

Abstract
Background
An inflatable lunar/Mars analog habitat (ILMAH), simulated closed system isolated by HEPA filtration, mimics International Space Station (ISS) conditions and future human habitation on other planets except for the exchange of air between outdoor and indoor environments. The ILMAH was primarily commissioned to measure physiological, psychological, and immunological characteristics of human inhabiting in isolation, but it was also available for other studies such as examining its microbiological aspects. Characterizing and understanding possible changes and succession of fungal species is of high importance since fungi are not only hazardous to inhabitants but also deteriorate the habitats. Observing the mycobiome changes in the presence of human will enable developing appropriate countermeasures with reference to crew health in a future closed habitat.
Results
Succession of fungi was characterized utilizing both traditional and state-of-the-art molecular techniques during the 30-day human occupation of the ILMAH. Surface samples were collected at various time points and locations to observe both the total and viable fungal populations of common environmental and opportunistic pathogenic species. To estimate the cultivable fungal population, potato dextrose agar plate counts method was utilized. The internal transcribed spacer region-based iTag Illumina sequencing was employed to measure the community structure and fluctuation of the mycobiome over time in various locations. Treatment of samples with propidium monoazide (PMA; a DNA intercalating dye for selective detection of viable microbial populations) had a significant effect on the microbial diversity compared to non-PMA-treated samples. Statistical analysis confirmed that viable fungal community structure changed (increase in diversity and decrease in fungal burden) over the occupation time. Samples collected at day 20 showed distinct fungal profiles from samples collected at any other time point (before or after). Viable fungal families like Davidiellaceae, Teratosphaeriaceae, Pleosporales, and Pleosporaceae were shown to increase during the occupation time.
Conclusions
The results of this study revealed that the overall fungal diversity in the closed habitat changed during human presence; therefore, it is crucial to properly maintain a closed habitat to preserve it from deteriorating and keep it safe for its inhabitants. Differences in community profiles were observed when statistically treated, especially of the mycobiome of samples collected at day 20. On a genus level Epiccocum, Alternaria, Pleosporales, Davidiella, and Cryptococcus showed increased abundance over the occupation time.
Electronic supplementary material
The online version of this article (doi:10.1186/s40168-017-0280-8) contains supplementary material, which is available to authorized users.
Keywords: Closed habitat, Surface, Mycobiome, Succession
Background
Planning future space explorations, involving potential human missions to Mars, would require constructing a safe closed habitat [1, 2]. An inflatable lunar/Mars analog habitat (ILMAH) is a unique, simulated closed environment (isolated by HEPA filtration) that can be utilized to overcome challenges associated with both technical and scientific issues [3]. Because the ILMAH mimics International Space Station (ISS) conditions and is treated as a prototype habitat for future space explorations, microbiological characteristics of such a closed environment is of high interest to the National Aeronautics and Space Administration (NASA). The environmentally controlled ILMAH is an easily accessible system that enables samples to be collected and analyzed at multiple times at relatively low cost. Understanding the microbiome of a closed system and its association with human inhabitation will help to assess the correlation between human health and microbiome of the habitat as well as the influence of microorganism on the habitat deterioration [4–6].
The highly specialized structure of the simulated ILMAH keeps its inhabitants in isolation from the outside environment. Except for the exchange of the air between outdoor and indoor environments via an advanced environmental control system, the ILMAH mimics the ISS and other future habitats of human explorers on the other planets [3]. This unique feature of the ILMAH allows observing the changes in the microbiome during human occupation. The bacteriome of the ILMAH was recently reported [7], as in the case of most of the studies reporting on bacterial microbiomes [8]. The molecular fungal diversity of Japanese Experimental Module—Kibo, on the ISS, revealed abundance of fungi associated with astronauts, but succession of viable fungal population in their habitat was not addressed [9]. The skin fungal microbiota of 10 Japanese astronauts showed temporal changes before, during, and after their stay on the ISS. The molecular fungal diversity associated with various body parts was reduced during the spaceflight when compared to pre-flight data. However, the ratio of Malassezia genetic signatures to all fungal gene copies (including dead fungal cells) increased during their stay at the ISS—but the viability of these fungi was not confirmed [10]. This is the first report that thoroughly characterizes the mycobiome of a simulated habitat meant for the future human habitats on other planets.
Utilization of next generation sequencing (NGS) techniques enables more in-depth analysis of indoor microbiomes [11]. Many studies focus on the bacterial microbiome of intensive care units [8, 12–14], pharmaceutical clean rooms [15–17], or tissue banks [18] since their microbial composition has an impact on human health and life. Nosocomial infections acquired in hospitals and other health care facilities remain the sixth leading cause of death in the hospitals in USA [19, 20]. Nosocomial infections are mostly caused by various fungal species that belong to the Candida genus and filamentous fungus Aspergillus fumigatus [21–23]. Therefore, it remains important to screen future closed habitats for the presence of opportunistic pathogens that can affect health of immunocompromised astronauts. So far, majority of the indoor microbiome studies have focused on the bacterial microbiome without analyzing the mycobiome. In addition, those few studies that characterized fungi of indoor environments focused on culture-based populations [24–27]. In those cases, where new molecular techniques were implemented [28–31] viable fungi were not differentiated from the total population (viable and dead) [32]. The internal transcribed spacer region-based iTag Illumina sequencing coupled with the propidium monoazide (PMA) treatment used in this study can determine the viable mycobiome.
Fungi are extremophiles that can survive harsh conditions such as low nutrient [33], desiccation [34], high/low temperatures [35, 36], acidic/alkaline [37, 38], radiation [39, 40], and other environments [41, 42]. Fungal species not only have been isolated from all known environments on Earth, including barren lands like deserts, caves, or nuclear accident sites, but also are known to be difficult to eradicate from other types of environments including indoor and closed spaces [8, 36, 42, 43].
Characterizing and understanding possible changes to, and succession of, fungal species in the ILMAH is of high importance since some of the fungi are extremophiles that are not only potentially hazardous to inhabitants but also can deteriorate the habitat itself [25, 44, 45]. It was previously reported that people spending a significant amount of time indoors might suffer from so called “sick building syndrome” (SBS). SBS is characterized by health- and comfort-related syndromes (e.g., headache, tiredness) that ease after leaving a building. Fatigue and discomfort might be caused not only by physical characteristics of the closed system (humidity, temperature, lighting) but also by biological contamination from both bacteria and fungi [46, 47]. Fungal pathogens’ presence in indoor areas might pose health hazards for people exhibiting immunodeficiency [48]. Pathogenic fungi produce a range of secondary metabolites (SMs) that influence their virulence (e.g., melanins, siderophores, or species-specific toxins), induce allergies, and cause diseases (e.g., aspergillosis, candidiasis, or cryptococcosis) [48, 49]. Prolonged stays in closed habitats (e.g., ILMAH, ISS, etc.) might be stressful for inhabitants and lead to a decrease in immune response; therefore, assessing the presence of any opportunistic pathogens is vital [50].
Previous reports on the mycobiome in NASA clean rooms and on the ISS documented NGS results from samples collected from various locations, but none of the studies focused on the analysis of the microbial succession of systematically collected samples [28, 32]. The bacterial and archaeal microbiome succession of the ILMAH using NGS has been carried out [7]. This is the first report characterizing the succession of fungi in a simulated closed system meant for human habitation on other planets utilizing both traditional and state-of-the-art molecular techniques. In addition, attempts were made during this study to elucidate the temporal and spatial distribution of the fungal population and diversity in a closed human habitat.
Methods
The ILMAH habitat
The physical characteristics of the ILMAH, along with detailed sampling procedures and periodicity, were previously described [7]. In brief, the ILMAH is located in Grand Forks, ND (47.9222 N, 97.0734 W) and has dimensions of 12 m by 10 m by 2.5 m. It contains a sleeping compartment, kitchen, toilet, and laboratory area. The ILMAH has its own ventilation system that pressurizes the habitat, provides breathing air for the crew, and removes unwanted material from the air stream. The ILMAH uses a blower that takes ambient air for pressurization and breathing air provision. It provides a positive pressure at 1 PSID (pressure differential) above normal atmosphere inside the habitat. The ambient air is pressurized by an industrial fan and sent to a standard HEPA flat panel particulate filter (Bryant GAPBBCAR2025, Honeywell, Morris Plains, NJ). This filter is replaced before each mission lasting up to 30 days. Maintenance personnel changes the filter from the outside. Since the humidity inside the habitat is not removed, water vapor during the months when the analog missions takes place range from 35 to 55%.
Three student crews inhabited the ILMAH for 30 days. During that period of time, there was no exchange between the interior and exterior environment except pumping the air filtered via ILMAH’s advanced controlling system [3]. In addition, students did not leave the ILMAH at any time for 30 days, and nothing came in or out, including food, people, water, or any supplies, except filtered air. Surface samples were collected consecutively during four sampling events (day 0 [T0], day 13 [T13], day 20 [T20], and day 30 [T30]) from eight designated sampling locations. Prior to inhabitation, the ILMAH surfaces were cleaned with 10% bleach and during the experiment period, it was cleaned weekly with antibacterial wipes.
Sampling materials and procedure
Samples were collected using biological sampling kits (BiSKits, Quicksilver Analytics Inc., Abingdon, MD) previously documented as an efficient sampling device for surfaces [51]. All the BiSKits were prepared following the procedure described elsewhere [7, 51]. Briefly, the sterile phosphate buffer saline (PBS) provided by the distributor was discarded from the bottle and replaced with 15 mL of sterile UltraPure DNA free PBS (MoBio Laboratories Inc, Carlsbad, CA). Each BiSKit was rinsed once with PBS that was later collected into a sterile 15-mL falcon tube and kept at 4 °C as a background measure for biological materials associated with macrofoam (sampling device control). This precautionary step was required to overcome, if encountered, microbial contamination associated with sampling devices and other processing reagents. BiSKits prepared as described above were then packed into sterile zip lock bags and shipped at 4 °C to the University of North Dakota where they were kept at 4 °C till the experiment was carried out (within 2 to 3 days).
The ILMAH architecture was previously described [7] (see Additional file 1: Figure SF1, Fig. 1). Surface samples were collected from eight locations at four consecutive samplings: day 0 (prior to inhabitation), day 13, day 20, and day 30 during the inhabitation and right before ending the experiment. Originally, sampling activities were scheduled at regular intervals days 0, 10, 20, and 30 of human occupation; however, day 10 sampling scheduled on Thursday was delayed to day 13 (Sunday) to avoid risk related to shipping the samples over the weekend. The work schedule of the ILMAH crew members was regulated and is detailed in Additional file 2. Each location (surface area 1 m2) was sampled with one BiSKit in three directions following the same steps, horizontally from the left to the right, vertically from the bottom to the top, and diagonally from the right bottom corner to the left upper corner. After sampling, the sampled BiSKit device was extracted with sterile PBS and sampling fluids were collected in sterile 50-mL falcon tubes. Each BiSKit was washed and extracted twice with the sterile PBS giving approximately 45 mL of unconcentrated sample. Likewise, the BiSKit left open in the sampling area for the time necessary for sampling one location was treated as a field control; the unopened BiSKit was treated as a sampling device (BiSKit) control. Collected samples along with the controls were stored at 4 °C and sent overnight to JPL via cold shipping for further analysis. Sample processing for the microbiological analyses was started within 24 h from the sample collection.
Fig. 1.

Picture of the closed habitat from outside
Sample processing
The extruded liquid samples from the BiSKit sampler (~45 mL per sample) were concentrated using an InnovaPrep Concentrating Pipette (Innova Prep LLC, Drexel, MO) to a final volume of ~4 mL. Appropriate aliquots of concentrated samples were further used for cultivation (200 μL) and molecular analyses (3 mL).
-
Cultivable fungal burden and diversity
For cultivation assay, samples were serially diluted by 10 and 100 times. One hundred microliters of each dilution was pour plated in duplicates on potato dextrose agar (PDA) and grown at room temperature (~25 °C). Colony-forming units (CFUs) were counted after 7 days of incubation, and cultivable fungal population was calculated per square meter of the sampling area. Simultaneously, up to 5 colonies exhibiting different morphologies were picked and stored as stab cultures in one-tenth semi-solid PDA medium. Cultivable isolates were identified using primers ITS 1F (5′-CTT GGT CAT TTA GAG GAA GTA A-3′) and Tw13 (5′-GGT CCG TGT TTC AAG ACG-3′) that target small and large subunit rRNA gene-coding regions of the small and large ribosomal subunit, including the internal transcribed spacers ITS1 and ITS2 [52, 53]. DNA was extracted using freezing (−80 °C) and thawing (+80 °C) cycle of fungal suspension in PBS for 15 min. This process was repeated 3 times. In some cases when DNA was not extracted by the freezing-thawing method, a PowerSoil® DNA Isolation Kit (MoBio) was used according to manufacturer’s instructions. PCR conditions were as follows: 95 °C for 3 min followed by 25 cycles of 95 °C for 30 s, 58 °C for 30 s, 72 °C for 2 min, and final elongation at 72 °C for 10 min. Amplified products were visualized by gel electrophoresis. PCR products were then enzymatically purified by using 40 IU of Exonuclease I (E. coli 20,000 IU/mL New England BioLabs, Inc. Ipswich, MA) and 8 IU of Antarctic Phosphatase (5,000 IU/mL, New England BioLabs, Inc.) per 20 μL amplification product. Heat reactions were carried out in a thermocycler as follows: 37 °C for 30 min, 80 °C for 15 min. Traditional Sanger sequencing was performed at Macrogen (Rockville, MD). The sequences were merged using DNAStar (Madison, WI), identified using UNITE fungal database [54], and aligned using ClustalW. A phylogenetic tree was constructed using MEGA6.06-mac applying neighbor-joining method [55]. Sequences from one representative of each strain and corresponding type strain were used to create the phylogenetic tree.
-
DNA extraction
The concentrated environmental samples (3 mL) were split into two equal parts. One half of the sample (1.5 mL) was treated with 12.5 μL of 2 mM PMA dye (Biotum, Inc., Hayward, CA), and the other half was left untreated. The final concentration of PMA in each treated sample was 25 μM. After PMA addition, both treated and untreated samples were kept in the dark for 5 min at room temperature and subsequently exposed to light in the PHaST Blue-Photo activation system for tubes (GenIUL, S.L, Terrassa, Spain) for 15 min. PMA-treated samples represent viable microorganisms whereas the non-PMA-treated samples represent the total number of viable and dead microorganisms [56]. After photo activation, each sample was split into two aliquots of 0.75 mL each. One aliquot of each sample was subjected to bead beating for 60 s at 5 m/s on the Fastprep-24 bead-beating instrument (MP Biomedicals, Santa Ana, CA). The solution after bead beating was combined with not bead-beated aliquots (1.5 mL), and then used for DNA extraction using the Maxwell-16 MDx automated system following manufacturer’s instructions (Promega, Madison, WI). Purified DNA was eluted into a final volume of 50 μL of Ultra Pure molecular water and divided into 4 aliquots that were stored at −80 °C.
-
Molecular fungal community analysis using Illumina sequencing
To determine fungal populations, a two-step amplification process was applied prior to MiSeq Illumina sequencing at Research and Testing Laboratory (RTL, Lubbock, TX). The forward primer was constructed with the Illumina i5 sequencing primer (5′-TCG TCG GCA GCG TCA GAT GTG TAT AAG AGA CAG-3′) and the ITS1F primer (5′-CTT GGT CAT TTA GAG GAA GTA A-3′) [57]. The reverse primer was constructed with the Illumina i7 sequencing primer (5′-GTC TCG TGG GCT CGG AGA TGT GTA TAA GAG ACA G-3′) and the ITS2aR primer (5′-GCT GCG TTC TTC ATC GAT GC-3′) [58]. Amplifications were performed in 25 μL reactions with Qiagen HotStar Taq master mix (Qiagen Inc, Valencia, CA), 1 μL of each 5 μM primer, and 1 μL of template. Reactions were performed on ABI Veriti thermocyclers (Applied Biosytems, Carlsbad, CA) under the following thermal profile: 95 °C for 5 min, then 25 cycles of 94 °C for 30 s, 54 °C for 40 s, 72 °C for 1 min, followed by one cycle of 72 °C for 10 min and 4 °C hold.
Products from the first stage amplification were added to a second PCR based on qualitatively determined concentrations. Primers for the second PCR were designed based on the Illumina Nextera PCR primers as follows: Forward –AAT GAT ACG GCG ACC ACC GAG ATC TAC AC [i5index] TCG TCG GCA GCG TC and Reverse – CAA GCA GAA GAC GGC ATA CGA GAT [i7index] GTC TCG TGG GCT CGG. The second stage amplification was run the same as the first stage except for 10 cycles.
Amplification products were visualized with eGels (Life Technologies, Grand Island, NY). Products were then pooled equimolar, and each pool was size selected in two rounds using Agencourt AMPure XP (BeckmanCoulter, Indianapolis, IN) in a 0.7 ratio for both rounds. Size-selected pools were then quantified using the Quibit 2.0 fluorometer (Life Technologies) and loaded on an Illumina MiSeq (Illumina, Inc. San Diego, CA) 2 × 300 flow cell at 10 pM.
-
Bioinformatic and statistical analysis of fungal cultivable counts and Illumina sequences
To assess the difference between fungal abundances in cultivable sample categories (based on time and location), the following univariate statistical analyses were carried out (https://www.r-project.org/). The normal distribution of the populations was tested using the Shapiro-Wilk normality test, and as most were not normally distributed (p value <0.05), we used a Kruskal-Wallis test coupled to a Dunn’s test (https://cran.r-project.org/web/packages/dunn.test/index.html) to investigate differences in the tested populations. Resulting p values were corrected using the Benjamini-Hochberg correction [59].
A total of 8,426,774 raw paired reads were processed with mothur v.1.36.1 [60]. The 250 bp paired reads were merged by aligning the reads and correcting discordant base calls by requiring one of the base calls to have a Phred quality score at least 6 points higher than the other. Sequences shorter than 200 bp, having more than 8 homopolymers or containing ambiguous base pairs were excluded from the dataset. Subsequently, reads were pre-clustered [61] by combining low-abundant sequences that differed by 3 or less bases of a more abundant sequence. Chimeric sequences in each sample were identified by UCHIME [62] and also excluded from the dataset. Reads were classified using the ribosomal database project (RDP) classifier II [63] implementation of mothur and UNITE fungal rDNA database [54]. Non-fungal sequences were subsequently removed. ITSx was used to exclude non-ITS sequences from the dataset prior to clustering unaligned sequences into operational taxonomic units (OTUs) with a distance of 0.03 [64] using mother v.1.36.1. The nearest neighbor (single-linkage) algorithm was used for this. OTUs were classified by using the consensus taxonomy of all sequences assigned [60].
An in-house R-script employing the libraries vegan, ape, gplots, mgcv, and GUniFrac was used to compare the fungal Illumina data (Additional file 3) [65, 66]. Each dataset consisting of the OTU abundances per sample was rarefied 1000 times to the lowest number of reads, and an average Bray-Curtis distance was calculated. This distance was then utilized to calculate nonmetric multidimensional scaling (NMDS) or principal coordinate analysis (PCoA), PERMANOVA (Adonis test), and multi-response permutation procedure (MRPP). In addition, the OTU abundances per sample of each dataset were sum normalized and used to employ either an analysis of variance (ANOVA) or a Spearman rank correlation on the statistically significant changing parameters and to generate a heat map (p value 0.05). The change of diversity was measured via the Shannon-Wiener diversity index. OTUs that were unclassified at phylum level were removed. Heat maps were presented at family level.
Results
Cultivable fungal burden and diversity
The culture-based fungal abundance of the ILMAH was estimated for each sampling location with colony-forming unit (CFU) values ranging from below detection limit (BDL) to 104 CFU/m2 (Additional file 4: Table ST1a). The highest abundance of the cultivable fungi was observed during the first sampling (T0) followed by ~1 log decrease in CFU during consecutive sampling events (T13, T20, T30) for each living compartment. High fungal population was noticed in samples collected at T0 (Fig. 2A). When statistically treated, the change in fungal abundance within different time points was significant (T0–T20 p = 0.008 and T0–T30 p = 0.0125) (Additional file 5: Table ST2a). The cultivable fungal population for the lab area was higher than observed in other compartments (Fig. 2B, Additional file 4: Table ST1b). A statistically significant difference was observed between the lab area and the bedroom (p = 0.0021) (Additional file 5: Table ST2b).
Fig. 2.
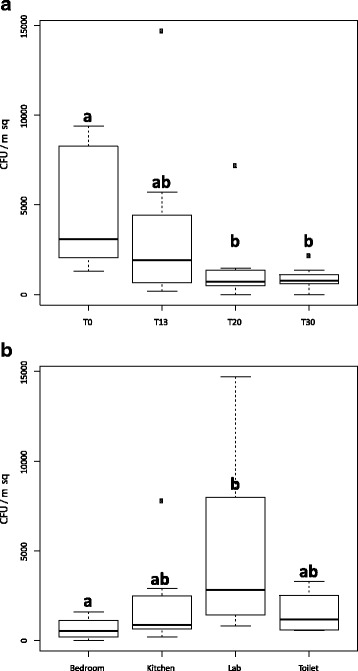
Statistical analysis of cultivable fungal diversity detected through the 30-day habitation period at all the locations based on colony-forming unit (CFU) counts. To assess the difference between fungal abundances in cultivable sample categories (based on time—A and location—B), we applied the following univariate statistics. The normal distribution of the populations were tested using Shapiro-Wilk normality test, and as most of them were not normally distributed (p value <0.05), we used a Kruskal-Wallis test coupled to a Dunn’s test to investigate differences in the tested populations. Resulting p values were corrected using the Benjamini-Hochberg correction. A CFU counts before crew occupation T0—a were statistically different from CFU counts at T20 and T30—b, but no statistical difference was observed between T0 and T13 counts—ab. Additionally, no statistical differences were observed between any other time points. B CFU counts in the bedroom—a, differed significantly from the CFU counts in lab—b, but no statistical differences were observed between bedroom and kitchen or toilet—ab. No statistical differences were observed between any other locations
One hundred seventeen cultivable isolates were collected and identified targeting the ITS region. Screening sequences against the UNITE fungal database enabled identification of 32 species (Fig. 3). Among the cultivable isolates, only five strains had similarity lower than 97% that did not allow identification to the species level. All of the identified species but one—Hydnopolyporus fimbriatus—belong to the Ascomycota division (Fig. 3). The most abundant cultivable species were Cladosporium cladosporioides (16), Epicoccum nigrum (15), Aspergillus tubingensis (13), Aspergillus fumigatus (8), Alternaria tenuissima (7), and Pennicillium brevicompactum (7). P. brevicompactum was the only, out of the most commonly identified species, that was not present at multiple time points but only at T13 sampling (Fig. 3). Abundance of C. cladosporioides colonies increased during the ILMAH occupation whereas the abundance of E. nigrum, A. tubingensis, and A. tenuissima decreased. Another commonly isolated species, A. fumigatus, was not isolated during the last sampling event. Neither field nor sampling device controls showed cultivable isolates.
Fig. 3.
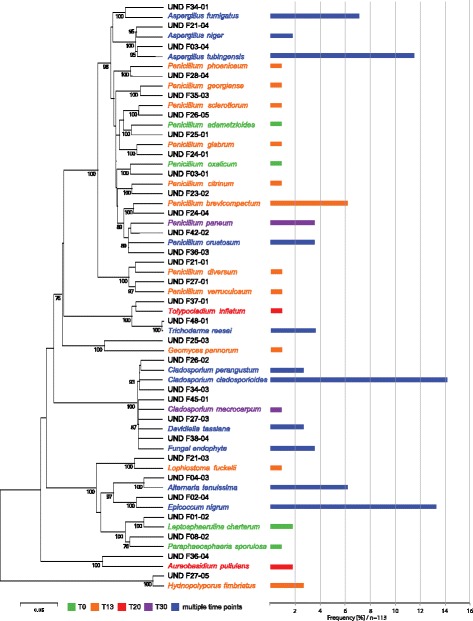
Cultivable fungal diversity detected through the 30-day habitation period at all the locations based on internal transcribed spacer (ITS) sequences. The phylogenetic tree was constructed using neighbor-joining method (bootstrap 1000). In total, 117 isolates were collected 113 of which were successfully sequenced (4 strains either did not show growth or did not respond to the sequencing methods attempted). The numbering of the isolates is explained as follows: F = fungi, first number (0–4) will be the sample collection day (0 = T0, 2 = T13, 3 = T20, 4 = T30), second number (1–8) will be sampling location, and the third number (1–5) is the replicate number of the isolate. For example, F23-02 will be a fungal strain, isolated from T20, at location number 3 and a second isolate. Frequency of isolates is given as a frequency bar after the name of fungus. Colors of the bars correspond to the collection time (single or multiple)
Viable and total mycobiome (iTag Illumina-based analysis)
The fungal richness of PMA-treated (viable) samples decreased when compared to untreated samples (dead and alive). In PMA-untreated samples, 98 families were detected, whereas OTUs belonging to 41 of these families were not viable. Moreover, both PMA-treated and PMA-untreated samples differed significantly in community relationships (NMDS analysis in Fig. 4, Adonis p value = 0.006 and MRPP, significance of delta = 0.002; A = 0.0419) and their Shannon diversity index indicated a significant reduction (paired T test p = 0.0000012) in viable fungal diversity. Observed differences in the PMA-treated (Fig. 4a, c) and PMA-untreated samples (Fig. 4b, d) indicate that untreated samples are overestimating the observed fungi. This observation was further confirmed when alpha diversity of cultivable, viable, and total mycobiome was plotted over time (Fig. 5). In this research communication, only viable mycobiome was considered and discussed in detail. Illumina-based reads of sampling device and field controls showed negligible signal from DNA contamination and hence not included in the following analysis.
Fig. 4.
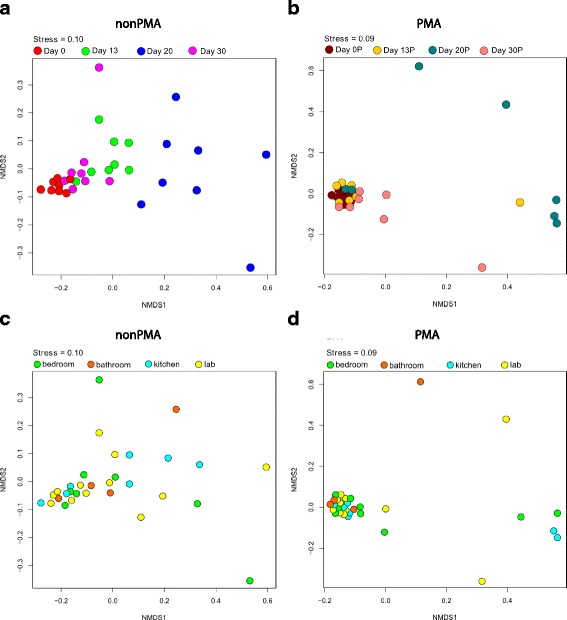
NMDS ordinations based on Bray-Curtis distances between all samples. a Ordination displaying the distance between non-PMA-treated samples taken at the different time points. b The distance between PMA-treated samples taken at the different time points. c The distance between non-PMA-treated samples taken at the different locations. d NMDS ordination displaying the distance between PMA-treated samples taken at the different locations. A “P” after the respective variable indicates that these are the samples treated with PMA. Plots a, c, and b, d represent the same data but differ in colors to underscore the focus on distribution over time and location, respectively
Fig. 5.
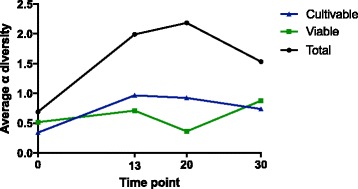
Linear representation of alpha diversity averages change over time for cultivable, viable and total mycobiome
Viable fungal community structure
The most abundant phylum that dominated the viable mycobiome of the ILMAH was Ascomycota (90% of all characterized OTUs) followed by Basidiomycota and unclassified fungi (~4 and ~5%, respectively). Incidence of the fungal OTUs at the family level for various time points and locations are presented in Table 1 and Table 2, respectively. The dominant Pleosporaceae (75% of all OTUs) along with unclassified fungi (5%) and Davidiellaceae (4%) constituted 84% of all OTUs present in PMA-treated samples (Tables 1 and 2 and Fig. 6). A closer look into the genus level of Pleosporaceae family indicated the domination of Epicoccum (92.95% of all OTUs) and Alternaria (6.8%) sequences. The dominant fungal OTUs in the ILMAH biome correspond with the most frequently isolated cultivable fungi (Figs. 3 and 6).
Table 1.
Incidence of the fungal OTUs at the family level for various time points
| Fungal phylum | Fungal family | Number of sequences | |||||||
|---|---|---|---|---|---|---|---|---|---|
| T0 | T13 | T20 | T30 | ||||||
| Total | Viable | Total | Viable | Total | Viable | Total | Viable | ||
| Ascomycota | Botryosphaeriales | 115 | 31 | 1620 | 176 | 96 | |||
| Capnodiales | 2 | 185 | 121 | 61 | 4353 | ||||
| Chaetomiaceae | 2415 | 1089 | 5533 | 1505 | 32981 | ||||
| Cucurbitariaceae | 451 | 221 | 2298 | 750 | 1879 | 2272 | 1256 | ||
| Davidiellaceae | 2840 | 1423 | 42496 | 6099 | 129620 | 28495 | 55079 | 38510 | |
| Dothioraceae | 32 | 104 | 510 | 289 | 12864 | 14785 | 4726 | 688 | |
| Hypocreaceae | 174 | 68 | 2357 | 197 | 5510 | 3897 | 10952 | ||
| Hypocreales | 73 | 86 | 9247 | 402 | 8566 | 1 | 7989 | 5638 | |
| Leptosphaeriaceae | 16 | 23 | 564 | 215 | 518 | 1 | 1639 | 1 | |
| Lophiostomataceae | 56 | 43 | 56 | 1850 | 1 | 26 | |||
| Microascaceae | 326 | 27007 | 9 | 735 | 6 | ||||
| Montanulaceae | 627 | 232 | 11273 | 621 | 22582 | 12697 | 77 | ||
| Nectriaceae | 82 | 121 | 2890 | 1287 | 8479 | 804 | 2242 | 49 | |
| Phaeosphaeriaceae | 343 | 222 | 3551 | 105 | 6082 | 1 | 6969 | 30695 | |
| Pleosporaceae | 295115 | 380533 | 273232 | 251840 | 172919 | 61791 | 648825 | 619794 | |
| Pleosporales | 5202 | 3071 | 6876 | 4475 | 10056 | 13 | 12085 | 8859 | |
| Saccharomycetales | 17 | 33 | 346 | 3 | 45461 | 10414 | 17840 | 2092 | |
| Sclerotiniaceae | 52 | 16 | 355 | 1920 | 99 | ||||
| Teratosphaeriaceae | 35 | 28 | 805 | 1 | 2 | 2 | 583 | 44749 | |
| Trichocomaceae | 68 | 32 | 10496 | 3461 | 14625 | 10495 | 12147 | 518 | |
| Trichosphaeriales | 3 | 122 | 1750 | 688 | |||||
| Unclassified | 712 | 686 | 5082 | 640 | 4161 | 4450 | 6317 | ||
| Basidiomycota | Filobasidiaceae | 1164 | 419 | 5997 | 8 | 11570 | 1925 | 2550 | 4425 |
| Peniophoraceae | 1 | 10916 | |||||||
| Polyporales | 1 | 1339 | |||||||
| Sporidiobolales | 2563 | 1236 | 6556 | 315 | 22900 | 2 | 35692 | 24352 | |
| Tremellales | 7916 | 4158 | 42068 | 1030 | 72870 | 9 | 34932 | 18439 | |
| Unclassified | 409 | 272 | 2151 | 2171 | 481 | 504 | 2445 | 279 | |
| Unclassified | Unclassified | 2973 | 10475 | 38015 | 17311 | 30832 | 10320 | 41608 | 53950 |
Table 2.
Incidence of the fungal OTUs at the family level for locations
| Fungal phylum | Fungal family | Number of sequences | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Bedroom | Kitchen | Bathroom | Lab | ||||||
| Total | Viable | Total | Viable | Total | Viable | Total | Viable | ||
| Ascomycota | Capnodiales | 215 | 26 | 121 | 5 | 4355 | |||
| Chaetomiaceae | 171 | 32858 | 1668 | 2073 | 5541 | 1212 | |||
| Cucurbitariaceae | 1186 | 16 | 2782 | 739 | 1248 | 57 | 1545 | 1415 | |
| Davidiellaceae | 71145 | 17039 | 40120 | 35104 | 53460 | 496 | 58969 | 21888 | |
| Dothioraceae | 4821 | 5774 | 161 | 286 | 172 | 9708 | 12970 | 98 | |
| Herpotrichiellaceae | 68 | 4 | 93 | 4 | 134 | 7 | 1407 | 28 | |
| Hypocreaceae | 979 | 11 | 4622 | 93 | 2095 | 10909 | 4133 | 204 | |
| Hypocreales | 9471 | 5617 | 8792 | 408 | 1712 | 12 | 5169 | 90 | |
| Lophiostomataceae | 30 | 21 | 71 | 19 | 9 | 1878 | 4 | ||
| Microascaceae | 3086 | 3 | 6642 | 3 | 18317 | 3 | 23 | 6 | |
| Montanulaceae | 9231 | 116 | 5112 | 520 | 24292 | 68 | 7141 | 226 | |
| Nectriaceae | 216 | 1374 | 5618 | 3 | 1542 | 18 | 6177 | 866 | |
| Phaeosphaeriaceae | 1360 | 131 | 4209 | 55 | 6150 | 18 | 4318 | 30819 | |
| Pleosporaceae | 223517 | 357893 | 195638 | 299314 | 247789 | 199402 | 626137 | 457349 | |
| Pleosporales | 5933 | 4284 | 6768 | 590 | 7339 | 644 | 10668 | 10900 | |
| Saccharomycetales | 1904 | 1658 | 18978 | 10681 | 10035 | 49 | 29496 | 154 | |
| Teratosphaeriaceae | 94 | 55 | 9 | 13 | 8 | 932 | 44763 | ||
| Trichocomaceae | 5453 | 1 | 11611 | 1167 | 8735 | 2236 | 10860 | 11102 | |
| Others (20) | 1835 | 5 | 494 | 12 | 530 | 16 | 365 | 26 | |
| Unclassified | 2007 | 426 | 5203 | 468 | 668 | 3105 | 5831 | 3644 | |
| Basidiomycota | Filobasidiaceae | 3624 | 2198 | 935 | 120 | 8171 | 4412 | 8259 | 47 |
| Peniophoraceae | 45 | 9922 | 79 | 1 | 994 | ||||
| Polyporales | 1 | 1339 | |||||||
| Sporidiobolales | 33860 | 24869 | 15193 | 173 | 10398 | 299 | 7923 | 564 | |
| Tremellales | 47729 | 17080 | 26291 | 2479 | 9002 | 1165 | 69144 | 2912 | |
| Unclassified | 1999 | 34 | 525 | 2172 | 1144 | 67 | 1459 | 953 | |
| Unclassified | Unclassified | 37959 | 13825 | 17047 | 26040 | 22401 | 6260 | 29713 | 45931 |
Fig. 6.
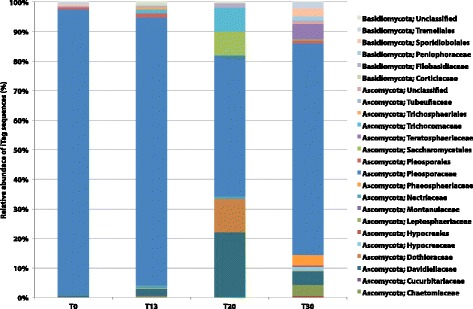
Dominant fungal population and succession patterns observed in 30-day occupation period of the ILMAH system. The OTUs presented in the bar graph are the most abundant. T20 surface samples show different fungal profile when compared to other time points. T30 samples show increase in fungal diversity when compared to other time point
Differences in the fungal community between samples were analyzed by multivariate statistics using ordination analyses and Monte Carlo-based permutation tests. Viable fungal communities showed similar mycobiome profiles throughout the sampling period except from the samples collected at T20, which were distinct (Fig. 4; Adonis p value = 0.001 and MRPP, significance of delta = 0.001 and A = 0.1168, Additional file 6: Figure SF2 MRPP: chance corrected within-group agreement A: 0.1626, significance of delta 0.001; Adonis: delta = 0.019). Interestingly, community profiles were similar between samples collected before crew occupation (T0) and at T13 and T30 (Additional file 7: Figure SF3; chance corrected within-group agreement A: 0.01802, significance of delta: 0.219, Adonis 0.228, Additional file 8: Table ST3).
Observed differences in multivariate statistics led to the investigation of mycobiome changes on a single-OTU level. First, an Anova test carried out on samples that did not cluster with the rest (Fig. 4) showed presence of representatives of Pleosporaceae, Pleosporales, Saccharomycetales, Tremellales, and Trichocomaceae families. Second, throughout the inhabitation, the level of Pleosporaceae showed significant fluctuation—from 96 to 47% and 70% at T0, T20, and T30, respectively (Fig. 7). Additionally, while Pleosporaceae presence decreased to 47%, a significant increase in the levels of Davidiellaceae (22%), Dothioraceae (11%), Saccharomycetales (8%), and Trichocomaceae (8%) was observed when compared to other time points (Fig. 6, Additional file 9: Table ST4). Interestingly, the presence of less abundant families observed before crew occupancy (T0) increased at (T30), i.e., Davidiellaceae (4.45%), Hypocreaceae (1.26%), Phaeosphaeriaceae (3.54%), Teratosphaeriaceae (5.17%), and Sporidiobolales (2.81%), as well as members of new fungal families were observed, i.e., Capnodiales (0.5%), Chaetomiaceae (3.81%), and Peniophoraceae (1.26%) (Table 1 and Fig. 6, Additional file 9: Table ST4).
Fig. 7.
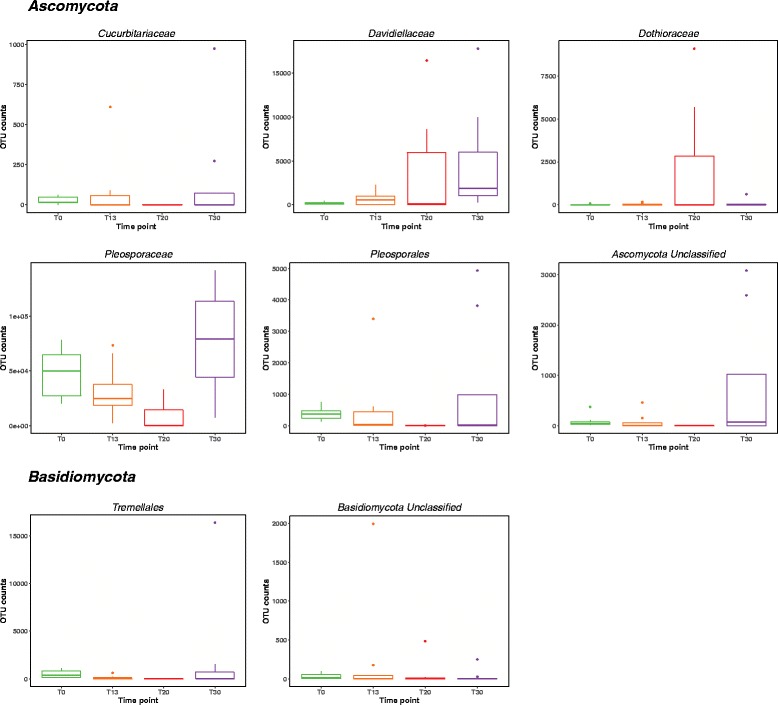
Box plots of viable dominant fungal families and their succession patterns observed in 30-day occupation period of the ILMAH system. The OTU counts presented in the boxplots are the most abundant. Each time point is represented in a different color: T0—green, T13—orange, T20—red, T30—purple
Spearman rank correlation was applied to each OTU’s abundance pattern and sampling event to determine significant correlation of viable fungal families with various time points. The results presented as a heat map contain 13 OTUs that showed a significant correlation with a p value of 0.01 and 32 OTUs with a p value of 0.05. All OTUs but Davidiellaceae, Teratosphaeriaceae, Tremellales_3, Pleosporales, and Pleosporaceae were more abundant during the sampling before the crew inhabited the ILMAH (Fig. 8).
Fig. 8.
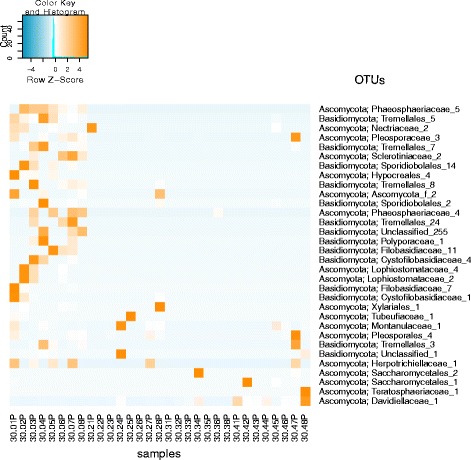
Heat map of the taxa that showed a significant correlation (p value 0.01) with the factor time in the PMA-treated sample set. The color blue indicates a low abundance of the single OTU in the respective sample, and orange indicates a high abundance of the single OTU in the respective sample. Each column represents one sample collected throughout the study. The numbering pattern is explained as follows 30 means 30-day study. The first number (0–4) will be the sample collection day (0 = T0, 2 = T13, 3 = T20, 4 = T30), second number (1–8) will be sampling location, and P stands for PMA-treated samples. For example, 30.06P will be a sample collected during the 30-day study from T0 at location 6
Further investigation of OTU abundances on the genus level revealed changes in OTU counts over the course of time. The numbers of OTUs identified as Epiccocum, Alternaria, Pleosporales, and Cryptococcus fluctuated from high abundance at T0 to significantly lower counts at T20 and then counts increased again at T30. OTUs identified as Davidiella increased throughout the occupation time whereas OTU counts for Aspergillus, Aureobasidium, and Candida were increasing over time (peak at T20) with a drastic decrease at T30. Location wise before the ILMAH occupation, all compartments exhibited high abundance of Epiccocum OTUs that decreased over time and again increased at T30. Alternaria OTUs were less abundant than the ones identified as Epiccocum genus. Nevertheless, they showed the same fluctuation pattern in all compartments but bedroom. While accumulation of Davidiella OTU counts was observed in bedroom and lab, the OTU counts in the bathroom did not differ between the T0 and T30. At the same time, accumulation of Davidiella OTUs was observed in the kitchen with the highest abundance during the T20 sampling event.
To sum up, statistical analysis revealed differences in community structure between time points in particular between T20 and any other previous or following time point. Davidiellaceae, Teratosphaeriaceae, Tremellales_3, Pleosporales, and Pleosporaceae families were shown to increase over the occupation time period. On a genus level, Epiccocum, Alternaria, Pleosporales, Davidiella, and Cryptococcus showed increased abundance.
Discussion
Understanding microbial characteristic of a controlled habitat, like the ILMAH, may facilitate discerning the microbial population dynamics as well as the development of appropriate countermeasures. Knowledge about the viable mycobiome will not only allow the development of required maintenance and cleaning procedures in the closed habitat but also prevent it from deteriorating and being a potential health hazard for its inhabitants [67]. Multiple studies have shown that use of propidium monoazide (PMA), a dye that can penetrate the compromised cell walls, enables more accurate analysis of the viable microbiome [68, 69]. It was also shown that PMA treatment might be successfully applied to determine dead and viable counts for various fungal species [70, 71]. As in this study, other report proved that DNA from dead cells, when not removed from samples, before molecular analyses, might overshadow the actual diversity since presence of less abundant microbial species were masked [32]. PMA-treated and PMA-untreated samples varied significantly in the ILMAH fungal community structures (p value = 0.006), and it had been reported before that low abundant species were detected in PMA-treated samples for bacteria, fungi, and viruses [66]. This approach validated the importance of PMA treatment to accurately determine the viable mycobiome of environmental samples. As a result, this study discussed only viable fungal communities to determine succession patterns and community structure over the course of time.
Various studies showed the positive correlation between changes in the indoor bacteriome and human presence [7, 28, 30, 66, 72] whereas such observation was not confirmed for the mycobiome [29, 30, 73]. Major sources of indoor microbiome were reported to be associated with human skin commensals transmitted via shoes, clothes, coughing, and talking [74, 75]. It was also shown that the indoor mycobiome was mostly influenced by airborne fungi rather than human presence and their shedding, despite the fact that fungi were associated with human skin, lungs, urogenital tract, oral nasal cavities, and the gut [30, 76–79]. In addition, few studies demonstrated that key determinants for mycobiome indoors might be the age of the built-in environments and the relative humidity, which can enhance fungal growth [80]. A recent 6-month survey on Japanese astronauts on the ISS revealed that the most abundant (dead or alive) fungal genus was Malassezia [10] whereas in this study, the most frequent viable fungal genera of closed habitat were Epiccocum, Alternaria, and Pleosporales, which are environmental organisms rather than human commensals.
The presence of fungi inside the closed habitat, houses, or any types of man-made buildings was correlated with the amount of the water (relative humidity; RH) present in the environment [80]. The environmental relative moldiness index (ERMI) increases with elevated amounts of water [81]. The most abundant genera in common households were Alternaria, Cladosporium, and Epicoccum whereas Aspergillus and Penicillium were predominant in water-damaged building [81]. Similarly, in this study (measured RH 32 to 55%), the most abundant fungal genera were Epiccocum, Alternaria, Pleosporales, and Cryptococcus, which could be compared with the previous observations for common households. Nevertheless, genera present in houses and closed habitats could have an impact on human health. These molds were associated with allergies and asthma [67, 82]. Elevated level of fungal allergenic molecules, such as enzymes, toxins, cell-wall components, and cross-reactive proteins could induce type I hypersensitivity [67]. Additionally, because of their ability to colonize the human body and produce toxins, volatile organic compounds, and proteases, the common molds (Alternaria, Cladosporium, Epicoccum) could damage airways of immunocompromised occupants, which make them more dangerous than any other allergenic source [82–85]. In this study, accumulation of Alternaria and Epicoccum genera over time was observed, which, in combination with reported decreased immunity in occupants of confined spaces, e.g., astronauts [50, 86] could lead to developing allergy and asthma symptoms.
Throughout this study, the abundance of dominant Pleosporaceae family members decreased (94% at T0 to 71% at T30), while other fungal families were observed to increase, possibly as a result of human presence. However, positive correlation between increased fungal diversity and human presence requires studying the mycobiome of the occupants. It might be possible that the implemented cleaning procedures including weekly dusting, sweeping, wet mopping the floor, and antibacterial wipes resulted in suppressed growth of Pleosporaceae members over the time. The community structure observed at T20 was distinct from other time points. Detailed logging data collected during 30-day mission did not show any abnormal accidents or cleaning activities preceding T20 sampling. The recorded data indicated that cleaning was conducted between the sampling events (~4–5 days prior to sampling). However, the inhabiting crew might have inadvertently cleaned just prior to T20 sampling and that might have removed both total and viable fungal populations (Table 1). The significant increase of Davidiellaceae (22%), Dothioraceae (11%), Saccharomycetales (8%), and Trichocomaceae (8%) sequences at T20 might be due to the fact that under-represented members became available for PCR reaction among the competing dominant fungal DNA.
Both Cladosporium sp. members of Davidiellaceae family and Aurobasidium sp. of Dothioraceae family have the capacity to survive extreme environments like the ice of the Antarctica [87] or radioactive explosion site of Chernobyl Power Plant accident [88]. Additionally, Cladosporium sp. and Aureobasidium pullulans were isolated from hypersaline waters with NaCl concentration reaching 25% indicating high osmotolerance [89, 90]. Penicillium sp. of Trichocomaceae family isolated from high-altitude soil in Indian Himalaya has been shown to tolerate a wide range of pH from 2 to 14 and a salt concentration between 10 and 20% [91]. Most of the Apergillus sp. of Trichocomaceae family is soil fungi or saprophytes [92], but there has been a recent report of isolation of A. fumigatus from ISS [32]. In-depth analysis of isolate ISSFT-021 and IF1SW-F4 revealed increased UV resistance (in preparation) and virulence in neutrophil-deficient larval zebrafish model of invasive aspergillosis [93]. All in all, the capacity to survive in such extreme environments might help the observed in the study Davidiellaceae, Dothioraceae, and Trichocomaceae species to adjust and survive hostile conditions of the ILMAH.
Conclusions
Accumulation of viable mycobiome during the experiment duration and differences in fungal community profiles were observed. On a genus level, Epiccocum, Alternaria, Pleosporales, Davidiella, and Cryptococcus showed increased abundance over the occupation time. Unlike results of molecular analyses, cultivable fungi counts decreased over time. Epicoccum nigrum was the most dominant cultivable isolate; however, most of the fungal species detected via molecular approach were not cultured. During human presence, the overall fungal diversity has changed, that in the long run, may lead to a conclusion that proper maintenance protocols of a closed habitat may be required to preserve it from deteriorating and keep it safe for its inhabitants.
Additional files
Schematic representation of the ILMAH architecture with dimensions. The sampling locations are indicated with stars and numbers (1, 2: Bedroom; 3, 4: Kitchen; 5: Bathroom; and 6, 7, 8: Laboratory). (PDF 911 kb)
Crew schedule assignments for the 30 days. (PDF 911 kb)
R code and script used as published in Weinmaier and Probst et al., 2015 Microbiome. (PDF 300 kb)
Cultivable fungal characteristics of ILMAH surface samples. Tables represent: a) CFU counts for each sampling area during consecutive sampling events (before crew occupation, Day 13, Day 20 and Day 30). Sampling areas are numbered 1-8. 1 and 2 correspond to bedroom area, 3, 4 – kitchen, 5 – bathroom and 6-8 lab. CFU counts are reported per meter square; b) CFU counts for specific ILMAH compartments. Table contains average CFU counts for each compartment: bedroom, kitchen, bathroom and lab area (for example CFU counts from area 1 and 2 at Day 13 were used to calculate the average CFU count for the bedroom compartment at Day 13). CFU counts are reported per meter square. (PDF 52 kb)
Statistical analysis to compare cultivable fungal populations of the different (a) time points and (b) locations. (a) CFU counts of cultivable fungal populations observed at various time point were compared to each other to asses if there are any statistically significant changes in CFU counts over the course of time; (b) CFU counts of cultivable fungal populations observed at different compartments were compared to each other to asses if there are any statistically significant changes in CFU counts between locations. (PDF 43 kb)
NMDS ordinations based on Bray-Curtis distances between non-PMA- and PMA-treated samples taken at different time points. The analysis shows a significant difference between the PMA treated and not treated samples and between the different time points but not between the different locations. A “P” after the respective variable indicates that these are the samples treated with PMA. (PDF 44 kb)
NMDS ordinations based on Bray-Curtis distances between PMA treated samples without T20 taken at different time points. A “P” after the respective variable indicates that these are the samples treated with PMA. (PDF 30 kb)
Statistical analysis of viable (PMA treated) samples to compare fungal populations of the different a) time points and b) locations. (a) Community profiles of viable fungal populations observed at various time point were compared to each other to asses if there are any statistically significant changes over the course of time; (b) Community profiles of viable fungal populations observed at different compartments were compared to each other to asses if there are any statistically significant changes between locations. Results marked with * are statistically significant. (PDF 44 kb)
Percent change of OTU counts at family level. Information about the percent changes of the OTU counts of selected families during consecutive time points is presented. (PDF 45 kb)
SRA numbers for individual samples. This file contains detailed information about accession number of each sample deposited within the study SPR069729. (TXT 12 kb)
OTU deduced from the PMA treated samples. This file contains OTU counts of all PMA treated samples collected during the study. The numbering pattern is explained as follows 30 means 30-day study. The first number (0–4) will be the sample collection day (0 = T0, 2=T13, 3=T20, 4=T30), second number (1–8) will be sampling location, and P stands for PMA-treated samples. For example, 30.06P will be a sample collected during the 30-day study from T0 at location 6. (CSV 334 kb)
OTU deduced from the PMA untreated samples. This file contains OTU counts of all samples not treated with PMA collected during the study. The numbering pattern is explained as follows 30 means 30-day study. The first number (0–4) will be the sample collection day (0 = T0, 2=T13, 3=T20, 4=T30), second number (1–8) will be sampling location. For example, 30.06 will be a sample collected during the 30-day study from T0 at location 6. (ZIP 35 kb)
OTU table sorted by location. This file contains OTU counts of all the families observed throughout the study for PMA treated (viable) and untreated (total) samples for various locations (bedroom, kitchen, bathroom and lab). (CSV 2 kb)
OTU table sorted by location. This file contains OTU counts of all the families observed throughout the study for PMA treated (viable) and untreated (total) samples for various time points (T0, T13, T20 and T30). (TXT 132 kb)
Acknowledgements
The authors are grateful to the three student crews that participated in this program. Our thanks to Mr. Tim Buli who physically collected the samples during occupation. AJ Probst is acknowledged for consulting and supporting in statistical analysis. Part of the research described in this publication was carried out at the Jet Propulsion Laboratory, California Institute of Technology, under a contract with NASA. We would also like to thank the Department of Space Studies, University of North Dakota, for allowing sampling in the ILMAH, and the members of the Planetary Protection group at JPL for their technical assistance. We appreciate M. Jones and S. Ozyildirim of JPL for critically reading the manuscript. © 2016 California Institute of Technology. Government sponsorship acknowledged.
Funding
This research was awarded to K. Venkateswaran and funded by a 2012 Space Biology NNH12ZTT001N Grant No. 19-12829-26 under Task Order NNN13D111T.
Availability of data and materials
The dataset supporting the results of this article is available in the NCBI SRA repository, under study accession # SRP069729. SRA numbers for individual samples are listed in attached Additional file 10. The ITS sequences of the cultivable fungi are available in the NCBI GenBank under accession # KX664307-KX664419. PMA-treated and untreated OTU tables are submitted as Additional files 11 and 12, respectively, along with the OTU tables sorted by location and time point (Additional files 12 and 13, respectively). ITS sequences used to create phylogenetic tree are included in Additional file 14.
Authors’ contributions
AB drafted the manuscript, coordinated sample collection, contributed to the sample processing and to the data analysis and interpretation, and carried out the identification of the fungal isolates in this study. TM helped with sample processing and statistical analysis and data interpretation. MB contributed to result processing. TRP contributed to data analysis (clinical perspective). PdL collected samples and sent them to JPL. KV designed the study, interpreted the data, and drafted the manuscript. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
The authors declare that our report did not contain data from any individual and hence there is no consent needed to publish these science data.
Ethics approval and consent to participate
The authors declare that our report did not collect data from human or animals. This research where ILMAH was inhabited with human subjects have been performed in accordance with the University of North Dakota (UND), ND, USA, and have been approved by the UND Institutional Review Board. Participating students have given their informed consent to publish the microbiological characteristics of the ILMAH where they have inhabited for 30 days.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (doi:10.1186/s40168-017-0280-8) contains supplementary material, which is available to authorized users.
References
- 1.Price H, Baker J, Naderi F. A minimal architecture for human journeys to Mars. New Space. 2015;3(2):73–81. doi: 10.1089/space.2015.0018. [DOI] [Google Scholar]
- 2.Wilhite AW, Chai P. Plan B for U.S. Human Space Exploration Program. 2014. [Google Scholar]
- 3.Swarmer TM, Anderson L, de León P: Performance review of a pressurized inflatable lunar habitat integrated with an electric rover and pressurized analog planetary suits during an initial ten day simulation. International Conference on Environmental Systems 2014.
- 4.Kelley ST, Gilbert JA. Studying the microbiology of the indoor environment. Genome Biol. 2013;14(2):202. doi: 10.1186/gb-2013-14-2-202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lax S, Hampton-Marcell JT, Gibbons SM, Colares GB, Smith D, Eisen JA, Gilbert JA. Forensic analysis of the microbiome of phones and shoes. Microbiome. 2015;3:21. doi: 10.1186/s40168-015-0082-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Meadow JF, Altrichter AE, Kembel SW, Moriyama M, O'Connor TK, Womack AM, Brown GZ, Green JL, Bohannan BJ. Bacterial communities on classroom surfaces vary with human contact. Microbiome. 2014;2(1):7. doi: 10.1186/2049-2618-2-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mayer T, Blachowicz A, Probst AJ, Vaishampayan P, Checinska A, Swarmer T, de Leon P, Venkateswaran K. Microbial succession in an inflated lunar/Mars analog habitat during a 30-day human occupation. Microbiome. 2016;4(1):22. doi: 10.1186/s40168-016-0167-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Oberauner L, Zachow C, Lackner S, Hogenauer C, Smolle KH, Berg G. The ignored diversity: complex bacterial communities in intensive care units revealed by 16S pyrosequencing. Sci Rep. 2013;3:1413. doi: 10.1038/srep01413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Satoh K, Nishiyama Y, Yamazaki T, Sugita T, Tsukii Y, Takatori K, Benno Y, Makimura K. Microbe-I: fungal biota analyses of the Japanese experimental module KIBO of the International Space Station before launch and after being in orbit for about 460 days. Microbiol Immunol. 2011;55(12):823–829. doi: 10.1111/j.1348-0421.2011.00386.x. [DOI] [PubMed] [Google Scholar]
- 10.Sugita T, Yamazaki T, Makimura K, Cho O, Yamada S, Ohshima H, Mukai C. Comprehensive analysis of the skin fungal microbiota of astronauts during a half-year stay at the International Space Station. Med Mycol. 2016;54(3):232–239. doi: 10.1093/mmy/myv121. [DOI] [PubMed] [Google Scholar]
- 11.Hewitt KM, Gerba CP, Maxwell SL, Kelley ST. Office space bacterial abundance and diversity in three metropolitan areas. PLoS One. 2012;7(5):e37849. doi: 10.1371/journal.pone.0037849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Poza M, Gayoso C, Gomez MJ, Rumbo-Feal S, Tomas M, Aranda J, Fernandez A, Bou G. Exploring bacterial diversity in hospital environments by GS-FLX Titanium pyrosequencing. PLoS One. 2012;7(8):e44105. doi: 10.1371/journal.pone.0044105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hewitt KM, Mannino FL, Gonzalez A, Chase JH, Caporaso JG, Knight R, Kelley ST. Bacterial diversity in two neonatal intensive care units (NICUs) PLoS One. 2013;8(1):e54703. doi: 10.1371/journal.pone.0054703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brooks B, Firek BA, Miller CS, Sharon I, Thomas BC, Baker R, Morowitz MJ, Banfield JF. Microbes in the neonatal intensive care unit resemble those found in the gut of premature infants. Microbiome. 2014;2(1):1. doi: 10.1186/2049-2618-2-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gupta RK: role of environmental monitoring and microbiological testing during manufacture of sterile drugs and biologics* Am Pharm Rev. 2014. http://www.americanpharmaceuticalreview.com/Featured-Articles/169384-Role-of-Environmental-Monitoring-and-Microbiological-Testing-During-Manufacture-of-Sterile-Drugs-and-Biologics/.
- 16.Park HK, Han JH, Joung Y, Cho SH, Kim SA, Kim SB. Bacterial diversity in the indoor air of pharmaceutical environment. J Appl Microbiol. 2014;116(3):718–727. doi: 10.1111/jam.12416. [DOI] [PubMed] [Google Scholar]
- 17.Ingle PV, Chatap VK, Bhatia NM. Design considerations for parenteral production facility. Int J Pharma Res Rev. 2014;3(8):15–28. [Google Scholar]
- 18.Klykens J, Pirnay JP, Verbeken G, Giet O, Baudoux E, Jashari R, Vanderkelen A, Ectors N. Cleanrooms and tissue banking how happy I could be with either GMP or GTP? Cell Tissue Bank. 2013;14(4):571–578. doi: 10.1007/s10561-012-9355-8. [DOI] [PubMed] [Google Scholar]
- 19.Huang SS, Datta R, Platt R. Risk of acquiring antibiotic-resistant bacteria from prior room occupants. Arch Intern Med. 2006;166(18):1945–1951. doi: 10.1001/archinte.166.18.1945. [DOI] [PubMed] [Google Scholar]
- 20.Peleg AY, Hooper DC. Hospital-acquired infections due to gram-negative bacteria. N Engl J Med. 2010;362(19):1804–1813. doi: 10.1056/NEJMra0904124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Symoens F, Burnod J, Lebeau B, Viviani MA, Piens MA, Tortorano AM, Nolard N, Chapuis F, Grillot R. Hospital-acquired Aspergillus fumigatus infection: can molecular typing methods identify an environmental source? J Hosp Infect. 2002;52(1):60–67. doi: 10.1053/jhin.2002.1263. [DOI] [PubMed] [Google Scholar]
- 22.Fernanda NdSP: a mini review of Candida species in hospital infection: epidemiology, virulence factor and drugs resistance and prophylaxis. Trop Med Surg 2013, 01(05). https://www.esciencecentral.org/journals/a-mini-review-of-candida-species-in-hospital-infection-2329-9088.1000141.php?aid=18547.
- 23.Sheevani, Sharma P, Aggarwal A. Nosocomial Candida infection in a rural tertiary care hospital. J Clin Diagn Res. 2013;7(2):405–406. doi: 10.7860/JCDR/2013/4574.2759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pitkaranta M, Meklin T, Hyvarinen A, Paulin L, Auvinen P, Nevalainen A, Rintala H. Analysis of fungal flora in indoor dust by ribosomal DNA sequence analysis, quantitative PCR, and culture. Appl Environ Microbiol. 2008;74(1):233–244. doi: 10.1128/AEM.00692-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Andersen B, Frisvad JC, Sondergaard I, Rasmussen IS, Larsen LS. Associations between fungal species and water-damaged building materials. Appl Environ Microbiol. 2011;77(12):4180–4188. doi: 10.1128/AEM.02513-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sandle T. A review of cleanroom microflora: types, trends, and patterns. PDA J Pharm Sci Technol. 2011;65(4):392–403. doi: 10.5731/pdajpst.2011.00765. [DOI] [PubMed] [Google Scholar]
- 27.Mandal J, Brandl H: Bioaerosols in indoor environment—a review with special reference to residential and occupational locations. Open Environ Biol Monit J. 2011; 4(1):83–96.
- 28.La Duc MT, Vaishampayan P, Nilsson HR, Torok T, Venkateswaran K. Pyrosequencing-derived bacterial, archaeal, and fungal diversity of spacecraft hardware destined for Mars. Appl Environ Microbiol. 2012;78(16):5912–5922. doi: 10.1128/AEM.01435-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Adams RI, Miletto M, Taylor JW, Bruns TD. Dispersal in microbes: fungi in indoor air are dominated by outdoor air and show dispersal limitation at short distances. ISME J. 2013;7(7):1262–1273. doi: 10.1038/ismej.2013.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Adams RI, Miletto M, Lindow SE, Taylor JW, Bruns TD. Airborne bacterial communities in residences: similarities and differences with fungi. PLoS One. 2014;9(3):e91283. doi: 10.1371/journal.pone.0091283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chase J, Fouquier J, Zare M, Sonderegger DL, Knight R, Kelley ST, Siegel J, Caporaso JG. Geography and location are the primary drivers of office microbiome composition. mSystems. 2016;1(2):e00022-00016. [DOI] [PMC free article] [PubMed]
- 32.Checinska A, Probst AJ, Vaishampayan P, White JR, Kumar D, Stepanov VG, Fox GE, Nilsson HR, Pierson DL, Perry J, et al. Microbiomes of the dust particles collected from the International Space Station and Spacecraft Assembly Facilities. Microbiome. 2015;3:50. doi: 10.1186/s40168-015-0116-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Onofri S, Selbmann L, Zucconi L, Pagano S. Antarctic microfungi as models for exobiology. Planet Space Sci. 2004;52(1-3):229–237. doi: 10.1016/j.pss.2003.08.019. [DOI] [Google Scholar]
- 34.Barnard RL, Osborne CA, Firestone MK. Responses of soil bacterial and fungal communities to extreme desiccation and rewetting. ISME J. 2013;7(11):2229–2241. doi: 10.1038/ismej.2013.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Onofri S, Selbmann L, de Hoog GS, Grube M, Barreca D, Ruisi S, Zucconi L. Evolution and adaptation of fungi at boundaries of life. Adv Space Res. 2007;40(11):1657–1664. doi: 10.1016/j.asr.2007.06.004. [DOI] [Google Scholar]
- 36.McKay CP, Friedmann EI, Gomez-Silva B, Caceres-Villanueva L, Andersen DT, Landheim R. Temperature and moisture conditions for life in the extreme arid region of the Atacama desert: four years of observations including the El Nino of 1997-1998. Astrobiology. 2003;3(2):393–406. doi: 10.1089/153110703769016460. [DOI] [PubMed] [Google Scholar]
- 37.Gonzalez-Toril E, Llobet-Brossa E, Casamayor EO, Amann R, Amils R. Microbial ecology of an extreme acidic environment, the Tinto river. Appl Environ Microbiol. 2003;69(8):4853–4865. doi: 10.1128/AEM.69.8.4853-4865.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Valerie G. Halophilic fungi in a polyhaline estuarine habitat. J Yeast Fungal Res. 2012;3(3):30–36.
- 39.Zhdanova NN, Tugay T, Dighton J, Zheltonozhsky V, Mcdermott P. Ionizing radiation attracts soil fungi. Mycol Res. 2004;108(09):1089–1096. doi: 10.1017/S0953756204000966. [DOI] [PubMed] [Google Scholar]
- 40.Dadachova E, Casadevall A. Ionizing radiation: how fungi cope, adapt, and exploit with the help of melanin. Curr Opin Microbiol. 2008;11(6):525–531. doi: 10.1016/j.mib.2008.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gessler NN, Egorova AS, Belozerskaya TA. Melanin pigments of fungi under extreme environmental conditions (review) Appl Biochem Microbiol. 2014;50(2):105–113. doi: 10.1134/S0003683814020094. [DOI] [PubMed] [Google Scholar]
- 42.Belozerskaya T, Aslanidi K, Ivanova A, Gessler N, Egorova A, Karpenko Y, Olishevskaya S. Characteristics of extremophylic fungi from chernobyl nuclear power plant. Curr Res Technol Educ Topics Applied Microbiol Microbial Biotechnol. 2010;1:88–94. [Google Scholar]
- 43.Onofri S, Barreca D, Selbmann L, Isola D, Rabbow E, Horneck G, de Vera JP, Hatton J, Zucconi L. Resistance of Antarctic black fungi and cryptoendolithic communities to simulated space and Martian conditions. Stud Mycol. 2008;61:99–109. doi: 10.3114/sim.2008.61.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sterflinger K. Fungi: their role in deterioration of cultural heritage. Fungal Biol Rev. 2010;24(1-2):47–55. doi: 10.1016/j.fbr.2010.03.003. [DOI] [Google Scholar]
- 45.Elumalai P, Elumalai E, David E. Fungi associated with deteriorations of painted wall surfaces: isolation and identification. Eur J Acad Essays. 2014;1(3):48–50. [Google Scholar]
- 46.Joshi SM. The sick building syndrome. Indian J Occup Environ Med. 2008;12(2):61–64. doi: 10.4103/0019-5278.43262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Haleem Khan AA, Mohan Karuppayil S. Fungal pollution of indoor environments and its management. Saudi J Biol Sci. 2012;19(4):405–426. doi: 10.1016/j.sjbs.2012.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Brown GD, Denning DW, Gow NA, Levitz SM, Netea MG, White TC. Hidden killers: human fungal infections. Sci Transl Med. 2012;4(165):165rv113. doi: 10.1126/scitranslmed.3004404. [DOI] [PubMed] [Google Scholar]
- 49.Scharf DH, Heinekamp T, Brakhage AA. Human and plant fungal pathogens: the role of secondary metabolites. PLoS Pathog. 2014;10(1):e1003859. doi: 10.1371/journal.ppat.1003859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mehta SK, Cohrs RJ, Forghani B, Zerbe G, Gilden DH, Pierson DL. Stress-induced subclinical reactivation of varicella zoster virus in astronauts. J Med Virol. 2004;72(1):174–179. doi: 10.1002/jmv.10555. [DOI] [PubMed] [Google Scholar]
- 51.Kwan K, Cooper M, La Duc MT, Vaishampayan P, Stam C, Benardini JN, Scalzi G, Moissl-Eichinger C, Venkateswaran K. Evaluation of procedures for the collection, processing, and analysis of biomolecules from low-biomass surfaces. Appl Environ Microbiol. 2011;77(9):2943–2953. doi: 10.1128/AEM.02978-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lai X, Cao L, Tan H, Fang S, Huang Y, Zhou S. Fungal communities from methane hydrate-bearing deep-sea marine sediments in South China Sea. ISME J. 2007;1(8):756–762. doi: 10.1038/ismej.2007.51. [DOI] [PubMed] [Google Scholar]
- 53.Taylor DL, Bruns TD. Community structure of ectomycorrhizal fungi in a Pinus muricata forest: minimal overlap between the mature forest and resistant propagule communities. Mol Ecol. 1999;8(11):1837–1850. doi: 10.1046/j.1365-294x.1999.00773.x. [DOI] [PubMed] [Google Scholar]
- 54.Abarenkov K, Henrik Nilsson R, Larsson KH, Alexander IJ, Eberhardt U, Erland S, Hoiland K, Kjoller R, Larsson E, Pennanen T, et al. The UNITE database for molecular identification of fungi—recent updates and future perspectives. New Phytol. 2010;186(2):281–285. doi: 10.1111/j.1469-8137.2009.03160.x. [DOI] [PubMed] [Google Scholar]
- 55.Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol. 2013;30(12):2725–2729. doi: 10.1093/molbev/mst197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Brescia CC, Griffin SM, Ware MW, Varughese EA, Egorov AI, Villegas EN. Cryptosporidium propidium monoazide-PCR, a molecular biology-based technique for genotyping of viable Cryptosporidium oocysts. Appl Environ Microbiol. 2009;75(21):6856–6863. doi: 10.1128/AEM.00540-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gardes M, Bruns TD. ITS primers with enhanced specificity for basidiomycetes—application to the identification of mycorrhizae and rusts. Mol Ecol. 1993;2(2):113–118. doi: 10.1111/j.1365-294X.1993.tb00005.x. [DOI] [PubMed] [Google Scholar]
- 58.White TJ, Bruns T, Lee S, Taylor J. Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. PCR Protocols. 1990;18(1):315–322. [Google Scholar]
- 59.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser B Methodol. 1995;57(1):289–300. [Google Scholar]
- 60.Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ, et al. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol. 2009;75(23):7537–7541. doi: 10.1128/AEM.01541-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Huse SM, Welch DM, Morrison HG, Sogin ML. Ironing out the wrinkles in the rare biosphere through improved OTU clustering. Environ Microbiol. 2010;12(7):1889–1898. doi: 10.1111/j.1462-2920.2010.02193.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics. 2011;27(16):2194–2200. doi: 10.1093/bioinformatics/btr381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wang Q, Garrity GM, Tiedje JM, Cole JR. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol. 2007;73(16):5261–5267. doi: 10.1128/AEM.00062-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bengtsson-Palme J, Ryberg M, Hartmann M, Branco S, Wang Z, Godhe A, De Wit P, Sánchez-García M, Ebersberger I, de Sousa F et al. Improved software detection and extraction of ITS1 and ITS2 from ribosomal ITS sequences of fungi and other eukaryotes for analysis of environmental sequencing data. Methods Ecol Evol 2013:n/a-n/a.
- 65.La Duc MT, Satomi M, Agata N, Venkateswaran K. gyrB as a phylogenetic discriminator for members of the Bacillus anthracis-cereus-thuringiensis group. J Microbiol Methods. 2004;56(3):383–394. doi: 10.1016/j.mimet.2003.11.004. [DOI] [PubMed] [Google Scholar]
- 66.Weinmaier T, Probst AJ, La Duc MT, Ciobanu D, Cheng JF, Ivanova N, Rattei T, Vaishampayan P. A viability-linked metagenomic analysis of cleanroom environments: eukarya, prokaryotes, and viruses. Microbiome. 2015;3:62. doi: 10.1186/s40168-015-0129-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sharpe RA, Bearman N, Thornton CR, Husk K, Osborne NJ. Indoor fungal diversity and asthma: a meta-analysis and systematic review of risk factors. J Allergy Clin Immunol. 2015;135(1):110–122. doi: 10.1016/j.jaci.2014.07.002. [DOI] [PubMed] [Google Scholar]
- 68.Nocker A, Sossa-Fernandez P, Burr MD, Camper AK. Use of propidium monoazide for live/dead distinction in microbial ecology. Appl Environ Microbiol. 2007;73(16):5111–5117. doi: 10.1128/AEM.02987-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Vaishampayan P, Probst AJ, La Duc MT, Bargoma E, Benardini JN, Andersen GL, Venkateswaran K. New perspectives on viable microbial communities in low-biomass cleanroom environments. ISME J. 2013;7(2):312–324. doi: 10.1038/ismej.2012.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Vesper S, McKinstry C, Hartmann C, Neace M, Yoder S, Vesper A. Quantifying fungal viability in air and water samples using quantitative PCR after treatment with propidium monoazide (PMA) J Microbiol Methods. 2008;72(2):180–184. doi: 10.1016/j.mimet.2007.11.017. [DOI] [PubMed] [Google Scholar]
- 71.Crespo-Sempere A, Estiarte N, Marin S, Sanchis V, Ramos AJ. Propidium monoazide combined with real-time quantitative PCR to quantify viable Alternaria spp. contamination in tomato products. Int J Food Microbiol. 2013;165(3):214–220. doi: 10.1016/j.ijfoodmicro.2013.05.017. [DOI] [PubMed] [Google Scholar]
- 72.Lax S, Smith DP, Hampton-Marcell J, Owens SM, Handley KM, Scott NM, Gibbons SM, Larsen P, Shogan BD, Weiss S, et al. Longitudinal analysis of microbial interaction between humans and the indoor environment. Science. 2014;345(6200):1048–1052. doi: 10.1126/science.1254529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Adams RI, Miletto M, Taylor JW, Bruns TD. The diversity and distribution of fungi on residential surfaces. PLoS One. 2013;8(11):e78866. doi: 10.1371/journal.pone.0078866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Noble WC. Dispersal of skin microorganisms*. Br J Dermatol. 1975;93(4):477–485. doi: 10.1111/j.1365-2133.1975.tb06527.x. [DOI] [PubMed] [Google Scholar]
- 75.Reinmüller B, Ljungqvist B. Modern cleanroom clothing systems: people as a contamination source. PDA J Pharm Sci Technol. 2003;57(2):114–125. [PubMed] [Google Scholar]
- 76.Cui L, Morris A, Ghedin E. The human mycobiome in health and disease. Genome Med. 2013;5(7):63. doi: 10.1186/gm467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Findley K, Oh J, Yang J, Conlan S, Deming C, Meyer JA, Schoenfeld D, Nomicos E, Park M, NIHISCCS Program et al. Topographic diversity of fungal and bacterial communities in human skin. Nature. 2013;498(7454):367–370. doi: 10.1038/nature12171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Huffnagle GB, Noverr MC. The emerging world of the fungal microbiome. Trends Microbiol. 2013;21(7):334–341. doi: 10.1016/j.tim.2013.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Richard ML, Lamas B, Liguori G, Hoffmann TW, Sokol H. Gut fungal microbiota: the Yin and Yang of inflammatory bowel disease. Inflamm Bowel Dis. 2015;21(3):656–665. doi: 10.1097/MIB.0000000000000261. [DOI] [PubMed] [Google Scholar]
- 80.Kettleson EM, Adhikari A, Vesper S, Coombs K, Indugula R, Reponen T. Key determinants of the fungal and bacterial microbiomes in homes. Environ Res. 2015;138:130–135. doi: 10.1016/j.envres.2015.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Vesper S, Wymer L. The relationship between environmental relative moldiness index values and asthma. Int J Hyg Environ Health. 2016;219(3):233–238. doi: 10.1016/j.ijheh.2016.01.006. [DOI] [PubMed] [Google Scholar]
- 82.Simon-Nobbe B, Denk U, Poll V, Rid R, Breitenbach M. The spectrum of fungal allergy. Int Arch Allergy Immunol. 2008;145(1):58–86. doi: 10.1159/000107578. [DOI] [PubMed] [Google Scholar]
- 83.Salo PM, Arbes SJ, Jr, Sever M, Jaramillo R, Cohn RD, London SJ, Zeldin DC. Exposure to Alternaria alternata in US homes is associated with asthma symptoms. J Allergy Clin Immunol. 2006;118(4):892–898. doi: 10.1016/j.jaci.2006.07.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kuna P, Kaczmarek J, Kupczyk M. Efficacy and safety of immunotherapy for allergies to Alternaria alternata in children. J Allergy Clin Immunol. 2011;127(2):502–508. doi: 10.1016/j.jaci.2010.11.036. [DOI] [PubMed] [Google Scholar]
- 85.Knutsen AP, Bush RK, Demain JG, Denning DW, Dixit A, Fairs A, Greenberger PA, Kariuki B, Kita H, Kurup VP, et al. Fungi and allergic lower respiratory tract diseases. J Allergy Clin Immunol. 2012;129(2):280–291. doi: 10.1016/j.jaci.2011.12.970. [DOI] [PubMed] [Google Scholar]
- 86.Crucian B, Stowe R, Quiriarte H, Pierson D, Sams C. Monocyte phenotype and cytokine production profiles are dysregulated by short-duration spaceflight. Aviat Space Environ Med. 2011;82(9):857–862. doi: 10.3357/ASEM.3047.2011. [DOI] [PubMed] [Google Scholar]
- 87.Seifert KA: Compendium of soil fungi—by K.H. Domsch, W. Gams & T.-H. Anderson. Eur J Soil Sci. 2009;59:1007.
- 88.Zhdanova NN, Zakharchenko VA, Vember VV, Nakonechnaya LT. Fungi from Chernobyl: mycobiota of the inner regions of the containment structures of the damaged nuclear reactor. Mycol Res. 2000;104(12):1421–1426. doi: 10.1017/S0953756200002756. [DOI] [Google Scholar]
- 89.Gunde-Cimerman N, Zalar P, Hoog S, Plemenitasd A. Hypersaline waters in salterns a “ natural ecological niches for halophilic black yeasts. FEMS Microbiol Ecol. 2000;32(3):235–240. doi: 10.1111/j.1574-6941.2000.tb00716.x. [DOI] [PubMed] [Google Scholar]
- 90.Butinar L, Santos S, Spencer-Martins I, Oren A, Gunde-Cimerman N. Yeast diversity in hypersaline habitats. FEMS Microbiol Lett. 2005;244(2):229–234. doi: 10.1016/j.femsle.2005.01.043. [DOI] [PubMed] [Google Scholar]
- 91.Dhakar K, Sharma A, Pandey A. Cold, pH and salt tolerant Penicillium spp. inhabit the high altitude soils in Himalaya, India. World J Microbiol Biotechnol. 2013;30:1315–1324. doi: 10.1007/s11274-013-1545-4. [DOI] [PubMed] [Google Scholar]
- 92.Wilson DM, Mubatanhema W, Jurjevic Z. Biology and ecology of mycotoxigenic Aspergillus species as related to economic and health concerns. Adv Exp Med Biol. 2002;504:3–17. doi: 10.1007/978-1-4615-0629-4_2. [DOI] [PubMed] [Google Scholar]
- 93.Knox BP, Blachowicz A, Palmer JM, Romsdahl J, Huttenlocher A, Wang CC, Keller NP, Venkateswaran K: Characterization of Aspergillus fumigatus isolates from air and surfaces of the International Space Station. mSphere 2016, 1(5). http://msphere.asm.org/content/1/5/e00227-16. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Schematic representation of the ILMAH architecture with dimensions. The sampling locations are indicated with stars and numbers (1, 2: Bedroom; 3, 4: Kitchen; 5: Bathroom; and 6, 7, 8: Laboratory). (PDF 911 kb)
Crew schedule assignments for the 30 days. (PDF 911 kb)
R code and script used as published in Weinmaier and Probst et al., 2015 Microbiome. (PDF 300 kb)
Cultivable fungal characteristics of ILMAH surface samples. Tables represent: a) CFU counts for each sampling area during consecutive sampling events (before crew occupation, Day 13, Day 20 and Day 30). Sampling areas are numbered 1-8. 1 and 2 correspond to bedroom area, 3, 4 – kitchen, 5 – bathroom and 6-8 lab. CFU counts are reported per meter square; b) CFU counts for specific ILMAH compartments. Table contains average CFU counts for each compartment: bedroom, kitchen, bathroom and lab area (for example CFU counts from area 1 and 2 at Day 13 were used to calculate the average CFU count for the bedroom compartment at Day 13). CFU counts are reported per meter square. (PDF 52 kb)
Statistical analysis to compare cultivable fungal populations of the different (a) time points and (b) locations. (a) CFU counts of cultivable fungal populations observed at various time point were compared to each other to asses if there are any statistically significant changes in CFU counts over the course of time; (b) CFU counts of cultivable fungal populations observed at different compartments were compared to each other to asses if there are any statistically significant changes in CFU counts between locations. (PDF 43 kb)
NMDS ordinations based on Bray-Curtis distances between non-PMA- and PMA-treated samples taken at different time points. The analysis shows a significant difference between the PMA treated and not treated samples and between the different time points but not between the different locations. A “P” after the respective variable indicates that these are the samples treated with PMA. (PDF 44 kb)
NMDS ordinations based on Bray-Curtis distances between PMA treated samples without T20 taken at different time points. A “P” after the respective variable indicates that these are the samples treated with PMA. (PDF 30 kb)
Statistical analysis of viable (PMA treated) samples to compare fungal populations of the different a) time points and b) locations. (a) Community profiles of viable fungal populations observed at various time point were compared to each other to asses if there are any statistically significant changes over the course of time; (b) Community profiles of viable fungal populations observed at different compartments were compared to each other to asses if there are any statistically significant changes between locations. Results marked with * are statistically significant. (PDF 44 kb)
Percent change of OTU counts at family level. Information about the percent changes of the OTU counts of selected families during consecutive time points is presented. (PDF 45 kb)
SRA numbers for individual samples. This file contains detailed information about accession number of each sample deposited within the study SPR069729. (TXT 12 kb)
OTU deduced from the PMA treated samples. This file contains OTU counts of all PMA treated samples collected during the study. The numbering pattern is explained as follows 30 means 30-day study. The first number (0–4) will be the sample collection day (0 = T0, 2=T13, 3=T20, 4=T30), second number (1–8) will be sampling location, and P stands for PMA-treated samples. For example, 30.06P will be a sample collected during the 30-day study from T0 at location 6. (CSV 334 kb)
OTU deduced from the PMA untreated samples. This file contains OTU counts of all samples not treated with PMA collected during the study. The numbering pattern is explained as follows 30 means 30-day study. The first number (0–4) will be the sample collection day (0 = T0, 2=T13, 3=T20, 4=T30), second number (1–8) will be sampling location. For example, 30.06 will be a sample collected during the 30-day study from T0 at location 6. (ZIP 35 kb)
OTU table sorted by location. This file contains OTU counts of all the families observed throughout the study for PMA treated (viable) and untreated (total) samples for various locations (bedroom, kitchen, bathroom and lab). (CSV 2 kb)
OTU table sorted by location. This file contains OTU counts of all the families observed throughout the study for PMA treated (viable) and untreated (total) samples for various time points (T0, T13, T20 and T30). (TXT 132 kb)
Data Availability Statement
The dataset supporting the results of this article is available in the NCBI SRA repository, under study accession # SRP069729. SRA numbers for individual samples are listed in attached Additional file 10. The ITS sequences of the cultivable fungi are available in the NCBI GenBank under accession # KX664307-KX664419. PMA-treated and untreated OTU tables are submitted as Additional files 11 and 12, respectively, along with the OTU tables sorted by location and time point (Additional files 12 and 13, respectively). ITS sequences used to create phylogenetic tree are included in Additional file 14.