

Abstract
Investigation of Alternaria sp. AST0039, an endophytic fungus obtained from the leaf tissue of Astragalus lentiginosus, led to the isolation of (−)-(10E,15S)-4,6-dichloro-10(11)-dehydrocurvularin (1), (−)-(10E,15S)-6-chloro-10(11)-dehydrocurvularin (2), (−)-(10E,15S)-10(11)-dehydrocurvularin (3), and alterperylenepoxide A (4) together with scytalone and α-acetylorcinol. Structures of 1 and 4 were established from their spectroscopic data, and the relative configuration of 4 was determined with the help of nuclear Overhauser effect difference data. All metabolites were evaluated for their cytotoxic activity and ability to induce heat-shock and unfolded protein responses. Compounds 2 and 3 exhibited cytotoxicity to all five cancer cell lines tested and increased the level of the pro-apoptotic transcription factor CHOP, but only 3 induced the heat-shock response and caused a strong unfolded protein response.
Graphical abstract
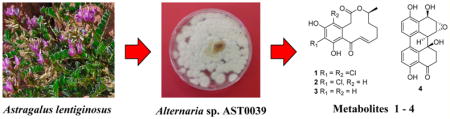
Recent studies have demonstrated that plant-associated fungi are prolific producers of structurally diverse small-molecule natural products with interesting biological activities.1 In continuing our search for metabolites with potential anticancer activity from endosymbiotic fungi of the Sonoran Desert bioregion we have employed an ecologically relevant strategy to identify metabolites targeting protein homeostasis by inducing the heat-shock and unfolded protein responses. The heat-shock response (HSR), in addition to being an important component of innate adaptive responses to counteract ecological challenges, plays a key role in enabling cells to accommodate drastic alterations in physiology that accompany malignant transformation.2 During our investigation of rhizosphere-associated ectosymbiotic fungi of the Sonoran desert plants, we have developed and used a moderate-throughput phenotypic screen to discover potential anticancer natural products that target protein homeostasis using the cellular HSR as monitored by the expression of enhanced green fluorescent protein (EGFP) as the end point. Our previous work using this heat-shock induction assay (HSIA) led to the identification of monocillin I as a potential anticancer agent3a and a fungal metabolite capable of conferring thermotolerance to Arabidopsis seedlings.3b We have now used HSIA to evaluate extracts of endosymbiotic fungi associated with plants and lichens of the Sonoran Desert region. One of those having promising activity was derived from the endophytic fungal strain Alternaria sp. AST0039, isolated from a healthy leaf tissue of Astragalus lentiginosus (spotted locoweed, Fabaceae) collected in central Arizona. Fractionation of this extract led to the isolation and characterization of two new metabolites, (−)-(10E,15S)-4,6-dichloro-10(11)-dehydrocurvularin (1) and the epoxyperylene alterperylenepoxide A (4), together with the known dehydrocurvularins 2 and 3, scytalone,4 and α-acetylorcinol.5 Although curvularin analogues have previously been encountered in several endophytic6 and marine fungi,7 and (−)-(10E,15S)-6-chloro-10(11)-dehydrocurvularin (2) was reported once before from a soil-borne fungus,8 this constitutes the first report of the natural occurrence of a curvularin with two chlorine substituents in the aromatic ring. Curvularins are resorcylic acid lactones of considerable biological interest because of their cytotoxic,9 phytotoxic,10 antimicrobial,7b nematicidal,11 antimycobacterial,12 and antitrypanosomal13 activities, their ability to act as spindle poisons,14 and their role as inhibitors of cell division,15 microtubule assembly,16 and components of the ubiquitin proteasome system including the ATPase p97 and the proteasome.17 Occurrence of altertoxins, epoxyperylenes, and perylenequinones with anti-HIV,18 anti-leishmanial,19 antimalarial,19 and cytotoxic19 activities has been recently documented in endophytic Alternaria sp. It is noteworthy that the fungal metabolite thioperylenol (5), structurally related to alterperylenepoxide A (4) and bearing a 2-hydroxy-3-mercaptopropionic acid moiety at C-6, has been converted into hydroperylene derivatives with therapeutic applications for platelet aggregation.20
RESULTS AND DISCUSSION
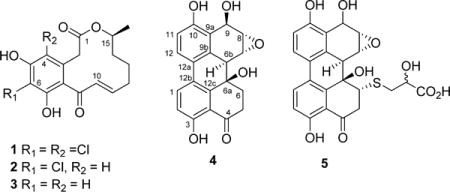
Metabolite 1, obtained as a pale yellow, amorphous solid, was determined to have the molecular formula C16H16Cl2O5 by a combination of HRESIMS and 13C NMR data indicating eight degrees of unsaturation. The LRMS peaks at m/z 359 (M + 1), 361 (M + 3), and 363 (M + 5) in the ratio of 9:6:1 provided additional support for the presence of two chlorine atoms in 1.21 Comparison of the spectroscopic data of 1, especially 1H and 13C NMR data (Table 1), with those of (−)-(10E,15S)-6-chloro-10(11)-dehydrocurvularin (2)8 and (−)-(10E,15S)-10(11)-dehydrocurvularin (3),9 which were found to co-occur in this fungal strain (see below), indicated that 1 contained the same basic structure as 2 but without the aromatic proton at C-4, suggesting that the additional chlorine atom is attached to this position. These together with its COSY and HMBC NMR data (Figure 1) established the gross structure of 1 as 4,6-dichloro-10(11)-dehydrocurvularin. The geometry of the 10(11)-double bond was determined as 10E from its 1H–1H coupling constant (J10,11 = 15.7 Hz). The absolute configuration of 3 at its only chiral center C-15 has been established as 15S by its total synthesis,6c,22 and the optical rotation data have been used to determine the absolute configuration of other curvularins.8 Comparison of [α]D of 1 (−47) with those reported for 2 (−51)8 and 3 (−82)6c suggested that the chiral center (C-15) of 1 had the same absolute configuration as those of 2 and 3. Thus, the structure of 1 was established as (−)-(10E,15S)-4,6-dichloro-10(11)-dehydrocurvularin.
Table 1.
1H NMR (400 MHz) and 13C NMR (100 MHz) Data for Compounds 1 and 2 in Acetone-d6a
| position | 1
|
2
|
||
|---|---|---|---|---|
| δC (type) | δH (J in Hz) | δC (type) | δH (J in Hz) | |
| 1 | 169.1, C | 171.6, C | ||
| 2 | 38.7, CH2 | 3.55, d (17.5) 3.67, d (17.5) |
42.2, CH2 | 3.63, d (17.6) 4.07, d (17.6) |
| 3 | 130.8, C | 136.9, C | ||
| 4 | 115.5, C | 113.2, CH | 6.57, s | |
| 5 | 151.5, C | 158.2, C | ||
| 6 | 109.6, C | 107.9, C | ||
| 7 | 151.9, C | 160.5, C | ||
| 8 | 122.0, C | 116.6, C | ||
| 9 | 196.4, C | 197.3, C | ||
| 10 | 132.5, CH | 6.28, d (15.7) | 132.3, CH | 6.77, d (15.4) |
| 11 | 156.6, CH | 6.74, ddd (15.7, 9.9, 5.9) | 151.3, CH | 6.63, ddd (15.4, 8.9, 5.0) |
| 12 | 34.3, CH2 | 2.21, m 2.34, m |
33.3, CH2 | 2.35, m 2.42, m |
| 13 | 25.6, CH2 | 1.29, m 1.98, m |
25.0, CH2 | 1.64, m 1.99, m |
| 14 | 34.1, CH2 | 1.53, m 1.85, m |
34.8, CH2 | 1.63, m 1.85, m |
| 15 | 73.4, CH | 4.90, qdd (6.3, 6.3, 2.4) | 73.1, CH | 4.75, m |
| 16 | 20.5, CH3 | 1.2, d (6.3) | 20.4, CH3 | 1.16, d (6.4) |
All signals were assigned by the analysis of 1H–1H COSY, HSQC, and HMBC spectra.
Figure 1.
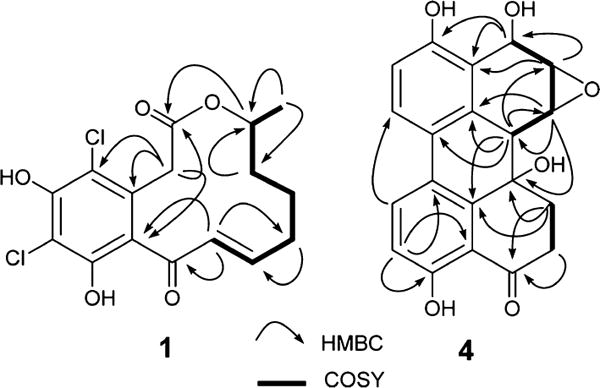
Selected 1H–1H COSY and HMBC correlations for 1 and 4.
Alterperylenepoxide A (4), obtained as a pale yellow, amorphous solid, was determined to have the molecular formula C20H16O6 by a combination of HRESIMS and NMR data and indicated 13 degrees of unsaturation. Comparison of its 1H and 13C NMR spectroscopic data with altertoxins I–III and V–VI, which we have encountered previously in an endophytic A. tenuissima,18 suggested that 4 contained an epoxyperylene skeleton. The presence of a [1,1′-biphenyl]-4,4′-diol moiety in 4 was further supported by its UV spectrum, which exhibited absorptions in the range typical for this chromophore of related perylenes.23 The 1H and 13C NMR spectra of 4 assigned with the help of 1H–1H COSY and HMBC data (Figure 1) displayed, in addition to those characteristic of the [1,1′-biphenyl]-4,4′-diol moiety of altertoxins I, II, V, and VI, signals due to an oxygenated benzylic methine [δH 5.25 (brs); δC 62.1], two methines of an oxirane ring [δH 3.87 (d, J = 4.0 Hz); δC 51.3, and δH 3.55 (brd, J = 4.0 Hz); δC 56.9], and an oxygenated and nonprotonated carbon (δC 69.2). The presence of only one carbonyl carbon (δC 206.8) in 4 suggested that the above oxygenated benzylic methine was derived as a result of the reduction of one of the two carbonyl groups typical of altertoxins.23 Detailed analysis of HMBC data of 4 (Figure 1) was helpful in locating the carbonyl group at C-4, the OH at C-6a, the oxirane moiety at C-7(8), and an OH-bearing methine at C-9. The relative configurations of chiral centers in 4 were determined by the application of nuclear Overhauser effect difference (NOEDIFF) data (Figure 2 and Figure S13, Supporting Information). A strong NOE observed for one of the methylene protons at C-6 (δH 2.40) upon irradiation of H-6b (δH 3.22) was indicative of a 1,3-cis relationship between these protons, confirming the trans orientation of H-6b and OH-6a. Irradiation of the oxymethine proton at δ 3.87 (H-7) caused enhancement of H-6β (δH 2.82), suggesting a β-configuration for H-7. Therefore, the oxirane ring must have an α-configuration. The absence of vicinal coupling between H-6b and H-7 further confirmed the α-configuration of the oxirane ring.18 Irradiation of the oxymethine proton at δ 3.55 (H-8) caused enhancement of H-7, H-9 (δH 5.25), and 9-OH (δH 4.90). Since H-7 showed NOE relationships with both H-9 and OH-9, it was not possible to use these data to establish the relative configuration of OH-9. Therefore, the relative configuration of OH-9 was deduced with the aid of the 1H NMR coupling constants and energy-minimized Chem3D models. Since H-9 appeared as a broad singlet, the dihedral angle between H-8 and H-9 should be closer to 90°. Thus, the hydroxy group at C-9 must be in a pseudoaxial orientation, and hence it should have a β-configuration. The 1H NMR coupling constants for H-6α and CH2-5 (J6α,5β = 13.5 and J6α,5α = 4.0 Hz) accounted for the preferred half-chair conformation of the cyclohexanone ring.23b The CD spectrum of 4 in MeOH showed a positive Cotton effect at λ = 240–260 nm (Figure S18, Supporting Information). Although the sign of the Cotton effect and the positive value for [α]D indicated an Sa configuration for the biphenyl moiety of 4, these data could not be used definitively to discern its absolute configuration.24 Application of the modified Mosher’s ester method for the determination of the absolute configuration of 4 failed, as it resulted in the formation of a complex mixture of MTPA esters, leading to interactions in ΔδS−R effects among the modification sites, presumably due to their close proximity.25 Thus, the structure of alterperylenepoxide A was determined as (+)-(6aβ,6bα,7α,8α,9β)-3,6a,9,10-tetrahydroxy-4-oxo-4,5,6,6a,6b,7,8,9-octahydro-7,8-epoxyperylene (4).
Figure 2.
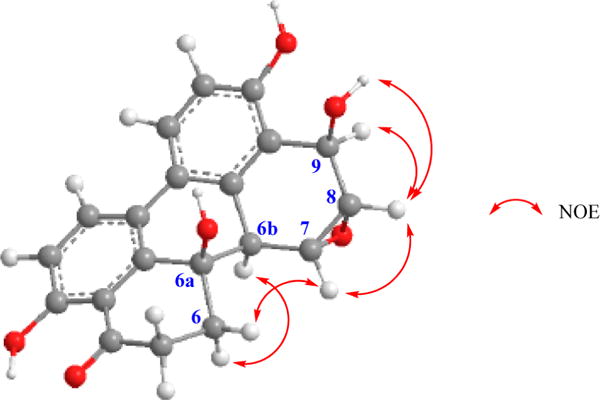
Selected NOE correlations for 4.
Metabolites 1–4 were evaluated for their in vitro inhibition of cell proliferation/survival using a panel of five cancer cell lines, NCI-H460 (human non-small-cell lung cancer), SF-268 (human CNS glioma), MCF-7 (human breast cancer), PC-3 M (human metastatic prostate adenocarcinoma), and MDA-MB-231 (human metastatic breast adenocarcinoma). Cells were exposed to serial dilutions of test compounds for 72 h in RPMI-1640 media supplemented with 10% fetal bovine serum, and relative viable cell number was measured by a standard dye reduction assay using resazurin (AlamarBlue).26 Only 2 and 3 were found to be cytotoxic below a concentration of 5.0 μM with no apparent selectivity for any of the cell lines tested (Table 2). Metabolites 1–4 were also evaluated for their heat-shock induction activity using a heat-shock reporter cell line as previously described.3a Interestingly, only the 10(11)-dehydrocurvularins 1–3 showed activity in this assay, with an activity order of 3 > 2 > 1 (Figure 3). Thus, from a structure–activity perspective, it appears that the stepwise substitution of hydrogens in the aromatic ring of (10,11)-dehydrocurvularins with chlorine causes reduction of both cytotoxic and heat-shock induction activities, which may be attributed to electronic and/or steric effects caused by the chlorine substituents. We have recently shown that 1–3 inhibited the ATPase activity of p97, a AAA+ chaperone known for its diverse cellular functions, and that chlorine substitution enhanced specificity for p97 relative to the proteasome.17 Of these, 1 showed only p97 inhibition in our cellular assays, 2 showed some proteasome inhibition, but mainly p97 inhibition, and 3 was capable of inhibiting both p97 and the proteasome to a similar extent. Given this observation and the known relationship between heat-shock induction and the unfolded protein response (UPR),27 the effects of 1–3 on UPR were explored as a potential mechanism of the observed heat-shock induction. Inhibition of p97 or proteasome activities have also been associated with the accumulation of misfolded proteins in the endoplasmic reticulum, promoting the activation of the UPR.28 In order to investigate UPR activation, MDB-MA-231 cells were treated with compounds 1–3 for 8 h (note: assay for cytotoxicity involved treatment for 72 h). The known p97 inhibitor CB-508329 was used as positive control, and the proteasome inhibitor MG13230 was included for comparison purposes. It was found that 10(11)-dehydrocurvularin analogues 1–3 induced activation of the UPR, as assessed by increased C/EBP homologous protein (CHOP) (Figure 4), but to differing extents. Of these, the analogue 1 had an effect on UPR similar to CB-5083 and the analogue 3 had an effect more similar to MG132 (Figure 4). This is most explicitly seen by the increase in the levels of x-box protein 1 (XBP1)31 and activating transcription factor 4 (ATF4)32 for 3 (Figure 4). These data also explain the order of heat-shock response observed for these 10(11)-dehydrocurvularins 1–3 and support the relationship between heat-shock and unfolded protein responses for this class of compounds.
Table 2.
Cytotoxicity Data for Compounds 2 and 3a
| compound | cell lineb
|
||||
|---|---|---|---|---|---|
| NCI-H460 | SF-268 | MCF-7 | PC-3M | MDA-MB-231 | |
| 2 | 1.45 ± 0.09 | 1.41 ± 0.20 | 1.99 ± 0.12 | 1.71 ± 0.06 | 2.95 ± 0.40 |
| 3 | 3.57 ± 0.26 | 4.73 ± 0.33 | 3.98 ± 0.14 | 4.51 ± 0.37 | >5.0 |
| doxorubicin | 0.42 ± 0.30 | 0.62 ± 0.07 | 0.42 ± 0.13 | 0.24 ± 0.09 | 0.58 ± 0.23 |
Results are expressed as IC50 value ± standard deviation in μM. Doxorubicin and DMSO were used as positive and negative controls.
Key: NCI-H460 = human non-small-cell lung cancer; SF-268 = human CNS cancer (glioma); MCF-7 = human breast cancer; PC-3M = metastatic human prostate adenocarcinoma; MDA-MB-231 = metastatic human breast adenocarcinoma.
Figure 3.
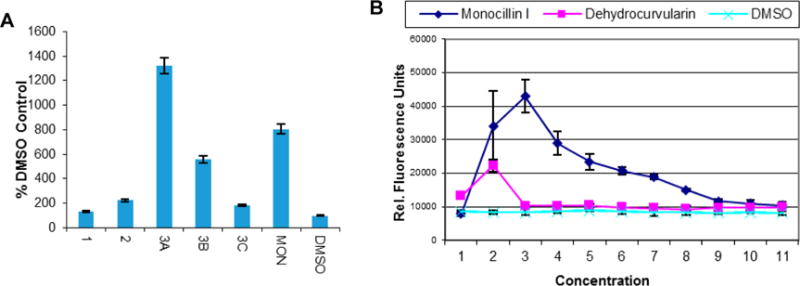
Cell-based heat-shock induction assay data for dehydrocurvularins (1–3), monocillin I (MON, positive control), and DMSO (negative control). (A) Data for 1 and 2 at 5.00 μM, 3 at 5.00 μM (3A), 2.50 μM (3B), and 1.25 μM (3C), and MON at 0.50 μM expressed as a percentage of the negative control (DMSO). (B) Concentration-dependent heat-shock induction for MON and 3 [dehydrocurvularin]; relative fluorescence units per well were determined as a measure of heat-shock reporter activation. For both experiments, the mean and standard deviation of triplicate determinations are presented, and the results are representative of three independent experiments.
Figure 4.
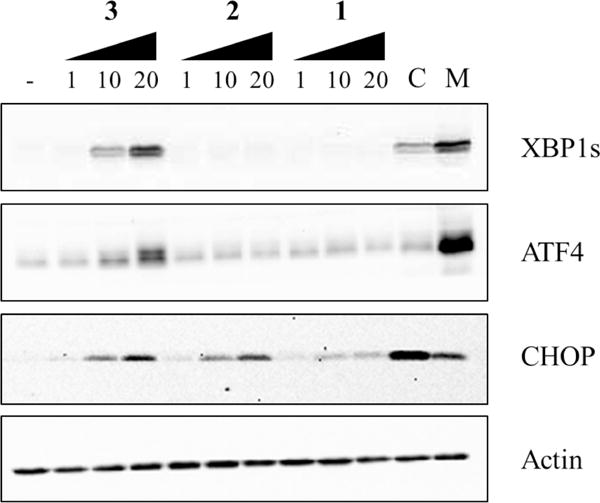
Induction of UPR by dehydrocurvularins 1–3. MDB-MA-231 cells were treated with indicated concentrations (μM) of 1–3 for 8 h. The proteins XBP1, ATF4, and CHOP were analyzed by immunoblot. DMSO and CB-5083 (C; 1.0 μM) were used as negative and positive controls, respectively. The known proteasome inhibitor MG132 (M; 5.0 μM) was included for comparison purposes, and β-actin (Actin) was used as a loading control.
EXPERIMENTAL SECTION
General Experimental Procedures
Optical rotations were measured in MeOH with a JASCO Dip-370 digital polarimeter. UV spectra were recorded on a Shimadzu UV-1601 UV–vis spectrophotometer. IR spectra were recorded on a Shimadzu FTIR-8300 spectrometer using KBr disks. 1D and 2D NMR spectra were recorded in CDCl3, methanol-d4, or acetone-d6 with a Bruker Avance III 400 spectrometer at 400 MHz for 1H NMR and 100 MHz for 13C NMR using residual solvent resonances as internal references. Low-resolution and high-resolution MS were recorded on Shimadzu LCMS-QP8000α and JEOL HX110A spectrometers, respectively. Analytical thin-layer chromatography (TLC) was performed on precoated 0.25 mm thick plates of silica gel 60 F254 for normal and RP-18 F254 S for reversed-phase and spraying with a solution of anisaldehyde in EtOH followed by heating to visualize the spots. Preparative HPLC was performed on a Waters Delta Prep 4000 preparative chromatography system equipped with a Waters 996 photodiode array detector and a Waters Prep LC controller utilizing Empower Pro software and using an RP column (Phenomenex Luna 5 μm, C18, 100 Å, 250 × 10 mm) with a flow rate of 2.0 mL/min; chromatograms were acquired at 254 and 208 nm.
Fungal Isolation and Identification
In June 2008, a healthy individual of Astragalus lentiginosus (spotted locoweed) was collected from a roadside area in central Arizona (34°34′ N, 111°51′ W, 960 m.a.s.l.). Healthy leaves were washed in tap water and cut into ca. 2 mm2 segments, which were surface-sterilized by agitating sequentially in 95% EtOH for 30 s, 0.5% NaOCl for 2 min, and 70% EtOH for 2 min.33 A total of 32 tissue segments were surface-dried under sterile conditions and then placed onto 2% malt extract agar (MEA) in 100 mm Petri plates. Plates were sealed with Parafilm and incubated under ambient light/dark conditions at room temperature (ca. 21.5 °C) for four months. Emergent fungi were isolated into pure culture on 2% MEA, vouchered in sterile water, and deposited as living vouchers at the Robert L. Gilbertson Mycological Herbarium at the University of Arizona. One fungus of interest was used for the present study: isolate AST0039, which has been accessioned at the Robert L. Gilbertson Mycological Herbarium (accession AST0039). AST0039 did not produce reproductive structures in culture and therefore was placed taxonomically using molecular phylogenetic analysis.
Total genomic DNA was isolated from fresh mycelium,33 and the nuclear ribosomal internal transcribed spacers and 5.8s gene (ITS rDNA; ca. 600 base pairs [bp]) and an adjacent portion of the nuclear ribosomal large subunit (LSU rDNA; ca. 500 bp) were amplified as a single fragment by PCR.32 Positive amplicons were sequenced bidirectionally as described previously.33 A consensus sequence was assembled, and basecalls were made by phred34 and phrap35 with orchestration by Mesquite,36 followed by manual editing in Sequencher (Gene Codes Corp.). The sequence was submitted to GenBank under accession KY236016. Two methods were used to tentatively identify isolate AST0039. First, the LSUrDNA portion of the sequence was evaluated using the naïve Bayesian classifier for fungi37 available through the Ribosomal Database Project (http://rdp.cme.msu.edu/). The Bayesian classifier estimated placement within the Pleosporales (Dothideomycetes) with high support, but placement at finer taxonomic levels was not possible. Therefore, we compared the entire sequence against the GenBank database using BLAST.38 The top BLAST matches were to diverse Alternaria. To clarify the phylogenetic placement and taxonomic assignment of AST0039, we downloaded ITS-LSUrDNA sequences for well-vouchered strains from the top 50 BLAST matches. We aligned these data with AST0039 automatically using MUSCLE (http://www.ebi.ac.uk/Tools/msa/muscle/) with default parameters. The alignment was trimmed so that starting and ending points were generally consistent with the sequence length for AST0039, and the alignment was verified by eye prior to analysis.39 The final data set consisted of 14 sequences and 1081 characters. The data set was analyzed using maximum likelihood in PAUP 4.0a14740 followed by a bootstrap analysis with 100 replicates. The analysis unequivocally placed the sequence with strong support within Alternaria. The isolate was placed with strong support in a clade consisting of Alternaria burnsii, A. tenuissima, and A. alternata, which are largely invariant over their lengths. Therefore, this strain was designated as Alternaria sp. AST0039, pending morphological description.
Fungal Culturing, Extraction, and Isolation of Metabolites
A seed culture of Alternaria sp. AST0039 grown in potato dextrose agar for 2 weeks was used for inoculation. Mycelia were scraped out, mixed with sterile water, and filtered through a 100 μM filter to separate spores from the mycelia. UV absorbance of the spore solution was measured (at 600 nm) and adjusted to between 0.3 and 0.5. This spore solution was used to inoculate 5 × 2.0 L Erlenmeyer flasks, each containing 1.0 L of potato dextrose broth (Difco, Plymouth, MN, USA) medium, and incubated in a shaker at 160 rpm and 28 °C for 13 days. The fermentation broth (5.0 L) was filtered, the filtrate was extracted with EtOAc (3 × 5.0 L), and the resulting extract was evaporated under reduced pressure to afford a viscous solid (593.0 mg), which was applied to a column of silica gel (20.0 g) equilibrated with hexanes. The column was eluted with 100 mL each of hexanes–CH2Cl2 (70:30), CH2Cl2 (100%), and CH2Cl2–MeOH (98:2) and then with 50 mL each of CH2Cl2–MeOH (97:3), CH2Cl2–MeOH (96:4), CH2Cl2–MeOH (95:5), CH2Cl2–MeOH (94:6), CH2Cl2–MeOH (93:7), CH2Cl2–MeOH (92:8), CH2Cl2–MeOH (91:9), and CH2Cl2–MeOH (90:10). Ninety-five fractions (8.0 mL each) were collected and combined based on their TLC profiles to yield 11 combined fractions [A (11.3 mg), B (1.4 mg), C (47.8 mg), D (225.8 mg), E (7.6 mg), F (16.8 mg), G (18.9 mg), H (27.8 mg), I (8.0 mg), J (218.0 mg), K (5.6 mg)]. Fraction C (47.8 mg) was further separated using reversed-phase HPLC with a solvent gradient of 20% to 100% acetonitrile in water over 100 min (flow rate 2 mL/min) to afford 3 (8.1 mg, tR = 42.1), 1 (3.5 mg, tR = 46.2), and 2 (25.4 mg, tR = 48.9). Fraction D contained 3 as the major constituent. Fraction G (18.9 mg) was further purified using reversed-phase HPLC with a solvent gradient of 20% to 100% acetonitrile in water over 40 min (flow rate 2 mL/min) to yield α-acetylorcinol (7.0 mg, tR = 13.8). Further purification of the fraction H (27.8 mg) using reversed-phase HPLC with a solvent gradient of 20% to 100% acetonitrile in water over 40 min (flow rate 2 mL/min) afforded an additional quantity of α-acetylorcinol (3.0 mg, tR = 13.5), scytalone (3.0 mg, tR = 15.3), and alterperylenepoxide A (4) (2.8 mg, tR = 21.5).
(−)-(10E,15S)-4,6-Dichloro-10(11)-dehydrocurvularin (1): white, amorphous solid; −47 (c 0.18, CH3OH); UV (CH3OH) λmax (log ε) 363.5 (3.89), 208.0 (4.68) nm; IR (KBr) νmax 3421, 2929, 2858, 1724, 1637, 1587, 1434, 1379, 1267, 1263, 1184, 1082, 1008, 952, 852, 765 cm−1; for 1H and 13C NMR data, see Table 1; LRMS m/z 359 (M + 1)+, 361 (M + 3)+, and 363 (M + 5)+ in the ratio of 9:6:1; HRESIMS m/z 357.0297 [M – H]− (calcd for C16H15Cl2O5, 357.0302).
Alterperylenepoxide A (4): yellow, amorphous solid; (c 0.28, CH3OH); UV (CH3OH) λmax (log ε) 291.0 (4.44), 259.0 (4.47), 218.0 (4.58) nm; IR (KBr) νmax 3419, 2923, 2856, 1639, 1600, 1460, 1332, 1232, 1184, 1056, 1028, 993, 952, 823, 785, 615 cm−1; 1H NMR (methanol-d4, 400 MHz) δ 8.04 (1H, d, J = 8.8 Hz, H-1), 7.59 (1H, d, J = 8.5 Hz, H-12), 7.01 (1H, d, J = 8.8 Hz, H-2), 6.85 (1H, d, J = 8.5 Hz, H-11), 5.25 (1H, brs, H-9), 3.87 (1H, d, J = 4.0 Hz, H-7), 3.55 (1H, brd, J = 4.0 Hz, H-8), 3.22 (1H, brs, H-6b), 3.18 (1H, ddd, J = 17.8, 13.5, 5.0 Hz, H-5β), 2.82 (1H, ddd, J = 13.5, 5.0, 2.6 Hz, H-6β), 2.73 (1H, ddd, J = 17.8, 4.0, 2.6 Hz, H-5α), 2.40 (1H, ddd, J = 13.5, 13.5, 4.0 Hz, H-6α); 13C NMR (methanol-d4, 100 MHz) δ 206.8 (C, C-4), 163.1 (C, C-3), 158.0 (C, C-10), 141.0 (C, C-12c), 134.1 (CH, C-1), 128.5 (C, C-9b), 126.0 (C, C-12b), 125.8 (C, C-12a), 125.7 (CH, C-12), 123.4 (C, C-9a), 119.7 (CH, C-2), 115.4 (CH, C-11), 115.2 (C, C-3a), 69.2 (C, C-6a), 62.1 (CH, C-9), 56.9 (CH, C-8), 51.3 (CH, C-7), 46.2 (CH, C-6b), 34.5 (CH2, C-5), 34.4 (CH2, C-6); HRESIMS m/z 351.0879 [M – H]− (calcd for C20H15O6, 351.0874).
Cytotoxicity Assay
Relative cell growth and survival were measured in 96-well microplate format using fluorescence detection of resazurin (AlamarBlue)26 dye reduction as the end point. Compounds were tested against human non-small-cell lung cancer (NCI-H460), CNS glioma (SF-268), breast cancer (MCF-7), metastatic prostate adenocarcinoma (PC-3M), and human metastatic breast adenocarcinoma (MDA-MB-231). Serial dilutions of the test sample or vehicle control (DMSO) were added to triplicate wells and incubated at 37 °C. After 72 h the dye solution was added to each well (1:10 dilution), agitated briefly, and incubated for an additional 4 h at 37 °C before data acquisition using a microplate fluorometer (Ex/Em: 560/590). Mean fluorescence intensity per well as a measure of relative viable cell number in compound-treated wells was compared to that of the DMSO-treated wells. The conventional chemotherapeutic drug doxorubicin served as the positive control.
Heat-Shock Induction Assay
Mouse fibroblasts stably transfected with a reporter construct encoding enhanced green fluorescent protein under the transcriptional control of a minimal consensus heat-shock element were used to measure the heat-shock-inducing activity of extract and pure compounds as previously described.3a Reporter cells were seeded into flat-bottomed 96-well plates (Falcon; Becton Dickinson, Lincoln Park, NJ, USA) at a density of 20 000 cells/well and allowed to attach overnight (one column of wells was left empty to serve as a blank control). On the following day, serial 2-fold dilutions of pure compounds or single dilutions of extracts or fractions were added in triplicate to cell-containing wells. DMSO vehicle alone (volume not to exceed 0.1%) served as a negative control. Cells were incubated overnight, the medium was removed, and wells were rinsed once with phosphate-buffered saline (PBS), followed by addition of 150 μL of PBS to each well. Fluorescence was determined on an Analyst AD (LJL Biosystems) plate reader equipped with filter sets for excitation at 485 nm and emission at 525 nm. Mean florescence and standard deviations of triplicate determinations were calculated and plotted.
Western Blot Analysis
MDB-MA-231 cells were cultured with the ascribed compound in Dulbecco’s modified Eagle medium supplemented with 10% fetal bovine serum for 8 h in 1.9 cm2 culture dishes (Greiner Bio-One). The medium was removed, and the cells were harvested in sample buffer (50 mM Tris-HCl pH 6.8, 2% sodium dodecyl sulfate (SDS), 10% glycerol, 100 mM dithiothreitol, 0.1% bromophenol blue) and lysed via sonication followed by clearance of the cell debris by centrifugation. These were then applied to 4–20% gradient SDS PAGE gels (Invitrogen) and transferred to Nitrobind Nitrocellulose transfer membrane (Maine Manufacturing) using a Bolt Miniblot Module gel box (Life Technologies). The blots were blocked in 5% milk for 1 h. Primary antibodies were applied in 5% milk at 1:1000 anti-XBP 1s (Cell Signaling Technologies), 1:1000 anti-ATF4 (Cell Signaling Technologies), 1:1000 anti-CHOP (Cell Signaling Technologies), or 1:1000 anti-actin (Santa Cruz). The blots were washed three times at 10 min intervals with wash buffer (1× PBS, 0.1% Tween 20). Secondary antibodies were applied in 5% milk at 1:3000 goat anti-mouse (Santa Cruz) or 1:3000 goat anti-rabbit (Santa Cruz). The blots were washed three times at 10 min intervals with wash buffer, incubated in Supersignal West Pico, Dura, or Femto Substrate (Thermo Scientific), and imaged using ChemiDoc XRS (Bio-Rad) and were analyzed using Quantity One 1-D Analysis software (Bio-Rad).
Acknowledgments
Financial support for this work was provided by Grant R01 CA090265 funded by the National Cancer Institute (NCI, NIH) and Grant P41 GM094060 funded by National Institute of General Medical Sciences (NIGMS, NIH). We thank Drs. L. Whitesell and S. Santagata of Whitehead Institute for some heat-shock induction assay data and useful discussions, Prof. M. C. F. Oliveira of Universidade Federal do Ceará, Brazil, for some HRMS data, and Ms. P. Espinosa-Artiles and Ms. M. X. Liu for their help with large-scale culturing of the fungus and for conducting heat-shock induction and cytotoxicity assays.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jnatprod.6b00960.
Details of phylogenetic majority consensus tree indicating placement of Alternaria sp. AST0039; 1H and 13C NMR spectra of 1–4; 2D NMR (1H–1H COSY, HSQC, and HMBC) spectra of 1 and 4; NOEDIFF and CD spectra of 4 (PDF)
ORCID
A. A. Leslie Gunatilaka: 0000-0001-9663-3600
Notes
The authors declare no competing financial interest.
References
- 1.(a) Gunatilaka AAL. J Nat Prod. 2006;69:509–526. doi: 10.1021/np058128n. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kusari S, Spiteller M. Nat Prod Rep. 2011;28:1203–1207. doi: 10.1039/c1np00030f. [DOI] [PubMed] [Google Scholar]
- 2.(a) Dai C, Whitesell L, Rogers AB, Lindquist S. Cell. 2007;130:1005–1018. doi: 10.1016/j.cell.2007.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Santagata S, Xu Y, Wijeratne EMK, Kontnik R, Rooney C, Perley CC, Kwon H, Clardy J, Kesari S, Whitesell L, Lindquist S, Gunatilaka AAL. ACS Chem Biol. 2012;7:340–349. doi: 10.1021/cb200353m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.(a) Turbyville TJ, Wijeratne EMK, Liu MX, Burns AM, Seliga CJ, Luevano LA, David CL, Feath SH, Whitesell L, Gunatilaka AAL. J Nat Prod. 2006;69:178–184. doi: 10.1021/np058095b. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) McLellan CA, Turbyville TJ, Wijeratne EMK, Kerschen A, Vierling E, Queitsch C, Whitesell L, Gunatilaka AAL. Plant Physiol. 2007;145:174–182. doi: 10.1104/pp.107.101808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.(a) Findlay JA, Kwan D. Can J Chem. 1973;51:1617–1619. [Google Scholar]; (b) Jordan DB, Zheng YJ, Lockett BA, Basarab GS. Biochemistry. 2000;39:2276–2282. doi: 10.1021/bi991839y. [DOI] [PubMed] [Google Scholar]
- 5.Nukina M, Marumo S. Agric Biol Chem. 1977;41:717. [Google Scholar]
- 6.(a) Munro HD, Musgrave OC, Templeton R. J Chem Soc C. 1967;0:947–948. [Google Scholar]; (b) Starratt AN, White GA. Phytochemistry. 1968;7:1883–1884. [Google Scholar]; (c) Hyeon SB, Ozaki A, Suzuki A, Tamura S. Agric Biol Chem. 1976;40:1663–1664. [Google Scholar]; (d) Caputo O, Viola F. Planta Med. 1977;31:31–32. doi: 10.1055/s-0028-1097485. [DOI] [PubMed] [Google Scholar]; (e) Lai S, Shizuri Y, Yamamura S, Kawai K, Terada Y, Furukawa H. Tetrahedron Lett. 1989;30:2241–2244. [Google Scholar]; (f) Arai K, Rawlings BJ, Yoshizawa Y, Vederas JC. J Am Chem Soc. 1989;111:3391–3399. [Google Scholar]; (g) Lai S, Shizuri Y, Yamamura S, Kawai K, Furukawa H. Bull Chem Soc Jpn. 1991;64:1048–1050. [Google Scholar]; (h) Gutierrez M, Theoduloz C, Rodriguez J, Lolas M, Schmeda-Hirschmann G. J Agric Food Chem. 2005;53:7701–7708. doi: 10.1021/jf051021l. [DOI] [PubMed] [Google Scholar]
- 7.(a) Greve H, Schupp PJ, Eguereva E, Kehraus S, Kelter G, Maier A, Fiebig H-H, König GM. Eur J Org Chem. 2008;2008:5085–5092. doi: 10.1002/ejoc.200800522. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Xie LW, Ouyang YC, Zou K, Wang GH, Chen MJ, Sun HM, Dai SK, Li X. Appl Biochem Biotechnol. 2009;159:284–293. doi: 10.1007/s12010-009-8591-2. [DOI] [PubMed] [Google Scholar]
- 8.Ghisalberti EL, Rowland CY. J Nat Prod. 1993;56:2175–2177. doi: 10.1021/np50100a020. [DOI] [PubMed] [Google Scholar]
- 9.(a) He J, Wijeratne EMK, Bashyal BP, Zhan J, Seliga CJ, Liu M, Pierson EE, Pierson LS, VanEtten HD, Gunatilaka AAL. J Nat Prod. 2004;67:1985–1991. doi: 10.1021/np040139d. [DOI] [PubMed] [Google Scholar]; (b) Aly AH, Debbab A, Edrada ER, Muller WEG, Kubbutat MHG, Wray V, Ebel R, Proksch P. Mycosphere. 2010;1:153–162. [Google Scholar]
- 10.(a) Robeson DJ, Strobel GA. J Biosci. 1981;36:1081–1083. [Google Scholar]; (b) Jiang SJ, Qiang S, Zhu YZ, Dong YF. Ann Appl Biol. 2008;152:103–111. [Google Scholar]
- 11.Kusano M, Nakagami KJ, Fujoka S, Kawano T, Shimada A, Kimura Y. Biosci, Biotechnol, Biochem. 2003;67:1413–1416. doi: 10.1271/bbb.67.1413. [DOI] [PubMed] [Google Scholar]
- 12.De Souza AO, Galetti FCS, Silva CL, Bicalho B, Parma MM, Fonseca SF, Marsaioli AJ, Trindade ACLB, Gil RPF, Bezerra FS, Neto MA, De Oliveira MCF. Quim Nova. 2007;30:1563–1566. [Google Scholar]
- 13.Aly AH, Debbab A, Clements C, Edrada-Ebel R, Orlikova B, Diederich M, Wray V, Lin W, Proksch P. Bioorg Med Chem. 2011;19:414–421. doi: 10.1016/j.bmc.2010.11.012. [DOI] [PubMed] [Google Scholar]
- 14.Kobayashi A, Hino T, Yata S, Itoh TJ, Sato H, Kawazu K. Agric Biol Chem. 1988;52:3119–3123. [Google Scholar]
- 15.Ghisalberti EL, Hockless DCR, Rowland CY, White AH. Aust J Chem. 1993;46:571–575. [Google Scholar]
- 16.Almassi F, Ghisalberti EL, Skelton BW, White AH. Aust J Chem. 1994;47:1193–1197. [Google Scholar]
- 17.Tillotson J, Bashyal BP, Kang MJ, De La Cruz F, Gunatilaka AAL, Chapman E. Org Biomol Chem. 2016;14:5918–5921. doi: 10.1039/c6ob00560h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bashyal BP, Wellensiek BP, Ramakrishnan R, Faeth SH, Ahmad N, Gunatilaka AAL. Bioorg Med Chem. 2014;22:6112–6116. doi: 10.1016/j.bmc.2014.08.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Idris A, Tantry MA, Ganai BA, Kamili AN. Phytochem Lett. 2015;11:264–269. [Google Scholar]
- 20.Kurz M, Herrmann M, Toti L, Vertesy L. 045905. Worldwide Patent. 2003
- 21.Bouchonnet S. Introduction to GC-MS Coupling. CRC Press; Boca Raton: 2013. p. 193. [Google Scholar]
- 22.Gerlach H. Helv Chim Acta. 1977;60:3039–3044. [Google Scholar]
- 23.(a) Stack ME, Mazzola EP, Page SW, Pohland AE. J Nat Prod. 1986;49:866–871. doi: 10.1021/np50047a017. [DOI] [PubMed] [Google Scholar]; (b) Arnone A, Nasini G, Merlini L, Assante G. J Chem Soc, Perkin Trans. 1986;1:525–530. [Google Scholar]; (c) Fleck SC, Burkhardt B, Pfeiffer E, Metzler M. Toxicol Lett. 2012;214:27–32. doi: 10.1016/j.toxlet.2012.08.003. [DOI] [PubMed] [Google Scholar]
- 24.Podlech J, Fleck SC, Metzler M, Bürck J, Ulrich AS. Chem - Eur J. 2014;20:11463–11470. doi: 10.1002/chem.201402567. [DOI] [PubMed] [Google Scholar]
- 25.(a) Wagoner RMV, Satake M, Bourdelais AJ, Baden DG, Wright JLC. J Nat Prod. 2010;73:1177–1179. doi: 10.1021/np100159j. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Wang X-N, Bashyal BP, Wijeratne EMK, U’Ren JM, Liu MX, Gunatilaka MK, Arnold AE, Gunatilaka AAL. J Nat Prod. 2011;74:2052–2061. doi: 10.1021/np2000864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wijeratne EMK, Bashyal BP, Liu MX, Rocha DD, Gunaherath GMKB, U’Ren JM, Gunatilaka MK, Arnold AE, Whitesell L, Gunatilaka AAL. J Nat Prod. 2012;75:361–369. doi: 10.1021/np200769q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.(a) Liu Y, Chang A. EMBO J. 2008;27:1049–1059. doi: 10.1038/emboj.2008.42. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Hou J, Tang H, Liu Z, Österlund T, Nielsen J, Petranovic D. FEMS Yeast Res. 2014;14:481–494. doi: 10.1111/1567-1364.12125. [DOI] [PubMed] [Google Scholar]; (c) Weindling E, Bar-Nun S. Biochem Biophys Res Commun. 2015;457:473–478. doi: 10.1016/j.bbrc.2015.01.021. [DOI] [PubMed] [Google Scholar]
- 28.Wójcik C, Rowicka M, Kudlicki A, Nowis D, McConnell E, Kujawa M, DeMartino GN. Mol Biol Cell. 2006;17:4606–4618. doi: 10.1091/mbc.E06-05-0432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Anderson DJ, Le Moigne R, Djakovic S, Kumar B, Rice J, Wong S, Wang J, Yao B, Valle E, Kiss, von Soly S, Madriaga A, Soriano F, Menon MK, Wu ZY, Kampmann M, Chen Y, Weissman JS, Aftab BT, Yakes FM, Shawver L, Zhou HJ, Wustrow D, Rolfe M. Cancer Cell. 2015;28:653–665. doi: 10.1016/j.ccell.2015.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Goldberg AL. J Cell Biol. 2012;199:583–588. doi: 10.1083/jcb.201210077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K. Cell. 2001;107:881–891. doi: 10.1016/s0092-8674(01)00611-0. [DOI] [PubMed] [Google Scholar]
- 32.Han J, Back SH, Hur J, Lin YH, Gildersleeve R, Shan J, Yuan CL, Krokowski D, Wang S, Hatzoglou M, Kilberg MS, Sartor MA, Kaufman RJ. Nat Cell Biol. 2013;15:481–491. doi: 10.1038/ncb2738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.U’Ren JM, Lutzoni F, Miadlikowska J, Laetsch AD, Arnold AE. Am J Bot. 2012;99:898–914. doi: 10.3732/ajb.1100459. [DOI] [PubMed] [Google Scholar]
- 34.Ewing B, Green P. Genome Res. 1998;8:186–194. [PubMed] [Google Scholar]
- 35.Ewing B, Hillier L, Wendl MC, Green P. Genome Res. 1998;8:175–185. doi: 10.1101/gr.8.3.175. [DOI] [PubMed] [Google Scholar]
- 36.Maddison WP, Maddison DR. Mesquite. 2011 www.mesquiteproject.org.
- 37.Liu KL, Porras-Alfaro A, Kuske CR, Eichorst SA, Xie G. Appl Environ Microbiol. 2012;78:1523–1533. doi: 10.1128/AEM.06826-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 39.Maddison DR, Maddison WP. MacClade v 4.08a. 2005 http://macclade.org.
- 40.Swofford DL. Phylogenetic Analysis Using Parsimony (*and other methods), Version 4. Sinauer Associates; Sunderland, MA: 2002. [Google Scholar]