Elucidating the functions of specific channels in distinct EC flow responses therefore offers considerable promise for solving important problems in vascular biology.
Acknowledgments
We thank Dr. Irena Levitan for helpful comments, recommendations, and critical reading of this manuscript.
Footnotes
This work was supported by National Heart, Lung, and Blood Institute Grant PO1 HL-107235 to M.A.S.
No conflicts of interest, financial or otherwise, are declared by the author(s).
Author contributions: K.A.G. prepared figures; K.A.G. and M.A.S. drafted, revised, and edited the manuscript; K.A.G. and M.A.S. approved the final version of the manuscript.
References
- 1.AbouAlaiwi WA, Takahashi M, Mell BR, Jones TJ, Ratnam S, Kolb RJ, Nauli SM. Ciliary polycystin-2 is a mechanosensitive calcium channel involved in nitric oxide signaling cascades. Circ Res 104: 860–869, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alesutan I, Munoz C, Sopjani M, Dermaku-Sopjani M, Michael D, Fraser S, Kemp BE, Seebohm G, Foller M, Lang Inhibition of Kir2 F.1. (KCNJ2) by the AMP-activated protein kinase . Biochem Biophys Res Commun 408: 505–510, 2011. [DOI] [PubMed] [Google Scholar]
- 3.Ando J, Komatsuda T, Kamiya A. Cytoplasmic calcium response to fluid shear stress in cultured vascular endothelial cells. In vitro. Cell Dev Biol 24: 871–877, 1988. [DOI] [PubMed] [Google Scholar]
- 4.Ando J, Ohtsuka A, Korenaga R, Kawamura T, Kamiya A. Wall shear stress rather than shear rate regulates cytoplasmic Ca++ responses to flow in vascular endothelial cells. Biochem Biophys Res Commun 190: 716–723, 1993. [DOI] [PubMed] [Google Scholar]
- 5.Ando J, Yamamoto K. Flow detection and calcium signalling in vascular endothelial cells. Cardiovasc Res 99: 260–268, 2013. [DOI] [PubMed] [Google Scholar]
- 6.Barakat AI, Leaver EV, Pappone PA, Davies PF. A flow-activated chloride-selective membrane current in vascular endothelial cells. Circ Res 85: 820–828, 1999. [DOI] [PubMed] [Google Scholar]
- 7.Baratchi S, Almazi JG, Darby W, Tovar-Lopez FJ, Mitchell A, McIntyre P. Shear stress mediates exocytosis of functional TRPV4 channels in endothelial cells. Cell Mol Life Sci 73: 649–666, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Barauna VG, Magalhaes FC, Campos LC, Reis RI, Kunapuli SP, Costa-Neto CM, Miyakawa AA, Krieger JE. Shear stress-induced Ang II AT1 receptor activation: G-protein dependent and independent mechanisms. Biochem Biophys Res Commun 434: 647–652, 2013. [DOI] [PubMed] [Google Scholar]
- 9.Berg J, Yang H, Jan LY. Ca2+-activated Cl− channels at a glance. J Cell Sci 125: 1367–1371, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bernier LP, Ase AR, Chevallier S, Blais D, Zhao Q, Boue-Grabot E, Logothetis D, Seguela P. Phosphoinositides regulate P2X4 ATP-gated channels through direct interactions. J Neurosci 28: 12938–12945, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Beyder A, Rae JL, Bernard C, Strege PR, Sachs F, Farrugia G. Mechanosensitivity of Nav1.5 a voltage-sensitive sodium channel. J Physiol 588: 4969–4985, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bhagyalakshmi A, Berthiaume F, Reich KM, Frangos JA. Fluid shear stress stimulates membrane phospholipid metabolism in cultured human endothelial cells. J Vasc Res 29: 443–449, 1992. [DOI] [PubMed] [Google Scholar]
- 13.Boehlke C, Janusch H, Hamann C, Powelske C, Mergen M, Herbst H, Kotsis F, Nitschke R, Kuehn EW. A cilia independent role of Ift88/polaris during cell migration. PLos One 10: e0140378, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Borovina A, Ciruna B. IFT88 plays a cilia- and PCP-independent role in controlling oriented cell divisions during vertebrate embryonic development. Cell Rep 5: 37–43, 2013. [DOI] [PubMed] [Google Scholar]
- 15.Brahler S, Kaistha A, Schmidt VJ, Wolfle SE, Busch C, Kaistha BP, Kacik M, Hasenau AL, Grgic I, Si H, Bond CT, Adelman JP, Wulff H, de Wit C, Hoyer J, Kohler R. Genetic deficit of SK3 and IK1 channels disrupts the endothelium-derived hyperpolarizing factor vasodilator pathway and causes hypertension. Circulation 119: 2323–2332, 2009. [DOI] [PubMed] [Google Scholar]
- 16.Cao Q, Zhong XZ, Zou Y, Murrell-Lagnado R, Zhu MX, Dong XP. Calcium release through P2X4 activates calmodulin to promote endolysosomal membrane fusion. J Cell Biol 209: 879–894, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Carlini RG, Gupta A, Liapis H, Rothstein M. Endothelin-1 release by erythropoietin involves calcium signaling in endothelial cells. J Cardiovasc Pharmacol 26: 889–892, 1995. [DOI] [PubMed] [Google Scholar]
- 18.Chachisvilis M, Zhang YL, Frangos JA. G protein-coupled receptors sense fluid shear stress in endothelial cells. Proc Natl Acad Sci USA 103: 15463–15468, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chatterjee S, Al-Mehdi AB, Levitan I, Stevens T, Fisher AB. Shear stress increases expression of a KATP channel in rat and bovine pulmonary vascular endothelial cells. Am J Physiol Cell Physiol 285: C959–C967, 2003. [DOI] [PubMed] [Google Scholar]
- 20.Chatterjee S, Browning EA, Hong N, DeBolt K, Sorokina EM, Liu W, Birnbaum MJ, Fisher AB. Membrane depolarization is the trigger for PI3K/Akt activation and leads to the generation of ROS. Am J Physiol Heart Circ Physiol 302: H105–H114, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chatterjee S, Levitan I, Wei Z, Fisher AB. KATP channels are an important component of the shear-sensing mechanism in the pulmonary microvasculature. Microcirculation 13: 633–644, 2006. [DOI] [PubMed] [Google Scholar]
- 22.Chiu JJ, Chien S. Effects of disturbed flow on vascular endothelium: pathophysiological basis and clinical perspectives. Physiol Rev 91: 327–387, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cinar E, Zhou S, DeCourcey J, Wang Y, Waugh RE, Wan J. Piezo1 regulates mechanotransductive release of ATP from human RBCs. Proc Natl Acad Sci USA 112: 11783–11788, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Clapham DE. Calcium signaling. Cell 131: 1047–1058, 2007. [DOI] [PubMed] [Google Scholar]
- 25.Cooke JP, Rossitch E Jr, Andon NA, Loscalzo J, Dzau VJ. Flow activates an endothelial potassium channel to release an endogenous nitrovasodilator. J Clin Invest 88: 1663–1671, 1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Coste B, Mathur J, Schmidt M, Earley TJ, Ranade S, Petrus MJ, Dubin AE, Patapoutian A. Piezo1 and Piezo2 are essential components of distinct mechanically activated cation channels. Science 330: 55–60, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dahal GR, Rawson J, Gassaway B, Kwok B, Tong Y, Ptacek LJ, Bates E. An inwardly rectifying K+ channel is required for patterning. Development 139: 3653–3664, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.dela Paz N, Melchior B, Frangos J. Shear stress induces G protein-coupled receptor (GPCR)-independent heterotrimeric G protein activation in endothelial cells. FASEB J 29: 1029–7, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dinsmore C, Reiter JF. Endothelial primary cilia inhibit atherosclerosis. EMBO Rep 17: 156–166, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Du J, Ma X, Shen B, Huang Y, Birnbaumer L, Yao X. TRPV4, TRPC1, and TRPP2 assemble to form a flow-sensitive heteromeric channel. FASEB J 28: 4677–4685, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dull RO, Davies PF. Flow modulation of agonist (ATP)-response (Ca2+) coupling in vascular endothelial cells. Am J Physiol Heart Circ Physiol 261: H149–H154, 1991. [DOI] [PubMed] [Google Scholar]
- 32.Earley S, Heppner TJ, Nelson MT, Brayden JE. TRPV4 forms a novel Ca2+ signaling complex with ryanodine receptors and BKCa channels. Circ Res 97: 1270–1279, 2005. [DOI] [PubMed] [Google Scholar]
- 33.Emerson GG, Segal SS. Electrical coupling between endothelial cells and smooth muscle cells in hamster feed arteries: role in vasomotor control. Circ Res 87: 474–479, 2000. [DOI] [PubMed] [Google Scholar]
- 34.Fakler B, Brandle U, Glowatzki E, Zenner HP, Ruppersberg Kir2 JP.1. inward rectifier K+ channels are regulated independently by protein kinases and ATP hydrolysis. Neuron 13: 1413–1420, 1994. [DOI] [PubMed] [Google Scholar]
- 35.Falcone JC, Kuo L, Meininger GA. Endothelial cell calcium increases during flow-induced dilation in isolated arterioles. Am J Physiol Heart Circ Physiol 264: H653–H659, 1993. [DOI] [PubMed] [Google Scholar]
- 36.Fang Y, Mohler ER 3rd, Hsieh E, Osman H, Hashemi SM, Davies PF, Rothblat GH, Wilensky RL, Levitan I. Hypercholesterolemia suppresses inwardly rectifying K+ channels in aortic endothelium in vitro and in vivo. Circ Res 98: 1064–1071, 2006. [DOI] [PubMed] [Google Scholar]
- 37.Fang Y, Schram G, Romanenko VG, Shi C, Conti L, Vandenberg CA, Davies PF, Nattel S, Levitan I. Functional expression of Kir2.x in human aortic endothelial cells: the dominant role of Kir22. Am J Physiol Cell Physiol 289: C1134–C1144, 2005. [DOI] [PubMed] [Google Scholar]
- 38.Feaver RE, Gelfand BD, Blackman BR. Human haemodynamic frequency harmonics regulate the inflammatory phenotype of vascular endothelial cells. Nat Commun 4: 1525, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Garcia-Elias A, Mrkonjic S, Jung C, Pardo-Pastor C, Vicente R, Valverde MA. The TRPV4 channel. Hand Exp Pharmacol 222: 293–319, 2014. [DOI] [PubMed] [Google Scholar]
- 40.Garcia-Roldan JL, Bevan JA. Flow-induced constriction and dilation of cerebral resistance arteries. Circ Res 66: 1445–1448, 1990. [DOI] [PubMed] [Google Scholar]
- 41.Garcia JG, Davis HW, Patterson CE. Regulation of endothelial cell gap formation and barrier dysfunction: role of myosin light chain phosphorylation. J Cell Physiol 163: 510–522, 1995. [DOI] [PubMed] [Google Scholar]
- 42.Garty H, Palmer LG. Epithelial sodium channels: function, structure, and regulation. Physiol Rev 77: 359–396, 1997. [DOI] [PubMed] [Google Scholar]
- 43.Gautam M, Shen Y, Thirkill TL, Douglas GC, Barakat AI. Flow-activated chloride channels in vascular endothelium. Shear stress sensitivity, desensitization dynamics, and physiological implications. J Biol Chem 281: 36492–36500, 2006. [DOI] [PubMed] [Google Scholar]
- 44.Geiger RV, Berk BC, Alexander RW, Nerem RM. Flow-induced calcium transients in single endothelial cells: spatial and temporal analysis. Am J Physiol Cell Physiol 262: C1411–C1417, 1992. [DOI] [PubMed] [Google Scholar]
- 45.Goetz JG, Steed E, Ferreira RR, Roth S, Ramspacher C, Boselli F, Charvin G, Liebling M, Wyart C, Schwab Y, Vermot J. Endothelial cilia mediate low flow sensing during zebrafish vascular development. Cell Rep 6: 799–808, 2014. [DOI] [PubMed] [Google Scholar]
- 46.Griffith TM. Endothelium-dependent smooth muscle hyperpolarization: do gap junctions provide a unifying hypothesis? Br J Pharmacol 141: 881–903, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Guo D, Liang S, Wang S, Tang C, Yao B, Wan W, Zhang H, Jiang H, Ahmed A, Zhang Z, Gu Y. Role of epithelial Na+ channels in endothelial function. J Cell Sci 129: 290–297, 2016. [DOI] [PubMed] [Google Scholar]
- 48.Hartmannsgruber V, Heyken WT, Kacik M, Kaistha A, Grgic I, Harteneck C, Liedtke W, Hoyer J, Kohler R. Arterial response to shear stress critically depends on endothelial TRPV4 expression. PLos One 2: e827, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hassane S, Claij N, Jodar M, Dedman A, Lauritzen I, Duprat F, Koenderman JS, van der Wal A, Breuning MH, de Heer E, Honore E, DeRuiter MC, Peters DJ. Pkd1-inactivation in vascular smooth muscle cells and adaptation to hypertension. Lab Invest 91: 24–32, 2011. [DOI] [PubMed] [Google Scholar]
- 50.Heckel E, Boselli F, Roth S, Krudewig A, Belting HG, Charvin G, Vermot J. Oscillatory flow modulates mechanosensitive klf2a expression through trpv4 and trpp2 during heart valve development. Curr Biol 25: 1354–1361, 2015. [DOI] [PubMed] [Google Scholar]
- 51.Helmlinger G, Berk BC, Nerem RM. Calcium responses of endothelial cell monolayers subjected to pulsatile and steady laminar flow differ. Am J Physiol Cell Physiol 269: C367–C375, 1995. [DOI] [PubMed] [Google Scholar]
- 52.Helmlinger G, Berk BC, Nerem RM. Pulsatile and steady flow-induced calcium oscillations in single cultured endothelial cells. J Vasc Res 33: 360–369, 1996. [DOI] [PubMed] [Google Scholar]
- 53.Hoger JH, Ilyin VI, Forsyth S, Hoger A. Shear stress regulates the endothelial Kir2.1 ion channel. Proc Natl Acad Sci USA 99: 7780–7785, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Iomini C, Tejada K, Mo W, Vaananen H, Piperno G. Primary cilia of human endothelial cells disassemble under laminar shear stress. J Cell Biol 164: 811–817, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jacobs ER, Cheliakine C, Gebremedhin D, Birks EK, Davies PF, Harder DR. Shear activated channels in cell-attached patches of cultured bovine aortic endothelial cells. Pflügers Arch 431: 129–131, 1995. [DOI] [PubMed] [Google Scholar]
- 56.Jafarnejad M, Cromer WE, Kaunas RR, Zhang SL, Zawieja DC, Moore JE Jr. Measurement of shear stress-mediated intracellular calcium dynamics in human dermal lymphatic endothelial cells. Am J Physiol Heart Circ Physiol 308: H697–H706, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jaffe EA, Grulich J, Weksler BB, Hampel G, Watanabe K. Correlation between thrombin-induced prostacyclin production and inositol trisphosphate and cytosolic free calcium levels in cultured human endothelial cells. J Biol Chem 262: 8557–8565, 1987. [PubMed] [Google Scholar]
- 58.James NL, Harrison DG, Nerem RM. Effects of shear on endothelial cell calcium in the presence and absence of ATP. FASEB J 9: 968–973, 1995. [DOI] [PubMed] [Google Scholar]
- 59.Jin ZG, Ueba H, Tanimoto T, Lungu AO, Frame MD, Berk BC. Ligand-independent activation of vascular endothelial growth factor receptor 2 by fluid shear stress regulates activation of endothelial nitric oxide synthase. Circ Res 93: 354–363, 2003. [DOI] [PubMed] [Google Scholar]
- 60.Jow F, Numann R. Fluid flow modulates calcium entry and activates membrane currents in cultured human aortic endothelial cells. J Membr Biol 171: 127–139, 1999. [DOI] [PubMed] [Google Scholar]
- 61.Jung B, Obinata H, Galvani S, Mendelson K, Ding BS, Skoura A, Kinzel B, Brinkmann V, Rafii S, Evans T, Hla T. Flow-regulated endothelial S1P receptor-1 signaling sustains vascular development. Dev Cell 23: 600–610, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kallakuri S, Yu JA, Li J, Li Y, Weinstein BM, Nicoli S, Sun Z. Endothelial cilia are essential for developmental vascular integrity in zebrafish. J Am Soc Nephrol 26: 864–875, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kessler S, Clauss WG, Fronius M. Laminar shear stress modulates the activity of heterologously expressed P2X(4) receptors. Biochim Biophys Acta 1808: 2488–2495, 2011. [DOI] [PubMed] [Google Scholar]
- 64.Kim K, Drummond I, Ibraghimov-Beskrovnaya O, Klinger K, Arnaout MA. Polycystin 1 is required for the structural integrity of blood vessels. Proc Natl Acad Sci USA 97: 1731–1736, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kohler R, Heyken WT, Heinau P, Schubert R, Si H, Kacik M, Busch C, Grgic I, Maier T, Hoyer J. Evidence for a functional role of endothelial transient receptor potential V4 in shear stress-induced vasodilatation. Arterioscler Thromb Vasc Biol 26: 1495–1502, 2006. [DOI] [PubMed] [Google Scholar]
- 66.Kwan HY, Leung PC, Huang Y, Yao X. Depletion of intracellular Ca2+ stores sensitizes the flow-induced Ca2+ influx in rat endothelial cells. Circ Res 92: 286–292, 2003. [DOI] [PubMed] [Google Scholar]
- 67.Langille BL, O'Donnell F. Reductions in arterial diameter produced by chronic decreases in blood flow are endothelium-dependent. Science 231: 405–407, 1986. [DOI] [PubMed] [Google Scholar]
- 68.Lansman JB, Hallam TJ, Rink TJ. Single stretch-activated ion channels in vascular endothelial cells as mechanotransducers? Nature 325: 811–813, 1987. [DOI] [PubMed] [Google Scholar]
- 69.le Noble F, Moyon D, Pardanaud L, Yuan L, Djonov V, Matthijsen R, Breant C, Fleury V, Eichmann A. Flow regulates arterial-venous differentiation in the chick embryo yolk sac. Development 131: 361–375, 2004. [DOI] [PubMed] [Google Scholar]
- 70.Lee HJ, Koh GY. Shear stress activates Tie2 receptor tyrosine kinase in human endothelial cells. Biochem Biophys Res Commun 304: 399–404, 2003. [DOI] [PubMed] [Google Scholar]
- 71.Li J, Hou B, Tumova S, Muraki K, Bruns A, Ludlow MJ, Sedo A, Hyman AJ, McKeown L, Young RS, Yuldasheva NY, Majeed Y, Wilson LA, Rode B, Bailey MA, Kim HR, Fu Z, Carter DA, Bilton J, Imrie H, Ajuh P, Dear TN, Cubbon RM, Kearney MT, Prasad KR, Evans PC, Ainscough JF, Beech DJ. Piezo1 integration of vascular architecture with physiological force. Nature 515: 279–282, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Li J, McLerie M, Lopatin AN. Transgenic upregulation of IK1 in the mouse heart leads to multiple abnormalities of cardiac excitability. Am J Physiol Heart Circ Physiol 287: H2790–H2802, 2004. [DOI] [PubMed] [Google Scholar]
- 73.Liedtke W, Choe Y, Marti-Renom MA, Bell AM, Denis CS, Sali A, Hudspeth AJ, Friedman JM, Heller S. Vanilloid receptor-related osmotically activated channel (VR-OAC), a candidate vertebrate osmoreceptor. Cell 103: 525–535, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Liedtke W, Friedman JM. Abnormal osmotic regulation in trpv4−/− mice. Proc Natl Acad Sci USA 100: 13698–13703, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lieu DK, Pappone PA, Barakat AI. Differential membrane potential and ion current responses to different types of shear stress in vascular endothelial cells. Am J Physiol Cell Physiol 286: C1367–C1375, 2004. [DOI] [PubMed] [Google Scholar]
- 76.Liu B, Lu S, Zheng S, Jiang Z, Wang Y. Two distinct phases of calcium signalling under flow. Cardiovasc Res 91: 124–133, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ma X, Qiu S, Luo J, Ma Y, Ngai CY, Shen B, Wong CO, Huang Y, Yao X. Functional role of vanilloid transient receptor potential 4-canonical transient receptor potential 1 complex in flow-induced Ca2+ influx. Arterioscler Thromb Vasc Biol 30: 851–858, 2010. [DOI] [PubMed] [Google Scholar]
- 78.Matrougui K, Loufrani L, Levy BI, Henrion D. High NaCl intake decreases both flow-induced dilation and pressure-induced myogenic tone in resistance arteries from normotensive rats: involvement of cyclooxygenase-2. Pharmacol Toxicol 89: 183–187, 2001. [DOI] [PubMed] [Google Scholar]
- 79.Mattson MP, Chan SL. Calcium orchestrates apoptosis. Nat Cell Biol 5: 1041–1043, 2003. [DOI] [PubMed] [Google Scholar]
- 80.Mendoza SA, Fang J, Gutterman DD, Wilcox DA, Bubolz AH, Li R, Suzuki M, Zhang DX. TRPV4-mediated endothelial Ca2+ influx and vasodilation in response to shear stress. Am J Physiol Heart Circ Physiol 298: H466–H476, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Miki T, Seino S. Roles of KATP channels as metabolic sensors in acute metabolic changes. J Mol Cell Cardiol 38: 917–925, 2005. [DOI] [PubMed] [Google Scholar]
- 82.Miura H, Liu Y, Gutterman DD. Human coronary arteriolar dilation to bradykinin depends on membrane hyperpolarization: contribution of nitric oxide and Ca2+-activated K+ channels. Circulation 99: 3132–3138, 1999. [DOI] [PubMed] [Google Scholar]
- 83.Mizuno A, Matsumoto N, Imai M, Suzuki M. Impaired osmotic sensation in mice lacking TRPV4. Am J Physiol Cell Physiol 285: C96–C101, 2003. [DOI] [PubMed] [Google Scholar]
- 84.Mo M, Eskin SG, Schilling WP. Flow-induced changes in Ca2+ signaling of vascular endothelial cells: effect of shear stress and ATP. Am J Physiol Heart Circ Physiol 260: H1698–H1707, 1991. [DOI] [PubMed] [Google Scholar]
- 85.Nakache M, Gaub HE. Hydrodynamic hyperpolarization of endothelial cells. Proc Natl Acad Sci USA 85: 1841–1843, 1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Nakao M, Ono K, Fujisawa S, Iijima T. Mechanical stress-induced Ca2+ entry and Cl− current in cultured human aortic endothelial cells. Am J Physiol Cell Physiol 276: C238–C249, 1999. [DOI] [PubMed] [Google Scholar]
- 87.Nauli SM, Alenghat FJ, Luo Y, Williams E, Vassilev P, Li X, Elia AE, Lu W, Brown EM, Quinn SJ, Ingber DE, Zhou J. Polycystins 1 and 2 mediate mechanosensation in the primary cilium of kidney cells. Nat Gen 33: 129–137, 2003. [DOI] [PubMed] [Google Scholar]
- 88.Nauli SM, Kawanabe Y, Kaminski JJ, Pearce WJ, Ingber DE, Zhou J. Endothelial cilia are fluid shear sensors that regulate calcium signaling and nitric oxide production through polycystin-1. Circulation 117: 1161–1171, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Nollert MU, Eskin SG, McIntire LV. Shear stress increases inositol trisphosphate levels in human endothelial cells. Biochem Biophys Res Commun 170: 281–287, 1990. [DOI] [PubMed] [Google Scholar]
- 90.Ohno M, Cooke JP, Dzau VJ, Gibbons GH. Fluid shear stress induces endothelial transforming growth factor beta-1 transcription and production. Modulation by potassium channel blockade. J Clin Invest 95: 1363–1369, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ohno M, Gibbons GH, Dzau VJ, Cooke JP. Shear stress elevates endothelial cGMP. Role of a potassium channel and G protein coupling. Circulation 88: 193–197, 1993. [DOI] [PubMed] [Google Scholar]
- 92.Olesen SP, Clapham DE, Davies PF. Haemodynamic shear stress activates a K+ current in vascular endothelial cells. Nature 331: 168–170, 1988. [DOI] [PubMed] [Google Scholar]
- 93.Outeda P, Huso DL, Fisher SA, Halushka MK, Kim H, Qian F, Germino GG, Watnick T. Polycystin signaling is required for directed endothelial cell migration and lymphatic development. Cell Rep 7: 634–644, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Petrilli V, Papin S, Dostert C, Mayor A, Martinon F, Tschopp J. Activation of the NALP3 inflammasome is triggered by low intracellular potassium concentration. Cell Death Diff 14: 1583–1589, 2007. [DOI] [PubMed] [Google Scholar]
- 95.Peyronnet R, Martins JR, Duprat F, Demolombe S, Arhatte M, Jodar M, Tauc M, Duranton C, Paulais M, Teulon J, Honore E, Patel A. Piezo1-dependent stretch-activated channels are inhibited by Polycystin-2 in renal tubular epithelial cells. EMBO Rep 14: 1143–1148, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Pohl U, Holtz J, Busse R, Bassenge E. Crucial role of endothelium in the vasodilator response to increased flow in vivo. Hypertension 8: 37–44, 1986. [DOI] [PubMed] [Google Scholar]
- 97.Praetorius HA, Spring KR. Bending the MDCK cell primary cilium increases intracellular calcium. J Membr Biol 184: 71–79, 2001. [DOI] [PubMed] [Google Scholar]
- 98.Prasad AR, Logan SA, Nerem RM, Schwartz CJ, Sprague EA. Flow-related responses of intracellular inositol phosphate levels in cultured aortic endothelial cells. Circ Res 72: 827–836, 1993. [DOI] [PubMed] [Google Scholar]
- 99.Qian Q, Hunter LW, Du H, Ren Q, Han Y, Sieck GC. Pkd2+/− vascular smooth muscles develop exaggerated vasocontraction in response to phenylephrine stimulation. J Am Soc Nephrol 18: 485–493, 2007. [DOI] [PubMed] [Google Scholar]
- 100.Ranade SS, Qiu Z, Woo SH, Hur SS, Murthy SE, Cahalan SM, Xu J, Mathur J, Bandell M, Coste B, Li YS, Chien S, Patapoutian A. Piezo1, a mechanically activated ion channel, is required for vascular development in mice. Proc Natl Acad Sci USA 111: 10347–10352, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Ranade SS, Woo SH, Dubin AE, Moshourab RA, Wetzel C, Petrus M, Mathur J, Begay V, Coste B, Mainquist J, Wilson AJ, Francisco AG, Reddy K, Qiu Z, Wood JN, Lewin GR, Patapoutian A. Piezo2 is the major transducer of mechanical forces for touch sensation in mice. Nature 516: 121–125, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Sathanoori R, Rosi F, Gu BJ, Wiley JS, Muller CE, Olde B, Erlinge D. Shear stress modulates endothelial KLF2 through activation of P2X4. Purinergic Signal 11: 139–153, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Satlin L.M, Sheng S, Woda CB, Kleyman TR. Epithelial Na+ channels are regulated by flow. Am J Physiol Renal Physiol 280: F1010–F1018, 2001. [DOI] [PubMed] [Google Scholar]
- 104.Schierling W, Troidl K, Apfelbeck H, Troidl C, Kasprzak PM, Schaper W, Schmitz-Rixen T. Cerebral arteriogenesis is enhanced by pharmacological as well as fluid-shear-stress activation of the Trpv4 calcium channel. Eur J Vasc Endovasc Surg 41: 589–596, 2011. [DOI] [PubMed] [Google Scholar]
- 105.Schilling WP, Mo M, Eskin SG. Effect of shear stress on cytosolic Ca2+ of calf pulmonary artery endothelial cells. Exp Cell Res 198: 31–35, 1992. [DOI] [PubMed] [Google Scholar]
- 106.Schwarz G, Callewaert G, Droogmans G, Nilius B. Shear stress-induced calcium transients in endothelial cells from human umbilical cord veins. J Physiol 458: 527–538, 1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Schwarz G, Droogmans G, Nilius B. Shear stress induced membrane currents and calcium transients in human vascular endothelial cells. Pflügers Arch 421: 394–396, 1992. [DOI] [PubMed] [Google Scholar]
- 108.Sharif-Naeini R, Folgering JH, Bichet D, Duprat F, Lauritzen I, Arhatte M, Jodar M, Dedman A, Chatelain FC, Schulte U, Retailleau K, Loufrani L, Patel A, Sachs F, Delmas P, Peters DJ, Honore E. Polycystin-1 and -2 dosage regulates pressure sensing. Cell 139: 587–596, 2009. [DOI] [PubMed] [Google Scholar]
- 109.Shen J, Luscinskas FW, Connolly A, Dewey CF Jr, Gimbrone MA Jr. Fluid shear stress modulates cytosolic free calcium in vascular endothelial cells. Am J Physiol Cell Physiol 262: C384–C390, 1992. [DOI] [PubMed] [Google Scholar]
- 110.Sigurdson WJ, Sachs F, Diamond SL. Mechanical perturbation of cultured human endothelial cells causes rapid increases of intracellular calcium. Am J Physiol Heart Circ Physiol 264: H1745–H1752, 1993. [DOI] [PubMed] [Google Scholar]
- 111.Strotmann R, Harteneck C, Nunnenmacher K, Schultz G, Plant TD. OTRPC4, a nonselective cation channel that confers sensitivity to extracellular osmolarity. Nat Cell Biol 2: 695–702, 2000. [DOI] [PubMed] [Google Scholar]
- 112.Syeda R, Xu J, Dubin AE, Coste B, Mathur J, Huynh T, Matzen J, Lao J, Tully DC, Engels IH, Petrassi HM, Schumacher AM, Montal M, Bandell M, Patapoutian A. Chemical activation of the mechanotransduction channel Piezo1. eLife 4: 07369, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Takai J, Santu A, Zheng H, Koh SD, Ohta M, Filimban LM, Lemaitre V, Teraoka R, Jo H, Miura H. Laminar shear stress upregulates endothelial Ca2+-activated K+ channels KCa2.3 and KCa31 via a Ca2+/calmodulin-dependent protein kinase kinase/Akt/p300 cascade. Am J Physiol Heart Circ Physiol 305: H484–H493, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Traub O, Ishida T, Ishida M, Tupper JC, Berk BC. Shear stress-mediated extracellular signal-regulated kinase activation is regulated by sodium in endothelial cells. Potential role for a voltage-dependent sodium channel. J Biol Chem 274: 20144–20150, 1999. [DOI] [PubMed] [Google Scholar]
- 115.Tsuda M, Masuda T, Tozaki-Saitoh H, Inoue K. P2X4 receptors and neuropathic pain. Front Cell Neurosci 7: 191, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Tzima E, Irani-Tehrani M, Kiosses WB, Dejana E, Schultz DA, Engelhardt B, Cao G, DeLisser H, Schwartz MA. A mechanosensory complex that mediates the endothelial cell response to fluid shear stress. Nature 437: 426–431, 2005. [DOI] [PubMed] [Google Scholar]
- 117.Vriens J, Watanabe H, Janssens A, Droogmans G, Voets T, Nilius B. Cell swelling, heat, and chemical agonists use distinct pathways for the activation of the cation channel TRPV4. Proc Natl Acad Sci USA 101: 396–401, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Wang S, Iring A, Strilic B, Albarran Juarez J, Kaur H, Troidl K, Tonack S, Burbiel JC, Muller CE, Fleming I, Lundberg JO, Wettschureck N, Offermanns S. P2Y(2) and Gq/G(1)(1) control blood pressure by mediating endothelial mechanotransduction. J Clin Invest 125: 3077–3086, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Watanabe H, Vriens J, Janssens A, Wondergem R, Droogmans G, Nilius B. Modulation of TRPV4 gating by intra- and extracellular Ca2+. Cell Calcium 33: 489–495, 2003. [DOI] [PubMed] [Google Scholar]
- 120.Woo SH, Ranade S, Weyer AD, Dubin AE, Baba Y, Qiu Z, Petrus M, Miyamoto T, Reddy K, Lumpkin EA, Stucky CL, Patapoutian A. Piezo2 is required for Merkel-cell mechanotransduction. Nature 509: 622–626, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Wu G, Markowitz GS, Li L, D'Agati VD, Factor SM, Geng L, Tibara S, Tuchman J, Cai Y, Park JH, van Adelsberg J, Hou H Jr, Kucherlapati R, Edelmann W, Somlo S. Cardiac defects and renal failure in mice with targeted mutations in Pkd2. Nat Gen 24: 75–78, 2000. [DOI] [PubMed] [Google Scholar]
- 122.Yamamoto K, Furuya K, Nakamura M, Kobatake E, Sokabe M, Ando J. Visualization of flow-induced ATP release and triggering of Ca2+ waves at caveolae in vascular endothelial cells. J Cell Sci 124: 3477–3483, 2011. [DOI] [PubMed] [Google Scholar]
- 123.Yamamoto K, Korenaga R, Kamiya A, Ando J. Fluid shear stress activates Ca2+ influx into human endothelial cells via P2X4 purinoceptors. Circ Res 87: 385–391, 2000. [DOI] [PubMed] [Google Scholar]
- 124.Yamamoto K, Korenaga R, Kamiya A, Qi Z, Sokabe M, Ando J. P2X(4) receptors mediate ATP-induced calcium influx in human vascular endothelial cells. Am J Physiol Heart Circ Physiol 279: H285–H292, 2000. [DOI] [PubMed] [Google Scholar]
- 125.Yamamoto K, Sokabe T, Matsumoto T, Yoshimura K, Shibata M, Ohura N, Fukuda T, Sato T, Sekine K, Kato S, Isshiki M, Fujita T, Kobayashi M, Kawamura K, Masuda H, Kamiya A, Ando J. Impaired flow-dependent control of vascular tone and remodeling in P2X4-deficient mice. Nat Med 12: 133–137, 2006. [DOI] [PubMed] [Google Scholar]
- 126.Yamamoto K, Sokabe T, Ohura N, Nakatsuka H, Kamiya A, Ando J. Endogenously released ATP mediates shear stress-induced Ca2+ influx into pulmonary artery endothelial cells. Am J Physiol Heart Circ Physiol 285: H793–H803, 2003. [DOI] [PubMed] [Google Scholar]
- 127.Zaritsky JJ, Eckman DM, Wellman GC, Nelson MT, Schwarz TL. Targeted disruption of Kir2.1 and Kir22 genes reveals the essential role of the inwardly rectifying K+ current in K+-mediated vasodilation. Circ Res 87: 160–166, 2000. [DOI] [PubMed] [Google Scholar]
- 128.Zarychanski R, Schulz VP, Houston BL, Maksimova Y, Houston DS, Smith B, Rinehart J, Gallagher PG. Mutations in the mechanotransduction protein PIEZO1 are associated with hereditary xerocytosis. Blood 120: 1908–1915, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Zhang H, Inazu M, Weir B, Daniel E. Endothelin-1 inhibits inward rectifier potassium channels and activates nonspecific cation channels in cultured endothelial cells. Pharmacology 49: 11–22, 1994. [DOI] [PubMed] [Google Scholar]
- 130.Zhang XD, Timofeyev V, Li N, Myers RE, Zhang DM, Singapuri A, Lau VC, Bond CT, Adelman J, Lieu DK, Chiamvimonvat N. Critical roles of a small conductance Ca2+-activated K+ channel (SK3) in the repolarization process of atrial myocytes. Cardiovasc Res 101: 317–325, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]