

Abstract
Basic fibroblast growth factor (FGF2) is a highly pleiotropic member of a large family of growth factors with a broad range of activities, including mitogenesis and angiogenesis (Ornitz, et al. 1996, Zhang, et al. 2006), and it is known to be essential for maintenance of balance between survival, proliferation, and self-renewal in human pluripotent stem cells (Eiselleova, et al. 2009, Zoumaro-Djayoon, et al. 2011). A single FGF2 transcript can be translated into five FGF2 protein isoforms, an 18kDa low molecular weight (LMW) isoform and four larger high molecular weight (HMW) isoforms (Arese, et al. 1999, Arnaud, et al. 1999). As they are not generally secreted, high molecular weight (HMW) FGF2 isoforms have predominantly been investigated intracellularly; only a very limited number of studies have investigated their activity as extracellular factors. Here we report over-expression, isolation, and biological activity of all recombinant human FGF2 isoforms. We show that HMW FGF2 isoforms can support self-renewal of human embryonic stem cells (hESCs) in vitro. Exogenous supplementation with HMW FGF2 isoforms also activates the canonical FGFR/MAPK pathway and induces mitogenic activity in a manner similar to that of the 18kDa FGF2 isoform. Though all HMW isoforms, when supplemented exogenously, are able to recapitulate LMW FGF2 activity to some degree, it appears that certain isoforms tend to do so more poorly, demonstrating a lesser functional response by several measures. A better understanding of isoform-specific FGF2 effects will lead to a better understanding of developmental and pathological FGF2 signaling.
1. Introduction
The fibroblast growth factors (FGFs) are an important class of cellular signaling molecules. The FGF family has 23 members (22 identified in humans), many of which express several isoforms, and four FGF receptors (FGFRs), three of which express several splice variants (Miki, et al. 1992, Root, et al. 2000). FGF ligands and their receptors are expressed in a developmental stage-dependent manner and in tissue-specific patterns (Ornitz, et al. 2002, Powers, et al. 2000, Spivak-Kroizman, et al. 1994). The large number of FGF ligands and their varying levels of promiscuity for FGFR variant targets provide a very high level of ligand-receptor diversity, resulting in many distinct functions during mammalian development (Givol, et al. 1992, Ornitz, et al. 2002, Presta, et al. 2005, Spivak-Kroizman, et al. 1994). Aberrant expression of FGF ligands and FGFR variants has been implicated in various cellular processes and diseases [reviewed in (Eswarakumar, et al. 2005)]. However, a complete understanding of the roles that FGF ligands and their receptors play in development and disease requires a better understanding of FGF/FGFR interactions, and of the specificity with which each FGF ligand interacts with particular FGFR variants.
FGF2 is a member of the FGF family known as heparin binding factors, and it was first purified as a heparin-binding molecule from bovine pituitary gland (Gospodarowicz, et al. 1984). Five isoforms of FGF2, ranging in molecular weight from 18kDa to 34kDa, exist as a result of translation initiation at alternative start sites (Arnaud, et al. 1999, Florkiewicz, et al. 1989). The high molecular weight (HMW) isoforms with molecular weights 22kDa, 22.5kDa, 24kDa, and 34 kDa are co-linear, N-terminal extensions of the 18kDa isoform (Arnaud, et al. 1999, Florkiewicz, et al. 1989, Touriol, et al. 2003). The HMW isoforms are predominantly intracellular and localize to the nucleus, while the 18kDa isoform is mainly cytoplasmic and is secreted from cells by an unconventional process that is not fully understood, but appears to depend at least in part on interaction with cell surface-bound heparin sulfate proteoglycans (Florkiewicz, et al. 1998, Nickel 2011, Piotrowicz, et al. 1997, Zehe, et al. 2006).
Unlike the LMW FGF2 isoform, which is well-studied, the HMW isoforms of FGF2 are not nearly as well-understood, but recent reports have investigated the effects of HMW FGF2 signaling in various contexts. Localization of FGF2 isoforms can vary depending on cellular conditions and stimuli (Arese, et al. 1999). HMW isoforms have been described in some occasions to be present in the extracellular space, released through a process known as vesicle shedding (Greber, et al. 2007, Taverna, et al. 2003). The vesicles have the potential to stimulate plasminogen activator production and influence chemotaxis (Taverna, et al. 2003), suggesting a role for HMW FGF2 in this process, and previous studies have shown that the 24kDa FGF2 isoform administered exogenously to endothelial cells induces cell proliferation with a similar dose response to the 18kDa isoform (Taverna, et al. 2003), suggesting a potential role for extracellular, canonical FGFR signaling by HMW FGF2 isoforms. In HEK293 cells, overexpression of HMW FGF2 led to chromatin compaction and apoptosis in a manner dependent on its localization to the nucleus, further supporting the important role of subcellular localization in the biology of HMW FGF2 (Ma, et al. 2007). Mice specifically overexpressing HMW FGF2 isoforms quickly develop osteoarthropathy, associated with dysregulated expression of inflammatory proteins and cytokines (Meo Burt, et al. 2016), and HMW FGF2 has also recently been implicated in cardiac fibrosis and cardiomyocyte hypertrophy in rats (Jiang, et al. 2007, Santiago, et al. 2014). Previously, it had been demonstrated that, in neonatal rat cardiac non-myocytes, hypertrophic stimuli result in upregulation and subsequent secretion of HMW FGF2 to the extracellular space (Santiago, et al. 2011). Taken together, these reports suggest non-redundant functions of HMW FGF2 compared to LMW FGF2, as well as the potential of HMW FGF2 to be present and active not only intracellularly, but also in the extracellular milieu in certain scenarios.
Human dermal fibroblasts express multiple isoforms of FGF2 as well as select FGF receptors (FGFRs) (Dailey, et al. 2005, Dvorak, et al. 2005, Eiselleova, et al. 2009, Grose, et al. 2005, Quarto, et al. 1994, Root, et al. 2000). The LMW (18kDa) isoform of FGF2 has been studied most robustly and is reported to act in autocrine, intracrine, and paracrine manners, through interactions with its cell-surface receptors, the FGFRs (Ornitz, et al. 1996, Zhang, et al. 2006). Four distinct genes that encode for FGF receptors have been identified, FGFR1, FGFR2, FGFR3, and FGFR4 (Arese, et al. 1999, Arnaud, et al. 1999, Dvorak, et al. 2005, Eiselleova, et al. 2009, Florkiewicz, et al. 1989, Ornitz, et al. 1996, Sperger, et al. 2003, Taverna, et al. 2003). Furthermore, alternative mRNA splicing produces several receptor variants, which exhibit varied binding kinetics and affinities for different FGF ligands (Champion-Arnaud, et al. 1991, Miki, et al. 1992, Root, et al. 2000). The variety of FGF ligands and FGFR variants provides a high level of diversity in ligand-binding specificity and biological function, depending on which FGFRs are expressed by various cell types and which FGFs are present in the surrounding milieu.
Binding of the FGF2 ligand to its receptor triggers receptor dimerization, phosphorylation of its kinase domain, and signal transduction via activation of several intracellular pathways that have been implicated in multiple aspects of vertebrate and invertebrate embryonic development, tumor growth, angiogenesis, wound healing, and physiology (Ornitz, et al. 2002, Powers, et al. 2000, Spivak-Kroizman, et al. 1994). Dysregulated expression of FGFs has also been implicated in cancer development and progression (Ezzat, et al. 2005, Givol, et al. 1992, Krejci, et al. 2012, Presta, et al. 2005, Zubilewicz, et al. 2001).
Investigation of the ability of each FGF isoform to bind to different FGF receptors and activate downstream signaling pathways, and identification of FGF-FGFR pair specificities, is critical for understanding the biological mechanisms involved in normal development and pathogenesis. Previous studies have identified the 18kDa FGF2 as an important factor for the maintenance of pluripotency in human stem cells (Eiselleova, et al. 2009, Zoumaro-Djayoon, et al. 2011), and our lab has demonstrated that the 18kDa FGF2 isoform, in combination with sub-atmospheric oxygen, induces expression of stem cell specific genes and proteins in human dermal fibroblasts cultured in vitro (Page, et al. 2009).
The pleiotropic nature of FGF2 and its variety of downstream effects make generation of highly pure active protein essential. Heparin chromatography has been used for purification of 18kDa FGF2 isoform. However, subsequent heparin contamination in purified FGF2 preparations has been previously described to interfere with the stability and biological activity of FGF2 (Eiselleova, et al. 2009, Gasparian, et al. 2009). To avoid heparin contamination and achieve high protein purity after a single chromatographic step, we had DNA constructs synthesized for all FGF2 isoforms as 6xHis tag fusion proteins. Overexpressed 6xHis tagged FGF2 isoforms demonstrated high affinity for Ni-NTA, and the biological activity of purified HMW FGF2 isoforms was compared to commercially available 18kDa FGF2 by monitoring activation of downstream kinase pathways, in order to ensure that the pure protein activity was comparable to that of the commercial formulation. Furthermore, we examined the ability of each isoform to support self-renewal of human embryonic stem cells in vitro, promote mitogenic activity via activation of several FGFR variants, and increase the rate of proliferation of human dermal fibroblasts.
2. Materials and Methods
2.1 Antibodies
The antibodies used include: anti-6xHis antibody (Millipore, Cat# 05-949), anti-OCT4 antibody (Santa Cruz Biotechnology, Cat# sc-5279), anti-NANOG antibody (Cell Signaling Technology, Cat# 4903), anti-Lin28 antibody (Cell Signaling Technology, Cat# 3695), anti-phospho FGFR1 antibody (Life Technologies, Cat# 44-11406), anti-phospho FRS2α antibody (Cell Signaling Technology, Cat# 3861), anti-phospho ERK1/2 antibody (Abcam, Cat# ab4819), anti-Actin antibody (Sigma-Aldrich, Cat# A2066), Phalloidin-AlexaFluor488 (Life Technologies), secondary AlexaFluor568 conjugated antibodies (Life Technologies), AP-conjugated anti-mouse secondary antibody and AP-conjugated anti-rabbit secondary antibody (both from Abcam).
2.2 Reagents
18kDa FGF2 protein and FGF1 used as positive controls were purchased from Peprotech. All other FGF2 isoforms were produced in our laboratory as explained in the following sections. DNA for all FGF2 isoforms was synthesized by Epoch Life Sciences (Missouri City, Texas).
2.3 Design and synthesis of pFastBac1 expression vectors
Each construct was designed as a 6xHis-tag fusion protein with a tobacco etch virus (TEV) recognition site between the 6xHis-tag and the protein to aid with tag removal after purification if necessary. NheI and EcoRI restriction sites were included on the N-terminal end, and a HindIII restriction site was included on the C-terminal end to simplify the cloning process. Constructs for all five isoforms were synthesized by Epoch labs in pBluescript (+) cloning vectors. Inserts for all five FGF2 isoforms were digested using EcoRI and HidIII (New England Biolabs) restriction and isolated by agarose gel electrophoresis. Each FGF2 isoform was subcloned in pFastBac1 (Life Technologies) vector using the same restriction sites as above, which ensured directional insertion of the genes for each FGF2 isoform. The correct orientation of the insert was confirmed by restriction digest.
2.4 Human FGF2 isoform expression
The Bac-to-Bac® baculovirus expression system (Life Technologies) was used to overexpress FGF2 isoforms in SF21 cells. pFastBac® I vectors containing individual constructs for each FGF2 isoform were transformed in DH10Bac competent cells (Life Technologies). Recombinant bacmid DNA was isolated following the protocol provided by the manufacturer (Life Technologies). Forty mL cultures of SF21 cells were transfected with each isoform using Cellfectin® II reagent (Life Technologies), following manufacturer’s protocol. P1 virus was harvested at 72–80 hours after transfection when cell viability was between 60–70%, according to the manufacturer’s protocol. Clarified supernatant containing the P1 viral stock for each FGF2 isoform was used to test expression in an 18 point small-scale expression optimization experiment. 24 hours prior to infection, 300 ml SF9 and SF21 cultures were seeded at 7.5 × 105 cells/ml, in SF900 II SFM media (Life Technologies). 18 to 24 hours later, 50 ml cultures were set up for infection for each viral dilution 1:100/1:1000/1:10,000, in both SF9 and SF21. Recombinant protein expression was analyzed by Western blot analysis at 24, 48 and 72 hours after infection. Based on Western blot results, the optimal expression conditions were identified as SF9 cells, with a 1:1000 viral dilution harvested at 72 hours post infection. Four 500ml cultures in 1L flasks were infected for each construct. Cells were seeded at 7.5×105 cells/ml and 24 hours later infected with P1 virus stock at a 1:1000 concentration. Cells were harvested by centrifugation 72 hours post infection, flash frozen in liquid nitrogen, and stored at −80°C.
2.5 SDS-PAGE and Immunoblotting
A 0.2 gram pellet from each transfected culture was lysed in 200μl cold lysis buffer with protease inhibitor cocktail. Cells were lysed on ice using a Missonix XL-2000 ultrasonic cell disruptor for 3 × 10 pulses at power 2. Total cell lysate was diluted in 5X Laemmli sample buffer. Clarified soluble fraction was generated by centrifugation at 14,000 × g for 30 minutes at 4°C and mixed with 5X Laemmli sample buffer. Samples were boiled for 5 minutes and separated electrophoretically on 4–20% SDS-PAGE gels (Bio-Rad) using a Bio-Rad mini protean system. For SDS-PAGE analysis the gel was stained with Coomassie brilliant blue stain. For Western blot analysis the proteins were transferred onto a PVDF membrane (Millipore) using a semi-dry transfer apparatus (GE Healthcare). Membrane was blocked in TBST buffer (25mM Tris-HCl pH7.5, 130mM NaCl, 0.1% Tween-20) with 5% fat-free dry milk for 30 minutes. The membrane was incubated in the indicated primary antibody overnight at 4°C. After washing the membrane 3 × 10 minutes in TBST, the membrane was incubated in alkaline phosphatase-conjugated secondary antibody for 2 hours at room temperature. The bands were visualized using Western blue reagent (Promega). Images were acquired with a Canon LiDE 200 scanner.
2.6 Immunocytochemistry
Cells were fixed with ice-cold methanol or 2% methanol-free formaldehyde. Samples were permeabilized with 1 N HCl or 0.1% Triton X-100 prior to blocking and labeling. Cells were blocked with 5% bovine serum albumin (BSA) in TBST. Indicated primary antibodies were diluted in TBST and 1% BSA. Cells were labeled for 2 hours at room temperature. Plates were washed three times with TBST and appropriate secondary antibodies conjugated to AlexaFluor568 diluted in TBST with serum added for 45 minutes. After being washed with PBS, the cells were incubated for 10 min at room temperature with phalloidin-AlexaFluor488 (Life Technologies) to stain cellular F-actin. Coverslips were mounted using Prolong Gold® (Life Technologies) and analyzed using an IX81 inverted microscope (Olympus), equipped with automatic stage (Prior), fluorescence, and a cooled CCD camera (Orca, Hamamatsu). Images were acquired using appropriate filters and processed using SlideBook (Olympus).
2.7 Recombinant Human FGF2 Isoform Purification
Cell pastes from 1L expression cultures were subjected to purification over Ni-NTA resin. Cells were lysed in 10mL ice cold lysis buffer (50mM Tris pH 7.5, 250mM NaCl, 1mM DTT, EDTA free protease inhibitor cocktail) per 1 gram of cell paste using a Missonix XL-2000 ultrasonic cell disruptor, for 3 × 40 pulses at power 15. Clarified soluble fraction was generated by centrifugation at 12,000 × g for 2 hours at 4°C. 5ml Ni-NTA column (Qiagen) was equilibrated with 10 column volumes of lysis buffer. Clarified soluble fraction was loaded over the column using an AKTA Explorer FPLC at 5mL/min flow rate. The column was washed to baseline with lysis buffer. A second wash to baseline was done with lysis buffer containing 30mM imidazole. Protein was eluted with lysis buffer containing 300mM imidazole and 2.5mL fractions were collected and analyzed by Coomassie-stained SDS-PAGE. Ni-NTA resin was stripped with lysis buffer containing 1M imidazole. All starting material and fractions collected were analyzed by Coomassie-stained SDS-PAGE and Western blot analysis with anti-His antibody. Fractions containing over 90% pure protein, by Coomassie-stained SDS-PAGE, were pooled together and dialyzed in 4L of 1× PBS overnight. The dialyzed pool was centrifuged at 20,000 × g for 1 hour and then filtered through a 0.2μm filter. Final protein concentration was determined using Bradford protein assay (Thermo Scientific) using BSA as a standard. Final material was analyzed by Coomassie-stained SDS-PAGE gel and Western blot using monoclonal anti-His antibody.
2.8 Recombinant Human FGF2 Isoform Activity
CRL-2352 primary human dermal fibroblasts at passage 8 were plated in 6-well tissue culture plates as well as 12-well plates on coverslips. Cultures were grown to 80% confluence using standard procedures. On the day of the assay, serum was removed from cells for 2 hours. At the time of the assay, media was replaced with serum free DMEM:Ham’s F12 (50:50) media containing human FGF2 isoforms at a final molar concentration of 222.22 ± 0.4pM. Serum-free DMEM:Ham’s F12 (50:50) containing commercially available 18kDa human FGF2 (Peprotech) was used as a positive control, at the same concentration as the experimental treatments. Serum-free DMEM:Ham’s F12 (50:50) with no supplements was used as a negative control. All cultures were incubated for 30 minutes at 37°C, 5% O2, 5% CO2 and high humidity. At the end of the incubation period, cells were washed once in PBS. Coverslips were used for ICC analysis using the method described above. Cells from 6-well plates were washed once in PBS, harvested in 2X Laemmli buffer, lysed by sonication using a Missonix XL-2000 ultrasonic cell disruptor for 3 × 10 pulses at power 2 and subjected to Western blot analysis using the indicated antibodies.
2.9 Cell Culture
CRL-2352 primary human dermal fibroblasts (American Tissue Culture Collection) at passage 8 were cultured in DMEM:Ham’s F12 (50:50), supplemented with 10% fetal bovine serum (FBS, Hyclone), and 4 mM L-glutamine (Mediatech). Cultures were incubated at 37°C, 5% O2, 5% CO2 and high humidity, and grown to 80% confluence using standard procedures. Human embryonic stem cells (hESC) line WA09 (H9, WiCell) were cultured on in-house, primary derived mouse embryonic fibroblasts (MEF) plated at a density of 2.25 × 104 cells/cm2 on plates coated for 60 minutes with 1% gelatin solution (MP Biologicals). MEFs were inactivated with mitomycin-C (Sigma-Aldrich) for 3 hours before plating on gelatin-coated plates. hESC were cultured in Knockout DMEM (Life Technologies), supplemented with 15% Knockout serum (Life Technologies), 2mM Glutamax (Life Technologies), 50mM 2-Mercaptoethanol (Sigma-Aldrich), 1x MEM non-essential amino acids (Mediatech) and 8ng/mL FGF2 (Peprotech). Medium was replaced daily except for first day after plating to allow hESCs to attach. Cells were passaged when colonies had been growing for five days. hESC were dissociated using 0.01% trypsin (Mediatech) and split 1:12 onto new MEFs feeder layer prepared as described above. OCT4, NANOG and LIN28 protein expression was analyzed by Western blot.
2.10 RT-PCR analysis
RNA was extracted from cultured cells using TRIzol reagent (Life Technologies) according to the manufacturer’s protocol, and quantified by spectrophotometry using a Nanodrop 2000 (Thermo Scientific). One microgram of RNA was subjected to DNase digestion, followed by a reverse transcription using qSCRIPT™ cDNA Synthesis Kit (Quanta Biosciences), following the manufacturer’s protocol. RT-PCR was performed using GoTaq green master mix (Promega) with a 20μL final reaction volume, final primer concentrations of 250nM, and a final template concentration of 2ng/μL. PCR cycling was performed as follows: Initial denaturation at 95°C for 7 minutes; followed by 30 cycles of denaturation at 95°C for 30 seconds; annealing at primer-specific annealing temperature for 1 minute; and extension at 72°C for 20 seconds, followed by a final extension at 72°C for 7 minutes, and the samples held at 4°C until use. Amplification products were resolved on 2% agarose gels containing .5μg/mL ethidium bromide in 1X TAE buffer and imaged using a BioRad Gel Doc XR+ system (BioRad).
2.11 Preparation of Endotoxin Free MIRB-FGFR Plasmid for Transfection
FGFR variants in MIRB vectors were a kind gift from Dr. David Ornitz at the Washington University School of Medicine. Each FGFR variant was transformed into chemically competent E.coli. Cells were harvested by centrifugation at 3000 × g, 24 hours after growing at 37°C with shaking at 250 rpm. Cells were resuspended (1:10/weight:volume) in buffer P1 (50mM Tris-HCl pH 8.0, 10mM EDTA, 100μg/ml RNase A). Cells were lysed in an equal volume of lysis buffer (200mM NaOH, 1% SDS) for 30 minutes on ice. The lysate mixture was neutralized in neutralization buffer (3.0M potassium acetate pH 5.5) and centrifuged at 9,000 × g for 60 minutes. Supernatant was filtered to remove any particles and then incubated on ice for 15 minutes after addition of 1/10 volume of endotoxin removal buffer (1% Triton X-114). Ion exchange resin from Qiagen was hydrated and equilibrated in column loading buffer (750mM NaCl, 50mM MOPS, 15% isopropanol, 0.15% triton X-100, pH 7.0). The sample was loaded by gravity flow, and resin was washed with 10 column-volumes of wash buffer (1.0mM NaCl, 50mM MOPS, 15% Isopropanol, pH 7.0). Plasmid was eluted in 20 mL elution buffer (1.25mM NaCl, 50mM Tris-HCl, 15% isopropanol pH 8.5). Plasmid was precipitated by centrifugation at 9,000 × g with 0.7 volumes of isopropanol. The sample was washed twice in 70% endotoxin-free ethanol, air dried and resuspended in endotoxin free water. Prior to transfection, 50 μg of plasmid were linearized by digesting with 50 units of ClaI (New England Biolabs) restriction enzyme for 1 hour at 37°C. The final plasmid concentration was determined using a Nanodrop 2000 spectrophotometer (Thermo Fisher).
2.12 Overexpression of FGF receptor variants in BaF3 cells
Three receptor variants, FGFR1 IIIb, FGFR2 IIIb and FGFR3 IIIb, were transfected into wild type BaF3 cells, individually. BaF3 cells were grown in suspension following published protocols (Ornitz, et al. 1996) in RPMI 1640 media (Corning), supplemented with 10% FBS, L-glutamine (both from Cyclone), 50nM 2-Mercaptoethanol (Life Technologies), 10ng/mL IL-3 (Thermo Scientific) and 1x penicillin/streptomycin (Sigma Aldrich). Previously published electroporation protocols (Ornitz, et al. 1996, Root, et al. 2000) were used to electroporate 20μg of ClaI-linearized MIRB-FGFR plasmid into 106 cells (Dell, et al. 1992, Ornitz, et al. 1992, Ornitz, et al. 1996, Ornitz, et al. 1992) using a Gene Pulser Xcell™ Electroporation System (Bio Rad). Cells were grown in media without selection pressure for 16–18 hours, then selected in BaF3 complete growth media containing 600 μg/ml G418 (Life Technologies). Clonal cell lines for each receptor variant were established by limiting dilutions in a 96 well plate. The clonal cell lines used for the following experiments were evaluated for mitogenic responsiveness to FGF1 (Peprotech).
2.13 Mitogenic Assay
Stably transfected BaF3 cells and untransfected controls were grown in suspension in RPMI 1640 media (Corning), supplemented with 10% FBS, L-glutamine, 50nM 2-Mercaptoethanol, 10ng/ml IL-3 (Thermo Scientific) and 1 × penicillin/streptomycin (Sigma Aldrich). Labeling for 6 hours with bromodeoxyuridine (BrdU) after treatment with individual FGF2 isoforms was used to determine the percentage of cells progressing to S-phase for each FGF2 isoform-FGFR variant interaction.
Mitogenic activity was defined as the ability of FGF ligands to cause a cell to transition from G1 to S phase of the cell cycle, and was determined by BrdU incorporation visualized by staining with anti-BrdU antibody. For the mitogenic assay, wild type BaF3 cells and stably transfected BaF3 cells were washed twice in RPMI media devoid of IL-3. Wild type cells and stably transfected BaF3 cells were plated in 6-well plates at 2 × 105 cells per well with the growth factor concentrations indicated in Figure 7 and 2μg/mL heparin. This concentration of heparin has been shown to be optimal for FGF1 binding to the receptors (Ornitz, et al. 1995, Ornitz, et al. 1992, Santos-Ocampo, et al. 1996), and does not inhibit FGF2 activity (Ornitz, et al. 1996). All cultures were incubated at 37°C, 5% O2, 5% CO2 and high humidity for 36–48 hours. BrdU labeling reagent (Zymed) was added to each treatment following manufacturer’s suggestions, and cells were incubated at 37°C for 6 hours. Cells were centrifuged at 1,000 × g for 5 minutes, washed twice in Dulbecco’s phosphate buffered saline (DPBS), and then fixed in ice cold methanol for 20 minutes at 4°C. After rinsing with DPBS, cells were permeabilized with 1.5N HCl at 37°C for 10 minutes, and washed three times with PBS-Tween, with 5 minutes for each wash. Cells were incubated for 30 minutes at room temperature in primary anti-BrdU antibody (Developmental Studies Hybridoma Bank, Cat# G3G4) diluted 1:200 in PBS-Tween. Cells were washed three times 5 minutes each in PBS-1% Tween and then incubated for 30 minutes in secondary Alexafluor 488 goat anti-mouse antibody (Molecular Probes), diluted 1:500 in PBS–1% Tween. Cells were washed three times for 5 minutes each in PBS, resuspended in 250μl PBS then subjected to flow cytometry analysis using a BD Accuri C6 flow cytometer. Twenty thousand events were collected for each reaction. Transgenic BaF3 cells expressing FGFR variants treated with FGF1 at a concentration of 1250pM were used as a positive control, and the same cells without any growth factor supplements were used as negative controls.
Fig. 7.
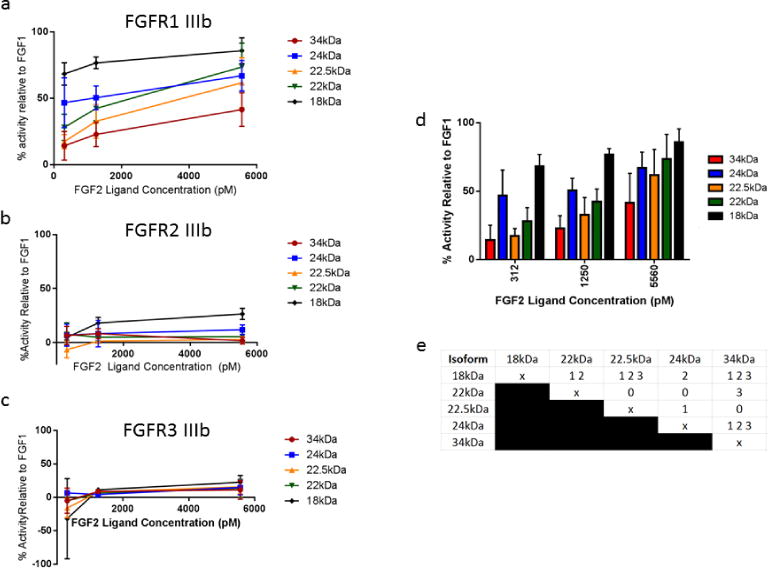
Relative mitogenic activity mediated by FGF2 isoforms. (a) Mitogenic activity was measured relative to 1250pM FGF1 for each FGF2 isoform interacting with receptor variant FGFR1 IIIb, FGFR2 IIIb, and FGFR3 IIIb at three different ligand concentrations and was quantified based on BrdU positive cells after flow cytometry analysis. (b) Mitogenic activity of individual isoforms of FGF2 for FGFR1 IIIb was compared within each ligand concentration. Error bars indicate Standard Deviation (SD). n=4 biological replicates per isoform, per ligand concentration. (c) The activity of each isoform was compared to each other isoform within each ligand concentration by an analysis of variance (ANOVA), followed by Tukey’s post-hoc pairwise comparison test. A “0” at the intersection of two isoforms in the table indicates statistical similarity between the two isoforms at all 3 ligand concentrations. A “1” indicates statistical difference at the 312pM ligand concentration. A “2” indicates statistical difference at the 1250pM ligand concentration. A “3” indicates statistical difference at the 5560pM ligand concentration. Multiple numbers in a box indicate statistical differences at more than one ligand concentration. Statistical differences were calculated with significance level α=.05.
2.14 Statistical Analysis of Mitogenic Activity
The relative mitogenic activity for all FGF2 isoforms on individual FGFR variants was assessed through comparison with FGF1 activity. FGF1 is considered a positive control for FGF mitogenic assays, as previous studies have shown that FGF1 has the ability to activate all FGFR variants (Ornitz, et al. 1996). Relative mitogenic activity was calculated for each FGF2 isoform for its activity when interacting with the FGFR1 IIIb receptor variant, as FGF2 is a native ligand for this receptor variant (Ornitz, et al. 1996). An ANOVA with Tukey’s post-hoc multiple comparisons test was performed in order to determine differences in relative activity between different FGF2 isoforms supplemented at the same concentration.
3. Results
3.1 Expression and Purification of Recombinant Human FGF2 Isoforms
We first focused on establishing an FGF2 expression and purification procedure for the production of highly pure, biologically active, recombinant FGF2 isoforms. As previously-described, heparin chromatography-based purification strategies have been shown to interfere with the biological activity of the purified product (Gasparian, et al. 2009), these strategies were forgone in favor of an approach using Ni-NTA affinity chromatography. Alternative translation start sites in the FGF2 sequence were mutated to ensure exclusive expression of a single isoform from each construct (Fig. 1a) and a 6xHis tag was engineered in each isoform to aid in the purification process by allowing for purification via affinity chromatography (Fig. 1b). The FGF2 genes to be inserted were confirmed by restriction digestion and purification via agarose gel electrophoresis before cloning (Fig 1c). We showed that all five FGF2 isoforms can be over-expressed as individual isoforms using the bac-to-bac baculovirus® expression system. One-liter expression cultures using the baculovirus® expression system, followed by single step immobilized metal affinity chromatography (IMAC) over Ni-NTA resin, resulted in final protein yields ranging from 4–10mg of purified protein per culture. Protein purity was estimated to be 85–90% by SDS-PAGE stained with Coomassie brilliant blue protein stain (Fig. 2a). Western blot analysis with anti-6xHis antibody confirmed the identity of the FGF2 isoform bands visible on the Coomassie stained SDS-PAGE gel (Fig. 2b) and the chromatogram of the FPLC demonstrates a single peak for the elution fraction of each isoform (Fig. 2c).
Fig. 1.
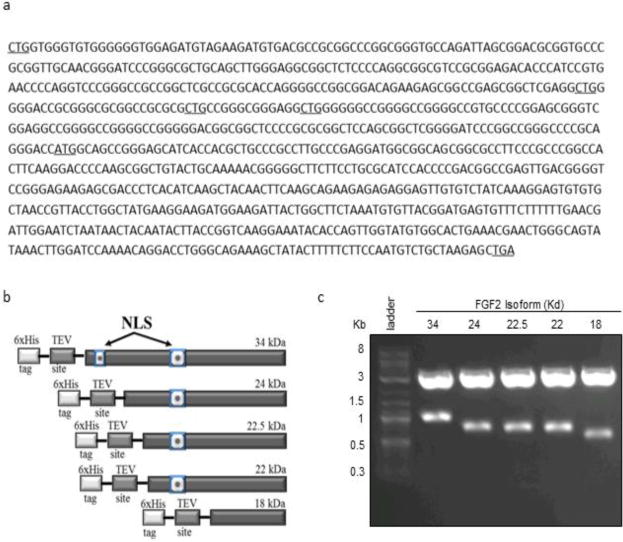
Design of human recombinant FGF2 isoforms. (a) Sequence of human FGF2 mRNA showing mutation of alternative translation start sites (underlined). (b) Design of FGF2 isoforms as fusion proteins with a 6xHistidine tag and a TEV cleavage site. (c) Gene inserts for all five FGF2 isoforms were isolated by restriction digest followed by agarose gel purification from a pBluescript cloning vector.
Fig. 2.
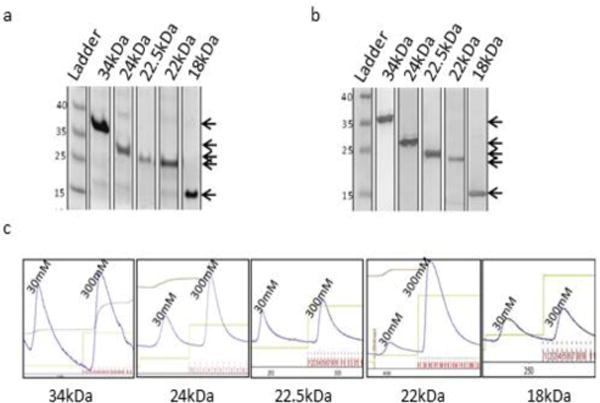
Purification of recombinant human FGF2 isoforms. (a) Coomassie stained SDS-PAGE gel of the final dialyzed prep for each rhFGF2 isoform. (b) Western blot analysis with anti-6xHis antibody. (c) Chromatograms of rhFGF2 isoforms purified over a Ni-NTA column. 30mM peak corresponds to 30mM imidazole wash fraction. 300mM peak corresponds to elution fractions.
3.2 Recombinant Human FGF2 Isoform Activity
Having purified each FGF2 isoform successfully, we next wished to determine whether these isoforms were able to recapitulate the signal transduction activity of the 18kDa isoform when added exogenously to cell culture media. Upon binding of the 18kDa FGF2 ligand to the receptor, one of the earliest known events is receptor phosphorylation, followed by phosphorylation of fibroblast growth factor receptor substrate 2α (FRS2α), resulting in a subsequent cascade of phosphorylation events (Schlessinger 2000). To qualitatively determine the activity of each purified recombinant HMW FGF2 isoform, their abilities to activate signal transduction of the ERK/MAPK canonical pathway were investigated. HMW FGF2 isoforms were added exogenously to the culture medium of human dermal fibroblasts in vitro. Western blot analysis with phospho-specific antibodies was used to determine whether recombinant human FGF2 isoforms were able to induce phosphorylation of FGFR1, FGF receptor substrate 2α (FRS2α), and subsequently activate the downstream ERK1/2 pathway. Our findings demonstrate that, when supplemented exogenously into the culture medium of human dermal fibroblasts, all purified HMW isoforms have the ability to activate FGFR1, as demonstrated by the changes in the phosphorylation state of the receptor (Fig. 3a), much in the way that 18 kDa FGF2 is able to activate FGFR1 when added exogenously. Moreover, Western blot analysis with an antibody specific to phosphorylated FRS2α protein showed that all HMW FGF2 isoforms supplemented into the human dermal fibroblast cultures were able to induce FRS2α phosphorylation with similar efficiency to that of both in-house affinity-purified and commercially available 18 kDa FGF2 (Fig. 3a). Western blot analysis also demonstrated that HMW FGF2 isoforms were able to activate the MAPK pathway by phosphorylation of ERK1/2, in a manner similar to 18 kDa FGF2, while levels of total ERK1/2 appear to be similar in cultures under all treatments (Fig. 3b). Immunocytochemistry confirmed the presence of phosphorylated FGF receptor 1 and phosphorylated ERK1/2 in samples treated with all isoforms of FGF2, including both the HMW and 18 kDa isoforms (Fig 3c). Despite detection of phosphorylated FGFR1 in control fibroblasts cultured in the absence of exogenous FGF2, which is likely due to autocrine or paracrine receptor-mediated signaling via secretion of endogenous FGF2, we did not detect phosphorylated ERK1/2 in these samples by immunocytochemistry.
Fig. 3.
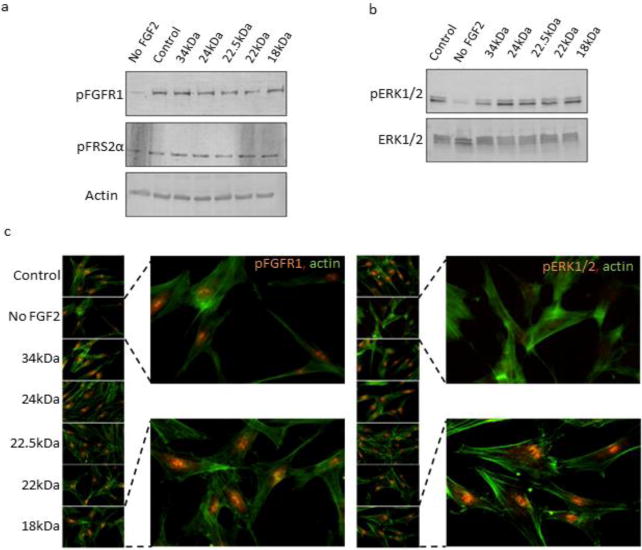
Receptor-mediated activity of recombinant human FGF2 isoforms. Western blot analysis of primary human dermal fibroblasts treated with recombinant human FGF2 isoforms for 7 days was performed for phosphorylated forms of (a) FGFR1, FRS2α, β-actin control, and (b) phospho-ERK1/2 and total ERK1/2. Individual isoforms of rhFGF2 are indicated by their respective molecular weights in kDa. Controls were treated with commercially available FGF2. (c) Immunocytochemistry analysis was performed on human dermal fibroblasts treated with rhFGF2 isoforms for pFGFR and pERK1/2. Signal was visualized with Alexafluor568-conjugated secondary antibody (orange). Filamentous actin was stained with Alexafluor488-conjugated phalloidin (green). Individual isoforms of rhFGF2 are indicated by their respective molecular weights in kDa. The “control” sample was treated with commercially available 18kDa FGF2 (Peprotech).
3.3 FGF2 Isoforms Support Self-Renewal of hES cells in vitro
Receptor-mediated activity of 18kDa FGF2 has been associated with its ability to support the maintenance of pluripotency and self-renewal capacity of human embryonic stem cells via activation of various downstream signal transduction pathways (Ding, et al. 2010, Eiselleova, et al. 2009, Gurdon 1962). Thus we decided to investigate whether exogenous supplementation with various HMW FGF2 isoforms provided similar support in vitro to hESCs as 18kDa FGF2.
Morphological analysis revealed that hESC colonies grown without FGF2 supplementation appear to begin differentiating by 8 days in culture, whereas hES cells supplemented exogenously with any of the FGF2 isoforms demonstrated little if any observable peripheral differentiation at this time point (Fig. 4a). Interestingly, Western blot analysis showed that expression of stem cell specific factors NANOG, OCT4 and LIN28 was maintained until day 8 with no major changes in protein expression level between all treatments and controls, suggesting that exogenous FGF2 may not be required for the maintenance of stem cell marker expression up to day 8 in culture under these conditions. For this reason, maintenance of stem cell-specific protein expression in hESCs treated with or without exogenously added FGF2 was analyzed at day 18.
Fig. 4.
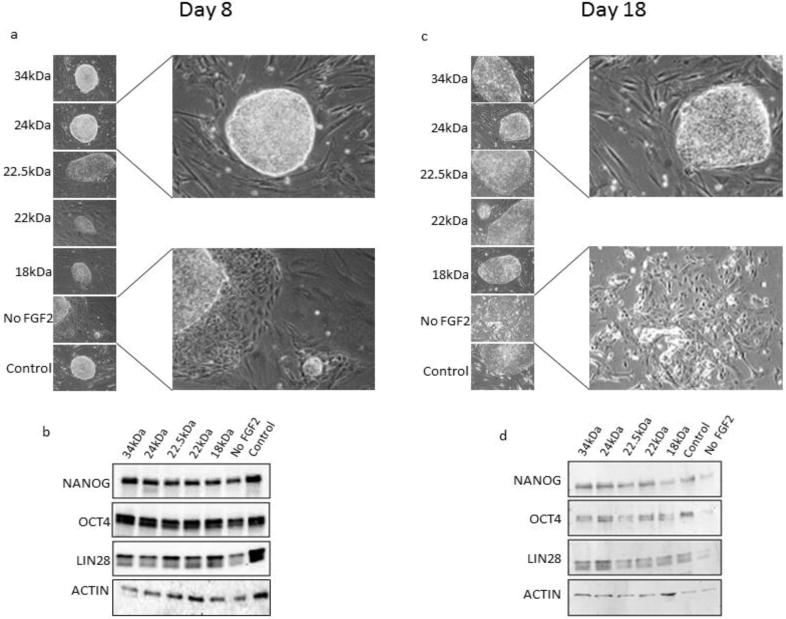
Maintenance of colony morphology and expression of stem cell genes in hESCs treated with recombinant FGF2 isoforms. Phase contrast microscopy was used in order to observe hESC colony morphology on (a) day 8 and (c) day 18 of culture. Expression of stem cell transcription factors was examined by Western blot on (b) day 8 and (d) day 18 of culture. Individual isoforms of rhFGF2 are indicated by their respective molecular weights in kDa. β-Actin was used as a loading control for Western blots. hESCs cultured in hESC cell medium without exogenous FGF2 (“No FGF2”) was used as a negative control. The “control” sample was treated with commercially available 18kDa FGF2 (Peprotech).
Treatment of hESCs for 18 days in the absence of exogenously supplemented FGF2 resulted in morphological changes characterized by the absence of the typical hESC colonies. The cells in treatments lacking exogenous FGF2 acquired a more fibroblastic morphology compared to the FGF2 treatments which maintained the typical in vitro hES morphology characterized by smaller cell size and small nuclear to cytoplasm ratio. Moreover, hES cultures maintained in the presence of exogenous FGF2 isoforms each maintained compact colony structure through the end of the 18 day treatment (Fig. 4c). Western blot analysis of these samples demonstrated that supplementation with exogenous FGF2 maintained expression of stem cell-specific markers NANOG, OCT4, and LIN28 out to day 18, while hES cells cultured in the absence of FGF2 lost the majority of NANOG, OCT4 and LIN28 protein expression by day 18 (Fig. 4d). These results suggest that expression of the stem cell-associated proteins analyzed can be supported by all FGF2 isoforms exogenously supplemented in the culture media of hESCs grown in vitro under these conditions. These findings also suggest that all FGF2 isoforms have the ability to maintain the colony morphology of undifferentiated hESCs in vitro.
3.4 Exogenously Supplemented FGF2 Isoforms Increase Proliferation Rate of Human Dermal Fibroblasts
Previous studies from our lab and others have demonstrated that 18 kDa FGF2 has the ability to increase the rate of cellular proliferation and extend proliferative lifespan in vitro when supplemented exogenously into the media of primary human dermal fibroblasts and mesenchymal cells (Bianchi, et al. 2003, Dolivo, et al. 2016, Page, et al. 2009). To examine whether exogenous addition of HMW FGF2 isoforms produces the same response on cellular proliferation we used primary adult human dermal fibroblasts (CRL-2352) grown in media supplemented with individual HMW FGF2 isoforms. The data demonstrate that primary human dermal fibroblasts underwent between 11.8 and 20.1 population doublings (PDs) over the course of 32 days of culture, depending on the isoform added. In comparison, the same primary adult human dermal fibroblasts grown in parallel and in the absence of exogenously supplemented FGF2 underwent only 8.9 PDs over the same time period in culture (Fig. 5), suggesting that all FGF2 isoforms were able to accelerate proliferation beyond that of untreated control fibroblasts to some degree. Addition of each FGF2 isoform to the media also resulted in a morphological change in cultured fibroblasts. HMW FGF2-treated fibroblasts acquired a smaller and more compact morphology compared to untreated fibroblasts, such that HMW FGF2-treated fibroblasts resembled 18kDa FGF2-treated fibroblasts morphologically (data not shown).
Fig. 5.
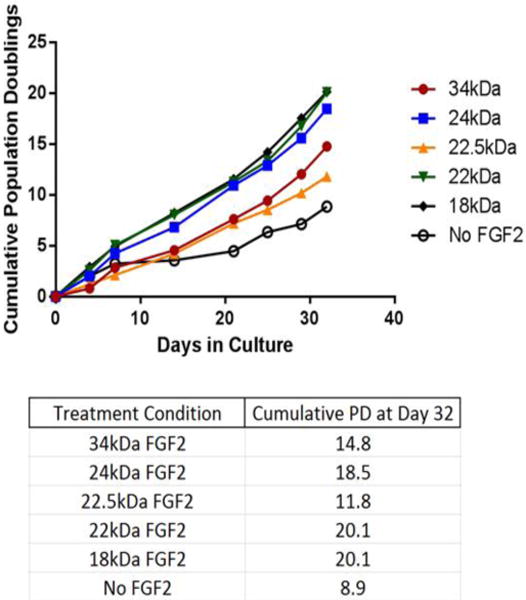
Fibroblast growth curves. CRL-2352 human dermal fibroblasts were cultured for 32 days in the presence of 4ng/mL of each FGF2 isoform, individually. Fibroblasts grown in the absence of FGF2, labeled “No FGF2,” were used as a negative control for fibroblast proliferation, to determine the proliferation-stimulating effects of FGF2 above this baseline rate. Cumulative population doublings at Day 32 are listed in the table beneath the growth curves.
3.5 Overexpression of FGF Receptor Variants in BaF3 Cells
In order to investigate specific FGF2 isoform/FGFR variant interactions, we utilized BaF3 cells, which lack native expression of FGFRs (Ornitz, et al. 1992, Wang, et al. 1994). Plasmids encoding for FGFR variants (a kind gift from Dr. David Ornitz) were used to transfect BaF3 cells by electroporation, following previously-published protocols (Ornitz, et al. 1996). The limiting dilution method was used to generate stable clones in media with antibiotic selection pressure. RT-PCR analysis with primers specific to FGF receptors (primer sequences listed in Supplementary Table 1) demonstrated the presence of the desired FGFR mRNAs in all stably-transfected cultures, and wild type BaF3 cells were used as a negative control and demonstrated no amplification (Fig. 6a). Western blot analysis of cell lysates shows a positive signal for FGFR1 IIIb and FGFR3 IIIb only for the stably-transfected cultures, and not in the untransfected wild type control cells (Fig. 6b). The presence of multiple bands in cells transfected with FGFRs is likely due to varying FGFR glycosylation states, which has been documented in previous studies (Duchesne, et al. 2006, Feige, et al. 1988).
Fig. 6.
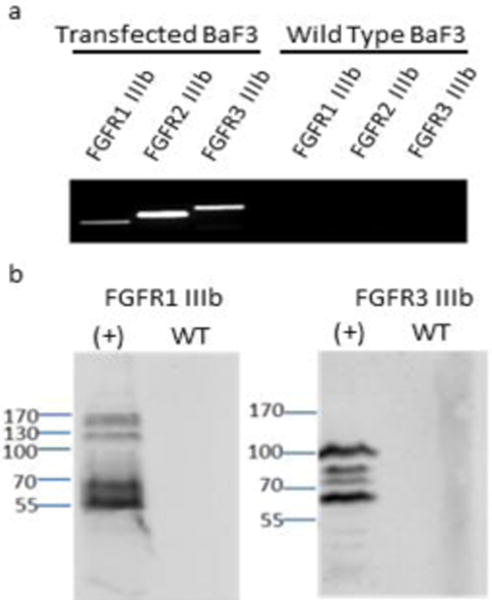
RT-PCR and Western blot analysis for overexpression of FGFR variants in BaF3 cells. (a) RT-PCR analysis with primers specific to FGFR1 IIIb, FGFR2 IIIb and FGFR3 IIIb, respectively. Transfected and wild type BaF3 cells were analyzed for each receptor variant. (b) Western blot analysis with antibodies specific to FGFR1 and FGFR3. (+) lane refers to the lane loaded with protein from transfected BaF3 cells, while WT lane refers to the lane loaded with protein from untransfected, wild-type BaF3 cells.
3.6 Mitogenic Activity in BaF3 cells
To directly compare the activity of individual FGF2 isoforms with select FGFR variants, increasing concentrations of purified FGF2 ligand were added to the culture of transgenic BaF3 cells expressing individual FGFR variants. Concurrently, the entry of cells into S-phase was monitored by observing BrdU incorporation 48 hours after treatment with individual FGF2 isoforms. Flow cytometry analysis for BrdU incorporation demonstrated that all three transgenic BaF3 cell lines expressing FGFR variants respond to FGF1 ligand. These results are in agreement with previously published work reporting FGF1 to be a universal ligand for FGFR variants (Arnaud, et al. 1999, Dell, et al. 1992, Ornitz, et al. 1995, Root, et al. 2000), and thus all subsequent FGF2 isoform activity is reported as a percentage of mitogenic activity induced by FGF1, which was supplemented at a constant concentration of 1250pM across all experiments.
Addition of FGF2 isoforms to BaF3 cells transfected with FGFR1 IIIb resulted in dose-dependent increases in proliferation, as determined by BrdU incorporation (Fig. 7a). As FGFR1 IIIb is known to have high affinity for FGF2 (Ornitz, et al. 1996), and thus FGF2 is considered a native ligand for FGFR1 IIIb, we observed substantially higher relative activity in BaF3 cells transduced with FGFR1 IIIb than in cells transduced with the other two receptor variants, FGFR2 IIIb and FGFR3 IIIb, for which FGF2 is not considered a native ligand (Fig. 7b,c). The relative mitogenic activity for the 18kDa isoform is comparable with findings previously reported in the literature (Ornitz, et al. 1996).
Though all FGF2 isoforms demonstrated dose-dependent increases in activity of its native receptor variant FGFR1 IIIb to some extent, the level of relative mitogenic activity differed for individual FGF2 isoforms (Fig 7d). Generally, 18kDa FGF2 demonstrated the highest activity; the difference between the mitogenic activity of 18kDa FGF2 and that of each other isoform was statistically significant for at least one of the three FGF2 ligand concentrations tested. In comparison, 34kDa and 22.5kDa FGF2 both demonstrated consistently lower FGF2 activity than all other isoforms, in a manner that was statistically significant for at least one of the three ligand concentrations tested. Thus, based on our data, and in terms of proliferative activity mediated by FGFR1 IIIb, the FGF2 isoforms seem to separate themselves roughly into three groups, the most potent activator (18kDa FGF2), the moderate activators (24kDa and 22kDa FGF2), and the least potent activators (22.5kDa and 34kDa FGF2).
4. Discussion
The study of FGF signaling is inherently important, as FGF signaling is ubiquitous and implicated in a myriad of important cellular and organismal processes, from embryogenesis to angiogenesis and beyond (Eiselleova, et al. 2009, Ornitz, et al. 1996, Zhang, et al. 2006, Zoumaro-Djayoon, et al. 2011). FGF signaling is mediated by specific ligand-receptor interactions, thus allowing for cell-specific tuning of FGF effects based on qualitative and quantitative expression patterns of FGFRs on the cell surface, and based on the presence of various FGFs in the milieu (Zhang, et al. 2006). Better understanding of FGF ligand biology and their interactions with specific FGFR variants would allow us to better predict the effects of particular FGFs on specific cells based on their FGFR expression patterns, and better enable us to understand the cellular physiology of pathologies associated with aberrant FGF/FGFR signaling.
The biological consequences of HMW FGF2 isoforms functioning as secreted ligands have not been extensively studied and are not well-understood. The data presented in this study demonstrate the ability of HMW FGF2 isoforms to act as extracellular ligands for select FGF receptor tyrosine kinases and to activate the canonical FGF2/FGFR/ERK pathway that is proven to mediate many different cellular responses in various cell types. We have demonstrated that HMW FGF2 isoforms can induce FGF receptor phosphorylation and lead to signal transduction at a level comparable to that induced by the 18kDa isoform. In addition to activation of the ERK/MAPK pathway (Fig. 3), the ability of HMW FGF2 isoforms to partake in canonical FGF2 signaling was also demonstrated by supporting hES cell growth in culture and maintenance of expression of stem cell-specific markers in embryonic stem cells (Fig. 4), mitogenic stimulation of fibroblasts (Fig. 5), and the ability to support proliferation of BaF3 cells expressing specific FGFR variants, in the absence of exogenously supplemented IL-3, conditions under which they generally will not otherwise proliferate (Fig. 7).
While all of the HMW FGF2 isoforms demonstrate the ability to recapitulate the activity of the 18kDa isoform qualitatively via the aforementioned metrics, activities differ between different FGF2 isoforms. The LMW 18kDa FGF2 isoform appears to be the most potent activator of receptor-mediated FGF2 signaling, which is unsurprising given that it is generally secreted from the cell and thus produces cellular responses via autocrine or paracrine signaling (Goetz, et al. 2013). Though all of the HMW isoforms appear to be less efficient than the 18kDa isoform at inducing canonical FGF2 signaling by at least one measure investigated in the work presented here, the least effective FGF2 isoforms by several measures appear to be the 22.5kDa and 34kDa isoforms.
Compared to treatments with other FGF2 isoforms, stimulation with 22.5kDa FGF2 results in lower stem cell gene expression in hESCs (Fig. 4d), weaker proliferative activity in human dermal fibroblasts (Fig. 5), and lower relative mitogenic activity in BaF3 cells ectopically expressing FGFR1 IIIb, one of the primary receptor variants that is known to be activated by 18kDa FGF2 (Fig. 7d,e) (Ornitz, et al. 1996). Similarly, stimulation with 34kDa FGF2 results in a lower level of ERK1/2 phosphorylation and weaker proliferative activity in human dermal fibroblasts (Fig. 3b, 5), and lower relative mitogenic activity in BaF3 cells ectopically expressing FGFR1 IIIb (Fig. 7d, e). Despite their lower activities by several measures, both 22.5kDa and 34kDa FGF2 still maintain the ability to support several FGF2-related phenotypes. In a manner qualitatively similar to other FGF2 isoforms, 22.5kDa and 34kDa FGF2 are able to induce phosphorylation of FGFR1, FRS2α, and ERK (Fig 3a, b), support embryonic stem cell gene expression and self-renewal in culture (Fig 4c, d), stimulate proliferation in human dermal fibroblasts (Fig 5), and induce mitogenesis to some degree in BaF3 cells transduced with FGFR1 IIIb (Fig 7).
There are several possible explanations for the differential efficiencies among the HMW isoforms, in particular for the 22.5kDa and 34kDa isoforms, to maintain canonical FGFR-mediated phenotypes compared to each other and to the 18kDa isoform. Since the HMW FGF2 isoforms are all co-linear, N-terminal extensions of the 18kDa isoform, it is possible that some portions of the N-terminal extensions of the HMW FGF2 isoforms affect the interaction of FGF2 with its receptor, resulting in quantitative differences in stimulation of downstream signaling cascades. Additionally, we cannot discount the possibility that the differential activity of FGF2 isoforms may be due to differences in proteolytic degradation or protein stability conferred by the N-terminal extensions to the 18kDa isoform, or due to differences in subcellular protein localization upon internalization of the isoform after interaction with its receptor (Więdłocha, et al. 2004). Further study into the biochemical details of canonical FGFR-mediated signal transduction by HMW FGF2 isoforms should be performed keeping in mind these paradigms as possibilities.
Though localization of HMW isoforms of FGF2 is primarily intracellular and generally considered to be contained to the nucleus (Arnaud, et al. 1999, Quarto, et al. 1991), it has also been proposed that HMW FGF2 isoforms can be released from FGF2-expressing cells by several processes including vesicle shedding, cell death, or wounding (McNeil, et al. 1989, Schweigerer, et al. 1987, Taverna, et al. 2003). Since we have determined that extracellular, HMW isoforms of FGF2 can cause many of the same phenotypic effects of 18kDa, it is plausible to suspect that release of these HMW FGF2 isoforms may potentiate canonical FGF2 signaling via FGFRs, allowing for them to act via transmembrane receptors in a similar manner to secreted, 18 kDa FGF2. Thus, conditions under which HMW FGF2 isoforms are released into the extracellular space could lead to potentiation of canonical autocrine or paracrine FGF2 signaling.
Improved understanding of the ability of different FGF2 isoforms to interact with different receptors will lead to a better understanding of the different functions of FGF2 not only on various cell types, which express different patterns of FGFs and FGFRs (Hughes 1997), but also in different cellular processes, such as angiogenesis, embryogenesis, differentiation, proliferation, self-renewal, and others involved in normal or pathological physiologies (Eswarakumar, et al. 2005). Future studies may reveal differences among FGF2 isoforms in binding kinetics or in ability to activate particular FGFR-mediated pathways, thus explaining some of the differences we see in cell phenotypes induced by treatment with different FGF2 isoforms.
Highlights.
All 5 FGF2 isoforms were individually expressed and purified.
Addition to culture media of high molecular weight FGF2 maintains FGF2 phenotypes.
All FGF2 isoforms activate FGFR1 IIIb, activate ERK, and induce proliferation.
All FGF2 isoforms maintain the ability of embryonic stem cells to self-renew.
Different FGF2 isoforms display differential abilities to mediate specific effects.
Acknowledgments
We would like to thank Dr. David Ornitz, Alumni Endowed Professor, Washington University School of Medicine for his generosity in providing the constructs for FGFR variants and the wild type BaF3 cell line. The anti-BrdU G3G4 antibody was obtained from the Developmental Studies Hybridoma Bank, created by the NICHD of the NIH, and maintained at the University of Iowa, Department of Biology (Iowa City, IA).
Funding: This work was funded by NIH R01GM085456 awarded to Tanja Dominko.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interest: The authors declare no conflicts of interest.
References
- Arese M, Chen Y, Florkiewicz RZ, Gualandris A, Shen B, Rifkin DB. Nuclear activities of basic fibroblast growth factor: potentiation of low-serum growth mediated by natural or chimeric nuclear localization signals. Mol Biol Cell. 1999;10:1429–1444. doi: 10.1091/mbc.10.5.1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnaud E, Touriol C, Boutonnet C, Gensac MC, Vagner S, Prats H, Prats AC. A new 34-kilodalton isoform of human fibroblast growth factor 2 is cap dependently synthesized by using a non-AUG start codon and behaves as a survival factor. Mol Cell Biol. 1999;19:505–514. doi: 10.1128/mcb.19.1.505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianchi G, Banfi A, Mastrogiacomo M, Notaro R, Luzzatto L, Cancedda R, Quarto R. Ex vivo enrichment of mesenchymal cell progenitors by fibroblast growth factor 2. Exp Cell Res. 2003;287:98–105. doi: 10.1016/s0014-4827(03)00138-1. [DOI] [PubMed] [Google Scholar]
- Champion-Arnaud P, Ronsin C, Gilbert E, Gesnel MC, Houssaint E, Breathnach R. Multiple mRNAs code for proteins related to the BEK fibroblast growth factor receptor. Oncogene. 1991;6:979–987. [PubMed] [Google Scholar]
- Dailey L, Ambrosetti D, Mansukhani A, Basilico C. Mechanisms underlying differential responses to FGF signaling. Cytokine & Growth Factor Reviews. 2005;16:233–247. doi: 10.1016/j.cytogfr.2005.01.007. [DOI] [PubMed] [Google Scholar]
- Dell KR, Williams LT. A novel form of fibroblast growth factor receptor 2. Alternative splicing of the third immunoglobulin-like domain confers ligand binding specificity. J Biol Chem. 1992;267:21225–21229. [PubMed] [Google Scholar]
- Ding VM, Ling L, Natarajan S, Yap MG, Cool SM, Choo AB. FGF-2 modulates Wnt signaling in undifferentiated hESC and iPS cells through activated I3-K/GSK3beta signaling. J Cell Physiol. 2010;225:417–428. doi: 10.1002/jcp.22214. [DOI] [PubMed] [Google Scholar]
- Dolivo D, Hernandez S, Dominko T. Cellular lifespan and senescence: a complex balance between multiple cellular pathways. BioEssays. 2016;38:S33–S44. doi: 10.1002/bies.201670906. [DOI] [PubMed] [Google Scholar]
- Duchesne L, Tissot B, Rudd TR, Dell A, Fernig DG. N-glycosylation of fibroblast growth factor receptor 1 regulates ligand and heparan sulfate co-receptor binding. J Biol Chem. 2006;281:27178–27189. doi: 10.1074/jbc.M601248200. [DOI] [PubMed] [Google Scholar]
- Dvorak P, Dvorakova D, Koskova S, Vodinska M, Najvirtova M, Krekac D, Hampl A. Expression and potential role of fibroblast growth factor 2 and its receptors in human embryonic stem cells. Stem Cells. 2005;23:1200–1211. doi: 10.1634/stemcells.2004-0303. [DOI] [PubMed] [Google Scholar]
- Eiselleova L, Matulka K, Kriz V, Kunova M, Schmidtova Z, Neradil J, Tichy B, Dvorakova D, Pospisilova S, Hampl A, Dvorak P. A complex role for FGF-2 in self-renewal, survival, and adhesion of human embryonic stem cells. Stem Cells. 2009;27:1847–1857. doi: 10.1002/stem.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eswarakumar VP, Lax I, Schlessinger J. Cellular signaling by fibroblast growth factor receptors. Cytokine Growth Factor Rev. 2005;16:139–149. doi: 10.1016/j.cytogfr.2005.01.001. [DOI] [PubMed] [Google Scholar]
- Ezzat S, Asa SL. FGF receptor signaling at the crossroads of endocrine homeostasis and tumorigenesis. Horm Metab Res. 2005;37:355–360. doi: 10.1055/s-2005-870151. [DOI] [PubMed] [Google Scholar]
- Feige JJ, Baird A. Glycosylation of the basic fibroblast growth factor receptor. The contribution of carbohydrate to receptor function. J Biol Chem. 1988;263:14023–14029. [PubMed] [Google Scholar]
- Florkiewicz RZ, Anchin J, Baird A. The inhibition of fibroblast growth factor-2 export by cardenolides implies a novel function for the catalytic subunit of Na+, K+-ATPase. J Biol Chem. 1998;273:544–551. doi: 10.1074/jbc.273.1.544. [DOI] [PubMed] [Google Scholar]
- Florkiewicz RZ, Sommer A. Human basic fibroblast growth factor gene encodes four polypeptides: three initiate translation from non-AUG codons. Proc Natl Acad Sci U S A. 1989;86:3978–3981. doi: 10.1073/pnas.86.11.3978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasparian ME, Elistratov PA, Drize I, Nifontova IN, Dolgikh DA, Kirpichnikov MP. Overexpression in Escherichia coli and purification of human fibroblast growth factor (FGF-2), Biochemistry. Biokhimiia. 2009;74:221–225. doi: 10.1134/s000629790902014x. [DOI] [PubMed] [Google Scholar]
- Givol D, Yayon A. Complexity of FGF receptors: genetic basis for structural diversity and functional specificity. FASEB J. 1992;6:3362–3369. [PubMed] [Google Scholar]
- Goetz R, Mohammadi M. Exploring mechanisms of FGF signalling through the lens of structural biology. Nature reviews Molecular cell biology. 2013;14:166–180. doi: 10.1038/nrm3528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gospodarowicz D, Cheng J, Lui GM, Baird A, Bohlent P. Isolation of brain fibroblast growth factor by heparin-Sepharose affinity chromatography: identity with pituitary fibroblast growth factor. Proc Natl Acad Sci U S A. 1984;81:6963–6967. doi: 10.1073/pnas.81.22.6963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greber B, Lehrach H, Adjaye J. Fibroblast growth factor 2 modulates transforming growth factor beta signaling in mouse embryonic fibroblasts and human ESCs (hESCs) to support hESC self-renewal. Stem Cells. 2007;25:455–464. doi: 10.1634/stemcells.2006-0476. [DOI] [PubMed] [Google Scholar]
- Grose R, Dickson C. Fibroblast growth factor signaling in tumorigenesis. Cytokine & Growth Factor Reviews. 2005;16:179–186. doi: 10.1016/j.cytogfr.2005.01.003. [DOI] [PubMed] [Google Scholar]
- Gurdon JB. Adult frogs derived from the nuclei of single somatic cells. Dev Biol. 1962;4:256–273. doi: 10.1016/0012-1606(62)90043-x. [DOI] [PubMed] [Google Scholar]
- Hughes SE. Differential expression of the fibroblast growth factor receptor (FGFR) multigene family in normal human adult tissues. Journal of Histochemistry & Cytochemistry. 1997;45:1005–1019. doi: 10.1177/002215549704500710. [DOI] [PubMed] [Google Scholar]
- Jiang ZS, Jeyaraman M, Wen GB, Fandrich RR, Dixon IM, Cattini PA, Kardami E. High-but not low-molecular weight FGF-2 causes cardiac hypertrophy in vivo; possible involvement of cardiotrophin-1. Journal of molecular and cellular cardiology. 2007;42:222–233. doi: 10.1016/j.yjmcc.2006.09.002. [DOI] [PubMed] [Google Scholar]
- Krejci P, Aklian A, Kaucka M, Sevcikova E, Prochazkova J, Masek JK, Mikolka P, Pospisilova T, Spoustova T, Weis M, Paznekas WA, Wolf JH, Gutkind JS, Wilcox WR, Kozubik A, Jabs EW, Bryja V, Salazar L, Vesela I, Balek L. Receptor tyrosine kinases activate canonical WNT/beta-catenin signaling via MAP kinase/LRP6 pathway and direct beta-catenin phosphorylation. PLoS One. 2012;7:e35826. doi: 10.1371/journal.pone.0035826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma X, Dang X, Claus P, Hirst C, Fandrich RR, Jin Y, Grothe C, Kirshenbaum LA, Cattini PA, Kardami E. Chromatin compaction and cell death by high molecular weight FGF‐2 depend on its nuclear localization, intracrine ERK activation, and engagement of mitochondria. Journal of cellular physiology. 2007;213:690–698. doi: 10.1002/jcp.21139. [DOI] [PubMed] [Google Scholar]
- McNeil PL, Muthukrishnan L, Warder E, D’Amore PA. Growth factors are released by mechanically wounded endothelial cells. The Journal of Cell Biology. 1989;109:811–822. doi: 10.1083/jcb.109.2.811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meo Burt P, Xiao L, Dealy C, Fisher MC, Hurley MM. FGF2 High Molecular Weight Isoforms Contribute to Osteoarthropathy in Male Mice. Endocrinology. 2016;157:4602–4614. doi: 10.1210/en.2016-1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miki T, Bottaro DP, Fleming TP, Smith CL, Burgess WH, Chan AM, Aaronson SA. Determination of ligand-binding specificity by alternative splicing: two distinct growth factor receptors encoded by a single gene. Proc Natl Acad Sci U S A. 1992;89:246–250. doi: 10.1073/pnas.89.1.246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickel W. The unconventional secretory machinery of fibroblast growth factor 2. Traffic. 2011;12:799–805. doi: 10.1111/j.1600-0854.2011.01187.x. [DOI] [PubMed] [Google Scholar]
- Ornitz DM, Herr AB, Nilsson M, Westman J, Svahn CM, Waksman G. FGF binding and FGF receptor activation by synthetic heparan-derived di- and trisaccharides. Science. 1995;268:432–436. doi: 10.1126/science.7536345. [DOI] [PubMed] [Google Scholar]
- Ornitz DM, Leder P. Ligand specificity and heparin dependence of fibroblast growth factor receptors 1 and 3. J Biol Chem. 1992;267:16305–16311. [PubMed] [Google Scholar]
- Ornitz DM, Marie PJ. FGF signaling pathways in endochondral and intramembranous bone development and human genetic disease. Genes Dev. 2002;16:1446–1465. doi: 10.1101/gad.990702. [DOI] [PubMed] [Google Scholar]
- Ornitz DM, Xu J, Colvin JS, McEwen DG, MacArthur CA, Coulier F, Gao G, Goldfarb M. Receptor specificity of the fibroblast growth factor family. J Biol Chem. 1996;271:15292–15297. doi: 10.1074/jbc.271.25.15292. [DOI] [PubMed] [Google Scholar]
- Ornitz DM, Yayon A, Flanagan JG, Svahn CM, Levi E, Leder P. Heparin is required for cell-free binding of basic fibroblast growth factor to a soluble receptor and for mitogenesis in whole cells. Mol Cell Biol. 1992;12:240–247. doi: 10.1128/mcb.12.1.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Page RL, Ambady S, Holmes WF, Vilner L, Kole D, Kashpur O, Huntress V, Vojtic I, Whitton H, Dominko T. Induction of stem cell gene expression in adult human fibroblasts without transgenes. Cloning Stem Cells. 2009;11:417–426. doi: 10.1089/clo.2009.0015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piotrowicz RS, Martin JL, Dillman WH, Levin EG. The 27-kDa heat shock protein facilitates basic fibroblast growth factor release from endothelial cells. J Biol Chem. 1997;272:7042–7047. doi: 10.1074/jbc.272.11.7042. [DOI] [PubMed] [Google Scholar]
- Powers CJ, McLeskey SW, Wellstein A. Fibroblast growth factors, their receptors and signaling. Endocr-Relat Cancer. 2000;7:165–197. doi: 10.1677/erc.0.0070165. [DOI] [PubMed] [Google Scholar]
- Presta M, Dell’Era P, Mitola S, Moroni E, Ronca R, Rusnati M. Fibroblast growth factor/fibroblast growth factor receptor system in angiogenesis. Cytokine Growth Factor Rev. 2005;16:159–178. doi: 10.1016/j.cytogfr.2005.01.004. [DOI] [PubMed] [Google Scholar]
- Quarto N, Amalric F. Heparan sulfate proteoglycans as transducers of FGF-2 signalling. J Cell Sci. 1994;107(Pt 11):3201–3212. doi: 10.1242/jcs.107.11.3201. [DOI] [PubMed] [Google Scholar]
- Quarto N, Finger FP, Rifkin DB. The NH2-terminal extension of high molecular weight bFGF is a nuclear targeting signal. J Cell Physiol. 1991;147:311–318. doi: 10.1002/jcp.1041470217. [DOI] [PubMed] [Google Scholar]
- Root LL, Shipley GD. Normal human fibroblasts produce membrane-bound and soluble isoforms of FGFR-1. Molecular cell biology research communications: MCBRC. 2000;3:87–97. doi: 10.1006/mcbr.2000.0199. [DOI] [PubMed] [Google Scholar]
- Santiago JJ, Ma X, McNaughton LJ, Nickel BE, Bestvater BP, Yu L, Fandrich RR, Netticadan T, Kardami E. Preferential accumulation and export of high molecular weight FGF-2 by rat cardiac non-myocytes. Cardiovascular research. 2011;89:139–147. doi: 10.1093/cvr/cvq261. [DOI] [PubMed] [Google Scholar]
- Santiago JJ, McNaughton LJ, Koleini N, Ma X, Bestvater B, Nickel BE, Fandrich RR, Wigle JT, Freed DH, Arora RC. High molecular weight fibroblast growth factor-2 in the human heart is a potential target for prevention of cardiac remodeling. PloS one. 2014;9:e97281. doi: 10.1371/journal.pone.0097281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos-Ocampo S, Colvin JS, Chellaiah A, Ornitz DM. Expression and biological activity of mouse fibroblast growth factor-9. J Biol Chem. 1996;271:1726–1731. doi: 10.1074/jbc.271.3.1726. [DOI] [PubMed] [Google Scholar]
- Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2000;103:211–225. doi: 10.1016/s0092-8674(00)00114-8. [DOI] [PubMed] [Google Scholar]
- Schweigerer L, Neufeld G, Friedman J, Abraham JA, Fiddes JC, Gospodarowicz D. Capillary endothelial cells express basic fibroblast growth factor, a mitogen that promotes their own growth. Nature. 1987;325:257–259. doi: 10.1038/325257a0. [DOI] [PubMed] [Google Scholar]
- Sperger JM, Chen X, Draper JS, Antosiewicz JE, Chon CH, Jones SB, Brooks JD, Andrews PW, Brown PO, Thomson JA. Gene expression patterns in human embryonic stem cells and human pluripotent germ cell tumors. Proc Natl Acad Sci U S A. 2003;100:13350–13355. doi: 10.1073/pnas.2235735100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spivak-Kroizman T, Lemmon MA, Dikic I, Ladbury JE, Pinchasi D, Huang J, Jaye M, Crumley G, Schlessinger J, Lax I. Heparin-induced oligomerization of FGF molecules is responsible for FGF receptor dimerization, activation, and cell proliferation. Cell. 1994;79:1015–1024. doi: 10.1016/0092-8674(94)90032-9. [DOI] [PubMed] [Google Scholar]
- Taverna S, Ghersi G, Ginestra A, Rigogliuso S, Pecorella S, Alaimo G, Saladino F, Dolo V, Dell’Era P, Pavan A, Pizzolanti G, Mignatti P, Presta M, Vittorelli ML. Shedding of membrane vesicles mediates fibroblast growth factor-2 release from cells. J Biol Chem. 2003;278:51911–51919. doi: 10.1074/jbc.M304192200. [DOI] [PubMed] [Google Scholar]
- Touriol C, Bornes S, Bonnal S, Audigier S, Prats H, Prats AC, Vagner S. Generation of protein isoform diversity by alternative initiation of translation at non-AUG codons. Biology of the cell/under the auspices of the European Cell Biology Organization. 2003;95:169–178. doi: 10.1016/s0248-4900(03)00033-9. [DOI] [PubMed] [Google Scholar]
- Wang JK, Gao G, Goldfarb M. Fibroblast growth factor receptors have different signaling and mitogenic potentials. Molecular and Cellular Biology. 1994;14:181–188. doi: 10.1128/mcb.14.1.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Więdłocha A, Sørensen V. Signalling from Internalized Growth Factor Receptors. Springer; 2004. Signaling, internalization, and intracellular activity of fibroblast growth factor; pp. 45–79. [DOI] [PubMed] [Google Scholar]
- Zehe C, Engling A, Wegehingel S, Schäfer T, Nickel W. Cell-surface heparan sulfate proteoglycans are essential components of the unconventional export machinery of FGF-2. Proceedings of the National Academy of Sciences. 2006;103:15479–15484. doi: 10.1073/pnas.0605997103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Ibrahimi OA, Olsen SK, Umemori H, Mohammadi M, Ornitz DM. Receptor specificity of the fibroblast growth factor family. The complete mammalian FGF family. J Biol Chem. 2006;281:15694–15700. doi: 10.1074/jbc.M601252200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoumaro-Djayoon AD, Ding V, Foong LY, Choo A, Heck AJ, Munoz J. Investigating the role of FGF-2 in stem cell maintenance by global phosphoproteomics profiling. Proteomics. 2011;11:3962–3971. doi: 10.1002/pmic.201100048. [DOI] [PubMed] [Google Scholar]
- Zubilewicz A, Hecquet C, Jeanny JC, Soubrane G, Courtois Y, Mascarelli F. Two distinct signalling pathways are involved in FGF2-stimulated proliferation of choriocapillary endothelial cells: a comparative study with VEGF. Oncogene. 2001;20:1403–1413. doi: 10.1038/sj.onc.1204231. [DOI] [PubMed] [Google Scholar]