

Abstract
Early maladaptive internalization of synaptic GABAA receptors (GABAA R) and externalization of NMDA receptors (NMDAR) may explain the time-dependent loss of potency of standard anti-epileptic drugs (AED) in refractory status epilepticus (SE). We hypothesized that correcting the effects of changes in GABAAR and NMDAR would terminate SE, even when treatment is delayed 40 minutes. SE was induced in adult Sprague-Dawley rats with a high dose of lithium and pilocarpine. The GABAAR agonist midazolam, the NMDAR antagonist ketamine and the AED valproate were injected 40 min after SE onset in combination or as monotherapy. The midazolam-ketamine-valproate combination was more efficient than triple-dose midazolam, ketamine or valproate monotherapy or higher-dose dual therapy in reducing several parameters of SE severity. Triple therapy also reduced SE-induced acute neuronal injury and spatial memory deficits. In addition, simultaneous triple therapy was more efficient than sequential triple therapy: giving the three drugs simultaneously was more efficient at stopping seizures than the standard practice of giving them sequentially. Furthermore, midazolam-ketamine-valproate therapy suppressed seizures far better than the midazolam-fosphenytoin-valproate therapy, which follows evidence-based AES guidelines. These results show that a treatment aimed at correcting maladaptive GABAAR and NMDAR trafficking can reduce the severity of SE and its long-term consequences.
Keywords: refractory status epilepticus, cholinergic seizures, midazolam, ketamine, valproate
INTRODUCTION
Pharmacoresistance to benzodiazepines and other drugs (Kapur and Macdonald, 1997; Mazarati et al., 1998) remains a challenge in the treatment of status epilepticus (SE), a life-threatening condition which affects 150,000–200,000 patients per year in the USA and is responsible for 22,000–42,000 deaths yearly (DeLorenzo et al., 1996). The incidence of SE increased from 3.5 to 12.5/100,000 between 1979 and 2010 (Dham et al., 2014). Benzodiazepine monotherapy, which is recommended for initial treatment of SE, fails to stop seizures in 35–69% of cases (Glauser et al., 2016; Holtkamp et al., 2005; Mayer et al., 2002; Treiman et al., 1998).
Studies in experimental models of SE show that early maladaptive internalization of synaptic GABAA receptors (GABAAR) may explain the loss of benzodiazepine potency (Goodkin et al., 2008; Goodkin et al., 2005; Kapur and Macdonald, 1997; Mazarati et al., 1998; Naylor et al., 2013; Naylor et al., 2005). The drugs may stop the seizures in the early stage of SE by binding to GABAAR, but progressively lose potency when GABAAR are inactivated by internalization into endosomes. At the same time, glutamatergic excitation, driven by migration of NMDAR subunits toward synapses (Naylor et al., 2013), is increasing runaway excitation and excitotoxicity. We hypothesized that polytherapy aimed at correcting the consequences of receptor trafficking should reduce SE severity (Niquet et al., 2016b). Indeed, combinations of a GABAAR agonist and an NMDAR antagonist, such as diazepam and ketamine (Martin and Kapur, 2008) or midazolam and ketamine (Niquet et al., 2016a) have been successful in treating experimental SE and may be synergistic. However, when treatment is delayed, the reduction of the number of synaptic GABAAR makes it difficult to fully restore inhibition with benzodiazepines, and another AED acting at a non-benzodiazepine site is needed to restore the balance between excitation and inhibition. In the present study, we treated 40 minutes after seizure onset, and combined midazolam and ketamine with the AED valproate. We also studied the timing of drug delivery, since recent studies suggest that it is a major determinant of pharmacoresistance (Silbergleit et al., 2012), and compared AES guideline-inspired combinations to our combination, which is based on the receptor-trafficking hypothesis. Our results show that the simultaneous administration of midazolam, ketamine and valproate is more efficient in stopping seizures than triple dose monotherapy, higher-dose dual therapy, sequential triple therapy, or the midazolam-fosphenytoin-valproate combination.
METHODS
Animals
Male Sprague-Dawley rats (200–300g, mean 249g; Charles River, MA) were housed in a temperature- and humidity- controlled room with 12 h light-dark cycles (7 am–7 pm) and had free access to food and water. All experiments were conducted with the approval and in accordance with the regulations of the Institutional Animal care and Use Committee of West Los Angeles VA Medical Center.
Induction of SE, Monotherapy and Dual Therapy
Rats were administered lithium chloride (5 mEq/kg; #L-0505 Sigma, St. Louis MO, USA) subcutaneously and, 16 h later, SE was induced with i.p. pilocarpine hydrochloride (320 mg/kg; #P6503 Sigma). Only lithium/pilocarpine-treated rats displaying behavioral /EEG seizures were used. All rats received scopolamine methyl bromide (1mg/kg; i.p., #S8502; Sigma), a muscarinic antagonist that does not cross the blood-brain barrier, at the same time as pilocarpine, to decrease peripheral cholinergic effects such as pulmonary secretions. Seizures occurred 7.6+/−2.7 min. after pilocarpine injection, so that time from pilocarpine injection to mono or dual therapy was approximately 48 min. All animals subsequently received scopolamine (10 mg/kg i.p.; #S1013; Sigma) to remove the original seizure trigger without stopping SE, and sham injection (control SE group), one drug (monotherapy), a combination of two drugs (dual therapy) or a combination of three drugs (triple therapy) i.p. 40 min after EEG seizure onset to make sure that pharmacoresistance and self-sustaining seizures were well established. Drugs for monotherapy groups included midazolam (9 mg/kg; Caraco Pharmaceutical Laboratories Ltd), ketamine (90 mg/kg; #RL3760 Hospira), sodium valproate (270 mg/kg; #P4543 Sigma). Dual therapy groups included combination of 4.5 mg/kg midazolam with 45 mg/kg ketamine. Triple therapy groups included combination of 3 mg/kg midazolam with 30 mg/kg ketamine and 90 mg/kg valproate, or 3 mg/kg midazolam with 30 mg/kg ketamine and 100 mg/kg levetiracetam (UCB Pharmaceuticals), or 3 mg/kg midazolam with 50 mg/kg fosphenytoin (Parke-Davis), and 90 mg/kg valproate. The doses used were determined in preliminary experiments when we delayed treatment to 40 min. after seizure onset, instead of after the second stage 3 seizure, see (Niquet et al., 2017), which required a higher therapeutic dose. The midazolam dose is lower than the anesthetic doses (25 mg/kg) used in other models of SE (Kofke et al., 1993) but similar to the 1–5 mg/kg used against sarin-induced SE (Chapman et al., 2015). The fosphenytoin dose was selected because of the effectiveness of phenytoin in the perforant path stimulation model of SE (Mazarati et al., 1998). The ketamine dose was higher than the dose (10 mg/kg) which stopped perforant path stimulation SE (Mazarati and Wasterlain, 1999), but similar to the monotherapy dose (100 mg/kg) which stopped hippocampal stimulation-induced or chemically-induced SE (Borris et al., 2000; Fujikawa, 1995) or the dose (50 mg/kg) which stopped SE when combined with diazepam (Martin and Kapur, 2008). Levetiracetam was previously used in performant path SE (Mazarati et al., 2004) groups, and triple dose of drugs compared to triple therapy groups to compensate for the number of drugs used. In the sequential monotherapy group (figure 4), 3 mg/kg midazolam, 90 mg/kg valproate and 30 mg/kg ketamine were injected sequentially 30 min apart. For long-term behavioral studies, a sham group, which did not receive drug treatment and was not exposed to SE, was added.
Figure 4. Midazolam-ketamine-valproate therapy reduces behavioral deficits in the Morris water maze.
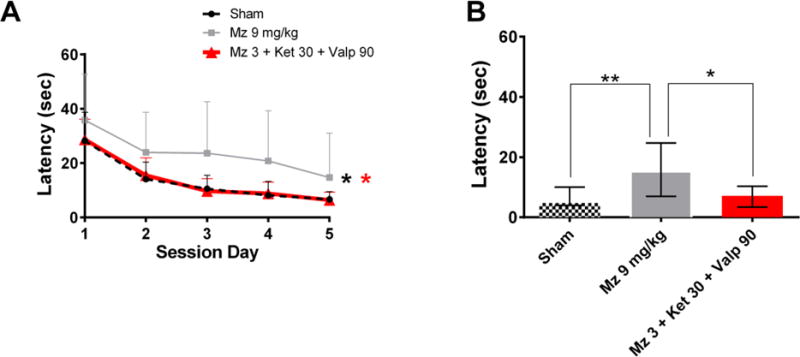
(A) Graph A shows the latency to reach the hidden platform (y-axis) on each testing day (x-axis). Data are presented as mean ± SEM. * p<0.05 vs Mz 9 mg/kg by 2-way-ANOVA.
(B) Graph B shows the latency during the retention test. * p<0.05 and ** p<0.01 vs Mz 9 mg/kg by Kruskal-Wallis followed by Dunn’s test.
Rats not capable of coordinated walking and movement 16 h after SE were injected SC (10ml/kg) with 5% glucose twice per day until capable of coordinated movement or until euthanasia at three days. Water moistened food pellets and or gelatin cubes were placed in the cage in Petri dishes. Euthanasia criteria consisted of failure to achieve coordinated movement three days after SE. Animals were euthanized if showing a weight loss of 5% sustained over two days after the coordination criterion had been achieved.
Implantation of electrodes
Under isoflurane anesthesia, the animals were implanted with stainless steel skull screws to serve as recording electrodes. Two Electrodes were used for bipolar recording and were located 3mm anterior to lambda and 4 mm left and right of the medial suture. The third electrode served as reference and was located 1mm anterior to bregma and 1mm to the right of the midline defined by the medial suture. The electrodes were connected to a tri-polar connector (Plastics One, VA) and dental cement was used to cover the electrodes so that only the connector was exposed. Animals were used one to two weeks after electrode implantation. The BioPac Systems MP150 was used to record digital EEG using a BioPac UM100A preamplifier. Sampling rate was 200Hz.
Acute video-EEG monitoring
Recording was started before pilocarpine injection and was continuous for 24 h which included an initial pre-pilocarpine segment of EEG, the development of SE, drug treatment, and the overnight recovery period (Fig. 1 A). The EEGs were processed offline to detect seizures and spikes using Stellate Systems Harmony software (Natus) with default parameters: amplitude threshold 2.7, minimum frequency 3 Hz, maximum coefficient of variation 40% for seizure detection, and a spike amplitude threshold of 6 for spike detection.
Figure 1. Midazolam-ketamine-valproate therapy is more effective than triple-dose midazolam, ketamine or valproate in reducing SE severity.
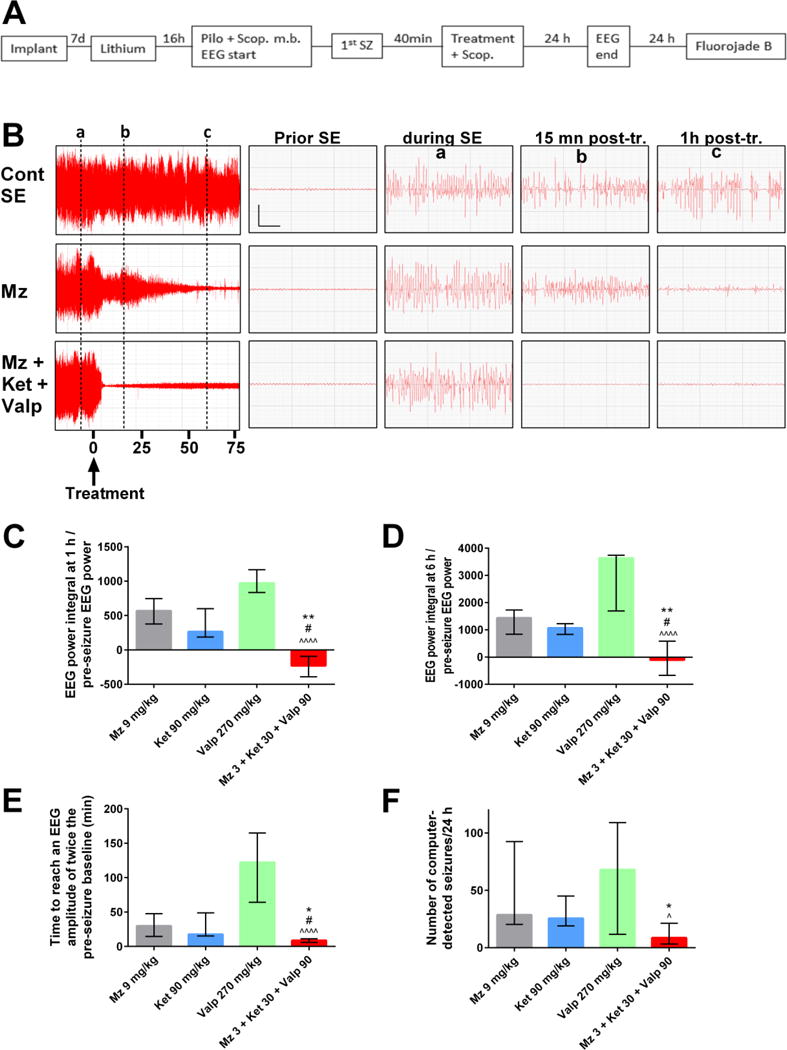
A) Experimental flow: A severe form of status epilepticus (SE) was induced by administration of a high dose of lithium + scopolamine methyl bromide (scop. m. b), followed by an injection of a high dose of pilocarpine. Drug(s) or vehicle, and scopolamine (scop.) were injected 40 minutes after seizure onset. Animals were implanted 1 week prior SE induction and sacrificed 48 h after SE onset to assess neuronal injury with fluoro-jade B staining.
B) The left panels show the compressed EEG from SE control, midazolam, or midazolam-ketamine-valproate animals up to 75 min following treatment. The right panels show magnified 6-sec EEG tracings prior to SE or following SE (marked by vertical lines a–c). Vertical bar = 0.5 mV; horizontal bar = 1 sec.
C) This graph shows the ratio of EEG power integral over the first hour to initial EEG power at baseline, before pilocarpine injection. The midazolam-ketamine-valproate group (n= 10), which displayed an EEG power that fell below the pre-pilocarpine baseline, is significantly different from the midazolam (n=10; ** p <0.01), ketamine (n=8, # p<0.05) and valproate (n=10, ^^^^ p <0.0001) groups by Kruskal-Wallis, followed by Dunn’s test.
D) This graph shows the ratio of EEG power integral over the first 6 hours following treatment to initial EEG power at baseline, before pilocarpine injection. The midazolam-ketamine-valproate group (n= 10), which lowered the EEG power below pre-pilocarpine baseline, is significantly different from midazolam (n=10; ** p <0.01), ketamine (n=8, # p<0.05) and valproate (n=7, ^^^^ p <0.0001) by Kruskal-Wallis, followed by Dunn’s test.
E) This graph shows the time needed after treatment to reach an EEG amplitude of twice the pre-seizure baseline. The midazolam-ketamine-valproate group (n=9), which has the shortest time, is significantly different from midazolam (n=10, * p <0.05), ketamine (n=8, # p < 0.05) and valproate (n=8, ^^^^ p <0.0001) by Kruskal-Wallis, followed by Dunn’s test.
F) This graph shows the number of computer-detected seizures per 24 hours. The midazolam-ketamine-valproate group (n=9), which has the lowest number of seizures, is significantly different from midazolam (n=10, * p <0.05) and valproate (n=6, ^ p <0.05) by Kruskal-Wallis, followed by Dunn’s test.
Outcome measures were the ratio of EEG power at T time divided by the average baseline EEG power before pilocarpine; the number of seizures per 24 h, the number of spikes per 24 h; the time needed for EEG amplitude to fall for the first time below 2 times the pre-pilocarpine EEG amplitude and be free of semi-periodic spikes or sharp waves for at least one minute, and the time in SE after treatment, as previously described (Niquet et al., 2016a; Suchomelova et al., 2006). EEG outcome measures for midazolam, ketamine, valproate, and midazolam-ketamine therapy have been previously published (Niquet et al., 2016a).
Tissue preparation for detection of acute neuronal injury
The animals were anesthetized with an overdose of pentobarbital (100 mg/kg i.p.) 48 h after induction of SE. Then, the animals underwent transcardiac perfusion with 4% phosphate-buffered formaldehyde (#P-6148 Sigma). Brains were kept in situ at 4 °C overnight, after which they were removed and postfixed in the same perfusate for 2–3 h. Subsequently, brains were kept in PB 0.1 M containing 30% sucrose for 48–72 h. Floating sections (30 μM thickness) were obtained using a sliding microtome. Coronal sections were mounted, dried, incubated in potassium permanganate solution (0.06%; w/v) for 15 min, washed and incubated in fluoro-Jade B staining solution for 30 min. After 3 rinses, slides were dried overnight at room temperature, cleared three times in xylene and coverslipped with Permount medium. In CA1 and CA3 areas and in the hilus of the dentate gyrus, the number of injured cells was counted by unbiased stereology using the optical dissector method. The first series of one in five sections were stained with fluoro-Jade B, and the analysis was performed using a microscope (Olympus AX70) with a motorized stage connected to a computer running the Stereo Investigator software (MBF Bioscience). A counting frame of 45 × 45 μm was randomly positioned in a sampling grid of 70 × 120 μm. In the other areas (frontoparietal, entorhinal, and piriform cortices, thalamus, and amygdala), distribution of fluoro-jade B-positive cells was scored as follows: 0, no injury; 1: 1–30 positive cells per field; 2: 31–60 positive cells per field; 3: 61–100 positive cells per field; 4: more than 100 positive cells per field, as previously described (Suchomelova et al 2006, Niquet et al 2016). Cell counts for midazolam, ketamine, valproate, and midazolam-ketamine therapy have been previously published (Niquet et al., 2016a).
Morris water maze paradigm
Spatial learning and memory were evaluated 6 weeks after SE recording with a modified Morris water maze paradigm, by requiring the rats to swim in a pool 170 cm in diameter, with the water kept at 20°C, to find a 12-cm diameter circular platform submerged 2 cm beneath the surface of the water, which was opacified by the addition of black non-toxic tempera paint. The platform was in a constant position during training, as there were a number of visual cues in the testing room. Experiments were monitored with a Sony CCD-IRIS high-resolution camera mounted above the pool and using indirect lighting from a 25 W bulb. A video-tracking system (Ethovision; Noldus, Inc. Wageningen, The Netherlands) was used for data acquisition. The rats were brought to the experimental room at least 30 min prior to an experiment. Each rat was trained to find the hidden platform kept in the same location for one session of eight trials per day, for 5 consecutive days. The start sequence was randomly selected and was different for each day. For training, the rat was released in the water from one of the four starting positions, facing the wall of the pool. It was given 60s to locate and climb onto the platform, where it stayed for 30s. If a rat did not find the platform within 60s, it was gently guided to it by experimenter. After the end of the last session, the rat was dried with absorbent paper and kept in a warm cage. Ten days later, acquisition test for long-term memory retention was performed.
The treatment protocols and dosage are summarized in the table 1.
Table 1.
Summary of drug dosage and time of administration.
| Treatments | Time of admin. | |
|---|---|---|
| Figure 1 | midazolam 9 mg/kg ketamine 90 mg/kg valproate 270 mg/kg midazolam 3 mg/kg + ketamine 30 mg/kg + valproate 90 mg/kg |
40 min after EEG seizure onset |
| Figure 2 | midazolam 4.5 mg/kg + ketamine 45 mg/kg midazolam 3 mg/kg + ketamine 30 mg/kg + valproate 90 mg/kg midazolam 3 mg/kg + ketamine 30 mg/kg + levetiracetam 100 mg/kg |
40 min after EEG seizure onset |
| Figure 3 and table 2 | midazolam 9 mg/kg ketamine 90 mg/kg valproate 270 mg/kg midazolam 4.5 mg/kg + ketamine 45 mg/kg midazolam 3 mg/kg + ketamine 30 mg/kg + valproate 90 mg/kg |
40 min after EEG seizure onset |
| Figure 4 | midazolam 9 mg/kg midazolam 3 mg/kg + ketamine 30 mg/kg + valproate 90 mg/kg |
40 min after seizure onset |
| Figure 5 | midazolam 9 mg/kg midazolam 3 mg/kg + ketamine 30 mg/kg + valproate 90 mg/kg |
40 min after EEG seizure onset or sequentially |
| Figure 6 | midazolam 3 mg/kg + fosphenytoin 50 mg/kg + valproate 90 mg/kg midazolam 3 mg/kg + ketamine 30 mg/kg + valproate 90 mg/kg |
40 min after EEG seizure onset |
Statistical analyses
When data showed a Gaussian distribution (Figure 5 B–C), they were presented as mean values ± standard deviation (SD) and analyzed with parametric statistical methods: ANOVA followed by Tukey’s multiple comparison (GraphPad version 6). When data showed a non-Gaussian distribution, they were presented as median values with the interquartile range, which is the difference between the 75th and 25th percentile, and analyzed with nonparametric statistical methods: Kruskal-Wallis test followed by Dunn’s multiple comparison test (Figure 1, 2A, 3, 5 D–E) or Mann-Whitney test (Figure 6). MWM data was analyzed by 2-way ANOVA (GraphPad version 6). Statistical significance was defined as p< 0.05.
Figure 5. Simultaneous polytherapy is far more effective in reducing EEG power and stopping SE than sequential monotherapies.
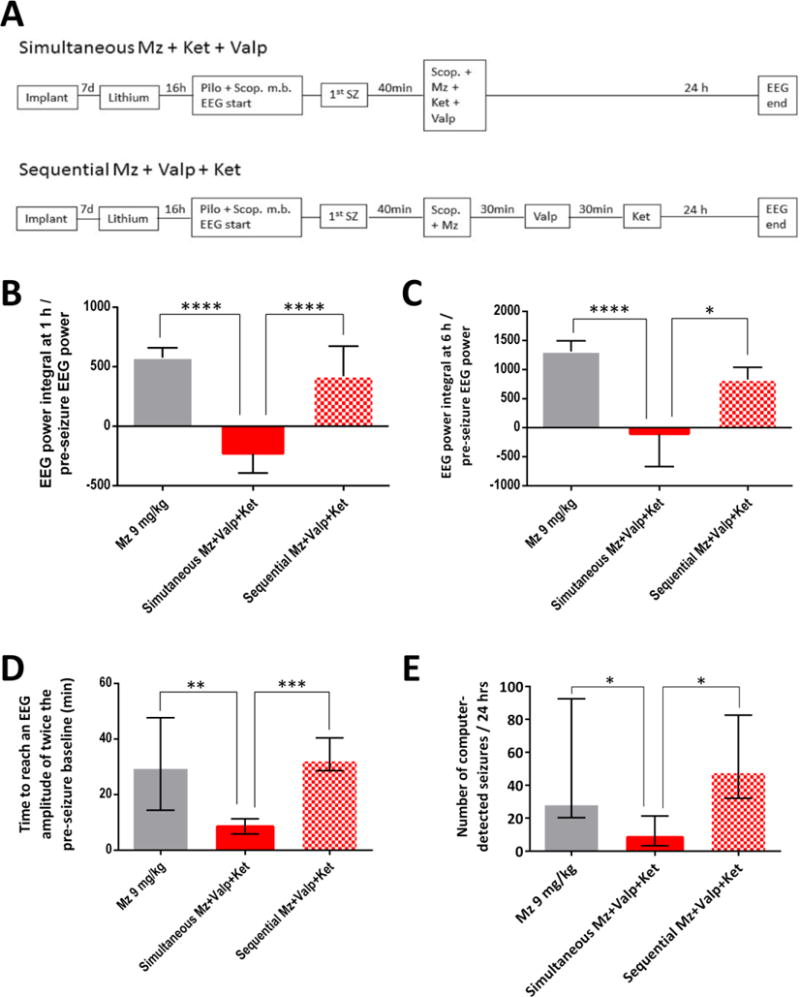
A) Experimental flow: In the simultaneous group, the combination of midazolam 3 mg/kg, ketamine 30 mg/kg and valproate 90 mg/kg were administered simultaneously 40 min after SE onset. In the sequential group, the same drugs at the same dose were injected 30 min apart.
B–E) The graphs show the ratio of EEG power integral to initial EEG power at baseline over the first hour (B) or the first six hours (C), the time needed for EEG amplitude to decline to twice the pre-seizure baseline (D) and the number of computer-detected seizures per 24 hours (E). Simultaneous polytherapy (n=10) was far more effective than sequential monotherapies (n=8–9) or higher-dose midazolam (n=10) in reducing EEG power, stopping SE (as indicated by EEG amplitude declining to twice pre-seizure baseline) and reducing the number of seizures. In graphs B–C, * p <0.05 or **** p <0.0001 by ANOVA followed by Tukey’s multiple comparison. In graphs D–E, * p <0.05, ** p <0.01, *** p <0.001 by Kruskal-Wallis analysis followed by Dunn’s test.
Figure 2.
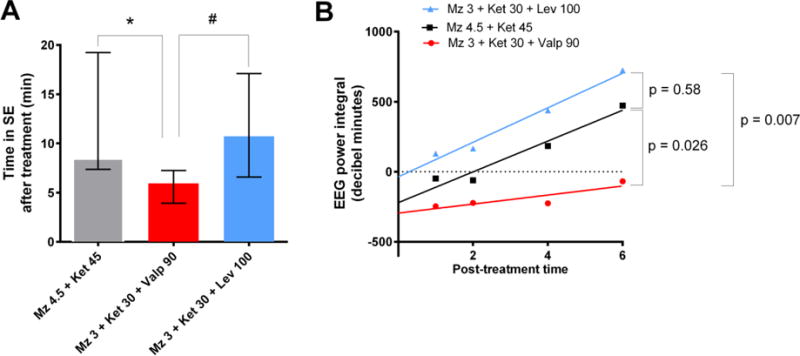
Midazolam-ketamine-valproate therapy is more effective than higher-dose dual therapy with midazolam and ketamine in reducing SE severity.
(A) This graph shows the time in SE after treatment. The midazolam-ketamine-valproate group (n=10), which has the lowest duration among all groups, is significantly different from midazolam-ketamine (n=9, * p <0.05) and midazolam-ketamine-levetiracetam (n=9, # p <0.05) by Kruskal-Wallis, followed by Dunn’s test.
(B) This graph shows the regression lines of the EEG power integral ratio over the first hour post-treatment. A p value smaller than 0.05 means that there is a significant difference between the slopes of the regression lines, with lower values indicating better seizure suppression. The slope of the midazolam-ketamine-valproate line is significantly lower than the slope of the midazolam-ketamine or midazolam-ketamine-levetiracetam lines.
Figure 3. Reduction of neuronal injury in animals treated with midazolam-ketamine-valproate therapy or triple-dose midazolam or ketamine.
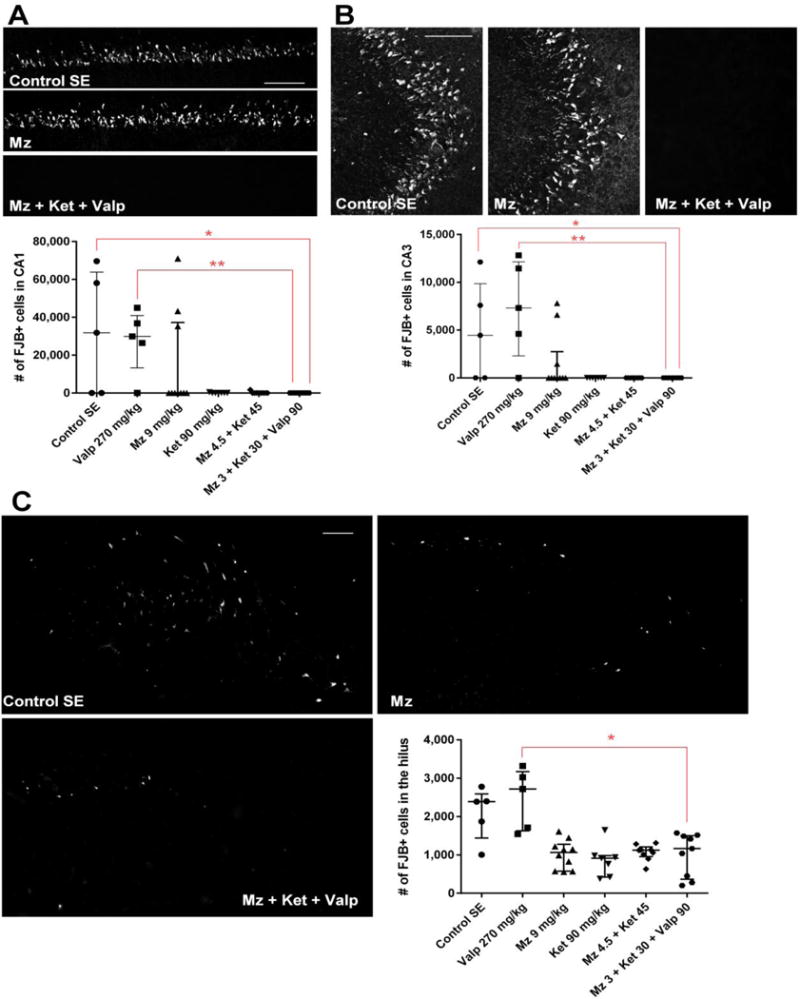
(A) The upper panels show confocal images of fluoro-jade B staining in CA1 48 h following SE in the SE control, midazolam and midazolam-ketamine-valproate groups. Bars = 100 microns. The graph shows the number of fluoro-jade B- positive cells counted by an unbiased stereological method in CA1. Midazolam-ketamine-valproate therapy reduced CA1 neuronal injury compared to SE control (* p <0.05) and valproate (** p <0.01) by Kruskal-Wallis analysis followed by Dunn’s test.
(B) The upper panels show confocal images of fluoro-jade B staining in CA3 48 h following SE in the SE control, midazolam and midazolam-ketamine-valproate groups. Bars = 100 microns. The graph shows the number of fluoro-jade B- positive cells counted by an unbiased stereological method in CA3. Midazolam-ketamine-valproate therapy reduced CA3 neuronal injury compared to SE control (* p <0.05) and valproate (** p <0.01) by Kruskal-Wallis analysis followed by Dunn’s test.
(C) The confocal images show fluoro-jade B staining in the hilus 48 h following SE in SE the control, midazolam and midazolam-ketamine-valproate groups. Bars = 100 microns. The graph shows the number of fluoro-jade B- positive cells counted by an unbiased stereological method in the hilus. Midazolam-ketamine-valproate therapy redced hilar neuronal injury compared to valproate (* p <0.05) by Kruskal-Wallis analysis followed by Dunn’s test.
Figure 6. The midazolam-ketamine-valproate combination, which targets seizure-induced changes in GABA and glutamate receptors, is more effective that the midazolam-fosphenytoin-valproate combination, which follows AES guidelines.
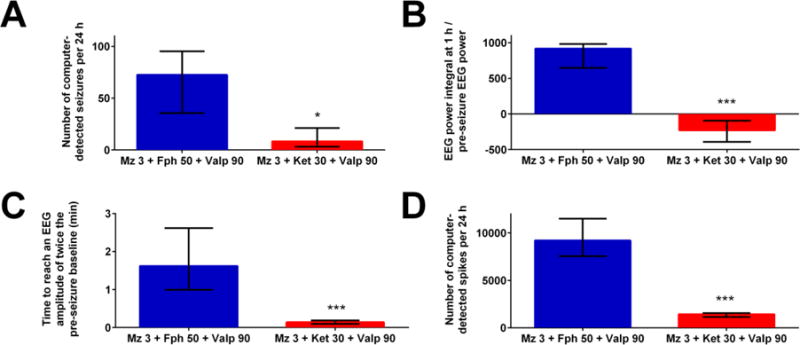
The graphs show the number of computer-detected seizures per 24 hours (A), the ratio of EEG power integral over the first hour to initial EEG power at baseline (B), the time needed to reach an EEG amplitude of twice the pre-seizure baseline (C) and the number of computer-detected spikes per 24 hours (D). The combination of 3 mg/kg midazolam, 30 mg/kg ketamine and 90 mg/kg valproate (n=10) was more potent than the combination of 3 mg/kg midazolam, 50 mg/kg fosphenytoin and 90 mg/kg valproate (n=6). * p <0.05, or *** p <0.001 by Mann-Whitney analysis.
RESULTS
Comparison of the midazolam-ketamine-valproate combination with triple-dose midazolam, ketamine or valproate monotherapy in reducing SE severity
In untreated (SE control) animals, SE started with discrete EEG seizures without behavioral manifestation, which increased in amplitude and duration and were accompanied by discrete behavioral seizures. The EEG seizures merged into nearly continuous polyspikes while behavioral seizures remained discrete and intermittent. Later, spike frequency slowed, and periods of low-amplitude EEG appeared between bursts of high-amplitude polyspikes which progressively decreased in amplitude and evolved into a burst-suppression pattern. Both mono- and polytherapy shortened the duration of seizures and reduced EEG power, but to different degrees. We compared the effect of triple therapy with that of triple-dose monotherapy on the EEG power integral over the first hour posttreatment. The midazolam-ketamine-valproate combination was the only treatment that decreased EEG power below the pre-pilocarpine baseline (Fig. 1C): Triple therapy (median = −228.5; interquartile range −391.8 to −93.75) decreased EEG power over the first hour post-treatment compared to midazolam (median = 567; interquartile range 375.3 to 746.5; p<0.01), ketamine (median = 263.5; interquartile range 188 to 600.5; p<0.05) and valproate monotherapy (median = 970; interquartile range 833.8 to 1165; p<0.0001). Triple therapy (median = −105.0; interquartile range −669.5 to 581.8) also significantly reduced the EEG power integral over the six hours posttreatment when compared to monotherapy with (Fig 1D) midazolam (median = 1440; interquartile range 834.8 to 1729; p<0.01), ketamine (median = 1070; interquartile range 830 to 1227; p<0.05) and valproate monotherapy (median = 3641; interquartile range 1692 to 3739; p<0.0001). In addition, this combination had the lowest time needed for EEG amplitude to decline to twice the pre-seizure baseline (median = 8.5 min; interquartile range 5.9 min – 11.25 min), suggesting that SE was terminated (Fig. 1E) compared to midazolam (median = 29.74 min; interquartile range 14.39 – 47.65; p<0.05), ketamine (median = 17.37min.; interquartile range 15.2 – 48.8; p<0.05) and valproate monotherapy (median = 122.2 min.; interquartile range 64.3 – 164.9; p<0.0001). Triple therapy also significantly reduced the number of computer-detected seizures (median = 8.5 seizures; interquartile range 3.25 – 21.25) (Fig. 1F) compared to midazolam (median = 28.5 seizures; interquartile range 20.25 – 92.5; p<0.05) and valproate monotherapy (median = 68 seizures; interquartile range 11.75 – 109; p<0.05) but not ketamine (median = 25.5; interquartile range 19 – 45; ns).
Furthermore, the midazolam 3 mg/kg-ketamine 30 mg/kg-valproate 90 mg/kg combination (median = 5.95 min; n=10) was more efficient in reducing the duration of SE than higher-dose midazolam 4.5 mg/kg-ketamine 45 mg/kg dual therapy (median = 8.45; n=9; p<0.05) or midazolam-ketamine-levetiracetam triple therapy (median = 10.73; n=9) (Fig 2 A). When we examined the slope of the regression lines of the EEG power integral ratio over the first hour post-treatment, the midazolam-ketamine-valproate group differed significantly from the dual therapy group and the midazolam-ketamine-levetiracetam group (Fig 2 B).
Altogether, these results suggest that the midazolam-ketamine-valproate combination suppresses seizures more efficiently to than triple dose monotherapy or higher dose dual therapy.
Midazolam-ketamine-valproate treatment and SE-induced neuronal injury
The distribution of neuronal injury was examined by fluoro-jade B staining in animals perfused 48 h after pilocarpine injection. The midazolam-ketamine-valproate combination, administered 40 minutes after seizure onset, completely prevented neuronal injury in CA1 and CA3, and this was significant compared to the untreated SE and valproate groups (Fig 3 A–B, Kruskall-Wallis with Dunn’s multiple comparisons). However, high-dose ketamine and the midazolam-ketamine combination were just as neuroprotective as triple therapy in those areas. Unbiased stereological counting in the hilus showed that triple therapy reduced neuronal injury compared to the valproate group (Fig. 3C). Semi-quantitative analysis showed that triple therapy reduced neuronal injury in the frontoparietal, entorhinal and piriform cortices, and in thalamus and amygdala compared to the untreated SE and valproate groups (table 2). Only the amygdala showed better protection by triple therapy over high-dose midazolam. The midazolam-ketamine-valproate combination was not significantly different from triple dose ketamine or higher dose dual therapy, which reduced neuronal injury in all areas studied.
Table 2.
Effect of monotherapies and polytherapies on neuronal injury assessed by fluoro-jade B staining. The first numbers represent the median damage score. The numbers in parentheses indicate the range of neuronal injury.
| SE control (n= 5) |
Valp 270 mg/kg (n=5) |
Mz 9mg/kg (n=10) |
Ket 90 mg/kg (n=7) |
Mz 4.5 + Ket 45 (n= 9) |
Mz 3 + Ket 30 + Valp 90 (n= 10) |
|
|---|---|---|---|---|---|---|
| Frontoparietal cortex | 4 (1–4) | 4 (4) | 2 (1–4) | 1 (1) | 1 (1–2) | 1 (0–2)**††† |
| Entorhinal cortex | 4 (4) | 4 (4) | 1 (1–4) | 1 (1) | 1 (1) | 1 (0–1)**** †††† |
| Piriform cortex | 4 (4) | 4 (4) | 1 (1–4) | 1 (1) | 1 (1–2) | 1 (0–2)***††† |
| Thalamus | 4 (1–4) | 4 (3–4) | 2 (1–4) | 1 (1–2) | 1 (1) | 1 (1)**††† |
| Amygdala | 4 (4) | 4 (4) | 2 (1–4) | 1 (1–2) | 1 (1) | 1 (0–1)***†††‡ |
p <0.01,
p<0.001,
p<0.0001 vs SE control;
p <0.001,
or p <0.0001 vs Valp 270 mg/kg;
p <0.05 vs Mz 9mg/kg.
Triple therapy reduced MWM deficits
Performance in the MWM 6 weeks after SE was impaired in the midazolam group (n=14) compared to sham controls (n=16). The midazolam-ketamine-valproate group (n=13) performed better than the high-dose midazolam group (p<0.05) and did not differ significantly from sham (no seizure) animals in both the acquisition (Figure 4A) and the retention tests (Figure 4B).
Sequential monotherapies versus simultaneous polytherapy
In order to mimic clinical situations where drugs are only injected after the previous treatment fails, we treated SE with the same drugs at the same dose in two groups of rats, except that in one group the second drug was injected 30 min after the first, and the third drug was delivered 30 min after the second drug (Fig. 5A). Simultaneous polytherapy was far more effective than sequential monotherapies in reducing the post-treatment EEG power integral during the first hour or the first six hours after treatment (Figure 5 B–C), in reducing the time needed for EEG amplitude to decline to twice the pre-seizure baseline (Fig 5D) and in reducing the number of post-treatment seizures per 24 h (Fig. 5E). In those measures, sequential monotherapies were not significantly different from high-dose benzodiazepine monotherapy.
The midazolam-ketamine-valproate combination versus the midazolam-fosphenytoin-valproate combination
We wanted to examine whether the combination we selected, based on the receptor trafficking hypothesis, offered any advantage over drug combinations suggested by current evidence-based clinical guidelines, which recommend initial benzodiazepine monotherapy followed by an AED (e.g. fosphenytoin), then by another AED (e.g. valproate) or anesthesia (Glauser et al 2016). We compared the effect of the midazolam 3 mg/kg-fosphenytoin 50 mg/kg-valproate 90 mg/kg combination (which follows AES guidelines) with the combination midazolam 3 mg/kg-ketamine 30 mg/kg-valproate 90 mg/kg, which targets seizure-induced changes in GABAA and glutamate receptors. Drugs were delivered simultaneously in both groups. The latter combination was far more effective in reducing the number of post-treatment seizures per 24 h (Fig. 6A), the EEG power integral over the first hour post-treatment (Figure 6B), the time needed for EEG amplitude to decline to twice the pre-seizure baseline (Fig 6C) and the number of post-treatment spikes for 24 h (Fig. 6D).
DISCUSSION
We treated at 40 min after seizure onset, in a model of severe SE, in order to test the therapeutic potential of drug combinations in SE at a time when it is strongly refractory to benzodiazepines. We compared three-drug combinations to single drugs at a dose three times higher than that used in combination. In this study, higher doses of monotherapy were not tested, but previous experiments showed that high doses of benzodiazepines do not stop SE in this model (Niquet et al., 2017). The combination of midazolam (a GABAA agonist) with ketamine (an NMDA antagonist) and valproate (an AED acting at a non-benzodiazepine site) was far more effective in stopping seizures than triple-dose monotherapy, suggesting that the effect of the three drugs may be synergistic and not just additive. This suggests that SE uses multiple networks, and when drugs which maximize GABAA inhibition, reduce glutamatergic excitation or block sodium channels no longer work when given alone, a combination of those treatments can still stop SE. Another combination of treatments (scopolamine and two types of GABAAR agonists) has recently been shown to stop SE in a different model (Brandt et al., 2015). The unquestioned benefits of treating chronic epilepsy with monotherapy (Reynolds and Shorvon, 1981) may not apply to SE, a brief and transient crisis with high morbidity and mortality (DeLorenzo et al., 1996).
These results support our hypothesis that reducing the consequences of receptor trafficking can stop benzodiazepine-refractory SE. Furthermore, not all combinations are effective. To mimic clinical situations, we followed evidence-based guidelines (Glauser et al., 2016) and combined a benzodiazepine (midazolam) with and AED (fosphenytoin) and another AED (valproate). This substitution reduced or eliminated the benefits of triple therapy, confirming that the GABAAR agonist-NMDAR antagonist-AED combination has specific, synergistic properties (Niquet et al., 2017). In a further attempt to model clinical situations, we compared the efficacy of delivering the three drugs sequentially, waiting for a drug to fail before injecting the next drug, versus simultaneously. The far greater efficacy of simultaneous polytherapy over sequential monotherapies at the same dose is compatible with increasing pharmacoresistance, associated with seizure-induced increases in receptor trafficking, during the delay between sequential drug injections. However, because of the short half-lives of drugs in rats, only 40–50% of injected midazolam would be expected to remain in plasma when valproate is injected (Bittner et al., 2003), and about 60% of injected valproate would be in circulation at the time of ketamine injection (Yoshioka et al., 2000) limiting interactions between sequential drugs. The improvement in outcome when adding low-dose valproate (which was ineffective when given alone) to ketamine and midazolam may reflect the inability of benzodiazepines to fully restore inhibition when treatment is delayed and many synaptic GABAAR are internalized, and the need to enhance inhibition at a non-benzodiazepine site when treatment is delayed and benzodiazepine pharmacoresistance is high.
In this study, combination therapy also reduced the long-term consequences of SE. Spatial memory tested at least 6 weeks after SE was significantly preserved compared with rats treated with triple-dose benzodiazepine monotherapy, and was not significantly different from untreated (no SE) controls. Neuronal injury was eliminated in CA1 and CA3, and was reduced in hilus and other regions compared to untreated SE. We do not know whether this was a true neuroprotective effect or the result of reducing the duration of SE. The efficacy of ketamine monotherapy, which was poor at stopping seizures, argues for the former. However, it may simply reflect the strong neuroprotective properties of ketamine, since all treatments that included that drug reduced neuronal injury. No treatment could eliminate all neuronal injury in hilus, suggesting a mechanism of injury distinct from CA1 and CA3. This is not surprising, since previous studies showed that SE-associated hilar neuronal injury involves at least two distinct mechanisms (Lopez-Meraz et al., 2010; Seo et al., 2009).
The relevance of these results in rodents to the treatment of SE in humans is unknown. While receptor properties are similar in both species, and benzodiazepine pharmacoresistance has been observed clinically (Silbergleit et al., 2012; Treiman et al., 1998), circuitry and brain size are quite different in rats and humans, and the heterogeneity of clinical SE may limit the applicability of conclusions drawn from any animal model to clinical situations. Drug dosage can be compared across species by using body surface area rather than body weight as a denominator (guidance, 2005; Reagan-Shaw et al., 2008). Comparing our 250–300 gm rats to 70 kg humans by that method suggests that the doses of valproate, fosphenytoin and levetiracetam used in this study are not above the “human equivalent dose” (HED) derived from the clinical literature (Chen and Wasterlain, 2006; Gezalian et al., 2014; Glauser et al., 2016), while the midazolam dose used is slightly higher than the HED and the ketamine dose is three times the recommended HED (Chen and Wasterlain, 2006) but lower than the dose used in patients with refractory SE who responded to ketamine (Gaspard et al., 2013).
With these limitations in mind, our results suggest that our strategy for treating SE should be re-evaluated. Our results confirm previous studies suggesting that early treatment is very effective (Silbergleit et al., 2012). They also suggest that an NMDA blocker should be given early in the course, rather than as a last resort and that the current guidelines recommending sequential delivery of AEDs may not be optimal (Fig. 5).
CONCLUSIONS
In conclusion, in this model of severe benzodiazepine-refractory SE, late treatment with the midazolam-ketamine-valproate combination was more potent than the midazolam-ketamine-levetiracetam or midazolam-fosphenytoin-valproate combinations, showing that not all triple therapies show synergism, and suggesting that simultaneously targeting GABAAR and glutamate receptor changes is a valid therapeutic strategy. The simultaneous administration of the three drugs is more efficient in stopping seizures than the standard practice of injecting the drugs sequentially (Glauser et al., 2016), suggesting that an early polytherapy arm should be included in future clinical trials.
Highlights.
Pharmacoresistance in SE may be due in part to GABAA and NMDA receptor trafficking.
We used combinations of drugs designed to counter these SE-induced changes.
Midazolam–ketamine-valproate therapy synergistically stopped SE.
This triple therapy reduced acute neuronal injury and Morris water maze deficits.
Simultaneous triple therapy was more effective than sequential triple therapy.
Acknowledgments
This work was supported in part by Merit Review Award # I01 BX000273-07 from the United States Department of Veterans Affairs (Biomedical Laboratory Research and Development Service), by NINDS (grant UO1 NS074926; CW) and by the Debbie and James Cho Foundation. We confirm that we have read the Journal’s position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosure of Conflicts of Interest
Jerome Niquet and Claude Wasterlain have a patent pending on polytherapy of cholinergic seizures (UC Case No. 2012-172-2). Other authors have no conflict of interest to disclose.
References
- Bittner B, et al. Impact of Solutol HS 15 on the pharmacokinetic behavior of midazolam upon intravenous administration to male Wistar rats. Eur J Pharm Biopharm. 2003;56:143–6. doi: 10.1016/s0939-6411(03)00041-9. [DOI] [PubMed] [Google Scholar]
- Borris DJ, et al. Ketamine controls prolonged status epilepticus. Epilepsy Res. 2000;42:117–22. doi: 10.1016/s0920-1211(00)00175-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandt C, et al. Effective termination of status epilepticus by rational polypharmacy in the lithium-pilocarpine model in rats: Window of opportunity to prevent epilepsy and prediction of epilepsy by biomarkers. Neurobiol Dis. 2015;75:78–90. doi: 10.1016/j.nbd.2014.12.015. [DOI] [PubMed] [Google Scholar]
- Chapman S, et al. Sarin-induced brain damage in rats is attenuated by delayed administration of midazolam. Neurotoxicology. 2015;49:132–8. doi: 10.1016/j.neuro.2015.05.001. [DOI] [PubMed] [Google Scholar]
- Chen JW, Wasterlain CG. Status epilepticus: pathophysiology and management in adults. Lancet Neurol. 2006;5:246–56. doi: 10.1016/S1474-4422(06)70374-X. [DOI] [PubMed] [Google Scholar]
- DeLorenzo RJ, et al. A prospective, population-based epidemiologic study of status epilepticus in Richmond, Virginia. Neurology. 1996;46:1029–35. doi: 10.1212/wnl.46.4.1029. [DOI] [PubMed] [Google Scholar]
- Dham BS, et al. The epidemiology of status epilepticus in the United States. Neurocrit Care. 2014;20:476–83. doi: 10.1007/s12028-013-9935-x. [DOI] [PubMed] [Google Scholar]
- Fujikawa DG. Neuroprotective effect of ketamine administered after status epilepticus onset. Epilepsia. 1995;36:186–95. doi: 10.1111/j.1528-1157.1995.tb00979.x. [DOI] [PubMed] [Google Scholar]
- Gaspard N, et al. Intravenous ketamine for the treatment of refractory status epilepticus: a retrospective multicenter study. Epilepsia. 2013;54:1498–503. doi: 10.1111/epi.12247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gezalian MM, et al. Management of Status Epilepticus. Federal Practitioner. 2014;31:24–29. [Google Scholar]
- Glauser T, et al. Evidence-Based Guideline: Treatment of Convulsive Status Epilepticus in Children and Adults: Report of the Guideline Committee of the American Epilepsy Society. Epilepsy Curr. 2016;16:48–61. doi: 10.5698/1535-7597-16.1.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodkin HP, et al. Subunit-specific trafficking of GABA(A) receptors during status epilepticus. J Neurosci. 2008;28:2527–38. doi: 10.1523/JNEUROSCI.3426-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodkin HP, et al. Status epilepticus increases the intracellular accumulation of GABAA receptors. J Neurosci. 2005;25:5511–20. doi: 10.1523/JNEUROSCI.0900-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FDA, editor. guidance F. Guidance for Industry: Estimating the Maximum Safe Starting Dose in Initial Clinical Trials for Therapeutics in Adult Healthy Volunteers. 2005. [Google Scholar]
- Holtkamp M, et al. Predictors and prognosis of refractory status epilepticus treated in a neurological intensive care unit. J Neurol Neurosurg Psychiatry. 2005;76:534–9. doi: 10.1136/jnnp.2004.041947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapur J, Macdonald RL. Rapid seizure-induced reduction of benzodiazepine and Zn2+ sensitivity of hippocampal dentate granule cell GABAA receptors. J Neurosci. 1997;17:7532–40. doi: 10.1523/JNEUROSCI.17-19-07532.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kofke WA, et al. Effect of anesthetics on neuropathologic sequelae of status epilepticus in rats. Anesth Analg. 1993;77:330–7. doi: 10.1213/00000539-199308000-00020. [DOI] [PubMed] [Google Scholar]
- Lopez-Meraz ML, et al. Vulnerability of postnatal hippocampal neurons to seizures varies regionally with their maturational stage. Neurobiol Dis. 2010;37:394–402. doi: 10.1016/j.nbd.2009.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin BS, Kapur J. A combination of ketamine and diazepam synergistically controls refractory status epilepticus induced by cholinergic stimulation. Epilepsia. 2008;49:248–55. doi: 10.1111/j.1528-1167.2007.01384.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer SA, et al. Refractory status epilepticus: frequency, risk factors, and impact on outcome. Arch Neurol. 2002;59:205–10. doi: 10.1001/archneur.59.2.205. [DOI] [PubMed] [Google Scholar]
- Mazarati AM, et al. Anticonvulsant effects of levetiracetam and levetiracetam-diazepam combinations in experimental status epilepticus. Epilepsy Res. 2004;58:167–74. doi: 10.1016/j.eplepsyres.2004.02.002. [DOI] [PubMed] [Google Scholar]
- Mazarati AM, et al. Time-dependent decrease in the effectiveness of antiepileptic drugs during the course of self-sustaining status epilepticus. Brain Res. 1998;814:179–85. doi: 10.1016/s0006-8993(98)01080-4. [DOI] [PubMed] [Google Scholar]
- Mazarati AM, Wasterlain CG. N-methyl-D-asparate receptor antagonists abolish the maintenance phase of self-sustaining status epilepticus in rat. Neurosci Lett. 1999;265:187–90. doi: 10.1016/s0304-3940(99)00238-4. [DOI] [PubMed] [Google Scholar]
- Naylor DE, et al. Rapid surface accumulation of NMDA receptors increases glutamatergic excitation during status epilepticus. Neurobiol Dis. 2013;54:225–38. doi: 10.1016/j.nbd.2012.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naylor DE, et al. Trafficking of GABA(A) receptors, loss of inhibition, and a mechanism for pharmacoresistance in status epilepticus. J Neurosci. 2005;25:7724–33. doi: 10.1523/JNEUROSCI.4944-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niquet J, et al. Midazolam-ketamine dual therapy stops cholinergic status epilepticus and reduces Morris water maze deficits. Epilepsia. 2016a;57:1406–15. doi: 10.1111/epi.13480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niquet J, et al. Treatment of experimental status epilepticus with synergistic drug combinations. Epilepsia. 2017 doi: 10.1111/epi.13695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niquet J, et al. Benzodiazepine-refractory status epilepticus: pathophysiology and principles of treatment. Ann N Y Acad Sci. 2016b;1378:166–173. doi: 10.1111/nyas.13147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reagan-Shaw S, et al. Dose translation from animal to human studies revisited. FASEB J. 2008;22:659–61. doi: 10.1096/fj.07-9574LSF. [DOI] [PubMed] [Google Scholar]
- Reynolds EH, Shorvon SD. Monotherapy or polytherapy for epilepsy? Epilepsia. 1981;22:1–10. doi: 10.1111/j.1528-1157.1981.tb04327.x. [DOI] [PubMed] [Google Scholar]
- Seo DW, et al. Contribution of a mitochondrial pathway to excitotoxic neuronal necrosis. J Neurosci Res. 2009;87:2087–94. doi: 10.1002/jnr.22035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silbergleit R, et al. Intramuscular versus intravenous therapy for prehospital status epilepticus. N Engl J Med. 2012;366:591–600. doi: 10.1056/NEJMoa1107494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suchomelova L, et al. Treatment of experimental status epilepticus in immature rats: dissociation between anticonvulsant and antiepileptogenic effects. Pediatr Res. 2006;59:237–43. doi: 10.1203/01.pdr.0000196333.16608.30. [DOI] [PubMed] [Google Scholar]
- Treiman DM, et al. A comparison of four treatments for generalized convulsive status epilepticus. Veterans Affairs Status Epilepticus Cooperative Study Group. N Engl J Med. 1998;339:792–8. doi: 10.1056/NEJM199809173391202. [DOI] [PubMed] [Google Scholar]
- Yoshioka H, et al. Effects of lithium on the pharmacokinetics of valproate in rats. J Pharm Pharmacol. 2000;52:297–301. doi: 10.1211/0022357001773986. [DOI] [PubMed] [Google Scholar]