

Abstract
Carbamazepine belongs to the class II biopharmaceutical classification system (BCS) which is characterized by a high per-oral dose, a low aqueous solubility and a high membrane permeability. The bioavailability of such a drug is limited by the dissolution rate. The present study deals with the formulations of immediate release tablets of poorly soluble carbamazepine. As model tablets for this investigation, two formulations (named “A” and “B” formulations) of carbamazepine tablets labeled to contain 200 mg were evaluated. The aim of this study was to establish possible differences in dissolution profile of these two formulations purchased from the local market.
The increased crystallinity together with enlarged particle size, enhanced aggregation and decreased wettability of the drug, resulted in insufficient dissolution rate for formulation “B’.’ From the dissolution point of view, this formulation was inferior to the formulation “A, due to the solubilization effect.
Keywords: carbamazepine, impairment, in vitro release
INTRODUCTION
Carbamazepine is a dibenzazepine derivative with antiepileptic and psychotropic properties. It is also used in the treatment of trigeminal neuralgia and pain associated with other neurological disorders (1). It belongs to Class II biopharmaceutical classification system (BCS) which is characterized by high membrane permeability, slow dissolution rate due to low aqueous solubility, and high peroral dose (2). The rate of oral absorption of poorly soluble drugs is often controlled by their dissolution rate in the gastrointestinal tract Therefore, bioavailability rate of this drug is limited by the dissolution rate (3). Thus solubility and dissolution rate are the key determinants of oral bioavailability, which is the concluding point drawn for fate of oral bioavailability (4, 5) Carbamazepine is a widely prescribed antiepileptic drug having poor water solubility (~170 mg/l at 25 °C) (6). The plasma half-life ranges from 18 to 60 hours following a single dose and from 10 to 35 hours during chronic therapy. Because of having poor water solubility, its absorption is dissolution rate limited, which often results in irregular and delayed absorption (7) Several attempts have been made to increase the dissolution rate or bioavailability of carbamazepine (8-14). Different methods have been exploited to enhance the dissolution of poorly water soluble drugs among which solid dispersion (SD) techniques, because of their simplicity and effectiveness have received considerable attentions (15)
The aim of this study was:
-to compare two formulations (named “A” and “B” formulations) of carbamazepine tablets labelled to contain 200 mg, purchased from the local market;
-to establish possible differences in dissolution profile of these two formulations.
MATERIAL AND METHODS
Reagents
The used reagents carbamazepine and sodium lauryl sulphate were of analytical grade and were provided by Sigma-Aldrich, St. Louis, USA, and by Merck, Darmstadt, Germany, respectively.
Tablet composition and preliminary comparison
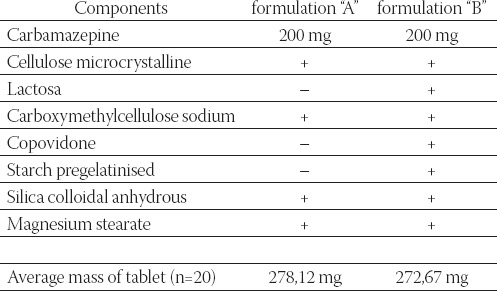
Two formulations (“A” and “B”) contained 200 mg of active substance, and excipients, as follows (Table 1.). Preliminary, disintegration testing (according to Ph. Eur. 2.9.1.) was performed: in each of six tubes, one tablet is placed. The assembly was suspended in the 1 litre beaker, containing water, and operated (without disks). A suitable device maintained temperature of the liquid at 37o±0,5°C The test was provided using Pharma Test disintegration tester Type PTZ-E 1/6E (Pharma Test, Hainburg, Germany). Disintegration time for both formulations indicated that there was no significant difference between productsthe tablets were disintegrated in less than 2 minutes (according to USP, up to 15 minutes). In accordance with the USP monograph for carbamazepine tablets (declared strength 200 mg), the rate of dissolution was determined for two products formulations (“A” and “B”).
TABLE 1.
Overview and comparison of formulation “A” and “B”
Dissolution test conditions and analysis procedure Preparation of standard solutions
A standard curve of absorbance versus concentration was constructed using solutions of carbamazepine in the dissolution medium (water containing 1% sodium lauryl sulphate) ranging in concentration from 0,0064 to 0,008 mg/ml. Absorbance versus concentration plot (y=51,42530x+0,00005; R2= 0,99998) was linear over this concentration range and was used to determine percent of drug dissolved in the dissolution experiments. UV absorbance of each standard solution was measured spectrophotometrically at 286 nm.
Dissolution test procedure
The dissolution tests of carbamazepine tablets (formulation “A” and “B”) were performed using USP apparatus 2 (n=6), ERWEKA DT 800 dissolution tester. The amount of drug in the tablet was 200 mg (declared strength) which was added to 900 ml of distilled water containing 1% sodium lauryl sulphate as dissolution medium. Under such circumstances a perfect sink condition was maintained to mimic the dynamic situation in GI tract. The mixture was stirred at 75 rpm at 37±0,5°C Samples in the amount of 10 ml were withdrawn at predetermined times (15th and 60th min), filtered and assayed at 286 nm spectrophotometrically. The drug concentration in the samples was corrected mathematically (correction for volume), considering the concentrations of the previous samples (withdrawn samples were not supplemented with an equal volume of fresh dissolution fluid to maintain a constant total volume). The dissolution apparatus was maintained at 37±0,5°C throughout the experiment. Prior to use, the dissolution media were equilibrated at 45°C for two hours and filtered using a 0,45 μm membrane filter (Sartorious GmbH, Goettingen, Germany) to deaerate the medium so that bubble formation during the test, due to escape of dissolved gases, was minimized. Dissolution samples were collected for analysis. These samples were filtered using a 0,45 μm membrane filter (Sartorious GmbH, Goettingen, Germany). The dissolution apparatus was connected with UV/VIS spectrophotometer Shimadzu UV-1700 (Shimadzu, Kyoto, Japan). Determination of dissolution rates for the active ingredient in tablets (formulations “A” and “B”) is carried out by the previously mentioned spectrophotometric method.
RESULTS AND DLSCUSSION
During dissolution testing, by visual examination, different behaviour of disintegrated, dispersed particles in dissolution medium (see Figures 1 and 2) can be noticed:
FIGURE 1.

Disintegrated partides of tablet formulation “K”
FIGURE 2.

Disintegrated partides of tablet formuktion “B”
Formulaton “A”: disintegrated fine particles are evenly dispersed in dissolution medium, without the presence of large agglomerates (Figure 1a, b and c):
formulation B: disintegrated particles bind together into larger agglomerates, which were formed at the bottoms of dissolution vessels, could be seen on the Figures 2a, b and c. This occurrence resulted in decreased dissolution surface, decreased stirring rate influence of dissolution medium and, consequently, lower results than those specified USP monograph:
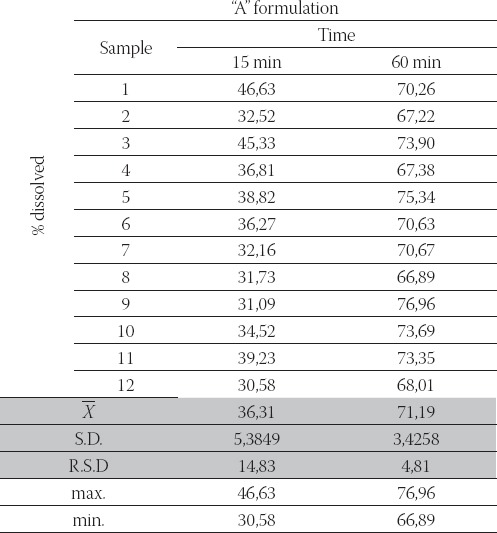
The results of dissolution studies for “A” and “B” formulations are summarized in Table 2., Table 3., which show the fraction of the dissolved drug as a function of time.
TABLE 2.
Fraction of dissolved carbamazepine from the “A” formulation as a function of time (stage I, n= 6).
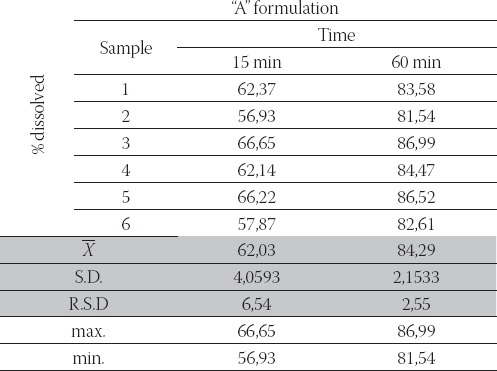
TABLE 3.
Fraction of dissolved carbamazepine from the “B” formulation as a function of time (stage II, n= 12).
According to the USP 31-NF 26, in vitro release of carbamazepine from tablets (labelled strength 200 mg) fulfilled requirements if: between 45% and 75% of the labeled amount of carbamazepine was dissolved in 15 minutes; not less than 75% (Q) of the declared contents was dissolved for a period of 60 minutes (stage I). Fractions of carbamazepine released in dissolution medium were calculated from calibration curve.
In our study, in vitro release of carbamazepine from:
-“A “ formulation Julfilled these requirements (obtained release profile: 56,93 %- 66,65 % and 81,54 %- 86,99 %, in 15 and 60 minutes, respectively);
-“B” formulation did not fulfil these requirements (obtained release profile: 32,52 %- 46,63 % and 67,22 %- 75,34 %, in 15 and 60 minutes, respectively).
For this reason, stage II on additional six tablets was performed for carbamazepine tablets (“B” formulation). According to the USP 31-NF 26, in vitro release of carbamazepine from tablets (labelled strength 200 mg), stage II fulfilled requirements if: average value (n=12) was in the range of 40 %- 80 % and no units were more than 5% outside the stated range within 15 minutes (40 %- 80 %); average value (n=12) was larger or equal to 75 % and no units were more than 5% outside the stated range within 60 minutes (< 70 %);
After performed stage II, “B” formulation did not fulfil these requirements (obtained release profile: 30,58 %- 46,63 % and 66,89 %- 76,96 %, in 15 and 60 minutes, respectively). Finally, because of differences in fulfilment the requirements at stage II, stage III (n= 24) would not give satisfactory results so, so this test (stage III) was not performed.
CONCLUSION
-regardless of similar composition of two tablet formulations, it could be assumed that the technological process of tablet production significantly affected dissolution rate;
-disintegration time could not be considered as a discriminatory test that would be able to point out the differences in the dissolution rate, due to the very short disintegration time for both formulations (< 2 minutes);
-inter-agglomeration of particles dispersed in dissolution medium could significantly reduce dissolution rate of carbamazepine tablets;
-visual examination that, eventually, could be able to confirm formation of agglomerate, during the dissolution test performed (as it was shown for tablet formulation “B” - Figure 2a, b and c), could point out the flaws in the development of tablets and point out to possible difficulties in the dissolution rate of the investigated products.
REFERENCES
- 1.Sweetman S.C Martindale. The complete drug reference. 34th ed. London: Pharmaceutical Press; 2005. [Google Scholar]
- 2.Rinaki E, Valsami G, Macheras P. Quantitative biopharmaceutics classification system: The central role of dose/solubility ratio. Pharm. Res. 2003;20:1917–1925. doi: 10.1023/b:pham.0000008037.57884.11. [DOI] [PubMed] [Google Scholar]
- 3.Lobenberg R, Amidon GL. Modern bioavailability, bioequivalence and biopharmaceutics classification system: new scientific approaches to international regulatory standards. Eur. J. Pharm. Biopharm. 2000;50:3–12. doi: 10.1016/s0939-6411(00)00091-6. [DOI] [PubMed] [Google Scholar]
- 4.Desai KGH, Kulakarni AR, Aminbhavi TM. Solubility of Rofecoxib in presence of methanol, ethanol and sodium lauryl sulphate at (298.15, 303.15 and 308.15) KJ. Chem. Eng. Data. 2003;48:942–945. [Google Scholar]
- 5.Rawat S, Jain SK. Rofecoxib—cyclodextrin inclusion complex for solubility enhancement. Pharmazie. 2003;58:639–641. [PubMed] [Google Scholar]
- 6.Moneghini M, Voinovich D, Perissutti B, Princivalle F. Action of carriers on carbamazepine dissolution. Pharm. Dev. Technol. 2002;7:289–296. doi: 10.1081/pdt-120005725. [DOI] [PubMed] [Google Scholar]
- 7.Bertilsson L. Clinical pharmacokinetics of carbamazepine. Clin. Pharmacokinet. 1978;3:128–143. doi: 10.2165/00003088-197803020-00003. [DOI] [PubMed] [Google Scholar]
- 8.Lake O.A, Olling M, Barends DM. In vitro/in vivo correlations of dissolution data of carbamazepine immediate release tablets with pharmacokinetic data obtained in healthy volunteers. Eur. J. Pharm. Biopharm. 1999;48:9–13. doi: 10.1016/s0939-6411(99)00016-8. [DOI] [PubMed] [Google Scholar]
- 9.Zerrouk N, Chemtob C, Arnaud P, Toscani S, Dugue J. In vitro and in vivo evaluation of carbamazepine-PFG 6000 solid dispersions. Int. J. Pharm. 2001;225:49–62. doi: 10.1016/s0378-5173(01)00741-4. [DOI] [PubMed] [Google Scholar]
- 10.Moneghini M, Kikic I, Voinovich D, Perissutti B, Filipovic Grčić J. Processing of carbamazepine-PEG 4000 solid dispersions with supercritical carbon dioxide: Preparation, characterization, and in vitro dissolution. Int. J. Pharm. 2001;222:129–138. doi: 10.1016/s0378-5173(01)00711-6. [DOI] [PubMed] [Google Scholar]
- 11.Sarkari M, Brown J, Chen X, Swinnea S, Williams RO, III, Johnston KP. Enhanced drug dissolution using evaporative precipitation into aqueous solution. Int. J. Pharm. 2002;243:17–31. doi: 10.1016/s0378-5173(02)00072-8. [DOI] [PubMed] [Google Scholar]
- 12.Sethia S, Squilante E. Physicochemical characterization of solid dispersions of carbamazepine formulated by supercritical carbon dioxide and conventional solvent evaporation method. J. Pharm. Sci. 2002;91:1948–1957. doi: 10.1002/jps.10186. [DOI] [PubMed] [Google Scholar]
- 13.Perissutti B, Rubessa F, Moneghini M, Voinovich D. Formulation design of carbamazepine fast-release tablets prepared by melt granulation technique. Int. J. Pharm. 2003;256:53–63. doi: 10.1016/s0378-5173(03)00062-0. [DOI] [PubMed] [Google Scholar]
- 14.Leuner C, Dressman J. Improving drug solubility for oral delivery using solid dispersions. Eur. J. Pharm. Biopharm. 2000;50:47–60. doi: 10.1016/s0939-6411(00)00076-x. [DOI] [PubMed] [Google Scholar]
- 15.Ribardiere A, Tchoreoff P, Couarraze G, Puisieux F. Modification of ketoprofen bead structure produced by the spherical crystallization technique with a two-solvent system. Int. J. Pharm. 1996;144:195–207. [Google Scholar]