

Abstract
The oral multikinase inhibitor sorafenib undergoes extensive UGT1A9‐mediated formation of sorafenib‐β‐D‐glucuronide (SG). Using transporter‐deficient mouse models, it was previously established that SG can be extruded into bile by ABCC2 or follow a liver‐to‐blood shuttling loop via ABCC3‐mediated efflux into the systemic circulation, and subsequent uptake in neighboring hepatocytes by OATP1B‐type transporters. Here we evaluated the possibility that this unusual process, called hepatocyte hopping, is also operational in humans and can be modulated through pharmacological inhibition. We found that SG transport by OATP1B1 or murine Oatp1b2 was effectively inhibited by rifampin, and that this agent can significantly increase plasma levels of SG in wildtype mice, but not in Oatp1b2‐deficient animals. In human subjects receiving sorafenib, rifampin acutely increased the systemic exposure to SG. Our study emphasizes the need to consider hepatic handling of xenobiotic glucuronides in the design of drug–drug interaction studies of agents that undergo extensive phase II conjugation.
Study Highlights
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
✓ Sorafenib undergoes extensive UGT1A9‐mediated formation of sorafenib‐β‐D‐glucuronide (SG), which is a substrate of the hepatocellular transporter OATP1B1.
WHAT QUESTION DID THIS STUDY ADDRESS?
✓ We hypothesized that sorafenib‐β‐D‐glucuronide can undergo hepatocyte hopping in humans, and that this process can be modulated through pharmacological inhibition.
WHAT THIS STUDY ADDS TO OUR KNOWLEDGE?
✓ Our findings signify an unusual contribution of OATP1B1 in the hepatocellular handling of sorafenib in humans, whereby compromised OATP1B1 function leads to systemic accumulation of sorafenib‐β‐D‐glucuronide.
HOW THIS MIGHT CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE
✓ Our study emphasizes the need to consider hepatic handling of xenobiotic glucuronides in the design of drug–drug interaction studies of agents that undergo extensive phase II conjugation.
The multikinase inhibitor sorafenib is used as a chemotherapeutic agent in the treatment of multiple malignant diseases, including cancers of the liver, kidney, and thyroid.1, 2, 3 The pharmacokinetic properties of orally administered sorafenib are characterized by up to 90% variation in exposure between patients receiving the same therapeutic regimen.4 The high degree of interindividual pharmacokinetic variability observed with sorafenib has important toxicological ramifications. For example, it was recently demonstrated that levels of sorafenib in plasma are correlated with the incidence of skin rash,5 with dose reduction and study withdrawal due to adverse effects,6 and with the development of severe adverse reactions.7
The mechanisms underlying the unpredictable pharmacokinetic profile of sorafenib remain largely unexplained. After oral administration, sorafenib enters hepatocytes by incompletely defined mechanisms,8, 9 and then undergoes CYP3A4‐mediated oxidation10, 11 and UGT1A9‐mediated glucuronidation.11 A mass balance study of oral sorafenib in humans has shown that 15% of the dose was eliminated as sorafenib‐β‐D‐glucuronide (SG), compared with less than 5% as oxidative metabolites. Interestingly, SG was not detectable in feces, which may be due to its instability in the presence of bacterial glucuronidases present in the gut.12 Therefore, it has been suggested that the actual contribution of glucuronidation to sorafenib elimination may have been underestimated in the mass balance study,9 and that, because of its effective secretion into bile,13 the appearance of SG in the systemic circulation represents an overshoot mechanism that poorly reflects the actual extent of its formation. These observations suggest that a critical determinant of sorafenib's pharmacokinetic variability with possible consequences for clinical management may be associated with differential expression and function of SG transporters regulating its distribution and elimination.14
After its formation, SG is secreted into the bile through a process mediated by the adenosine triphosphate (ATP)‐binding cassette efflux transporter ABCC2 (MRP2).13 Under normal physiologic conditions, a fraction of the hepatocellular SG is secreted back into the bloodstream by ABCC3 (MRP3), from where it can be taken up again into downstream hepatocytes via the uptake carrier OATP1B1 (Oatp1b2 in mice) (Figure 1).13 This liver‐to‐blood shuttling loop, called hepatocyte‐hopping, may prevent saturation of ABCC2‐mediated biliary secretion of endogenous and xenobiotic glucuronides in upstream hepatocytes, thereby ensuring their efficient biliary elimination and hepatocyte detoxification. Once secreted into bile, SG enters the intestinal lumen, where it serves as a substrate for a bacterial β‐glucuronidase that produces sorafenib, which subsequently undergoes intestinal absorption and then reenters the systemic circulation.13 In the current proof‐of‐concept study, we tested the hypothesis that the hepatocyte hopping of SG can be interrupted by a clinical OATP1B1‐mediated drug–drug interaction based on the expectation that inhibition of a hepatic uptake mechanism will lead to acute increases in levels of SG in plasma.
Figure 1.
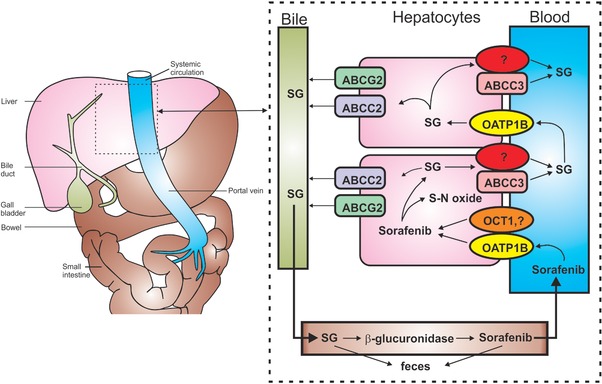
Hepatocyte hopping and recirculation of sorafenib‐β‐D‐glucuronide. After oral administration, sorafenib enters the hepatocytes by incompletely defined transporters mechanisms, including OATP1B‐type carriers and OCT1, and undergoes ABCG2‐mediated biliary secretion, CYP3A4‐mediated metabolism to sorafenib‐N‐oxide (S‐N‐oxide), or UGT1A9‐mediated glucuronidation to form sorafenib‐β‐D‐glucuronide (SG). After conjugation, SG is extensively secreted into the bile by a process that is mainly mediated by ABCC2. Under physiological conditions, a fraction of the intracellular SG is secreted by ABCC3 and at least one other transporter back to the blood, from where it can be taken up again into downstream hepatocytes via OATP1B‐type carriers. This secretion‐and‐reuptake loop may prevent the saturation of ABCC2‐mediated biliary excretion in the upstream hepatocytes, thereby ensuring efficient biliary elimination and hepatocyte detoxification. Once secreted into bile, SG enters the intestinal lumen, where it can either be excreted or serve as a substrate for an as‐yet unknown bacterial β‐glucuronidase that produces sorafenib, which is subsequently undergoing intestinal absorption and reenters the systemic circulation. This figure is a modified version of a figure from Vasilyeva et al.13 and is reprinted with permission.
MATERIALS AND METHODS
Cell lines and chemicals
A model of OATP1B1‐expressing cells was created by transfecting HEK293 cells with the pIRES2‐EGFP vector (Clontech, Mountain View, CA) containing SLCO1B1 cDNA. Similarly, HEK293 cells were transfected with the pDream2.1/MCS vector (GenScript, Piscataway, NJ) containing Slco1b2 cDNA. The HEK293 (ATCC CRL1573) cell line was obtained from American Type Culture Collection (ATCC, Manassas, VA). This cell line was exclusively used to study drug transport, and was not authenticated by the authors. All stable cell lines were selected and maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and G418 sulfate (500–1,000 μg/mL; AG Scientific, San Diego, CA) at 37°C under 5% CO2.
Sorafenib and rifampin were obtained from Chemie Tek (Indianapolis, IN). General tritium‐labeled sorafenib (specific activity, >1 Ci/mmol; radiochemical purity, >97.1%) was custom made by Moravek Biochemicals (Brea, CA), and [3H]estradiol‐17β‐D‐glucuronide (E2G; specific activity, 50.1 Ci/mmol; radiochemical purity, 99.0%), a positive control substrate for OATP1B1 and Oatp1b2, was obtained from American Radiolabeled Chemicals (St. Louis, MO).
Uptake studies
Cells were seeded in 6‐well plates in phenol red‐free DMEM media containing 10% FBS, and were incubated at 37°C for 24 h. Cells were then washed with warm phosphate‐buffered saline (PBS) and incubated with sorafenib or SG in phenol‐free DMEM media (without FBS and supplements) at 37°C. Uptake and inhibition studies were performed as outlined in detail elsewhere.13 The experiment was terminated by placing cells on ice and washing twice with ice‐cold PBS. Cells were collected and centrifuged at 1,050 r.p.m. for 5 min at 4°C. The cell pellet was lysed in 1 N NaOH by vortex‐mixing, incubated at 4°C overnight, and then the solution was neutralized with 2 M HCl. Total protein was measured using a Pierce BCA Protein Assay Kit (ThermoScientific, Waltham, MA) and total protein content was quantified using a Biotek μQuant microplate spectrophotometer (Winooski, VT). Intracellular E2G concentrations were determined in the remaining cell lysate by liquid scintillation counting using a LS 6500 Multipurpose Scintillation Counter (Beckman, Brea, CA). The experiments were performed in triplicate. Intracellular concentrations of SG were measured by liquid chromatography‐tandem mass spectrometry (LC‐MS/MS), as described previously.11
Transcellular transport
MDCKII cells were transduced with pIRES2 construct containing CFP‐ABCC2‐V5, GFP‐OAT1B1‐FLAG, or GFP‐OATP1B1‐FLAG/CFP‐ABCC2‐V5 (Supplementary Figure S1). The MDCKII (ATCC CRL2936) cell line was obtained from ATCC. This cell line was exclusively used to study drug transport, and was not authenticated by the authors. Transport of SG (1 μM) was performed in 6‐well plates (Corning, Corning, NY), as described,11 Trans‐epithelial electrical resistance was measured to confirm the integrity of cell monolayers.
Immunofluorescence
MDCKII cells transduced with CFP‐ABCC2‐V5 and/or GFP‐SLCO1B1‐FLAG were seeded at 2 × 105 cells/well into 6‐transwell plates. When they reached about 90% confluence, cells were washed with ice‐cold PBS, fixed with 4% paraformaldehyde‐PBS, permeabilized with 0.1% TritonX‐100‐PBS, and incubated in 3% BSA‐PBS blocking buffer. Then cells were stained with either anti‐V5 (Sigma‐Aldrich, St. Louis, MO) or anti‐FLAG M2 (Sigma‐Aldrich) followed by staining with Alexa488 and Alexa568 (Life Technologies, Camarillo, CA), respectively, along with DAPI (Invitrogen, La Jolla, CA) to stain the nuclei. Transwell membranes were cut out and placed on a slide, covered with a coverslip, and sealed. Imaging was done using a Marianas spinning disk confocal (SDC) imaging system (Intelligent Imaging Innovations/3i) based on AxioObserver Z1 inverted microscope (Carl Zeiss MicroImaging, Peabody, MA). Images were acquired with a Zeiss Plan‐Apochromat 63 × 1.4 NA DIC objective and Evolve 512 EMCCD camera (Photometrics, Tucson, AZ) using SlideBook 5 software (3i).
Murine pharmacokinetics
Female mice knockout for Oatp1b2 [Oatp1b2(‐/‐)] and age‐matched wildtype mice on a DBA1/lacJ background were bred in‐house. Mice were housed in a temperature‐controlled environment with a 12‐h light cycle and given a standard diet and water ad libitum. Experiments were approved by the Institutional Animal Care and Use Committee.
Sorafenib was formulated in 50% Cremophor EL (Sigma‐Aldrich) and 50% ethanol, and diluted 1:4 (vol/vol) with deionized water immediately before administration by oral gavage at a dose of 10 mg/kg. Mice were fasted for 3 h before and during the study, with unrestricted access to drinking water. In Oatp1b2(‐/‐) and wildtype mice (N = 4 per group), rifampin (20 mg/kg) was administered intravenously 5 min prior to the oral sorafenib administration. At select timepoints, blood samples (30 μL each) were taken from individual mice at 0.25, 0.5, and 1.5 h from the submandibular vein using a lancet, and at 3, and 4.5 h from the retro‐orbital venous plexus using a capillary. A final blood draw was obtained at 7.5 h by a cardiac puncture using a syringe and needle. For sampling via retro‐orbital bleeding, mice were anesthetized under 1–5% isoflurane through inhalation, and blood was collected using a heparinized capillary tube. The total blood volume collected during the procedure from each mouse was 150 μL. All blood samples were centrifuged at 3,000 × g for 5 min, and plasma was separated and stored at –80°C until analysis. At the terminal timepoints, liver samples were immediately collected and flash‐frozen on dry ice. Liver specimens were stored at –80°C until further processing. Plasma and liver concentrations of sorafenib, sorafenib‐N‐oxide, and SG were determined by LC‐MS/MS, as described previously.11 Pharmacokinetic parameters were calculated using WinNonlin 6.3 software (Pharsight, Princeton, NJ).
Clinical studies
Patients were enrolled and received standard of care treatment with sorafenib in an open‐label randomized crossover trial. The principle inclusion criteria were: age ≥18 years, confirmed diagnosis of cancer, WHO performance score 0–1, and adequate organ function, which was defined as absolute neutrophil count >1.0 × 109/L and platelet count ≥60 × 109/L for bone marrow function; as serum creatinine <1.25 × ULN (upper limit of normal) for renal function; and as total bilirubin <1.25 × ULN, aminotransferases <5 × ULN, prothrombin time ≤1.5 × ULN or international normalized ratio <1.5 for hepatic function. Also, patients were required to have reached steady state, which was defined as at least 14 days of treatment with the same sorafenib dose. The major exclusion criteria were: prior liver transplantation, contraindications for any of the study drugs, and use of any comedication or supplement that is known to cause interactions with the study drugs. The mean steady‐state (trough) concentration (Css) of SG used in the sample size calculation was 954 ng/mL, estimated from a group of 16 cancer patients receiving sorafenib at an oral dose of 90 mg/m2 that had sampling for pharmacokinetics on three consecutive occasions, namely, on days 7, 13, and 21.15 In this group of patients, the standard deviation (SD) of the expected differences of the two measurements (sorafenib given without and with rifampin) was estimated to be 223 ng/mL. It was assumed that the interval between treatments represents an adequate washout period, with no carryover or period effect. The clinical study was designed to detect an effect size of 239/223, where 239 is 25% of the mean Css of SG. Based on a pairwise (two‐sided) analysis, this results in a sample size of nine for the prospective evaluation, with a significance level of 0.05 and power of 0.8. All other pharmacokinetic end points were assessed exploratorily. Between August 2013 and March 2015, nine evaluable patients with hepatocellular carcinoma were included in the clinical study, of whom eight were male and one was female. The mean age was 71 years (range, 62–79) and all patients had a WHO performance score of 1. Patients were randomized by minimization using the web‐based application Trial Online Process (TOP). Patient characteristics were entered in TOP by one of the investigators and the patient's trial number and randomization arm were then sent to all investigators in an automatic email from TOP. Four patients were randomized to receive rifampin during the first sampling period (arm A), and five were randomized to rifampin during the second period (arm B). The administered sorafenib (Nexavar; Bayer, The Netherlands) dose was 200 mg b.i.d. for four patients (three in arm A, one in arm B) and 400 mg b.i.d. for five patients (one in arm A, four in arm B). The study was approved by the Institutional Review Board (Protocol number, MEC 2013–194), and registered in the Dutch Trial Registry (www.trialregister.nl; number NTR4110). All patients provided written informed consent, and the study was conducted in accordance with Good Clinical Practice guidelines and the Declaration of Helsinki (59th WMA General Assembly, Seoul, Republic of Korea; October 2008).
Subjects were admitted to the hospital for 2 separate days of blood sampling: once with prior rifampin administration and once without. Days of blood sampling were separated by a period of 9 days. Rifampin was taken without food as 600 mg tablets (Rifadin; Sanofi‐Aventis, The Netherlands) on the day before and on the day of sampling at 8 AM, exactly 1 h before sorafenib administration. Two and a half h after sorafenib intake, midazolam (2.5 mg; Actavis, The Netherlands) was administered intravenously as a probe for CYP3A4 activity.16 During both hospitalizations, blood samples (6 mL each) for the determination of sorafenib and metabolite levels were collected just before the administration of sorafenib, and 2, 4, and 7.5 h after the administration of sorafenib. Samples were prepared by centrifugation at 1,200 × g for 5 min to obtain plasma, which was stored at –80°C until analysis. Plasma concentrations of sorafenib, sorafenib‐N‐oxide, and SG were determined by LC‐MS/MS, as described and validated previously.11, 15, 17 Concentrations of midazolam and its metabolites were measured in three plasma samples taken 2, 4, and 6 h after midazolam administration during both sample periods. The midazolam samples were centrifuged and stored as described above. WinNonlin 6.3 (Pharsight) was used for calculating pharmacokinetic parameters.
Patients were seen in the outpatient clinic on a weekly basis for clinical examination, laboratory tests, and to evaluate possible side effects. Skin toxicity and diarrhea were managed according to local institutional guidelines. Adverse events were registered according to the National Cancer Institute's Common Terminology Criteria for Adverse Events (CTCAE), v. 4.03. During the study, dose changes of sorafenib and the use of comedication that is known to influence CYP3A4 function were not allowed.
Statistical analysis
Pharmacokinetic data are presented as geometric mean and 95% confidence interval. In patients receiving sorafenib at a dose of 200 mg b.i.d., dose‐dependent pharmacokinetic parameters such as the area under the curve (AUC) and absolute observed concentrations were normalized to a dose of 400 mg b.i.d.; that is, parameters were multiplied by a factor of 2. This procedure is justified as sorafenib is known to exhibit a linear pharmacokinetic profile within this dose range.18 Statistical analysis was performed using GraphPad Prism 5.0 (GraphPad Software, La Jolla, CA). Uptake rates in the in vitro experiments were compared using unpaired t‐tests. Geometric means of the pharmacokinetic parameters in mice were compared using analysis of variance (ANOVA), whereas those in humans were compared using two‐sided paired t‐tests. P < 0.05 was considered statistically significant.
RESULTS
Influence of rifampin on SG transport in vitro
Experiments were initially carried out with HEK293 cells engineered to express OATP1B1 or its murine equivalent Oatp1b2 using the recommended model substrate estradiol‐17β‐glucuronide (E2G),19 and the clinically relevant OATP1B‐type transporter inhibitor rifampin.20 Following a 15‐min incubation period, E2G uptake by human OATP1B1 and mouse Oatp1b2 was strongly inhibited by a preincubation with rifampin (30 μM) (P = 0.001 and P < 0.001, respectively; Figure 2 a,b). Since rifampin‐mediated inhibition of OATP1B1 can be substrate‐dependent, with up to 12‐fold variation in IC50 values,21 we next used SG as a test substrate in the same models. Similar to E2G, rifampin reduced the intracellular uptake of SG by both OATP1B1 and Oatp1b2 by 92% (P = 0.004) and 97% (P > 0.001), respectively (Figure 2 a,b). This process was dependent on the rifampin concentration (Figure 2 c). The resulting IC50 values of ∼1 μM for both OATP1B1 and Oatp1b2 were similar to those reported previously for uptake inhibition by rifampin of other substrates.22 As plasma Cmax of unbound rifampin is ∼1.3 μM (1.1 mg/L),23 the intrinsic likelihood of an OATP1B1‐mediated pharmacokinetic drug–drug interaction between rifampin and SG is high.
Figure 2.
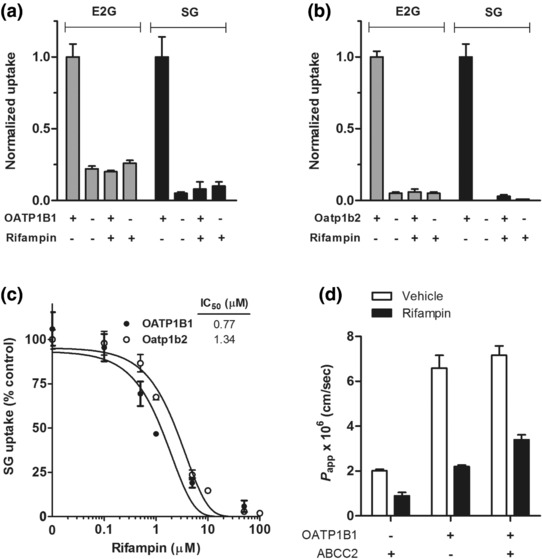
Transport of SG by OATP1B‐type transporters. Transport of estradiol‐17β‐glucuronide (E2G; 0.1 μM) and sorafenib‐β‐D‐glucuronide (SG; 10 μM) in HEK293 cells engineered to overexpress OATP1B1 (a) or Oatp1b2 (b) with or without rifampin (20 μM). All results are normalized to the transport rate in OATP1B transfected cells without rifampin, i.e., the experiments with unrestricted OATP1B effect, which were 4.77 pmol/mg protein (OATP1B1) and 26.73 pmol/mg protein (Oatp1b2) in 15 min for E2G, and 57.46 pmol/mg protein (OATP1B1) and 770.17 pmol/mg protein (Oatp1b2) in 15 min for SG. (c) Inhibition of OATP1B1 or Oatp1b2‐mediated transport of SG (10 μM) by different concentrations of rifampin (0–100 μM). Data are normalized to the relative uptake without rifampin, i.e., when the function of OATP1B is unrestricted, and represent the mean ± SE from 3–4 independent experiments (9–12 replicates). (d) Transcellular transport of SG in MDCKII cells expressing OATP1B1 and/or ABCC2. Cells were incubated with SG (1 μM), and 50‐μl aliquots were taken at 1, 2, 3, and 4 h from the compartment opposite to where the drug was added, in the presence or absence of rifampin (100 μM). Data are expressed as transporter‐mediated apparent permeability coefficient (P app) for the basolateral to apical direction (B‐to‐A). Data represent the mean ± SE (at least three replicates).
Because rifampin is also a known inhibitor of various ABC transporters,24 and can influence the transport of SG in inside‐out vesicles expressing ABCC2,13 we next evaluated the influence of rifampin on the flux of SG in MDCKII cells engineered to overexpress OATP1B1, ABCC2, or both OATP1B1 and ABCC2 (Supplementary Figure S1). Transfection of OATP1B1 (basolaterally localized) into MDCKII cells significantly increased the basal‐to‐apical transport of SG, and this translocation was diminished in the presence of rifampin (Figure 2 d). However, the basal‐to‐apical flux of SG was not substantially enhanced by co‐transfection of ABCC2 (apically localized), and not further reduced by rifampin in cells expressing both OATP1B1 and ABCC2 (Figure 2 d). This suggests that rifampin can be utilized for in vivo studies as a bona fide inhibitor of SG transport by OATP1B‐type carriers.
Effects of rifampin on SG disposition in mice
The in vivo role of rifampin (20 mg/kg) in the transport of SG was next evaluated in wildtype mice and Oatp1b2‐deficient [Oatp1b2(‐/‐)] littermates receiving a single oral dose of sorafenib (10 mg/kg). This experiment was based on the expectation that the systemic exposure to SG would increase by rifampin in an Oatp1b2‐dependent manner, as predicted from our in vitro transport experiments. In line with our previous findings,9 Oatp1b2‐deficiency in mice was associated with a significant 8‐fold increase in SG AUC0‐7.5h (Figure 3; Supplementary Table S1).
Figure 3.
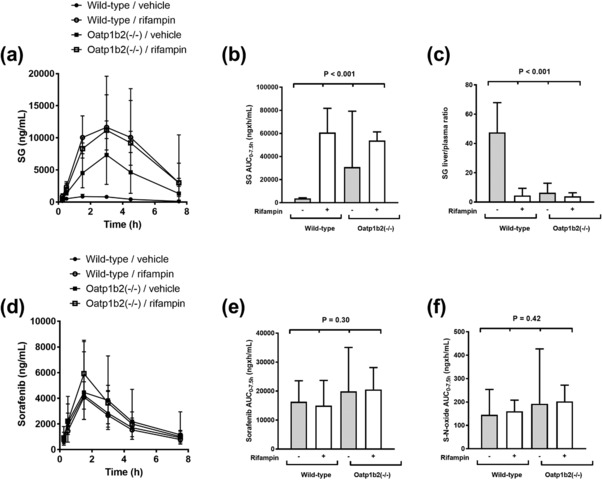
Pharmacokinetics of sorafenib and SG in wildtype and Oatp1b2(‐/‐) mice. Plasma concentration–time profiles of SG (a) and sorafenib (d) in wildtype mice and Oatp1b2(‐/‐) mice in the presence and absence of rifampin pretreatment. Corresponding area under the plasma concentration–time curves (AUC0‐7.5) of SG, sorafenib, and sorafenib‐N‐oxide (S‐N‐oxide) are shown in (b), (e), and (f). Sorafenib was administered orally at a dose of 10 mg/kg with or without pretreatment with rifampin (20 mg/kg). Livers were taken at 7.5 h after sorafenib administration (n = 4 per group), with results expressed as the liver‐to‐plasma concentration ratio of SG (c). Concentrations in liver were normalized to corresponding concentrations in plasma. All data represent the geometric mean and the 95% confidence interval.
The liver‐to‐plasma ratio of SG was reduced by ∼90% in wildtype mice pretreated with rifampin, and similar to that observed in Oatp1b2(‐/‐) mice receiving sorafenib either alone or when given in combination with rifampin (Figure 3 c). Oatp1b2‐deficiency and/or rifampin pretreatment did not substantially affect the plasma levels of sorafenib parent drug or of its primary oxidated metabolite sorafenib‐N‐oxide (Figure 3 d–f). This observation is consistent with our previous finding that sorafenib itself is not a transported substrate of Oatp1b2,9 and with the contention that the applied single dose of rifampin is unlikely to have artificially influenced other enzymes and transporters of relevance to the disposition of sorafenib or SG.
Effects of rifampin on SG disposition in humans
We next assessed the influence of pretreatment with rifampin (two daily oral doses of 600 mg) on the pharmacokinetics (PK) of SG in human subjects receiving oral sorafenib using an open‐label randomized crossover design. All patients met our criteria to be at steady state, as all patients were treated at the same dose for 14 days or more at the first day of PK sampling. As predicted from the murine pharmacokinetic studies, we found that concomitant rifampin administration resulted in acute, statistically significant increases in the systemic exposure to SG (32.5 μg × h/mL versus 14.6 μg×h/mL; P < 0.001) (Figure 4 a; Supplementary Table S2), and this was independent of the randomization sequence (Figure 4 b).
Figure 4.
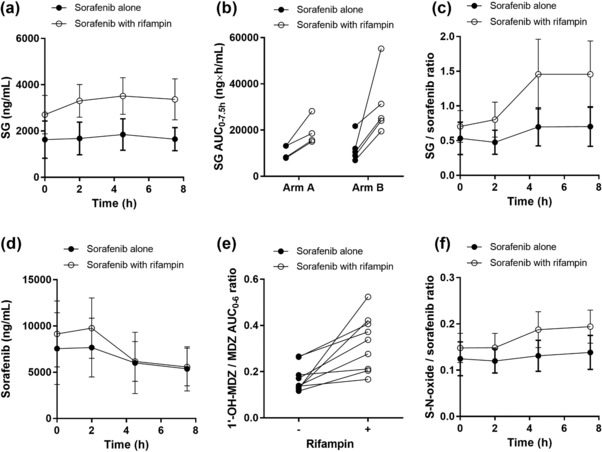
Influence of rifampin on the pharmacokinetics of sorafenib in humans. Plasma concentration–time profiles of SG (a) and sorafenib (d) in patients with hepatocellular carcinoma in the presence and absence of rifampin pretreatment. The corresponding area under the plasma concentration–time curve (AUC0‐7.5) of SG is shown as a function of the randomization sequence of the crossover trial (b). The metabolic ratios for SG to sorafenib and sorafenib‐N‐oxide (S‐N‐oxide) to sorafenib are shown in (c) and (f), respectively. The metabolic ratio for the AUC0‐6 of 1′‐hydroxy‐midazolam (1′‐OH‐MDZ) to the AUC0‐6 of midazolam (MDZ) is shown in (e). All data represent the geometric mean and the 95% confidence interval and all metabolic ratios were corrected for molecular weight.
The mean metabolic ratios of SG to sorafenib in the studied patient cohort were also significantly increased during rifampin administration (Figure 4 c). No statistically significant differences were observed in the PK parameter estimates of sorafenib (Figure 4 d). Although the mean ratio between 1’‐hydroxymidazolam and midazolam, used as a measure of CYP3A4 activity changes,25 was ∼2 times higher after rifampin intake (P = 0.005; Figure 4 e), the extent of sorafenib‐N‐oxide formation was not different between treatment cycles (Figure 4 f). During the course of the clinical study, no toxicities were observed that could be attributed to rifampin or midazolam. Liver function remained adequate during the study for all patients, although one experienced liver enzyme elevations due to cholestasis, for which previously placed biliary stents were replaced after the study. In another patient, bilirubin levels in plasma were elevated from 14–29 μM (upper limit of normal, 16 μM) on the second day of rifampin intake, but bilirubin levels normalized to baseline within 2 days.
DISCUSSION
This study shows that acute inhibition of the hepatic uptake transporter OATP1B1 by rifampin results in a PK interaction with SG at steady state in human subjects receiving oral sorafenib. This finding emphasizes the need to consider hepatic handling of xenobiotic glucuronides in the design of PK drug–drug interaction studies of agents that undergo extensive phase II conjugation, and can potentially have clinical relevance for the chemotherapeutic treatment with sorafenib.
It was previously suggested based on in vitro microsomal studies that the most prominent pathway of sorafenib elimination consists of CYP3A4‐mediated metabolism leading to the formation of sorafenib‐N‐oxide and several other metabolites.8 This finding suggested that sorafenib was potentially subject to a host of CYP3A4‐mediated drug interactions with commonly coprescribed medications.26 Here, the AUCs of the CYP3A4‐mediated metabolites of midazolam and sorafenib were increased after pretreatment with rifampin. As the ratio of sorafenib‐N‐oxide to sorafenib did not change between both cycles, the interaction of rifampin on sorafenib‐N‐oxide formation does not seem to be clinically relevant. In this context, it is noteworthy that the prototypical CYP3A4 inhibitor ketoconazole was previously found to have no influence on the PK of sorafenib in healthy male volunteers after single‐dose sorafenib administration, suggesting that the fraction of sorafenib that is metabolized by the phase I oxidative pathway is low.12 In contrast, although clinical data suggest that sorafenib glucuronidation accounts for only 15% of the dose, it is likely that this percentage is grossly underestimated,13 and may be increased further if the competing CYP3A4‐mediated pathway is inhibited.12 The long‐term clinical implications of such PK drug–drug interactions remain unstudied.
The use of rifampin as an OATP1B1 inhibitor in our studies was based on considerations published elsewhere.20 The relatively strong impact of rifampin on SG levels in mice (>ninefold increase) compared with humans (∼2‐fold) likely reflects a differential direct influence of rifampin on the uptake of sorafenib itself into hepatocytes, which process is partially dependent on OATP1B‐type transporters in humans but appears of less relevance in mice.9 In addition, SG is known to undergo substantial renal excretion in humans but not in mice,13 and the possible presence of a rifampin‐insensitive escape mechanism in the kidney may result in shunting of SG to urine when OATP1B1 function in humans is impaired.
It should also be pointed out that, although acute exposure to rifampin inhibits OATP1B1,27 extended daily administration of rifampin may induce enzymes and transporters of putative relevance to sorafenib. For example, exposure to rifampin for 5 days or more dramatically increased the clearance of the CYP3A4 substrate drugs midazolam,28 alfentanil,29 and erythromycin.30 Several recent studies have evaluated the effects of acute and extended exposure to rifampin on CYP3A4 activity in the same individuals. For example, rifampin given once daily for 1–2 doses of 600 mg each (acute exposure) and the same dose administered for 6.5 days (extended exposure) changed the systemic exposure to bosentan, a dual OATP1B1 and CYP3A4 substrate drug, by +500% and –58%, respectively.31 Thus, if induction of enzymes occurs, it is likely that rifampin exposure for 5 days or more is required to cause a clinically relevant, induced CYP3A4 phenotype. Because CYP3A4 induction seems to play only a modest role in our study, the present observations with sorafenib in conjunction with acute exposure to rifampin may not be extrapolated to the situation where the agent is coadministered with rifampin for an extended period of time. Moreover, the current study does not show whether prolonged OATP1B1 inhibition by other drugs leads to stronger effects on the PK of sorafenib and its metabolites than those observed here.
Interestingly, administered at a dose of 600 mg once daily for 5 days with a single oral dose of sorafenib in healthy volunteers, rifampin was previously found to cause a 37% decrease in the mean systemic exposure to sorafenib.32 The causal connection of this observation with altered CYP3A4 activity, however, remains uncertain and other plausible mechanisms could contribute to the reported observations. For example, prolonged exposure to rifampin can significantly upregulate OATP1B1 and ABCC2 in primary hepatocytes.33 It can also induce UGT1A9 mRNA and UGT1A9 activity in human subjects after 6 days of exposure to a once‐daily dose of 600 mg, although morinidazole was only minimally affected.34 Interestingly, prolonged treatment with rifampin may also affect the hepatic expression and function of the uptake transporter OCT1,35 which has been proposed as a possible hepatic uptake carrier of sorafenib.8, 36 However, this may not be of concern clinically, since recent studies in mice with a hepatic OCT1‐deficiency indicate this transporter plays only a relatively minor role in the overall elimination of sorafenib.37 As basolateral efflux via ABCC3 seems a prerequisite for hepatocyte hopping (Figure 1), differential functioning of this transporter—due to genetic polymorphisms or due to exogenous induction or inhibition—might also play a vital role in the processes described here. This study, however, was not designed to take that effect into account. Also OATP1B3, functioning identical to OATP1B1, is likely to have contributed to these findings, especially since it is also inhibited by rifampin. However, hepatocellular expression of OATP1B3 is lower than that of OATP1B1 and cells overexpressing the latter have higher uptake of SG than those overexpressing the former.9
The potential clinical ramifications of the hepatocyte‐hopping phenomenon of SG and the impact of interference in this process with transporter inhibitors such as rifampin requires additional investigation. For example, further study is required to determine if excessive systemic accumulation of SG can lead to increases in adverse events, as has been observed with morphine‐6‐glucuronide.38 This possibility is consistent with the notion that certain reduced function variants in SLCO1B1, the gene encoding OATP1B1, are associated with increased risk of sorafenib‐associated toxicity.39 Future research is also required to evaluate the consequences of a reduced biliary excretion of SG on maintaining plasma concentrations of sorafenib. This connection is plausible, considering that sorafenib undergoes enterohepatic recirculation40 following bacterial β‐glucuronidase‐mediated deconjugation of SG within the intestinal lumen,41 and would be consistent with the fact that intentional interference of this deconjugation by neomycin is associated with a decrease in the systemic exposure to sorafenib by more than 50% (Nexavar package insert).
Overall, our findings signify an important contribution of OATP1B1 in the hepatocellular handling of sorafenib in humans, whereby compromised OATP1B1 function leads to systemic accumulation of SG through a sinusoidal liver‐blood shuttling process known as hepatocyte hopping. We expect that the current observations with sorafenib may have relevance to other kinase inhibitors that undergo extensive phase II conjugation through glucuronidation, such as dasatinib, regorafinib, and trametinib (Supplementary Table S3). The PK of these drugs could also be altered in the presence of other OATP1B1‐interacting agents such as clarithromycin42 and ramipiril.22 Establishing the clinical significance of this interaction between kinase inhibitors and agents that potently inhibit OATP1B‐type transporters is warranted.
Supporting information
Table S1. Sorafenib pharmacokinetic parameters in mice.
Table S2. Sorafenib pharmacokinetic parameters in humans.
Table S3. Kinase inhibitors undergoing glucuronidation.
Figure S1. Transcellular transport of SG in transfected MDCKII cells.
Acknowledgments
We thank Richard Kim and Jeffrey Stock for providing the Oatp1b2(‐/‐) mice. This study was supported in part by ALSAC and the National Institutes of Health; Grants R01 CA138744 (to S.D.B.) and P30 CA021765 (to S.D.B.).
Author Contributions
S.B., A.S., R.H.J.M., and S.D.B. wrote the article; S.B., L.v.D., A.S., R.H.J.M., and S.D.B. designed the research; S.B., L.v.D., M.A.P., A.A.G., S.H., L.L., A.V., G.D., P.H., F.A.L.M.E., P.d.B., and R.H.J.M. performed the research; S.B., L.v.D., A.A.G., S.H., L.L., A.V., P.d.B., A.S., R.H.J.M., and S.D.B. analyzed the data.
Conflict of Interest
The authors declared no conflict of interest. The content is solely the responsibility of the authors and does not necessarily represent the official views of the funding agencies.
References
- 1. Escudier, B. et al Sorafenib in advanced clear‐cell renal‐cell carcinoma. N. Engl. J. Med. 356, 125–134 (2007). [DOI] [PubMed] [Google Scholar]
- 2. Brose, M.S. et al Sorafenib in radioactive iodine‐refractory, locally advanced or metastatic differentiated thyroid cancer: a randomised, double‐blind, phase 3 trial. Lancet 384, 319–328 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Llovet, J.M. et al Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med. 359, 378–390 (2008). [DOI] [PubMed] [Google Scholar]
- 4. Strumberg, D. et al Safety, pharmacokinetics, and preliminary antitumor activity of sorafenib: a review of four phase I trials in patients with advanced refractory solid tumors. Oncologist 12, 426–437 (2007). [DOI] [PubMed] [Google Scholar]
- 5. Kuczynski, E.A. , Lee, C.R. , Man, S. , Chen, E. & Kerbel, R.S. Effects of Sorafenib dose on acquired reversible resistance and toxicity in hepatocellular carcinoma. Cancer Res. 75, 2510–2519 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Shimada, M. et al Monitoring serum levels of sorafenib and its N‐Oxide is essential for long‐term sorafenib treatment of patients with hepatocellular carcinoma. Tohoku J. Exp. Med. 237, 173–182 (2015). [DOI] [PubMed] [Google Scholar]
- 7. Fucile, C. et al Measurement of sorafenib plasma concentration by high‐performance liquid chromatography in patients with advanced hepatocellular carcinoma: is it useful the application in clinical practice? A pilot study. Med. Oncol. 32, 335 (2015). [DOI] [PubMed] [Google Scholar]
- 8. Swift, B. et al Sorafenib hepatobiliary disposition: mechanisms of hepatic uptake and disposition of generated metabolites. Drug Metab. Dispos. 41, 1179–1186 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zimmerman, E.I. et al Contribution of OATP1B1 and OATP1B3 to the disposition of sorafenib and sorafenib‐glucuronide. Clin. Cancer Res 19, 1458–1466 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ghassabian, S. et al Role of human CYP3A4 in the biotransformation of sorafenib to its major oxidized metabolites. Biochem. Pharmacol. 84, 215–223 (2012). [DOI] [PubMed] [Google Scholar]
- 11. Zimmerman, E.I. et al Ontogeny and sorafenib metabolism. Clin. Cancer Res. 18, 5788–5795 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lathia, C. et al Lack of effect of ketoconazole‐mediated CYP3A inhibition on sorafenib clinical pharmacokinetics. Cancer Chemother. Pharmacol. 57, 685–692 (2006). [DOI] [PubMed] [Google Scholar]
- 13. Vasilyeva, A. et al Hepatocellular shuttling and recirculation of sorafenib‐glucuronide is dependent on Abcc2, Abcc3, and Oatp1a/1b. Cancer Res. 75, 2729–2736 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tlemsani, C. et al Effect of glucuronidation on transport and tissue accumulation of tyrosine kinase inhibitors: consequences for the clinical management of sorafenib and regorafenib. Expert Opin. Drug Metab. Toxicol. 11, 785–794 (2015). [DOI] [PubMed] [Google Scholar]
- 15. Inaba, H. et al phase I pharmacokinetic and pharmacodynamic study of the multikinase inhibitor sorafenib in combination with clofarabine and cytarabine in pediatric relapsed/refractory leukemia. J. Clin. Oncol. 29, 3293–3300 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mathijssen, R.H. & van Schaik, R.H. Genotyping and phenotyping cytochrome P450: perspectives for cancer treatment. Eur. J. Cancer 42, 141–148 (2006). [DOI] [PubMed] [Google Scholar]
- 17. Li, L. et al Quantitation of sorafenib and its active metabolite sorafenib N‐oxide in human plasma by liquid chromatography‐tandem mass spectrometry. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 878, 3033–3038 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Di Gion, P. et al Clinical pharmacokinetics of tyrosine kinase inhibitors: focus on pyrimidines, pyridines and pyrroles. Clin. Pharmacokinet. 50, 551–603 (2011). [DOI] [PubMed] [Google Scholar]
- 19. Izumi, S. et al Investigation of the impact of substrate selection on in vitro organic anion transporting polypeptide 1B1 inhibition profiles for the prediction of drug–drug interactions. Drug Metab. Dispos. 43, 235–247 (2015). [DOI] [PubMed] [Google Scholar]
- 20. International Transporter Consortium , Giacomini, K.M. et al Membrane transporters in drug development. Nat. Rev. Drug. Discov. 9, 215–236 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Izumi, S. et al Substrate‐dependent inhibition of organic anion transporting polypeptide 1B1: comparative analysis with prototypical probe substrates estradiol‐17beta‐glucuronide, estrone‐3‐sulfate, and sulfobromophthalein. Drug Metab. Dispos. 41, 1859–1866 (2013). [DOI] [PubMed] [Google Scholar]
- 22. Gui, C. , Obaidat, A. , Chaguturu, R. & Hagenbuch, B. Development of a cell‐based high‐throughput assay to screen for inhibitors of organic anion transporting polypeptides 1B1 and 1B3. Curr. Chem. Genomics. 4, 1–8 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. te Brake, L.H. et al Exposure to total and protein‐unbound rifampin is not affected by malnutrition in Indonesian tuberculosis patients. Antimicrob. Agents Chemother. 59, 3233–3239 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cui Y, Konig J, Keppler D. Vectorial transport by double‐transfected cells expressing the human uptake transporter SLC21A8 and the apical export pump ABCC2. Mol. Pharmacol. 60, 934–943 (2001). [DOI] [PubMed] [Google Scholar]
- 25. Kloth, J.S. et al Predictive value of CYP3A and ABCB1 phenotyping probes for the pharmacokinetics of sunitinib: the ClearSun study. Clin. Pharmacokinet. 53, 261–269 (2014). [DOI] [PubMed] [Google Scholar]
- 26. Bowlin, S.J. , Xia, F. , Wang, W. , Robinson, K.D. & Stanek, E.J. Twelve‐month frequency of drug‐metabolizing enzyme and transporter‐based drug–drug interaction potential in patients receiving oral enzyme‐targeted kinase inhibitor antineoplastic agents. Mayo Clin. Proc. 88, 139–148 (2013). [DOI] [PubMed] [Google Scholar]
- 27. Lau, Y.Y. , Huang, Y. , Frassetto, L. & Benet, L.Z. effect of OATP1B transporter inhibition on the pharmacokinetics of atorvastatin in healthy volunteers. Clin. Pharmacol. Ther. 81, 194–204 (2007). [DOI] [PubMed] [Google Scholar]
- 28. Backman, J.T. , Olkkola, K.T. & Neuvonen, P.J. Rifampin drastically reduces plasma concentrations and effects of oral midazolam. Clin. Pharmacol. Ther. 59, 7–13 (1996). [DOI] [PubMed] [Google Scholar]
- 29. Kharasch, E.D. et al The role of cytochrome P450 3A4 in alfentanil clearance. Implications for interindividual variability in disposition and perioperative drug interactions. Anesthesiology 87, 36–50 (1997). [DOI] [PubMed] [Google Scholar]
- 30. Paine, M.F. , Wagner, D.A. , Hoffmaster, K.A. & Watkins, P.B. Cytochrome P450 3A4 and P‐glycoprotein mediate the interaction between an oral erythromycin breath test and rifampin. Clin. Pharmacol. Ther. 72, 524–535 (2002). [DOI] [PubMed] [Google Scholar]
- 31. van Giersbergen, P.L. , Treiber, A. , Schneiter, R. , Dietrich, H. & Dingemanse, J. Inhibitory and inductive effects of rifampin on the pharmacokinetics of bosentan in healthy subjects. Clin. Pharmacol. Ther. 81, 414–419 (2007). [DOI] [PubMed] [Google Scholar]
- 32. Keating, G.M. & Santoro, A. Sorafenib: a review of its use in advanced hepatocellular carcinoma. Drugs 69, 223–240 (2009). [DOI] [PubMed] [Google Scholar]
- 33. Williamson, B. , Dooley, K.E. , Zhang, Y. , Back, D.J. & Owen, A. Induction of influx and efflux transporters and cytochrome P450 3A4 in primary human hepatocytes by rifampin, rifabutin, and rifapentine. Antimicrob. Agents Chemother. 57, 6366–6369 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Pang, X. et al Effects of rifampin and ketoconazole on pharmacokinetics of morinidazole in healthy chinese subjects. Antimicrob. Agents Chemother. 58, 5987–5993 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Cho, S.K. et al Rifampin enhances the glucose‐lowering effect of metformin and increases OCT1 mRNA levels in healthy participants. Clin. Pharmacol. Ther. 89, 416–421 (2011). [DOI] [PubMed] [Google Scholar]
- 36. Herraez, E. et al Expression of SLC22A1 variants may affect the response of hepatocellular carcinoma and cholangiocarcinoma to sorafenib. Hepatology 58, 1065–1073 (2013). [DOI] [PubMed] [Google Scholar]
- 37. Neul, C. et al Cellular uptake of sorafenib into hepatocellular carcinoma cells is independent of human organic cation transporter 1 (OCT1). Naunyn‐Schmied. Arch. Pharmacol. 388 (suppl. 1), S23 (2015). [Google Scholar]
- 38. Tiseo P.J. et al Morphine‐6‐glucuronide concentrations and opioid‐related side effects: a survey in cancer patients. Pain 61, 47–54 (1995). [DOI] [PubMed] [Google Scholar]
- 39. Bins, S. et al Polymorphisms in SLCO1B1 and UGT1A1 are associated with sorafenib‐induced toxicity. Pharmacogenomics Epub 2016 August 17. [DOI] [PubMed] [Google Scholar]
- 40. Jain, L. et al Population pharmacokinetic analysis of sorafenib in patients with solid tumours. Br. J. Clin. Pharmacol. 72, 294–305 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hilger, R.A. et al Pharmacokinetics of sorafenib in patients with renal impairment undergoing hemodialysis. Int. J. Clin. Pharmacol. Ther. 47, 61–64 (2009). [DOI] [PubMed] [Google Scholar]
- 42. Hirano, M. , Maeda, K. , Shitara, Y. & Sugiyama, Y. Drug‐drug interaction between pitavastatin and various drugs via OATP1B1. Drug Metab. Dispos. 34, 1229–1236 (2006). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Sorafenib pharmacokinetic parameters in mice.
Table S2. Sorafenib pharmacokinetic parameters in humans.
Table S3. Kinase inhibitors undergoing glucuronidation.
Figure S1. Transcellular transport of SG in transfected MDCKII cells.