

Abstract
The biochemical properties of the signal-induced multifunctional transcription factor TFII-I indicate that it is involved in a variety of gene regulatory processes. Although gene ablation in murine models and cell based assays show that it is encoded by an essential gene, GTF2I/Gtf2i, its physiologic role in human disorders was relatively unknown until recently. Novel studies show it is involved in an array of human diseases including neurocognitive disorders, Systemic Lupus Erythymatosus, and cancer. Here, I bring together these diverse observations to illustrate its multiple patho-physiological functions and further conjecture on how these could be related to its known biochemical properties. I expect that a better understanding of these “structure-function” relationships would lead to future diagnostic and/or therapeutic potentials.
Keywords: TFII-I, GTF2I, transcription, lymphoma, SLE, WBS
TFII-I—A Friend or Foe?
The prerequisite to switching on/activation of a gene or a set of genes is that the targeted transcriptional unit in the chromosome becomes “remodeled” and accessible for transcription via actions of chromatin modifiers, transcription factors/co-factors and epigenetic changes (1,2). Once the natural constraints imposed by chromatin are obviated, the next step in gene expression is the recruitment of gene/tissue/stage specific transcription factors to their cognate sites on a given promoter (1,2). These transcription factors, and a variety of co-factors, including mediators facilitate in turn, recruitment of the transcriptional machinery at the core promoter, the rate and/or stability of which dictates the rate of RNA synthesis (1). Normal cells proliferate and differentiate to acquire effector functions in a tightly regulated fashion in response to cell-extrinsic signals transduced through specific surface receptors to the nucleus in order to control gene expression (3). Consequently, a breakdown in transcriptional regulatory pathways results in abnormal cellular functions causing various malignancies, immune deficiencies and other human disorders.
While the biochemical properties of the multifunctional general transcription factor II-I (TFII-I), as well as its essential role in life have been known for some time, its clinical relevance in human diseases began to emerge only recently via patient studies. Therefore, it is important to bring these observations to light and evaluate our understanding of the role TFII-I plays in human health. Below I summarize these observations and classify them into three categories: i) pathologies associated with TFII-I gene dosage effects; ii) diseases correlated with single nucleotide polymorphisms (SNPs) in the gene encoding TFII-I; and iii) clinical manifestations associated with functional mutations in the coding region of TFII-I and its gene fusions/chromosomal translocations. Whenever appropriate, I provide my own perspectives as to how these patient-derived observations could be reconciled with its established biochemical properties and finally provide my views on future directions that could yield diagnostic and/or therapeutic potentials.
Biochemical Properties and Structural Features of TFII-I
TFII-I was biochemically discovered 25 years ago as a transcription factor that bound to the Adeno Virus Major Late core promoter Initiator (Inr) element and interacted with a sequence-specific DNA element (E-box element) with an E-box binding protein, USF (Upstrem Stimulatory Factor) in cell free systems (4,5). While binding and function of TFII-I via the Inr element has been noted in several instances both in vitro and in vivo (animal cells), it does not appear to be a general transcription factor required for all Inr-containing promoters, particularly when assayed in in vitro transcription systems with highly purified mammalian transcription factors (6). Whether there are functionally distinct classes of Inr elements and corresponding factors (TFII-I being one of them) or whether interaction of TFII-I with the Inr element is required for steps beyond transcription initiation remains currently unresolved (6). Subsequent studies indicated that TFII-I is a signal-induced transcription factor (6–8), and consequently, the biochemical function of TFII-I in mediating signal dependent transcription in various physiological contexts has drawn attention over the last few years.
TFII-I (GTF2I in humans and Gtf2i in mice) belongs to a family of vertebrate-specific transcription factors, encoded by three related genes that are closely located in human chromosome 7 (Fig 1A, ref 9). Genetic ablation studies in mice demonstrated that it is an essential gene as ablation causes early embryonic lethality with severe defects in angiogenesis and vasculogenesis most likely due to deregulation in the vascular endothelial growth factor receptor-2, VEGFR2 gene (10). TFII-I’s essential role in angiogenesis and transcriptional regulation of VEGFR2 has been further confirmed using both matrigel assays and silencing of TFII-I in murine neonatal retina (11). Recent analysis of high-quality exome (protein-coding region) DNA sequence data for 60,706 individuals of diverse ancestries, as well as extensive mutagenesis analysis of human haploid cells identified genes required for optimal fitness under culture conditions, including TFII-I, noted as being encoded by a cell-essential gene (12,13).
Each member of this family of transcription factors has multiple alternatively spliced isoforms (Fig 2). The best characterized is TFII-I, which has at least four alternatively spliced isoforms in humans (α, β, γ and Δ) (9, 14). However, the expressions patterns or transcriptional functions of the beta, gamma and delta isoforms are currently unknown (9). The Δ and β-isoforms were shown to have opposing transcription functions in NIH 3T3 cells (15). Expression of the γ-isoform appears to be restricted to neuronal cell types in mammals (14). GTF2I also harbors pseudogenes on chromosomes 9, 13, and 21 (9). An unusual structural characteristic of TFII-I family members is the presence of a repeated amino acid domain each containing a putative helix-loop-helix HLH-motif characterizing a protein–protein interaction module (5,6). TFII-I has six such repeated domains (Fig. 1A). However, there are differences between TFII-I HLH motifs and traditional HLH motifs, noted both by structure prediction and by actual determination of solution structure of one of the TFII-I repeats; this suggests that although TFII-I might harbor properties of typical HLH-containing transcription factors, it does not conform to a any known structural prototypes (6,16).
Fig 2. Gene Structure of Human (GTF2I) and Murine (Gtf2i).
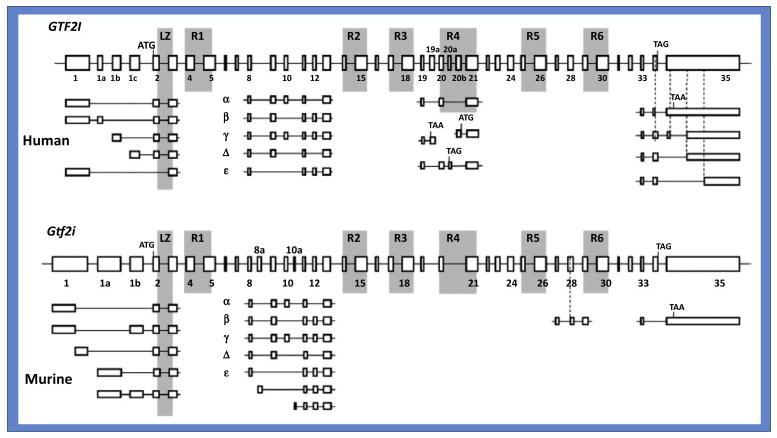
ATG is the initiation codon; TAA and TAG are potential stop codons; boxes with numbers 1–35 signifies different exons; α, β, γ, Δ and ε signifies various alternatively spliced isoforms with used or missing exons indicated. In mouse, two additional isoforms are predicted, although their actual physical existence remains unclear. (Figure Courtesy: Dashzeveg Bayarsaihan).
Fig 1. Biochemical Features of TFII-I.
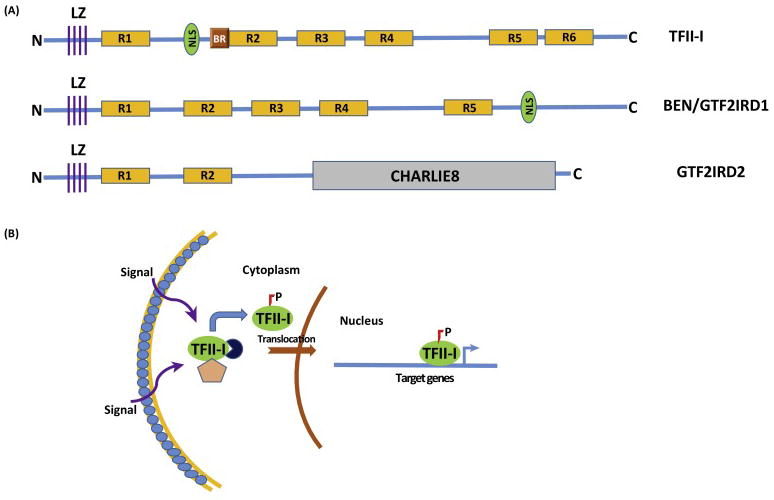
(A) Schematics of 3 TFII-I Family Proteins: The diagram shows TFII-I, GTF2IRD/BEN and GTF2IRD2 proteins. LZ-leucine zipper; R1-R6-TFII-I/GTF2I repeats; NLS-nuclear localization signal; BR-basic region, sequence important for DNA binding; N-N-terminal end and C-Carboxy-terminal end; CHARLIE8-a transposon-like element opf 534 residues (B) Mechanistic Model of Signal-Dependent Nuclear Translocation of TFII-I: Various cell extrinsic signals (such as B and T-cell receptor mediated signals, growth factor receptor stimulation etc) result in phosphorylation of TFII-I and its uncoupling from cytoplasmic tethers (designated by the pink pentagon and navy circle), with subsequent nuclear translocation. Phosphorylated and activated nuclear TFII-I can occupy sites on its target gene promoters (transcription start site is designated by the blue bent arrow) to induce transcription.
Signal-Induced Functions of TFII-I
TFII-I operates as a signal-induced transcription factor that is tyrosine phosphorylated (activated) in response to a variety of extracellular signaling pathways, including B- and T-cell receptor triggering as well as growth factor signaling in animal cells (6,7,17–20). TFII-I remains in the cytoplasm in a variety of cells in latent form, although the exact mechanism of its cytoplasmic localization remains unknown (Fig 1B). Several growth-promoting and mitogenic stimuli can enhance tyrosine phosphorylation and subsequent nuclear translocation as well as transcriptional activation of TFII-I (6, 17–20). Apart from its growth factor- and immune cell surface-receptor induced activation, TFII-I also appears to be an important regulator of the endoplasmic reticulum (ER) stress response pathway in biochemical assays (21). Additionally, TFII-I was found to specifically interact with G-kinase Iβ, and cGMP analogs enhanced the association between TFII-I and G-kinase Iβ in the nucleus when assayed in both murine and human cell lines (22).
Cytoplasmic Function of TFII-I
Engagement of cell surface receptor tyrosine kinases by their cognate ligands is associated with Ca2+ influx into the cell, resulting in the generation of IP3 (inositol 1,4,5-trisphosphate) and DAG (diacylglycerol) from PIP2 (phosphatidylinositol 4,5- bisphosphate) via the enzymatic action of phospholipase C (PLC) (23). However, a novel lipase-independent role of PLC-γ that is necessary for intracellular Ca2+ entry via transient receptor potential channel 3 (TRPC3) was reported (24). Specifically, TFII-I was identified as an interacting partner of PLC-γ in murine and human cell lines and because TRPC3 surface expression and function requires its binding to PLC-γ, competitive association of TFII-I with PLC-γ was shown to lower surface expression of TRPC3 and reduce its Ca2+ channeling activity (24). Moreover, this phenomenon did not require either the nuclear localization or transcriptional function of TFII-I (24, 25). Consistent with its cytoplasmic residency, these results suggest that TFII-I has a cytoplasmic function that is independent of its nuclear transcription function.
With these biochemical properties of TFII-I in mind, I will now turn to recent clinical observations that shed new light on its physiological role in human health and disease.
Role of TFII-I in Williams-Beuren syndrome (WBS) and other Neurodevelopmental Defects
Haploinsufficiency and other Dosage Effects
WBS is a multisystem dysfunction that includes supravalvar aortic stenosis (SVAS), hypercalcemia in infancy, mild to moderate mental retardation, cognitive defects, and characteristic craniofacial abnormalities (26). It is caused by a hemizygous deletion of 25–30 genes on chromosome 7q11.23 and the frequency of this genetic syndrome is estimated to be 1 in 20,000 live births (26). Although most cases are sporadic, WBS is inherited as an autosomal dominant trait in some families (26). All three TFII-I family genes are deleted in typical WBS patients (26, 27). However, rare individuals with smaller deletions in the WBS region have been identified, yielding significant insights (28, 29). For instance, hemizygosity of GTF2I in afflicted patients likely contributes to mental retardation and social behavior such as increased gaze and attention to strangers (28–30). In individuals with smaller microdeletions sparing GTF2I, a WBS cognitive profile but no mental retardation or intellectual difficulties was identified (30). GTF2I is also likely to be involved in other neurobehavioral impairments of subjects with WBS (31–33). Notably, one study focused on rs13227433, which is a single nucleotide polymorphism (SNP) within the GTF2I gene that has shown a functional association with higher engagement in social interaction but reduced social abilities (34). In a sample of 808 healthy individuals, they found that the AA genotype in rs13227433 indicated decreased threat-related amygdala reactivity, even when controlling for trait anxiety (34). This result perhaps highlights the concept that social anxiety could be dissociable from nonsocial fear even in the normal population through an amygdala-related mechanism (34,35). Because GTF2I is subject to alternative splicing, which generates isoforms with different activities and perhaps distinct biological roles (9,14), the particular risk haplotype (with a G/T SNP in an intron) could be linked to mutations associated with differential splicing of the gene, resulting in abnormal expression of TFII-I protein, which may be associated with autism vulnerability (36). However, these possibilities have not been rigorously tested yet.
Two neuropsychiatric disorders that have prominent social behavior abnormalities are autism spectrum disorders (ASD), characterized mainly by hyposociability, and WBS, where subjects exhibit hypersociability (37). While there is no clear overlap in transcriptional dysregulation between the two phenotypes, individuals with a duplication of GTF2I genes exhibit a phenotype with autistic traits—in some sense the opposite of hypersociality—implying a dosage-sensitive effect of GTF2I on affiliative behavior (36). Although it is currently unknown how transcriptional dysregulation resulting from GTF2I deletion can lead to the hypersocial (gregarious) phenotype in WBS, a recent study using human induced pluripotent stem cells (iPSCs) found that in the pluripotent state, GTF2I was responsible for 10–20% of the transcriptional dysregulation in disease-relevant pathways in WBS and 7q-microduplication syndrome (38). Furthermore, another report focused on a pair of genetic syndromes caused by symmetrical copy number variations (CNVs) at 7q11.23 involving the loss and gain of a number of genes, WBS and Williams-Beuren region duplication syndrome (also known as Somerville–van der Aa syndrome), respectively (26, 39); the latter includes an autistic spectrum disorder (7dupASD) (26, 39). The study concluded that 7q11.23 dosage imbalance could disrupt transcriptional circuits in disease-relevant pathways starting from the pluripotent state (Adamo et al., 2015). Hence it is likely that transcriptional deregulation, due to GTF2I deletion, could result in impaired development of neural circuits, potentially critical in conferring normal social behavior from early development stages (37). However, the transcriptional pathways and specific target genes involved in these two seemingly opposite “social” phenotypes are currently unknown. Regardless, the possibility that dosage effects of one transcription factor could be relevant for opposing social behaviors might technically lead to the elucidation of genes and pathways involved in these two clinical disorders in humans, in the near future (Fig 3). Of note, TFII-I has also been reported to transcriptionally inhibit reprogramming of murine induced pluripotent stem cells (iPSCs) (40); this is interesting because it might suggest that levels of TFII-I protein expression could potentially have dramatic effects on early developmental genes. However, this remains to be directly demonstrated. By employing ChIP-seq, a few specific target genes have been identified as potentially relevant for disease phenotypes,; for example, PDLIM1 (encoding a protein acting in neurites and which has been associated with cardiovascular defects), MYH14 (encoding a protein involved in the regulation of hearing) and, in particular, BEND4 (encoding a transcription factor involved in neural processes) were identified (38,41). Transcriptional targeting of BEND4 by TFII-I might also provide a rationale on how the effects of the large CNV might broadly impinge upon the molecular network controlling neuronal processes (38,41). However, given the complexity of TFII-I’s biochemical functions, to learn about its mechanism of action in controlling genes associated with social phenotypes, it would be important to characterize isoform specificity, phosphorylation status/signaling pathways and mechanism of regulation (e.g., co-factor association) of these genes by TFII-I. Moreover, in order to use it as a diagnostic tool it will also be important to determine whether the biochemical properties of TFII-I along with GTF2I dosage effects do indeed account for or directly contribute to these two opposing disease phenotypes (hypersocial vs hyposocial).
Fig 3. Human Chromosomal Location of GTF2I.

The diagram depicts the location of 7q11.23 band on human chromosome 7, which harbors all three GTF2I gene families. GTF2I gene dosage effects—either duplication or deletion—are correlated to Autism spectrum disorder (ASD with hyposociability) and WBS (with hypersociability), whereas single nucleotide polymorphism (SNPs) are associated with autoimmunity (SLE, Primary Sjögren’s syndrome and RA).
GTF2I/TFII-I Function in Immune Disorders
SNPs and Genome Wide Association Studies
Several recent Genome wide association studies (GWAS) revealed that TFII-I is involved in at least three autoimmune diseases--Primary Sjögren’s syndrome, Systemic Lupus Erythematosus (SLE) and rheumatoid arthritis (RA) (Fig 3). Primary Sjögren’s syndrome is one of the most common autoimmune diseases. To identify new genetic susceptibility loci for primary Sjögren’s syndrome, a three-stage genome-wide association study in a Han Chinese cohort was performed (42). The combined analysis identified GTF2I at 7q11.23 (rs117026326) as a new susceptibility locus for primary Sjögren’s syndrome. Fine mapping of the region around GTF2I showed that rs117026326 in GTF2I had the most significant association, with associated SNPs extending from GTF2I to GTF2IRD1-GTF2I (42, 43). Because of the close clinical relationship and shared pathophysiology of SLE and primary Sjögren’s, further studies were carried out. Indeed, among the new SLE susceptibility loci analyzed in a subsequent study in 4,478 SLE cases and 12,656 controls from six East Asian cohorts, the most significant locus was GTF2IRD1-GTF2I at 7q11.23 (rs73366469) (44). However, in multiple genotyping efforts, while rs117026326 showed the strongest association, given its proximity with rs73366469, it is difficult to separate the effects of these SNPs. Another study identified the largest-ever noted effects on Asian rheumatoid arthritis across human non-HLA regions at GTF2I by heterogeneity mapping followed by replication studies, and pinpointed a possible causal variant to the same SNP, rs73366469 (45).
To gain molecular insight, the authors carried out meta-data analysis. ENCODE data in CD4+ T cells and GM12878 lymphoblastoid cells indicated that the SNP rs73366469 lies within conserved enhancers, as well as active chromatin and transcription factor binding sites, also overlapping transcription start sites for GTF2I (44). Because the identified SNPs are located in the non-coding/intronic regions, these observations suggest that these nucleotide changes could alter transcription factor binding sites in enhancers and/or recruitment of the transcriptional machinery resulting in an aberrant expression of TFII-I in immune cells. In addition, altered splicing patterns could also occur, leading to expression of truncated isoforms of TFII-I that could potentially behave as dominant negatives. However, Further molecular analysis with both RNA and protein expression data is needed in primary human samples to decipher the molecular pathways affected by TFII-I in autoimmune disorders. Ultimately, combining GTF2I/TFII-I RNA-seq, with expression data as well as ChIP-seq could reveal target genes and/or pathways that are regulated by TFII-I in our immune system. Given certain similarities between the neural and the immune systems, and given the fact that both gene dosage effects (e.g. WBS and ASD) or expression levels (as in SLE) are related to alterations in TFII-I protein rather than its transcriptional activity, it would be interesting to determine whether there are overlaps of TFII-I target genes identified in immune disorders and neurocognitive disorders.
TFII-I in Cancer
Functional Mutations and Gene Fusions
Apart from signal-induced phosphorylation, TFII-I undergoes other post-translational modifications such as SUMOylation and Ubiquitylation (6,46,47). While the physiological significance of TFII-I SUMOylation is presently unclear, Ubiquitylation of TFII-I in response to DNA damaging ionizing radiation results in its degradation via proteosomal pathways in murine B cells (46). Although the exact mechanism of degradation has not been deciphered, potential sequences which could serve as destruction boxes/degrons were identified (47). In a recent fascinating study, 28 thymic epithelial tumors (TETs) were analyzed by using next-generation sequencing. A missense mutation (chromosome 7 c.74146970T>A) in GTF2I was identified at high frequency in type A thymomas, which are of a relatively indolent subtype (48). In a series of 274 TETs, the authors detected this GTF2I mutation in 82% of type A and 74% of type AB thymomas but rarely in the aggressive subtypes, where recurrent mutations of known cancer genes have been identified (48). Therefore, it appeared that this GTF2I mutation might correlate with better patient survival (48). This same GTF2I mutation (Leu404His) alters a residue within the amino acid sequence RILLAKE that may represent a non-canonical destruction box resembling the destruction box (RXXLXX[LIVM]) which is found in cyclins, PLK1 and Securin7 (47,48). Notably, although this mutation lies within repeat 2, structural predictions suggest that this residue is not conserved among the repeats and thus, is unlikely to significantly alter the structural integrity of TFII-I (48). However, it is possible that the RILLAKE to RILHAKE alteration may render TFII-I unrecognizable by the proteosomal degradation machinery (47,48). Consistent with this expectation, mutant tumors showed higher TFII-I expression than wild-type tumors at the protein level, but not the mRNA level (48), suggesting that that the Leu404His mutation may augment TFII-I expression post-transcriptionally by avoiding its turnover through the degradation machinery; this may in turn accelerate cell proliferation upon upregulation of TFII-I cell cycle–controlling target genes such as c-fos (7,48). Because TFII-I appears to be pro-proliferative in a variety of cell types based on its ability to regulate pro-proliferative genes, it’s protein turn-over could represent a normal regulatory program to control cellular growth. These observations beg the question: could GTF2I act as an oncogene?
Although a clear-cut answer is still lacking, two alternative scenarios are proposed: i) either a GTF2I mutation is present exclusively in a distinct subgroup of tumors with favorable prognosis independent of their histological appearance; or ii) a GTF2I mutation is necessary for the founder tumor clone, and is subsequently lost during clonal evolution to more aggressive histotypes (48). Per this second hypothesis, GTF2I mutation(s) might be initially necessary for a multistep process and may become superfluous in the presence of more dominant mutations, such as mutations in TP53 or CYLD, rendering GTF2I-mutant clones to be outnumbered in more aggressive tumors during clonal evolution (48). Whether this could be true only in TETs or in other tumors is currently unknown.
A recent study examined a cohort of 85 patients with Angioimmunoblastic T-cell lymphoma (AITL) (n572) or other follicular T-helper cells (TFH)-derived peripheral T-cell lymphomas (PTCLs) (n513), using targeted deep-sequencing of a gene panel enriched in T-cell receptor (TCR) signaling elements (49). These lymphomas represent a large proportion of tumors with poorly understood pathogenesis and unfavorable treatment results. In support of the notion that GTF2I could function as an oncogene in certain instances, apart from RHOA mutations that were identified in 51 of 85 cases (60%), half of the patients carried virtually mutually exclusive mutations in other TCR-related genes, most frequently in PLCG1 (14.1%), CD28 (9.4%, exclusively in AITL), PI3K elements (7%), CTNNB1 (6%), and GTF2I (6%) (49). These observations suggest that deregulated TCR activation may play a role in the pathogenesis of TFH-derived PTCL, which may further inform the development of novel targeted therapies (49). All five mutations identified in the coding region of TFII-I are new and supposedly all result in gain-of function, although they have not been tested molecularly (Fig 4A, ref 49). Only two of the five mutations are located in the repeat regions (R523S in repeat 2 and L607F in repeat 4) and two of the five mutations lie within regions close to the DNA binding and nuclear localization signals (Fig 4A). This is interesting in that It is possible that these latter two mutations might enhance the DNA binding and/or the nuclear localization properties of TFII-I, which could account for its potential gain-of-function activity in T cell tumors. Similarly to the mutation observed in TETs, the residues mutated within these repeats are also not conserved, suggesting that these mutations might not grossly alter the structural features of TFII-I. As noted earlier, because TFII-I physically and functionally interacts with PLCγ (24) and because both proteins lie downstream of TCR signaling, these TFII-I mutations could result in alterations in such interactions. However, Further molecular studies are warranted to test these possibilities.
Fig 4. TFII-I in Cancer.
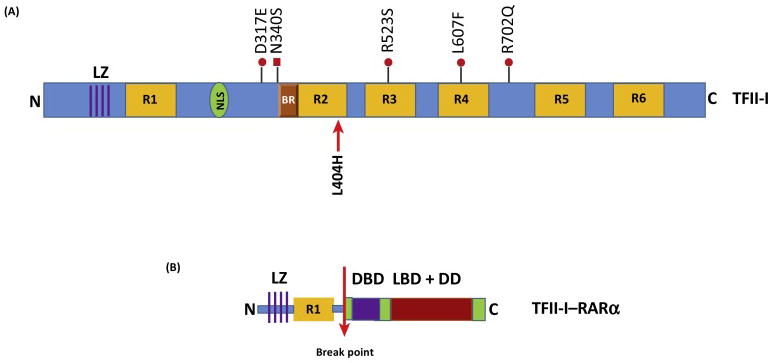
(A) Schematic of human TFII-I protein structure with proven (with red arrow) and purported (with red dots) gain-of-function mutations that are associated with various cancers. (B) Schematic of human TFII-I-RARα fusion protein. The N-terminal portion of TFII-I --containing the leucine zipper (LZ) and repeat 1 (R1) -- is fused to the C-terminal portion of RARα containing the DNA binding domain (DBD, purple box) as well as the ligand-binding domain (LBD) and dimerization domain (DD) (red box). The break point is shown with the red arrow.
In addition to gain of function mutations in TFII-I associated with various malignancies, a couple of reports have revealed the occurrence of fusion products with GTF2I. First, a novel GTF2I/NCOA2 fusion gene was identified in Soft Tissue Angiofibroma (fibrovascular tumor of unknown cellular origin) (50,51). A fusion product of t(7;8;14)(q11;q13;q31) resulted in two transcripts: one with an in-frame fusion of part of exon 14 of GTF2I with part of exon 15 of NCOA2 whereas the other one consisted of an out of-frame fusion of part of exon 11 of GTF2I with part of exon 14 of NCOA2 (51). The other report indicates a novel fusion transcript in a t(7;17) variant of acute promyelocytic leukemia (APL) with clinical resistance to retinoic acid (52). The fusion gene (GTF2I-RARA) results from the fusion between exon 6 of GTF2I and exon 3 of RARA (retinoic acid receptor alpha), and is predicted to encode a 599 amino acid protein. No alternative splicing variant was found and the reciprocal RARA-GTF2I fusion transcript was not detected (52). In the encoded TFII-I-RARα fusion protein, the first 195 amino acids of TFII-I, including the leucine zipper LZ and the first TFII-I-repeat (R1, 104–176), are retained (Fig 4B), providing the possibility of TFII-I-RARα dimerization or multimerization. Consistent with this expectation, biochemical interaction studies indicate both homodimerization as well as heterodimerization with TFII-I but not with RARα (52). Whether this fusion protein creates an unusually potent transcription factor or whether this aberrant protein results in multimerization and sequestration of TFII-I in foci is currently unknown, although an unusual cytoplasmic microgranule formation indicative of aggregation was observed in TFII-I-RARα-expressing patient-derived cells (52). Given the presence of multiple protein-protein interactions domains in TFII-I, it is not totally surprising that an aberrant fusion product of TFII-I could lead to aggregation and granule formation. However, further careful studies need to be undertaken to elucidate whether the fusion protein exhibits a gain of transcriptional function.
Concluding Remarks
Biochemical experiments, together with genetic ablation in mice, and more recent patient studies indicate that TFII-I is extremely important for human health and disease (see Box 1). Though patient studies are currently limited in availability of biospecimens amenable to molecular mechanistic studies, the biochemical primer that has been laid out over two decades should provide a clear path to decipher TFII-I’s mechanism of action in various clinical settings. These could include RNA-seq analysis (to identify transcriptomic changes), ChIP-seq (to identify target genes) and protein expression studies (to identify its expression and subcellular residency). In addition, the establishment of appropriate patient-derived lymphoblastoid cell lines would be warranted. it is not difficult to envisage how these might help in turn to exploit GTF2I/TFII-I (and its various functions) in molecular diagnostics (e.g., point mutations in TETs or AITLs), or in SNP analysis of autoimmune disorders. However, deciphering the consequences of its dosage effects (as in WBS or ASD) is going to be substantially more complicated as direct versus indirect, and/or overlapping effects could confound these studies (see Outstanding Questions). Nevertheless, once we have a better handle on our understanding of the relationships between specific functions of TFII-I and its involvement in different pathophysiologies, I fully anticipate that new therapeutic potentials targeting TFII-I could seriously emerge.
Box 1. Clinician’s Corner.
TFII-I is a signal-induced multifunctional transcription factor. Gene ablation studies in mice and high-quality exome (protein-coding region) DNA sequence data for 60,706 individuals of diverse ancestries demonstrates that it is encoded by an essential gene, GTF2I. Although biochemical properties of TFII-I have been known for over two decades, its role in human diseases is just beginning to be appreciated.
Three types of alterations in TFII-I are associated with various pathologies: 1) GTF2I gene dosage effects; 2) SNPs associated with non-coding regions that could alter TFII-I expression and/or its alternative splicing patterns; and 3) mutations in coding regions as well as GTF2I gene fusions/chromosomal translocations.
Gene dosage effects—haploinsufficiency or duplication in GTF2I at 7q11.23 are associated with WBS (hypersociability) and ASD (hyposociability) respectively, implying a dosage-sensitive effect of GTF2I on affiliative behavior.
SNPs identified in the non-coding regions of GTF2I are associated with autoimmune diseases such as Primary Sjögren’s syndrome, Systemic Lupus Erythematosus (SLE) and rheumatoid arthritis (RA).
Gain-of function mutations in the coding regions are found in indolent thymic epithelial tumors (TETs) and Angioimmunoblastic T-cell lymphoma (AITL), suggesting an oncogenic function of TFII-I. GTF2I/NCOA2 fusion gene in Soft Tissue Angiofibroma and GTF2I-RARA fusion transcript in a treatment-resistant variant of acute promyelocytic leukemia (APL) have also been identified.
Genetic alterations affecting TFII-I expression and mutations as well as fusions affecting its transcriptional activity could be used diagnostically. Identifying its target genes and pathways affected in these pathologies could ultimately result in therapeutic interventions.
Outstanding Questions Box.
What are the target genes and/or pathways that are affected by under- or over-expression of TFII-I that are causal to WBS and ASD? While some targets have been identified, it remains unclear how these genes affect social behaviors. Identification of transcriptional pathways and networks is required along with experimental demonstration of how TFII-I might regulate these genes.
How do specific SNPs affectGTF2I/TFII-I expression and/or function in such a way as to contribute to autoimmunity? While some SNPs are found to be in the enhancer and transcriptional start sites of GTF2I, it is unknown whether any SNPs alters its splicing patterns.
How do gain-of-function mutations alter the transcriptional activity ofGTF2I? Whereas one mutation appears to prevent its protein turn-over, it is unclear how the rest of the identified TFII-I mutations in T cell tumors affect function. It will be important to biochemically determine the functions of these mutants. Likewise, the mechanism of action of TFII-I fusion proteins remains to be elucidated.
Is there an overlap between TFII-I target genes involved in neurocognitive deficits and autoimmunity? Given certain similarities between the neuronal and immune systems, it would be interesting to determine whether there are commonalities in TFII-I target genes and/or pathways in these two different types of pathologies. Determining the mechanisms and pathways that TFII-I govern may help explain aspects of the molecular basis for these disease phenotypes, and hopefully provide novel diagnostic tools and/or therapeutic interventions.
Trends Box.
Vertebrate-specific transcription factor TFII-I is activated by various receptor signaling pathways, resulting in its nuclear translocation.
While gene ablation in mice and exome sequencing in humans indicates it is encoded by an essential gene, it precise role in human patho-physiology is just beginning to emerge.
Under-expression or over-expression of TFII-I is causal to opposing neuro-cognitive disorders such as WBS and ASD, respectively. Precise transcriptional targets are currently unknown but such determination should shed light on genes that are involved in these syndromes.
SNPs in enhancer and transcriptional start sites possibly affects its expression pattern as well as its alternative splicing patterns. These are causal to autoimmune disorders. It is currently unknown whether TFII-I targets involved in autoimmunity and neurocognition are shared.
Gain-of-function mutations in its protein-coding regions could affect its protein turnover, enhanced nuclear translocation and its DNA binding ability. Gene fusions involving GTF2I also appear to enhance TFII-I’s protein-protein interactions. This suggests that it could potentially function as an oncogene.
Acknowledgments
Work in my laboratory is generously supported by the Division of Program Coordination, Planning, and Strategic Initiatives, Office of the Director and the Intramural Research Program of the National Institutes of Health, National Institute on Aging. I sincerely thank Dr. Ranjan Sen for critically reading the manuscript.
Glossary
- E-box element
sequence-specific DNA element with consensus sequence CANNTG, recognized by HLH-containing transcription factors
- Initiator (Inr) element
DNA element present at the transcription start site of eukaryotic genes, critical for accurate transcription initiation
- Endoplasmic reticulum (ER) stress response pathway
Mammalian cells can be subjected to stress targeted to the endoplasmic reticulum (ER), such as depletion of the ER Ca(2+) store, resulting in the transcriptional induction of a family of glucose-regulated protein (GRP) genes encoding ER chaperones
- Supravalvar aortic stenosis (SVAS)
cardiovascular defect characteristic of WBS patients, caused by haploinsufficiency in the elastin gene
- Single nucleotide polymorphism (SNPs)
The most common type of genetic variation caused by a difference in a single nucleotide in an individual’s DNA. When SNPs occur within the coding region of a gene or in a regulatory region near a gene, they can alter the gene’s function
- Threat-related amygdala reactivity
neural biomarker of depression and anxiety risk in response to stress
- Symmetrical copy number variations (CNVs)
sections of the genome are duplicated or deleted affecting a considerable number of DNA base pairs, resulting in genomic variation between individuals in the human population. There are instances where both modifications can occur in within the same genome giving rise to symmetrical CNV
- Primary Sjögren’s syndrome
chronic and common inflammatory autoimmune disease characterized by chronic autoantibody production and infiltration in exocrine glands, particularly the salivary gland
- Systemic Lupus Erythematosus (SLE)
debilitating autoimmune disease with a strong genetic component, characterized by pathogenic autoantibody production that can affect virtually any organ
- Rheumatoid arthritis (RA)
persisting autoimmune disorder characterized by chronic joint inflammation and the presence of autoantibodies, although disease symptoms may appear or disappear unpredictably
- Heterogeneity mapping
Analyzes the presence of a variety of genetic defects that cause the same disease, often due to mutations at different loci on the same gene, and often common to many human diseases
- Replication studies
ensure that a genotype-phenotype association observed in a genome-wide association study represents a credible association and is not a chance finding or an artifact due to uncontrolled biases
- ENCODE
Encyclopedia of DNA elements—a large public consortium created to identify all functional elements in the human genome sequence
- Dominant negative
A genetic mutation that results in an altered allele or gene product acting antagonistically to the wild type allele or gene product
- Destruction boxes/degrons
region of a protein containing specific amino acid sequences (often comprising Lysine or Arginine) controlling the stability/turn over of the protein; structural alterations (masking or unmasking these regions via post-translational modifications) modify the protein levels
- RNA-seq
captures all transcripts (RNA) produced in a given biological sample at a specific time, and provides an analysis by massive parallel nucleotide sequencing
- Deep sequencing
automated process which can uncover the exisitence or changes in DNA and/or RNA species in biological samples by massive parallel nucleotide sequencing—this is also known as next generation sequencing (NGS)
- ChIP-seq
Chromatin immunoprecipitation (ChIP) coupled with deep sequencing—process by which regions of the entire genome bound by a transcription factor or histones is precipitated/isolated and identified by deep sequencing
- Soft tissue angiofibrioma
Fibrovascular tumor (often benign mesenchymal neoplasm) of unknown cellular origin—usually occurs in middle aged adults; with female predominance
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Bibliography
- 1.Malik S, Roeder RG. The metazoan Mediator co-activator complex as an integrative hub for transcriptional regulation. Nat Rev Genet 2010. 2010;11:761–72. doi: 10.1038/nrg2901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lee TI, Young RA. Transcriptional Regulation and Its Misregulation in Disease. Cell. 2013;152:1237–1251. doi: 10.1016/j.cell.2013.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jones KA. Transcription strategies in terminally differentiated cells: shaken to the core. Genes Dev. 2007;21:2113–2117. doi: 10.1101/gad.1598007. [DOI] [PubMed] [Google Scholar]
- 4.Roy AL, et al. Cooperative interaction of an initiator-binding transcription initiation factor and the helix-loop-helix activator USF. Nature. 1991;354:245–248. doi: 10.1038/354245a0. [DOI] [PubMed] [Google Scholar]
- 5.Roy AL, et al. Cloning of an inr- and E-box-binding protein, TFII-I, that interacts physically and functionally with USF1. EMBO J. 1997;16:7091–7104. doi: 10.1093/emboj/16.23.7091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Roy AL. Biochemistry and biology of the inducible multifunctional transcription factor TFII-I: 10 years later. Gene. 2012;492:32–41. doi: 10.1016/j.gene.2011.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yang W, Desiderio S. BAP-135, a target for Bruton’s tyrosine kinase in response to B cell receptor engagement. Proc Natl Acad Sci U S A. 1997;94:604–609. doi: 10.1073/pnas.94.2.604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Grueneberg DA, et al. A multifunctional DNA-binding protein that promotes the formation of serum response factor/homeodomain complexes: identity to TFII-I. Genes Dev. 1997;11:2482–2493. doi: 10.1101/gad.11.19.2482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Makeyev AV, Bayarsaihan D. Alternative splicing and promoter use in TFII-I genes. Gene 2009. 2009;433:16–25. doi: 10.1016/j.gene.2008.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Enkhmandakh B, et al. Essential functions of the Williams-Beuren syndrome-associated TFII-I genes in embryonic development. Proc Natl Acad Sci U S A. 2009;106:181–186. doi: 10.1073/pnas.0811531106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mammoto A, et al. A mechanosensitive transcriptional mechanism that controls angiogenesis. Nature. 2009;457:1103–1108. doi: 10.1038/nature07765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Blomen VA, et al. Gene essentiality and synthetic lethality in haploid human cells. Science. 2015;350:1092–1096. doi: 10.1126/science.aac7557. [DOI] [PubMed] [Google Scholar]
- 13.Lek M, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–291. doi: 10.1038/nature19057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cheriyath V, Roy AL. Alternatively spliced isoforms of TFII-I. Complex formation, nuclear translocation, and differential gene regulation. J Biol Chem. 2000;275:26300–26308. doi: 10.1074/jbc.M002980200. [DOI] [PubMed] [Google Scholar]
- 15.Hakre S, et al. Opposing functions of TFII-I spliced isoforms in growth factor-induced gene expression. Mol Cell. 2006;24:301–308. doi: 10.1016/j.molcel.2006.09.005. [DOI] [PubMed] [Google Scholar]
- 16.Doi-Katayama Y, et al. Solution structure of the general transcription factor 2I domain in mouse TFII-I protein. Protein Sci 2007. 2007;16:1788–1792. doi: 10.1110/ps.072792007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Novina CD, et al. Regulation of nuclear localization and transcriptional activity of TFII-I by Bruton’s tyrosine kinase. Mol Cell Biol. 1999;19:5014–5024. doi: 10.1128/mcb.19.7.5014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schmidt C, et al. Signalling of the BCR is regulated by a lipid rafts-localised transcription factor, Bright. EMBO J. 2009;28:711–724. doi: 10.1038/emboj.2009.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sacristán C, et al. Characterization of a novel interaction between transcription factor TFII-I and the inducible tyrosine kinase in T cells. Eur J Immunol. 2009;39:2584–2595. doi: 10.1002/eji.200839031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.August A. IL-2-inducible T-cell kinase (ITK) finds another (dance) partner...TFII-I. Eur J Immunol. 2009;39:2354–2357. doi: 10.1002/eji.200939813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hong M, et al. Transcriptional regulation of the Grp78 promoter by endoplasmic reticulum stress: role of TFII-I and its tyrosine phosphorylation. J Biol Chem. 2005;280:16821–16828. doi: 10.1074/jbc.M413753200. [DOI] [PubMed] [Google Scholar]
- 22.Casteel DE, et al. Identification of the interface between cGMP-dependent protein kinase Ibeta and its interaction partners TFII-I and IRAG reveals a common interaction motif. J Biol Chem. 2005;280:38211–38218. doi: 10.1074/jbc.M507021200. [DOI] [PubMed] [Google Scholar]
- 23.van Rossum DB, et al. Phospholipace Cγ1 controls surface expression of TRPC3 through an intermolecular PH domain. Nature. 2005;434:99–104. doi: 10.1038/nature03340. [DOI] [PubMed] [Google Scholar]
- 24.Caraveo G, et al. Action of TFII-I outside the nucleus as an inhibitor of agonist-induced calcium entry. Science. 2006;314:122–125. doi: 10.1126/science.1127815. [DOI] [PubMed] [Google Scholar]
- 25.Roy AL. Transcription factor TFII-I conducts a cytoplasmic orchestra. ACS Chem Biol. 2006;1:619–622. doi: 10.1021/cb6004323. [DOI] [PubMed] [Google Scholar]
- 26.Pober BR. Williams-Beuren syndrome. N Engl J Med. 2010;362:239–252. doi: 10.1056/NEJMra0903074. [DOI] [PubMed] [Google Scholar]
- 27.Pérez Jurado LA, et al. A duplicated gene in the breakpoint regions of the 7q11.23 Williams-Beuren syndrome deletion encodes the initiator binding protein TFII-I and BAP-135, a phosphorylation target of BTK. Hum Mol Genet. 1998;7:325–334. doi: 10.1093/hmg/7.3.325. [DOI] [PubMed] [Google Scholar]
- 28.Dai L, et al. Is it Williams syndrome? GTF2IRD1 implicated in visual-spatial construction and GTF2I in sociability revealed by high resolution arrays. Am J Med Genet A. 2009;149A:302–314. doi: 10.1002/ajmg.a.32652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Morris CA, et al. GTF2I hemizygosity implicated in mental retardation in Williams syndrome: genotype-phenotype analysis of five families with deletions in the Williams syndrome region. Am J Med Genet A. 2003;123A:45–59. doi: 10.1002/ajmg.a.20496. [DOI] [PubMed] [Google Scholar]
- 30.Hirota H, et al. Williams syndrome deficits in visual spatial processing linked to GTF2IRD1 and GTF2I on chromosome 7q11.23. Genet Med. 2003;5:311–321. doi: 10.1097/01.GIM.0000076975.10224.67. [DOI] [PubMed] [Google Scholar]
- 31.Antonell A, et al. Partial 7q11.23 deletions further implicate GTF2I and GTF2IRD1 as the main genes responsible for the Williams-Beuren syndrome neurocognitive profile. J Med Genet. 2010;47:312–320. doi: 10.1136/jmg.2009.071712. [DOI] [PubMed] [Google Scholar]
- 32.Sakurai T, et al. Haploinsufficiency of Gtf2i, a gene deleted in Williams Syndrome, leads to increases in social interactions. Autism Res. 2011;4:28–39. doi: 10.1002/aur.169. [DOI] [PubMed] [Google Scholar]
- 33.Mervis CB, et al. Duplication of GTF2I results in separation anxiety in mice and humans. Am J Hum Genet. 2012;90:1064–1070. doi: 10.1016/j.ajhg.2012.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Swartz JR, et al. A Common Polymorphism in a Williams Syndrome Gene Predicts Amygdala Reactivity and Extraversion in Healthy Adults. Biol Psychiatry. 2017;81:203–210. doi: 10.1016/j.biopsych.2015.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schweiger JI, Meyer-Lindenberg A. Common Variation in the GTF2I Gene: A Promising Neurogenetic Mechanism for Affiliative Drive and Social Anxiety. Biol Psychiatry. 2017;81:175–176. doi: 10.1016/j.biopsych.2016.11.008. [DOI] [PubMed] [Google Scholar]
- 36.Malenfant P, et al. Association of GTF2i in the Williams-Beuren syndrome critical region with autism spectrum disorders. J Autism Dev Disord. 2012;42:1459–1469. doi: 10.1007/s10803-011-1389-4. [DOI] [PubMed] [Google Scholar]
- 37.Barak B, Feng G. Neurobiology of social behavior abnormalities in autism and Williams syndrome. Nat Neurosci. 2016;19:647–655. doi: 10.1038/nn.4276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sanders SJ, et al. Multiple recurrent de novo CNVs, including duplications of the 7q11.23 Williams syndrome region, are strongly associated with autism. Neuron. 2011;70:863–885. doi: 10.1016/j.neuron.2011.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Adamo A, et al. 7q11.23 dosage-dependent dysregulation in human pluripotent stem cells affects transcriptional programs in disease-relevant lineages. Nat Genet. 2015;47:132–141. doi: 10.1038/ng.3169. [DOI] [PubMed] [Google Scholar]
- 40.Yang CS, et al. Genome-wide functional analysis reveals factors needed at the transition steps of induced reprogramming. Cell Rep. 2014;8:327–337. doi: 10.1016/j.celrep.2014.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Urban AE, Purmann C. Using iPSCs and genomics to catch CNVs in the act. Nat Genet. 2015;47:100–101. doi: 10.1038/ng.3204. [DOI] [PubMed] [Google Scholar]
- 42.Li Y, et al. A genome-wide association study in Han Chinese identifies a susceptibility locus for primary Sjögren’s syndrome at 7q11.23. Nat Genet. 2013;45:1361–1365. doi: 10.1038/ng.2779. [DOI] [PubMed] [Google Scholar]
- 43.Song IW, et al. Identification of susceptibility gene associated with female primary Sjögren’s syndrome in Han Chinese by genome-wide association study. Hum Genet. 2016;135:1287–1294. doi: 10.1007/s00439-016-1716-0. [DOI] [PubMed] [Google Scholar]
- 44.Sun C, et al. High-density genotyping of immune-related loci identifies new SLE risk variants in individuals with Asian ancestry. Nat Genet. 2016;48:323–330. doi: 10.1038/ng.3496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kim K, et al. Association-heterogeneity mapping identifies an Asian-specific association of the GTF2I locus with rheumatoid arthritis. Sci Rep. 2016;6:27563. doi: 10.1038/srep27563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Desgranges ZP, et al. Inhibition of TFII-I-dependent cell cycle regulation by p53. Mol Cell Biol. 2005;25:10940–10952. doi: 10.1128/MCB.25.24.10940-10952.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Desgranges ZP, Roy AL. TFII-I: connecting mitogenic signals to cell cycle regulation. Cell Cycle. 2006;5:356–359. doi: 10.4161/cc.5.4.2442. [DOI] [PubMed] [Google Scholar]
- 48.Petrini I, et al. A specific missense mutation in GTF2I occurs at high frequency in thymic epithelial tumors. Nat Genet. 2014;46:844–849. doi: 10.1038/ng.3016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vallois D, et al. Activating mutations in genes related to TCR signaling in angioimmunoblastic and other follicular helper T-cell-derived lymphomas. Blood. 2016;128:1490–1502. doi: 10.1182/blood-2016-02-698977. [DOI] [PubMed] [Google Scholar]
- 50.Yamada Y, et al. Histological spectrum of angiofibroma of soft tissue: histological and genetic analysis of 13 cases. Histopathology. 2016;69:459–469. doi: 10.1111/his.12943. [DOI] [PubMed] [Google Scholar]
- 51.Arbajian E, et al. A novel GTF2I/NCOA2 fusion gene emphasizes the role of NCOA2 in soft tissue angiofibroma development. Genes Chromosomes Cancer. 2013;52:330–331. doi: 10.1002/gcc.22033. [DOI] [PubMed] [Google Scholar]
- 52.Li J, et al. GTF2I-RARA is a novel fusion transcript in a t(7;17) variant of acute promyelocytic leukaemia with clinical resistance to retinoic acid. Br J Haematol. 2015;168:904–908. doi: 10.1111/bjh.13157. [DOI] [PubMed] [Google Scholar]