

Graphical abstract
1. Introduction
Alzheimer's disease (AD) challenges our society with an annual estimate cost of $1.08 trillion in the USA alone by 2050.(1) AD is a progressive irreversible neurological disorder with marked atrophy of cerebral cortex and loss of cortical and subcortical neurons, which is characterized pathologically by accumulation of amyloid plaques, and numerous neurofibrillary tangles formed from filaments of microtubule associated highly phosphorylated tau proteins.(2) The pathogenesis of AD includes other factors such as cholinergic malfunction and oxidative stress.(3)
The major constituents of the senile plaques are β-amyloid (Aβ) peptides of 39-43 amino acids. Aβ derives from cleavage of the transmembrane amyloid precursor protein (APP), located in chromosome 21, by β-secretase (BACE1) producing a 99 amino acid fragment (C99) that is further cleaved by the γ-secretase.(4) The human Aβ1-42 wild type (WT) sequence is DAEFRHDSGYEVHHQKLVFFAEDVGSNKGAIIGLMVGGVVIA. Five drugs are currently available for AD. These include four cholinesterase inhibitors: Donepezil, Reminyl, Razadyne and Rivastigmine, and the N-methyl-D-aspartate (NMDA) receptor antagonist, Memantine. However, they are only effective for 6 to 12 months, and for half of the patients with milder forms of Alzheimer's.(5) Scientists are developing novel benzopolycyclic amines with increased NMDA receptor antagonist activity,(6) and are targeting BACE1,(7) Tau and Aβ proteins.(8,9) Despite many in vitro and in vivo studies, drug after drug has failed to slow the progression of AD for several reasons.
First, while oligomers, such as dimers, trimers and 12-mers (Aβ*56), are the most critical players in the pathology of AD(10,11a-b) and larger aggregates and fibril fragmentation are toxic as well(12,13) there is currently little information on their rate and extent of formation. Experimental and theoretical studies showed that Aβ1-40/1-42 peptides self-assembly into amyloid fibrils by a nucleation-condensation polymerization mechanism. However, while master equations allow interpreting the experimental sigmoidal kinetic profiles of amyloid formation by means of primary and/or secondary (fragmentation or lateral) nucleation processes,(14-16) they do not provide any information on the 3D topology and size of the primary nucleus. Overall, probing the conformational changes of Aβ aggregation is challenging owing to the vast heterogeneity of the aggregates, the number of sub-states for each aggregate, and the sensitivity of the process to pH, agitation, temperature, concentration, ionic strength, surfactants, sample preparation and the sequence (Aβ1-40 vs. Aβ1-42).(17-19)
Second, standard tools of structural biology have failed to provide the 3D structures of the monomers and the oligomers of the Aβ1-40/1-42 peptides in aqueous solution. Aβ monomer is described as random coil by solution nuclear magnetic resonance, NMR,(20) and circular dichroism, CD.(21) Due to their heterogeneity and high propensity to aggregate, the low molecular weight Aβ oligomers are not amenable to NMR and X-ray crystallography. As a result, only low-resolution structural data from CD, ion-mobility mass spectrometry (IM-MS), electron microscopy (EM), transmission electron microscopy (TEM) and atomic force microscopy (AFM) measurements are available.(11,20-26) At the end of the reaction, the fibrils are insoluble and we are left with complicated experiments using isotopic labeling to propose models. These experiments revealed that fibrils of synthetic Aβ1-42 peptides have U-shaped conformations with β-strands at residues L17–F20 and I31–V40 with the 16 N-terminus residues disordered, while fibrils of synthetic Aβ1-40 peptides have β-strands at Y10-D23 and A30-G38 with the 9 N-terminus residues disordered.(27,28) Fibrils made of AD-brain derived Aβ1-40 peptides show, however, deformed U-shaped conformations, with a twist in residues F19-D23, a kink at G33 and a bend at G37-G38, and a more ordered N-terminus.(29) Overall the final products are very sensitive to the nature of the sample (synthetic or brain-derived Aβ peptides). Fibril formation is also under kinetics rather than thermodynamics, adding further complexity to the determination of the physical factors governing Aβ1-40/1-42 amyloid fibril formation.(17,30)
Third, because of their presence in the brain, the metal ions (Cu2+, Zn2+ and Fe3+ and the cell membrane have to be considered. A full dynamic and thermodynamic picture of the interactions of Aβ1-40/1-42 oligomers with metal ions or membrane is very difficult, but recent progress has been made.(31,32)
Fourth, it is important to better understand the molecular interactions of Aβ oligomers with the proteins co-localised in the brain, and notably human serum albumin,(33) the most abundant protein in cerebral spinal fluid, and the prion protein, PrP, concentrated at the synaptic terminals with a high affinity for Aβ.(34,35) Mapping all partners that bind to Aβ oligomers is a daunting task because disparate results can emerge from experiments depending on the initial state of the protein, its source, and its stoichiometry.(36) In addition, as functional genomics has taught us,(37) biomolecules are involved in a network of interactions, so toxicity is likely to be multifactorial and to result from interactions of Aβ with multiple partners. Three recent articles illustrate this feature.(38-40) Murine paired immunoglobulin-like receptor B and its human ortholog leukocyte immunoglobulin-like receptor B2 were identified as receptors for Aβ oligomers, with nanomolar affinity.(38) Aβ oligomers also induce synaptic damage via tau-dependent microtubule severing by Tubulin-Tyrosine-Ligase-Like-6 and spastin,(39) and Aβ oligomers-PrP generate metabotropic glutamate receptor 5-mediated increases of intracellular calcium.(40) Finally, among the apolipoprotein E (apoE) isoforms, apoE4 increases the risk of AD. While transporting cholesterol is its primary function, apoE regulates Aβ metabolism, aggregation, deposition, and competes with Aβ for cellular uptake through apoE receptors.(41) Overall, our current structural knowledge of Aβ oligomers with receptors is still in its infancy.
Fifth, the summer 2012 was plagued by the release of bad news from clinical trials of two drugs targeting Aβ, bapineuzumab and solanezumab.(42) The general consensus for a couple of years has been that the drugs are given too late.(43) Scientists have failed to provide the structures of Aβ monomers or Aβ oligomers with known inhibitors of Aβ aggregation in vitro and toxicity, a prerequisite to develop more specific drugs with optimal affinities for Aβ oligomers.(44)
In addition, working on the most abundantly produced species, Aβ1-40, and the far less abundant but more aggregation prone and toxic form, Aβ1-42, is a simplification as the γ-secretase generates peptides from Aβ1-36 to Aβ1-43. (45) Several truncated variants are also observed in the amyloid plaques with various populations. These include Aβ4-42 and Aβ5-42,(46) Aβ1-26 and Aβ1-30,(40) and post-translational modifications of Aβ peptides: isomerization at D1, phosphorylation at S26, dityrosine covalent bond at Y10 and proteolytic removal of D1 and A2 and the subsequent cyclizing of E3 and E11 to a pyroglutamate (designated Aβ3(pE) and Aβ11(pE)), among others.(47-49) The Aβ(pE) species in vivo consist of Aβ3(pE)-40/42 and Aβ11(pE)-40/42, with Aβ3(pE)-42 being most abundant. Aβ(pE) is more cytotoxic and aggregates more rapidly than conventional Aβ and a recent study raises the possibility that Aβ3(pE)-42 formation acts at a primary step in AD pathogenesis.(50)
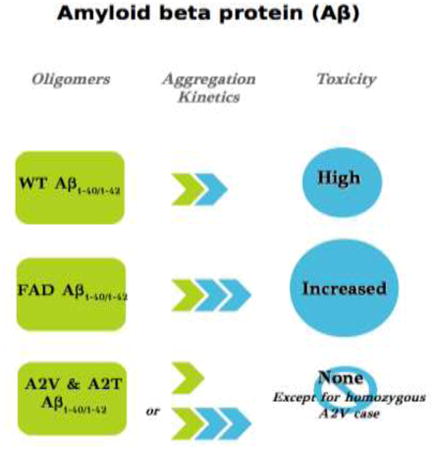
Finally, while most Alzheimer's disease is sporadic, i.e. not the result of inheritance, (familial AD (FAD) represents 5% of the cases), we have learned a large amount about the genetic risk factors that predispose an individual to contract the disease. One of the major risk factors for AD is mutation in the APP gene, though many mutations in two presenilin genes have also been reported and are constantly discovered.(51) Mutant APP may be more likely to be proteolytically cleaved into the Aβ form, which generates the amyloid plaques. Some familial mutations, including H6R (English), D7H (Taiwanese), D7N (Tottori), A21G (Flemish), E22Q (Dutch), E22G (Artic), E22Δ (Osaka) and D23N (Iowa), change aggregation and toxicity, and lead to different phenotypes.(52,53) Two recent FAD mutations, however, turn that inheritance pattern on its head. The A673V mutation in APP or A2V in Aβ is associated with AD, but the inheritance pattern is recessive, i.e., a patient needs two mutant alleles in order to acquire the disease risk. In combination with the WT allele, A673V does not cause AD. Furthermore, the presence of the mutant peptide prevents the WT peptide from forming amyloid fibrils, even under very favorable in vitro conditions.(54) The second striking result comes from the coding variants in APP in a set of whole-genome sequence data from 1,795 Icelanders and the discovery that the mutation A673T in APP or A2T in Aβ protects against AD in both homozygous and heterozygous patients. Though A2T reduces the cleavage of APP by 40%, how the mixing of Aβ1-40 A2T and Aβ1-40 WT protects patients from AD remains to be determined.(55)
Overall, a full understanding of AD within the amyloid cascade hypothesis requires the development and use of innovative biophysical techniques. Along with standard approaches, e.g. Fourier transform infrared spectroscopy (FTIR), CD, X-ray powder diffraction, TEM, AFM, solid-state nuclear magnetic resonance (ss-NMR), dynamic light scattering (DLS) and IM-MS, new techniques are being applied. These include, notably: pulsed hydrogen-deuterium exchange coupled with mass spectrometry analysis,(56) which unlike fluorescence methods, does not require labeling with a fluorophore, photonic crystal-based approaches,(57) single-molecule imaging techniques,(58) and specific isotope labelling with electron paramagnetic resonance (EPR), advanced hyperfine sub-level correlation (HYSCORE) and electron-nuclear double resonance (ENDOR) methods.(59,60)
Experimental studies alone are not sufficient, however, since they generally give time- and space-averaged properties. Computer simulations by exploring different time and length scales can complement experiments. Simulations are very challenging due to the inherent flexibility and heterogeneous ensemble of the Aβ1-40/1-42 monomers and oligomers, and the impact of a crowded environment. As a result, we need to develop and/or use various protein representations ranging from all-atom, coarse-grained (CG) to mesoscopic models and improve sampling techniques to converge rapidly to equilibrium and explore the dynamics over a wide range of time scales.(44,61)
In summary, we provide an in-depth review on the contribution of biophysical and biochemical studies and computer simulations to characterize the molecular structures of Aβ1-40/1-42 monomers, oligomers, protofibrils and amyloid fibrils in aqueous solution. We then focus on our current knowledge of the Aβ1-40/1-42 nucleus and the structures and dynamics of Aβ1-40/1-42 oligomers in or at proximity of the membrane. We summarize what is known about the interactions of Aβ monomer and oligomers with ion metals, cellular partners and potential inhibitors. We also report the main findings of the simulations on FAD mutations, and conclude by offering a perspective on the future of the field and the major questions that need to be addressed to discover drugs with much higher efficacy.
2. Molecular Structures of Aβ1-40 and Aβ1-42 Fibrils to Monomers from Experiments
Experimental characterization of amyloid fibril structures has been the topic of extensive research for decades, producing remarkable molecular-level insights.(62-65) Non-fibrillar monomer and oligomer structures, in contrast, are not well understood. We summarize the major findings on Aβ1-40/1-42 molecular structure from monomers to fibrils with emphasis on the most recent results. Structural understanding of Aβ fibrils and insights into the self-assembly process establish a basis for addressing the challenges associated with determining the structures of Aβ protofibrils and low molecular weight oligomers.
2.1 Fibrils
Due to the incompatibility of amyloid fibrils with X-ray crystallography and solution-state NMR, there is no single technique able to readily provide enough structural information to fully specify molecular structure within Aβ fibrils. Our structural knowledge of Aβ amyloid fibrils, therefore, is derived from the integration of complementary information from different experimental techniques. Fibril dimensions (nm length scale) have been probed by EM and AFM.(27,66-68) Fibril mass can be quantitatively measured by scanning TEM and, more recently, tilted beam TEM.(69) 2D structure (mostly β-strand) has been probed by FTIR.(70,71) This technique, along with diffraction based measurements, hydrogen/deuterium exchange, mutagenesis, proteolysis, EPR, and ss-NMR, can provide information on molecular fold and inter-molecular packing (β-sheet formation and organization).(25,27,71-79) Fibre diffraction studies established the “cross-β” structure, in which Aβ molecules assemble into β-sheets with β-strands oriented perpendicular to the long axis of the fibril.(72,80-82) The β-sheet structure was further confirmed by the binding of β-sheet-specific dyes such as Thioflavin-T and Congo red.(70) It should be noted that Sawaya et al. used X-ray diffraction to measure the detailed cross-β structures of microcrystals of several short peptides forming amyloid fibrils.(83) The data provide atomic details of “steric zippers” created by packing of interdigitated side chains between stacked β-sheets, described in terms of 8 possible symmetry classes. The free energies of different steric zipper configurations were also calculated using all-atom molecular dynamics (MD) simulations. Comparisons to experimental results suggest that the observed amyloid-like crystals are thermodynamically stable, although kinetic trapping can be driven by electrostatic side chain interactions.(84)
Among the experimental techniques mentioned here, ss-NMR has provided the most atomic-level detail of Aβ amyloid fibrils. This technique is well suited for amyloid fibrils because it provides information on local structure without requiring long-range orientation order.(85) In 1998, Benzinger used 13C-13C dipolar recoupling ss-NMR data on Aβ10-35 fibrils to challenge the then common belief that Aβ amyloid fibrils are composed of antiparallel β-sheets.(86) This preference for antiparallel β-sheets originates from earlier interpretations of FTIR data, the intuition that like-charged side chains are unlikely to be in close proximity, and NMR studies of the Aβ34-42 fibril.(71,87-89) Controversy over the arrangement of β-strands within Aβ amyloid fibrils further motivated the development of improved measurements for nuclear magnetic dipolar interactions,(90-92) and more analysis of Aβ fragments.(85,93,94a) It was found that the Aβ1-28 peptide forms in-register parallel β-sheets, in which β-strands are aligned for close proximity between like-residues.(95) The in-register parallel configuration which maximises overlap hydrophobic residues influenced the view that amyloid formation is driven by hydrophobic interactions.(68,85) A recent review on FTIR examines the experimental complications leading to incorrect assignment of antiparallel β-sheets, and describes more reliable approaches to data interpretation.(63)
In 2002, Petkova et al. reported a molecular model of an Aβ1-40 fibril based on constraints obtained from ss-NMR and EM.(27) This model refined subsequently with additional NMR constraints(79) reported an unstructured peptide for the first 10 residues with 2 β-strands (residues 11-24 and 30-40), as shown in Fig.1A. The first 10 residues were assigned to unstructured because isotopic labelling (13C and 15N) yielded NMR signals that were either broad (static disorder) or not observed (dynamic disorder). (27) The β-strands form in-register parallel β-sheets to produce the protofilament (Fig.1B). Protofilaments associate in pairs to form the two-fold topology for the striated fibrils (Fig.1C). The turn region (residues 25-29) is stabilised by a salt bridge between the charged D23 and K28 side chains. Note Nussinov et al. also proposed in 2002 the in-register parallel motif based on MD simulations using several topologies.(94b)
Figure 1.
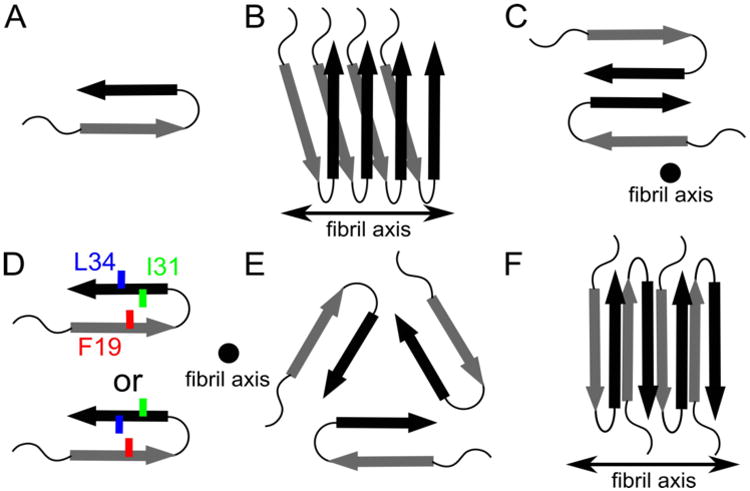
Schematics describing known structural motifs for Aβ1-40 WT and Aβ1-40 D23N fibrils. Arrows, thin lines, and colored symbols represent β-strand regions, non-β-strand regions, and selected residues, respectively. (A) Molecular conformation of Aβ molecules within fibrils, with arrows representing β-strand regions. (B) Organization of Aβ monomers into a protofilament. Each Aβ peptide contributes two β-strands to two stacked in-register parallel β-sheets, with hydrogen bonding interactions between equivalent β-strands along the fibril axis. (C) Cross-section of the Aβ1-40 fibril model of Petkova et al. composed of two protofilaments.(27,29) (D) Two distinct side chain arrangements experimentally observed for different Aβ1-40 fibrils. (E) Fibril cross-section predicted of the Aβ1-40 model determined by Paravastu et al. and composed of three protofilaments.(67) (F) The antiparallel β -sheet arrangement reported by Qiang et al. for fibrils of the Iowa Aβ1-40 D23N peptide.(29,101)
The structural model of Petkova does not describe, however, every Aβ1-40 fibril because fibrils are polymorphic. Polymorphism refers to the existence of multiple pathways for self-assembly, producing assemblies that differ in molecular structure. When observed by EM or AFM, distinct fibrils are observed with various width, twist, and cross-section dimensions.(27,66-68) Multiple fibril polymorphs usually co-exist within the same samples, although many samples are characterised by a dominant fibril polymorph. Petkova showed that subtle environmental factors as solution agitation can produce samples with different predominant fibril morphologies (two-fold symmetry under agitation and threefold symmetry under quiescent conditions) and that distinct NMR peak positions and line shapes indicate distinct underlying molecular structures.(27,29) Additional factors affecting fibril self-assembly are pH, the presence of metal ions, and interaction with interfaces.(64,67,97) Furthermore, there is an inheritance of structure when fibrils of one morphology are used to seed the self-assembly of Aβ monomers into new fibrils.(27) Aβ fibril polymorphism was further characterized using cryo-EM by Meinhardt(66) and Paravastu(67) using ss-NMR. An important structural difference between different Aβ fibril polymorphs lies in the orientations of the residues within the β-strands and the presence or absence of the D23-K28 salt bridge (Fig.1D). By taking advantage of observed differences in seeding efficiencies between different fibril polymorphs and using quiescent conditions, Paravastu isolated a new Aβ1-40 fibril with a 3-fold symmetric cross section (Fig.1E).(67) Bertini reported another Aβ1-40 fibril model with a topology similar to that of Petkova (Fig. 1A-C), but with different atomic details.(98) An Aβ1-42 fibril model with a similar configuration was also published by Luhrs, based primarily on hydrogen/deuterium exchange and mutagenesis data, but with different residues in the β-strand and turn regions.(28) While this model differs from structures observed for Aβ1-40, it is not clear how the ranges of possible Aβ1-40 and Aβ1-42 fibril structures could differ. The symmetries of all the experimentally constrained Aβ fibril structural models predict a single molecular conformation. Thus, detection of multiple NMR signals from each labelled site is normally assumed to imply polymorphism within the sample. Contrary to this interpretation, Lopez del Amo et al. recently published a NMR-derived fibril composed of Aβ molecules in two non-equivalent conformations.(99) An asymmetric fibril structure was also proposed by the theoretical work of Wu, but whether such Aβ fibril geometry is correct remains to be validated experimentally.(100)
The growing consensus that Aβ1-40 and Aβ1-42 fibrils are composed of in-register parallel β-sheets has recently been disrupted by recent reports on fibrils formed by the Iowa Mutant (D23N) of Aβ1-40 peptide.(29,101) This peptide forms fibrils composed of anti-parallel β-sheets as depicted in Fig.1F. It was suggested that substitution of the positively charged D23 side chain with the uncharged N23 side chain affects the nucleation rate of the parallel β-sheet structure, which is stabilized by the D23-K28 salt bridge.(101) Interestingly, the parallel β-sheet structure remains the thermodynamically preferred structure for the D23N mutant, but antiparallel β-sheet fibrils propagate more slowly in seeding experiments and dissolve at the expense of parallel β-sheet fibrils in mixtures.(29)
Experimental observations of environment-dependent self-assembly led to questions about the biological relevance of structural information from in vitro generated Aβ samples. The use of repeated seeding steps to amplify early nucleating or fast growing fibrils within a sample, for example, could result in a kinetically favoured structure, which may differ from the most thermodynamically stable structure. The theoretical work of Pellarin based on a mesoscopic model with one internal degree of freedom per peptide supports this notion, suggesting that less thermodynamically favoured fibril structures could nucleate more rapidly.(102) In addition, the microenvironment in vivo is likely to differ significantly from environments accessible in vitro, and may be affected by conditions promoted by Alzheimer's disease. Paravastu showed that amyloid plaques in the brains of deceased Alzheimer's patients could be isolated at concentrations high enough to seed the self-assembly of synthetic Aβ1-40 monomers, enabling the incorporation of isotopic labels into brain-derived fibril structures.(103) Lu analysed fibril samples from the brains of two deceased Alzheimer's patients with distinct clinical histories.(29) ss-NMR analysis of brain-seeded fibrils indicates that plaques from each brain are characterized by a single predominant fibril structure, though polymorphism was also observed.(29,103) Dominant structures differed, however, between the two Alzheimer's brains. Lu established a constrained structural model for the brain-derived Aβ1-40 fibril composed of in-register parallel β-sheets.(29) Fig.2 compares the all-atom pictures of the brain-derived Aβ1-40 fibril model and the fibril models of the synthetic Aβ1-40 peptides determined by Paravastu and Petkova. Lu observed NMR signals consistent with an ordered structure for every residue in Aβ1-40, and particularly a structured N-terminal region in contrast to the synthetic Aβ1-40 fibrils.(29) How this N-terminal structure will change with the binding of metal ions remains, however, to be determined.
Figure 2.
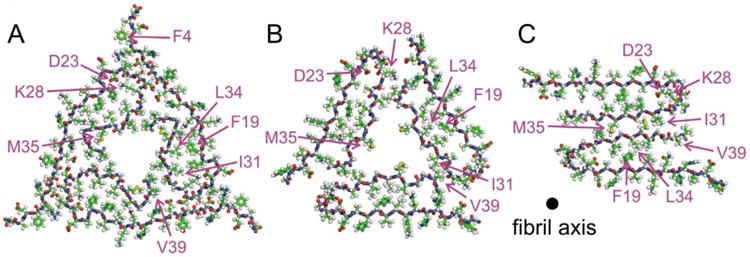
All-atom pictures of cross-sections of brain-derived Aβ1-40 fibrils modeled by Lu et al.(29) (A), compared to cross-sections of models for in vitro generated fibrils reported by Paravastu et al.(67) (B) and Petkova et al.(27) (C). The fibrils in (B) and (C) correspond to the diagrams shown in Figure 1C and 1E, respectively.
2.2 Protofibrils and Oligomers
Complexity in Aβ self-assembly was observed with the discovery of multiple soluble metastable Aβ oligomers at early and intermediate aggregation times.(104-108) While systematic classification of soluble Aβ species is difficult without more knowledge of structure and assembly pathways, soluble Aβ aggregates are generally referred to as protofibrils or oligomers. Protofibrils have elongated aspect ratios and a curvilinear appearance.(106) They are argued to be “on-pathway” intermediates to amyloid fibril formation and believed to convert to fibrillar structures without first dissociating to monomers.(106,108,109) Protofibrils have been reported to seed the growth of fibrils, have molecular weights as near 250 × 103 kDa (∼60 molecules), consist of β-sheets, and bind Thioflavin-T.(110) Oligomers are smaller species, with molecular weights ranging from 9 kDa (Aβ dimers) to hundreds of kDa (∼50 Aβ molecules). While oligomers exhibit β-strand secondary structure when probed by FTIR and CD, they do not necessarily bind Thioflavin-T or seed fibril formation.(63,111) These observations motivate the interpretation that at least some oligomers are “off-pathway” to fibril formation. Since fibril formation is accelerated by seed- or nucleus-dependent self-assembly, conversion of oligomers into fibrils is prolonged when oligomers are separated from fibrils and protofibrils; this separation is normally accomplished by size exclusion chromatography (SEC).(109) Species isolation and dynamics also increase structural homogeneity, which prevents structure elucidation. Oligomers have been further stabilized by crosslinking or interactions with engineered proteins.(112-114) Several protocols for production and isolation of oligomers have been reported and the different oligomer products have been named amyloid-derived diffusible ligands, globulomers, amylospheroids, Iβ, and annular protofibrils.(105,109,115-119)
Recent studies report structural characterization of protofibrils by ss-NMR. Scheidt stabilized Aβ1-40 protofibrils through interaction with an antibody-derived fusion protein (B10AP) and found 13C NMR chemical shifts which differ from those observed for amyloid fibrils and also indicate shorter β-strand regions.(120,121) The 13C NMR chemical shifts resemble reported values for Iβ oligomers, suggesting that protofibril structure more closely resembles oligomer structure than fibril structure.(121) Although Iβ oligomers were reported to be composed of in-register parallel β-sheets, Scheidt et al. proposed an intramolecular antiparallel β-hairpin within Aβ oligomers.(112-114) This β-hairpin model (Fig.3A) predicts hydrogen bonding between β-strands in the same molecule, which does not occur in the fibril structural models (Fig.1). Structural similarity between B10AP-stabilized Aβ1-40 oligomers and protofibrils was supported by intramolecular proximity between E22 and I31 observed for both structures.(120) Although these results do not specify how neighboring Aβ molecules are arranged within protofibrils or oligomers, they suggest that structural rearrangement is required for protofibrils to convert to fibrils. Further evidence for structural reorganization from protofibrils to fibrils was found by Doi using lyophilization to stabilize Aβ1-42 protofibrils and ss-NMR to show that protofibrils are not composed of in-register parallel β-sheets. (122)
Figure 3.
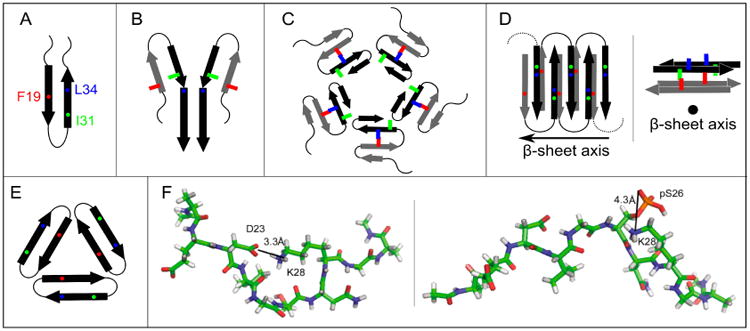
Proposed models for non-fibrillar Aβ aggregates. As in Figure 1, arrows, thin lines, and colored symbols represent β-strand and non-β-strand regions, and selected residues, respectively. (A) The antiparallel β-hairpin conformation predicted by Hoyer et al. for the monomer(114) and suggested by Scheidt et al. for protofibrils.(120,121) (B) The dimer structure proposed for preglobulomers by Yu et al.(123). (C) The disc-shaped pentamer model proposed by Ahmed et al.(124). (D) Two different views of the antiparallel β-sheet model for 150 kDa oligomers, reported by Tay et al.(111). (E) X-ray crystallographic structure of the trimer of the designed cyclic Aβ17-36 peptide.(127b) (F) Representative structures of highest populations in the MD ensembles of the Aβ21-30 WT peptide with the D23-K28 salt bridge (left) and the Aβ21-30 peptide with pS26 substitution (right).(47)
The diagrams in Fig.3A-3D depict alternative schemes for the inter-molecular organization based on experimental data of Aβ1-42 oligomer samples. Hoyer observed that Aβ1-40, which is normally an unstructured monomer in solution, adopts an antiparallel β-hairpin conformation (Fig.3A) upon binding to an antibody mimicking protein.(114) This result inspired the construction of a double cysteine Aβ mutant (at positions 21 and 30) that was constrained to adopt the β-hairpin conformation. This mutant was shown to form oligomers and protofibrils but not fibrils.(112,113) Yu performed solution NMR on preglobulomers produced through interactions between N-Met-Aβ42 (additional M residue on the N-terminus) and sodium dodecyl sulfate (SDS) micelles (0.2% SDS by weight), resulting in the proposed model shown in Fig.3B.(123) This model also predicts intramolecular hydrogen bonds to produce an antiparallel β-hairpin between β-strands spanning residues 17-23 and residues 28-33. In addition, inter-molecular parallel β-sheet contacts are predicted for pairs of β-strands formed by residues 34-42. While the preglobulomer molecular weight was measured to be 16 kDa (∼4 Aβ-molecules), it is not known how the dimer drawn in Fig. 3B would assemble into a larger structure. In another study, Ahmed prepared Aβ1-42 pentamers by incubation of monomers at low temperature, and ss-NMR provided the basis for the model of Fig.3C.(124) This model does not include hydrogen bonds between any of the β-strands (no β-sheets). Another ss-NMR study conducted by Tay was based on 150 kDa (∼33 Aβ molecules) oligomers produced using a procedure similar to that used for globulomers (removal of SDS from preglobulomer solutions by dialysis).(111) ss-NMR data were consistent with the molecular conformation found in fibrils (Fig.1A), but a significant distinction was reported in terms of intermolecular organization. Unlike fibrils, 150 kDa oligomers are not composed of in-register parallel β-sheets. Instead, Tay proposed the model shown on Fig.3D, with intermolecular antiparallel β-sheets.(111) Consistent with this interpretation, Gu used site-directed spin labelling and EPR on Aβ globulomers to show that their structures are not consistent with in-register parallel β-sheets, but rather antiparallel β-sheet structures.(59) Finally, Nowick et al. designed a macro-cycle peptide derived from Aβ17-36 in which residues 17-23 and 30-36 form the β-strands with two δ-linked ornithine β-turns connecting the side chains of D23 with A30, and the side chains of L17 with V36. X-ray structure shows that trimers consist of three highly twisted β-hairpins in a triangular arrangement (Fig.3E) and four trimers form a 12-mer Aβ*56 species with a central cavity, one important species to impair memory.(125)
Despite significant advances, we emphasize that it is not yet clear how Aβ oligomer and protofibril structures may be distinguished. None of the diagrams in Fig.3 correspond to widely accepted models for Aβ protofibril or oligomer structures. In contrast, the fibril structures in Figs. 1 and 2 benefitted from much more sample preparation experience and better optimized structural measurements. While the solution NMR-derived model of Yu is based on many constraints,(123) the high concentration of SDS in the final sample is known to affect Aβ structure. The models in Figs. 3A and 3D are works in progress, in need of many more structural constraints, including complete residue-specific information about secondary structure and experimental constraints on inter-molecular packing. In fact, a recent proline mutagenesis study conducted by Haupt reported that oligomers might be distinguished from protofibrils by the structure of the N-terminus.(126) Using fluorescence correlation spectroscopy (FCS) and Foster Resonance Energy Transfer (FRET), Maiti showed that the two hydrophobic regions (residues 10-21 and 30-40) have attained the β-strand conformation in both oligomers and fibrils. However the conformations of the turn region (residues 22-29) and the N-terminal tail (residues 1-9) are markedly different.(127a) The role of the turn region has also been emphasized by NMR and replica-exchange molecular dynamics (REMD) simulations which demonstrate that phosphorylation at S26, which interferes with formation of the D23-K28 salt bridge, impairs Aβ1-40 fibrilization while stabilizing its monomer and non-fibrillar aggregates (Fig.3F).(47) Using pulse hydrogen-deuterium exchange MS, the middle region of Aβ1-40 (residues 20-35) was found to be the first to aggregate, followed by the residues 36-42 and then the N-terminus (residues 1-19).(24)
It is interesting that these Aβ NMR-derived models from 4 to 33 peptides predict anti-parallel β-sheets. (111,120-121,123-124). In some cases, the oligomers take up a β-hairpin structure.(123) This conformation is, however, very different from that in the fibrils. The overall topology looks similar, but the orientation of the hydrogen bonding network and the side chain contacts are very different. This may be an important factor in the ability of the oligomers to insert into the membrane (because the only large family of membrane proteins with anti-paralllel β-sheets consists of porins), and in the formation of the nuclei as described in Section 5. Further support for antiparallel β-sheet arrangement of Aβ oligomers (Figs. 3A, 3B and 3D) comes from FTIR (22) and X-ray crystallography data on an hexamer of a segment of αB crystallin (K11V).(127b) These X-ray diffraction results suggest that it may be possible to study Aβ oligomer structure by crystallography without any chemical modification,(125) and Aβ oligomers could adopt a structure similar to the cylindrical antiparallel β-sheet structure (or cylindrin) of the K11V peptide.(127b) Indeed these β-barrels were predicted prior to the determination of the KV11 peptide structure on several peptides by computer simulations using coarse-grained and all-atom representations in explicit and implicit solvent.(128-133a)
Atomistic characterization of the small and large Aβ1-40/1-42 oligomers is very difficult as these oligomers are highly aggregation-prone, and degenerate by displaying multiple polymorphic structural variants analogous to strains in prion diseases. In a recent study, Glabe et al. designed 23 monoclonal antibodies against Aβ1-42 and showed that no single antibody is able to recognize the different states of Aβ1-42 in vitro and in AD brain.(133b) What is clear from various experiments is that synthetic Aβ1-40 and Aβ1-42 polymerize through distinct pathways. Photo-induced cross-linking of unmodified protein (PICUP) with a Y10-Y10 side-chain – side-chain bridge followed by SDS-PAGE, DLS and SEC has been used to unveil the oligomer size distribution of Aβ oligomers.(134-136) A first study showed that Aβ1-40 exists as monomers, dimers, trimers and tetramers in rapid equilibrium, while Aβ1-42 preferentially forms pentamer/hexamer units. This difference was linked to the specific roles of I41 and A42: I41 is essential to induce paranucleus formation while A42 enhances the self-association of these paranuclei.(135) Bowers et al. further used IM-MS to investigate the early oligomers of Aβ1-40 and Aβ1-42.(11) They confirmed that Aβ1-40 dominantly populates monomers, dimers and tetramers, while Aβ1-42 mostly forms dimers, tetramers, hexamers and dodecamers, and provided for each species a cross-collision section that can be used to validate the simulations. Moreover, they proposed an assembly mechanism in which the dimer plays a key role and they identified structural differences in the tetramer that rationalize the formation of higher order oligomers by Aβ1-42, but not by Aβ1-40.(11) However, using the same experiment with mutagenesis, Dadlez showed that Aβ1-40 oligomers consist of at least two families of conformers: compact and extended. The compact form resembles the fibril-like structure while the extended form resembles the globular form determined by Lu et al. with the C-terminal ends forming intermolecular parallel β-sheets.(137a-b) Note however that other globular structures could fit the cross collision section. Using fluorescence, Chen and Glabe found that the Aβ alloforms have different conformations and assembly states upon refolding from their unfolded conformations. Aβ1-40 is predominantly an unstable collapsed monomer, while Aβ1-42 samples a stable structured trimer or tetramer at concentrations > 12.5 μM.(137c)
Many experimental studies have revealed the importance of the central hydrophobic cluster (CHC, residues L17-A21) and the C-terminus. While these hydrophobic patches form intermolecular β sheets in fibrils, their role in aggregation is just beginning to become clear. Incubation of Aβ40 fragments with the full-length peptide show enhanced fibrilization rates only for the fragments containing residues L17-F20 or A30-M35.(138) Proline mutations of residues in the 17-20 or 30-35 regions in Aβ1-40 and Aβ1-42 are more disruptive to fibrilization than mutations in other regions.(139) Finally, tethered Aβ1-40 demonstrated decreased spin mobility at regions H14–V18, G29–A30, and G38-V40 when investigated with EPR spectroscopy.(140)
Other experimental studies have helped clarify the role of the two additional C-terminal residues in Aβ1-42-specific aggregation. The VPV substitution (G33V, V36P and G38V), promoting a β-hairpin at the C-terminus, increases Aβ aggregation rate and higher order oligomers, while the PP substitution (V36D-Pro, G37L-Pro), leading to a hairpin breaking motif, disrupts the Aβ1-42 aggregation kinetics and changes the oligomer size distribution to one more characteristic of Aβ1-40.(141) PICUP results suggest that a turn centered at residues V36 and G37 of Aβ1-42 and its absence in Aβ1-40 is responsible for the characteristic features of Aβ1-42 early oligomers.(136) These findings suggest that the formation of an additional β topology sampled at the C-terminus, driven by hydrophobic side chain interactions, may be responsible for Aβ1-42's unique assembly properties.
The central region, consisting of the hydrophilic residues E22-G29, has also been implicated for its unique properties and effects on Aβ assembly. This region was identified due to its inherent resistance to proteolysis, which is maintained when the Aβ21-30 fragment is isolated. Solution NMR of this fragment reveals that V24-K28 samples two turn-like structures that may be critical in the folding of the monomer(142) and this was confirmed by computer simulations using various force fields.(143-145) Further, substitution of contiguous pairs of residues in the V24-N27 region with a turn-nucleating D-ProGly motif largely accelerated fibril self-assembly of Aβ1-40.(146) Lastly, charge-altering point mutants of residues E22 and D23 that are implicated in FAD and cerebral amyloid angiopathy also demonstrate increased oligomerization orders and fibrilization rates when introduced into Aβ1-40. Further, the oligomerization propensities of each of these mutants in full length Aβ are directly correlated to both susceptibility of trypsin proteolysis and instability of the V24-K28 turn for the Aβ21-30 fragment.(147a) This suggests that these FAD mutants may destabilize turn like structures in the central region, possibly changing the ensemble and allowing the monomer to seed different types of aggregates. This change in turn confirmation has been confirmed by MD simulations on the Aβ21-30 peptide with the Arctic, Dutch and Iowa mutations and two biological relevant salts (CaCl2 and KCl).(147b)
Recently, various experiments have shed light on the role of the N-terminus in self-assembly. The D7N (Tottori) mutation accelerates the kinetics of transition from random coil states to β-sheet-rich configurations and promotes the early formation of higher order oligomers with more α/β structures that are significantly more toxic than WT Aβ1-40 and Aβ1-42 peptides.(148,149) The H6R mutation, the substitution of K16 by Ala and the substitution of D1 by Tyr also affect self-assembly and toxicity.(150-151a) Single-molecule AFM experiment shows many dimer configurations stabilized by N-terminal interactions, although there is a difference in the interaction patterns of Aβ1-42 and Aβ1-40 monomers within dimers.(26) Finally, two single mutations at position A2 protect from AD.(54,55)
In summary, in vitro and in AD brain experimental studies indicate polymorphism and inherent diversity of structures present in fibrils and aggregates. Many physical factors can contribute to the formation of strains, and in how the β-sheets pack or the strands hydrogen bond to each other: pH, temperature, concentration, supersaturation of the Aβ solution, ionic strength, sample, agitation conditions such as shear forces or sonication, interfaces, the presence of seeds.(18,29,151b-d) Other data indicate that the structures and polymorphism of Aβ fibrils critically depend on the oligomeric states of the starting materials, the ratio of monomeric-to-aggregated forms of Aβ1-42 (oligomers and protofibrils), and the probability of secondary nucleation.(16) A recent study investigated how local physical forces interfere with the fibrillation kinetics, the general morphology, and the local structure and dynamics of the fibrils formed from the Aβ1-40 peptide. The well described hydrophobic contact between F19 and L34 was rationally modified and the F19G, F19E, F19K, F19W and F19Y mutants were studied in order to understand the impact of the change in electrostatic (E and K mutations), hydrophobic interactions (W and Y mutation) between side-chains and larger flexibility of the backbone (G mutation). Local interactions were observed to influence the fibrillation kinetics, dynamics and structure (the register of the hydrogen bond pattern) of Aβ1-40, but leave the general fibril structure unchanged. These data also indicate the role of the non-local F19-L34 in the early oligomers.(151e) Overall a solid fundamental understanding of the principles underlying polymorpshism and strain behavior of fibrils remains to be determined.
2.3 Monomers
Although Aβ1-40 and Aβ1-42 monomers are described by disordered conformations, there is experimental evidence suggesting a bias toward β-strand character in the CHC core and the C-terminus and a propensity for turns at specific positions within the Aβ monomers. Solution NMR studies targeting the monomeric state best characterize Aβ with a collapsed coil ensemble and not by a unique structure.(152) Nevertheless, backbone Hα, Cα, and Cβ chemical shift indices suggest β-strand propensities in the CHC, I31-V36, and V39-I41, as well as turn character at D7-E11 and F20-S26 in Aβ1-42.(21) Residues V18-F20 (in Aβ1-40, Aβ1-42, and Aβ1-42-M35ox, i.e. with oxidation of the M35 side chain) and V39-I41 (in Aβ1-42 and Aβ1-42-M35ox) also possess experimental 3JHNHA > 7.5 Hz, indicative of a bias toward φ dihedrals characteristic of β-strands in these regions.(153,154) Far UV CD spectra for Aβ1-40 and Aβ1-42 monomers are also dominated by random coil, but suggest β-strand content.(149,135) Using different preparation methods, CD analysis reported a β-strand content between 12 % and 25% and an α-helix content between 3% to 9% at 295 K, pH 7.5 and day 0 (therefore for a mixture of aggregates), indicative of the dependence of the secondary structure on sample preparation.(20,149) 15N spin NMR relaxation data reveal that Aβ1-42 monomer demonstrates more rigidity at the C-terminus than Aβ1-40, both in terms of side chain and backbone dynamics,(155,156) suggesting residual secondary structure formation. Although these biases may characterize the Aβ monomeric ensemble in aqueous conditions, other individual structures may be possible. For example, an NMR structure of Aβ1-40 monomer forms a 3-10 helix from H13 to D23 at pH 7.3, even if exchange between the 3-10 helix and other conformations in this region cannot be ruled out. (157a) In contrast to previous NMR studies, this study was conducted at 50 mM NaCl and it is well established that salt shifts the ensemble from unstructured to more helical conformations.(157a) The structure of Aβ1-40 monomer with a N-terminus cysteine attached to silver nanoparticles has also been interrogated by surface plasmon enhanced Raman spectroscopy (SERS). This shows no change between pH 10.5 and 5.5, the presence of partial α-helical content, indicating the existence of short and transient α-helical conformations for WT Aβ1-40 monomer in physiological conditions.(157b) Finally phase-modulated CLEAN chemical exchange experiment with a fast HSQC detection scheme(158) on Aβ1-40 monomer shows that the residues 10-13, 17-22 and 30-36 are partially protected from exchange with solvent, while D23 and the region G25-G29 are susceptible to exchange,(159) consistent with the idea of a solvent exposed turn in the central region.
3. Simulations of Aβ1-40 and Aβ1-42 Monomers in Aqueous Solution
Characterizing the monomeric state of Aβ at atomic detail under physiological conditions can be key to understanding how Aβ assembles into disease-causing oligomers because they represent a base state common to all aggregation pathways.(2) This knowledge could be crucial in developing therapeutics that prevent nontoxic monomers from progressing into toxic species, one of the fundamental strategies in the ongoing effort to treat AD. It is well established that self-assembly is profoundly influenced by very subtle chemical changes, ranging from the two-residue difference between Aβ1-40 and Aβ1-42,(135,155) FAD mutations,(21,53) to the single atom modification caused by M35-ox.(160) The polymorphism of monomeric Aβ under physiological conditions may underlie this relationship. In the absence of unambiguous stable native states, simple chemical modifications could have a profound effect on the type of ensemble sampled by that particular Aβ peptide. This intrinsic disordered property, in addition to the high aggregation propensity, has frustrated experimental efforts to characterize the Aβ1-40/1-42 structures.
The challenges and limitations inherent to the current set of experimental techniques for studying these polymorphic, aggregation-prone Aβ monomers have encouraged many groups to use a wide variety of computational techniques to more thoroughly investigate the conformational properties of these peptides. Over the years, the ability to perform extensive MD simulations has improved. Today, simulations for Aβ extend over multiple microseconds using explicit and implicit solvent models. Additionally, REMD(161), simulated tempering(162) and metadynamics(163) simulations are used to escape energy minima and enhance sampling. In general, the results obtained in simulations of intrinsically disordered proteins (IDP) like Aβ depend strongly on the set of parameters (such as the force field) used to describe the energy of the peptide and their interactions with the aqueous solvent. Widely used force fields for biomolecular simulations are OPLS-AA,(164) AMBER99sb and their variants,(165) and CHARMM22*,(166) while frequently used water models are the three-site models TIP3P(167) and SPC/E,(168) and the four-site models TIP4P(167) and TIP4P-Ew.(169) These force fields have been calibrated against model compounds and peptides, and in most instances the force fields reproduce folded conformations of small globular proteins with root-mean-square deviations (RMSDs) within Ångstroms of the experimentally determined structures.(170a-d) However, experimental validation of the ensembles produced by these force fields, for either the unfolded states of globular proteins(170e) or IDPs(170f), remains essentially an unsolved problem. Here, we review some of the more recent simulation studies, which employ state-of-the-art strategies, ranging from all-atom REMD simulations to other approaches, to extensively characterize the equilibrium structures of Aβ1-40 and 1-42 monomers.
All-atom REMD simulations on the μs/replica timescale with OPLS-AA/TIP3P conducted by Rosenman et al.(154b) revealed that Aβ1-40 sampled β-hairpins between the L17-A21 and I31-V36 regions, while Aβ1-42 sampled a second β-hairpin spanning V39-A42, forming transient β-meander structures. The hairpin model of Aβ1-40 with the backbone of K16 hydrogen bonded to the backbone of G37 is found in both centroids 3 (4.2%) and 4 (3.9%) using a linkage clustering shown on Fig.4A (top). This conformation does not exist for Aβ1-42, as the centroid 1 of Aβ1-42 possesses a different CHC to C-terminal hairpin with a backbone H-bond register shifted by two residues. The double hairpin for Aβ1-42 is found in centroid 7 (Fig.4A, below). The predicted hairpin for Aβ1-40 is consistent with the NMR structure of Aβ1-40 in complex with a phage selected affibody(114) as shown in Fig.4B, and the intrapeptide model of Aβ1-40 fibrils as published by Bertini et al. (98) shown on Fig.4C. The predicted double hairpin for Aβ1-42 is consistent with the intrapeptide model of Aβ1-42 pentamers as published by Ahmed et al.(124) shown on Fig.4D. Taken together, these REMD-predicted hairpins, by demonstrating a structural similarity to models of higher order aggregates, suggest that they may act as the seeds for Aβ assembly.
Figure 4.
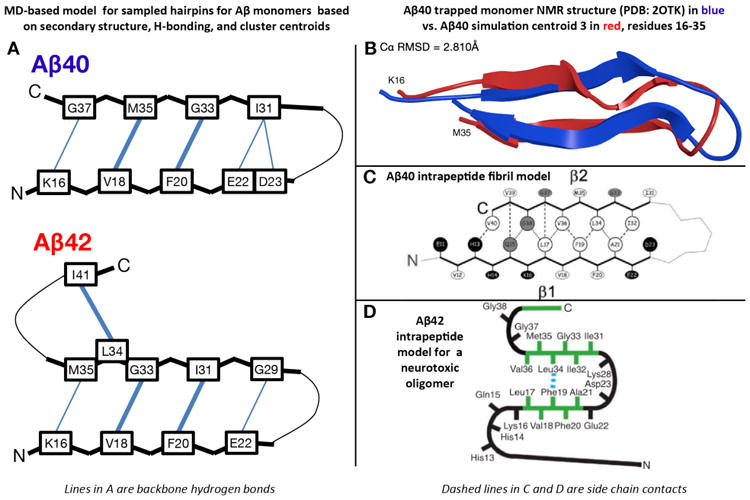
Transient REMD-sampled conformations of Aβ monomers bearing similarity to experimental intrapeptide models of higher order aggregates. (A) β-hairpin models for Aβ1-42 and Aβ1-40 monomers derived from the simulations of Rosenman et al.,(154b) based on the most populated cross-region backbone hydrogen bonds and secondary structure proclivities in the ensemble. Residues that have a high population for both donor to acceptor and acceptor to donor backbone hydrogen bonds are illustrated with a bold line. Sampled conformations matching these models exist as high-ranking centroid structures. (B) Ribbon overlay of residues 16-35 for centroid 3 derived from clustering analysis of the Aβ1-40 simulation(154b) and the solution NMR structure of monomeric Aβ1-40 in complex with a phage selected affibody (PDB entry 2OTK) published by Hoyer et al.(114). (C) Intrapeptide model for Aβ1-40 fibrils based on ss-NMR, as published by Bertini et al.(98). (D) Intrapeptide model for Aβ1-42 “on pathway” pentamers based on ss-NMR, as published by Ahmed et al.(124).
Previous characterizations of Aβ through REMD simulations with OPLS-AA/TIP3P and AMBER99sb/TIP4P-Ew also predicted conformations where Aβ was mostly flexible but possessed some structured segments; in particular, β-hairpins populated the C-terminus in Aβ1-42 but not Aβ1-40.(171) A different approach was taken by Ball et al.,(172) who had used 100 ns multiple reservoir replica exchange (MRRE) simulations with AMBER99sb/TIP4P-Ew to determine that Aβ1-42 was mostly disordered, with significant α character in residues Y10-F19 and E22-N27 and little to no β content. The same group then reported their analysis of multiple trajectories acquired with the same simulation method, this time processing the resulting ensemble using the ENSEMBLE package(173) to select structures that best match experimental chemical shifts, residual dipolar couplings, 3JHNHA couplings, and 1H-15N NOEs.(174) After refinement with ENSEMBLE, the aforementioned α-content is de-emphasized, and antiparallel β-hairpins between the K16-A21 and G29-V36 regions are promoted in Aβ1-42. Aβ1-40 after ENSEMBLE refinement, in contrast, is characterized by reduced C-terminal β propensity and sampling of a hairpin between the CHC and residues G9-H13.(174) The data suggest that the extra two residues in Aβ1-42 primarily promote hydrophobic clustering that direct the increase in β content in the CHC and G29-V36 region, rather than the direct formation of additional secondary structure, such as the second hairpin discussed above. Further, despite possessing similar overall biases, the ensembles generated by Ball(174) and Rosenman(154b) also differ in β % per residue, with the largest residue propensities in the range 20-30% in the former and 50-60% in the latter. It is worth noting a 100 ns/replica REMD simulation with AMBER99sb/TIP3P also revealed that Aβ1-42 forms contacts between L17-A21 and I31-V36 with a transient turn in the region D23-N26, consistent with a quasi-hairpin-like conformation.(175)
Other approaches beyond atomistic REMD in explicit solvent have also been used to investigate the properties of Aβ. The two alloforms were explored using the Folding@home platform and thousands of MD trajectories with AMBER99sb/TIP3P, each of average length ∼30 ns, for each species.(176,177) Both WT Aβs are described as mostly disordered ensembles, with some α-helical character from residue 10-20 and almost no β-content, in reasonable agreement with SERS experiment.(157b) Of the β content that exists, β-sheet propensity near the C-terminus is notably less in Aβ1-40 than in Aβ1-42. Granata et al.(178a) investigated the Aβ1-40 ensemble with NMR guided metadynamics, which uses experimental data as collective variables to drive metadynamics calculations rather than using them purely for simulation validation or as hard structural restraints.(178b) Simulations were carried out with CHARMM22*/TIP3P at 350 K and 8 replicas for 310 ns/replica. Each replica was biased by a history-dependent potential acting on a different collective variable, including two variables for the difference between predicted and experimental chemical shifts. The resulting unbiased free energy surface (FES) has many extended and highly disordered states and large radii of gyration.(178a) In terms of structured states, the FES displays structures with long α-helical content similar to the structure predicted by Vivekanadan,(157) and short α-helix content as predicted by Pande et al.,(176) and various β-hairpins spanning the L17-A21 and I31-V36 regions, similar to the structures predicted by Rosenman et al.(178) Further simulations showed the FES does not change at 300 K.
Aβ monomers have also been investigated by REMD(179) with the six-bead CG OPEPv3 model in implicit solvent(180a) and Monte Carlo simulated annealing with the all atom PROFASI force field.(181a) The OPEPv3 force field has been calibrated against non-amyloid peptides, and in most instances predicts folded conformations with 2-3 Ǻ RMSD from the NMR structures,(179,180a-e) although it cannot reproduce vibrational frequencies with high accuracy as all-atom models.(180f-g) OPEP has also been coupled to a greedy algorithm for structure prediction of peptides with 9-52 amino acids.(180h-i) The OPEP-REMD simulation revealed Aβ ensembles that were mostly turn/coil, but possessed substantial β-sheet propensity in the N-terminus.(182) It remains to be determined whether OPEPv5 with better electrostatic interactions leads to a different picture.(183) The PROFASI simulation characterized Aβ1-42 as possessing strong β probability in many of the residues over the peptide.(181b)
All-atom REMD simulation of both alloforms was performed for 110 ns per replica with AMBER99sb/Generalized Born (GB).(184a) Each monomer behaves as a unique statistical coil at 298 K with five relatively independent folding units comprising residues 1-5, 10-13, 17-22, 28-37, and 39-42, connected by four turns. The two turns predicted at positions 6-9 and 23-27 are in agreement with NMR and the residues I41 and A42 increase contacts within the C-terminus and between the CHC and the C-terminus leading to a more structured C-terminus.(184a) Finally, discrete MD (DMD) simulations, where all inter-particle interactions are expressed by square-well and step-like potentials, coupled to a four-bead CG model, find that Aβ1-42 displays a turn centered at G37-G38 and a β-hairpin spanning V36-A42 that are absent in Aβ1-40.(184b) DMD simulations also capture two other differences between the alloforms: a highly populated β-strand at A2-F4 in Aβ1-40 but not in Aβ1-42, and a β-hairpin centered at S8-Y10 in Aβ1-42 but not in Aβ1-40.
While the most recent and exhaustive all-atom studies in explicit solvent have started to show some consistent depictions of the properties of the Aβ ensemble, most characterizations of the Aβ1-40 and Aβ1-42 peptides, in our opinion, remain highly divergent. These variations may arise from differences in simulation conditions, extent of sampling, or trajectory analysis. IDPs like Aβ, or even the unfolded ensembles of well-folded proteins, remain difficult test cases for our current range of computational techniques because they lack non-ambiguous energy minima. The types of conformations sampled through simulation may be much more sensitive to simulation conditions than that of globular proteins, where parameter differences could still lead to similar final results. For the globular villin headpiece, for example, independent MD simulations using different all-atom force fields were able to recapture the experimental folded structure and folding rate of the protein, but the unfolded states and folding mechanism were highly dependent on force field choice.(185) Meanwhile, in the case of two intrinsically disordered proteins (a 50 residue peptide derived from a FG-nucleoporin and a 20 residue RS repeat peptide), μs length REMD simulations with four different all-atom force fields were found to adopt substantially different hydrogen bonds, secondary structure tendencies, and radii of gyration.(185) With this in mind, the force fields that are capable of reversibly folding globular proteins such as AMBER99sb(165a) with *(165b) and/or ILDN(165c) modifications and CHARMM22*(166) in the studies described in Refs. 170b and 170d may not necessarily be the most suitable for characterizing the metastable states of disordered ensembles.
Given these circumstances, we suggest that multiple simulation studies consistent with experimental data are likely to be much more valuable than a single study with one force field. To sort out un-generalizable findings, more stringent and sensitive experimental validations are necessary, particularly using better reporters on the tertiary structural biases. Many of the values commonly used for experimental comparison, such as NMR chemical shifts and scalar J-couplings are good “sanity checks” on sampling, but primarily report on local structure and are highly prone to sequence-specific bias. Full characterization of the structures sampled by intrinsically disordered proteins remains a major challenge. The development of new experimental techniques to probe the Aβ monomer structures in solution is also needed.
4. Simulations of Aβ1-40/1-42 Dimers and Higher Order Assemblies In Aqueous Solution From Random States
Soluble Aβ dimers are the smallest toxic species in AD(10) and isolated from Alzheimer cortex they directly induce tau hyperphosphorylation and neuritic degeneration.(186) Trimers and larger aggregates are also toxic. Knowledge of their key structural and dynamical features is of significant interest to design drugs inhibiting their formation and toxicity. The conformational stability of preformed Aβ assemblies of various oligomer sizes, inspired from ss-NMR derived fibril structures (5-mers and 10-mers of Aβ17-42),(187) fibril polymorphisms of other amyloid sequences (10-mers of Aβ19-42),(188) globulomers (12-mers of Aβ17-42)(189) and the design of triple-sheet motifs (24-mers and 60-mers of Aβ17-42)(190) was assessed by atomistic MD of 50-100 ns, proving only that these states are stable within the simulation times. In what follows, we describe the most recent simulations aimed at understanding the aggregation of Aβ peptides from random states (Table 1). Along with Aβ1-40 and Aβ1-42, we also report the results of three non-pathogenic truncated variants, Aβ9-40, Aβ10-40 and Aβ17-42. We recall the N-terminal, central and C-terminal regions cover residues 1-16, 22-29 and 30-40/42.
Table 1.
Summary of Aβ aggregation simulations from random states. In the timescale column, sim stands for the number of independent simulations, rep for the number of replicas for REMD or HT-REMD, and the time is that of one MD trajectory or one replica.
| Ref. | Force field | Solvent model | Method | Timescale | Aβ alloforms | Oligomer size |
|---|---|---|---|---|---|---|
| 195 | OPEP 3.2 | Implicit | HT-REMD | 1.25 μs × 26 rep | 1-40, 1-42 | 2 |
| 199 | CHARMM19 | SASA | T-REMD | 0.8 μs × 24 rep × 8 sim | 1-40 | 2 |
| 202 | PROFASI | Implicit | MC | 2×l010 steps × 40 sim | 1-42 | 2 |
| 204‡ | OPLS-AA | SPC/E | MD | 0.05 μs × 1000 sim | 1-40, 1-42 | 2 |
| OPLS-AA | TIP3P | MD | 0.05 μs × 1000 sim | 1-40, 1-42 | 2 | |
| 207 | OPLS-AA | TIP3P | T-REMD | 0.2 μs × 64 rep | 1-42 | 2 |
| 206 | AMBER99sb | TIP4P-Ew | T-REMD | 0.05 μs × 52 rep | 1-42, 1-43 | 2 |
|
| ||||||
| 205 | AMBER99 | TIP3P | MD | 0.1 μs | 1-42 | 2 |
| 208 | OPEP 3.2 | Implicit | T-REMD | 1.2 μs × 22 rep | 17-42 | 3 |
| 203† | CG | Implicit | DMD | 6×l07 steps × 8 sim | 1-40, 1-42 | 1–32 |
| 200 | CHARMM19 | SASA | T-REMD | 0.8 μs × 24 rep × 8 sim | 10-40 | 4 |
| 210† | OPLS-AA | GB/SA | T-REMD | 0.2 μs × 5 sim | 1-42 | 1–20 |
4.1. Dimer Simulations With Simplified Representations
Using the 6-bead CG OPEPv3 model with implicit solvent(61,183,191-194) the structures of Aβ1-40 and Aβ1-42 dimers were determined by HT-REMD simulations starting from randomly chosen conformations.(195) HT-REMD(196) combines standard REMD(179) with a Hamiltonian exchange procedure, where several replicas with reduced non-bonded energies are used at the highest temperature. Both alloforms populate mostly turn/random coil conformations with a β-sheet propensity at the C-terminal region higher than in the monomers. Dimerization is characterized by inter-peptide hydrophobic contacts between CHC/CHC, CHC/C-terminal region and C-terminal/C-terminal regions. However, the Aβ1-42 dimer has a higher propensity than the Aβ1-40 dimer to form β-strands at the CHC and in the C-terminal region. The free energy landscape of the Aβ1-42 dimer is also broader and more complex than that of the Aβ1-40 dimer.(195)
United-atom REMD simulations with CHARMM19(197) and a solvent-accessible surface area (SASA) implicit solvent(198) were performed on Aβ1-40 and Aβ10-40 dimers.(199,200) Truncation of the first 9 residues leads to minor changes in the structure of the dimer.(200) The conformational ensemble of the Aβ10-40 dimer can be described by three distinct basins differing with respect to the distribution of secondary structure and the amount of inter- and intrapeptide interactions. The interface is largely confined to the region 10–23, which forms the bulk of inter-peptide interactions and a few inter-peptide hydrogen bonds.(199) Random reshuffling of the amino acids, i.e. sequence permutation, does not impact the Aβ10-40 dimer globule-like states, suggesting that the Aβ10-40 peptides in the dimer behave as ideal chains in polymer melt, in which amino acids lose their identities.(201) These results run in contrast to MC simulations with the all-atom PROFASI model and implicit solvent(181a) where the Aβ1-42 dimer is mostly composed of a four-stranded anti-parallel β-sheet or two layers with mixed parallel/antiparallel arrangements and three major clearly identified turns at positions 13-16, 23-26 and 35-38.(202)
Finally, extensive DMD simulations coupled to a four-bead CG model found that the Aβ dimer conformations are collapsed and disordered with a small content of β-strands linked by loops and turns.(203) The Aβ1-42 dimer has a higher propensity of β-sheets at the CHC and C-terminal region than the Aβ1-40 dimer. Aβ1-40 dimer formation is mainly driven by intermolecular interactions between the CHC regions, while the C-terminal region plays a significant role for Aβ1-42.(203) 50-ns MD stability simulations with OPLS-AA/TIP3P or SPC/E starting from the dominant DMD-obtained CG structures confirm the main DMD results(203) and enable a precise analysis of secondary structures, salt-bridges and free energy landscapes.(204) Overall, the free-energy landscape of Aβ1-42 is much more complex than that of Aβ1-40.
4.2. All-Atom Dimers in Explicit Solvent
Solvation free energy analysis based on the integral-equation theory of liquids and MD trajectories of 100 ns suggests that dimerization occurs through a two-step nucleation-accommodation mechanism: decrease of the monomer solvation free-energy followed by structural reorganizations in the dimer leading to a decrease in the protein internal energy.(205) 50-ns per-replica REMD with AMBER99sb/TIP4P-Ew, followed by ab-initio energy calculations on selected poses, concluded that the stability of the water molecules solvating around the dimer mainly determines the relative stability for the different conformations of the Aβ1-42 dimer.(206a)
Recently, REMD simulations using OPLS-AA/TIP3P with 250 ns per replica were performed on Aβ1-42.(207) The Aβ1-42 dimer mostly populates coil/turn (80.4%), and then α-helix and β-strand with 11.1% and 8.4%. The latter values do not match exactly, but are consistent with the CD-derived values: a α-helix content varying from 3% to 10.5% and a β-strand content varying between 12% and 38%.(20,149b) The most β-rich signal is at the C-terminal region. Looking at the networks of inter-chain contacts, the interface of the 1-42 dimer is mainly composed of the C-terminal and CHC regions, as the regions of highest contact probability are C-terminal/C-terminal, CHC/CHC and CHC/C-terminal. The calculated collision cross sections of the three most populated dimer states nicely fit to IM-MS values.(11a) Using a general method to characterize oligomer structures,(206b) there is no evidence of well-formed intermolecular parallel and antiparallel β-sheet configurations. Rather the first 11 N-terminal residues are essentially disordered and the residues 12–17 have a non-negligible probability for α-helix.(207)
4.3 Towards Atomistic Structures For Dimers
As for the monomers, common trends start to emerge from the most recent dimer simulations: (1) the dimerization of Aβ is mainly driven by an hydrophobic collapse through intermolecular contacts involving CHC and the C-terminal region, agreeing with the importance of these regions during aggregation as observed experimentally,(149,124) (2) Aβ1-42 has a larger β-strand propensity than Aβ1-40 at the CHC and C-terminal regions, (3) the free-energy landscape of Aβ1-42 is more complex than that of Aβ1-40, (4) all possible salt-bridges are highly accessible to the solvent,(207) and (5) both alloforms have many structural differences already at the dimer level that can account for their very different oligomerization pathways and toxicity potencies as observed experimentally.(135-136,11,53) For each alloform the results between the simulations still diverge and we can identify qualitative differences in the total and per-residue propensities of secondary structure (Table 2, Fig. 5) and the tertiary/quaternary structures.
Table 2.
Secondary structure contents of Aβ1-40 and Aβ1-42 dimers using enhanced sampling techniques starting from randomly chosen states. Values from simulations are computed using STRIDE.
| Aβ1-40 | Aβ1-42 | |||||||
|---|---|---|---|---|---|---|---|---|
| Ref. | Helix % | β-Strand % | Turn % | Random Coil % | α-Helix % | β-Strand % | Turn % | Coil % |
| 149b† | 10.5 | 38.6 | – | 50.9 | – | – | – | – |
| 20† | 0 | 12 | 28 | 60 | 11 | 3 | 26 | 60 |
| 195 | 1.3 ± 0.1 | 12.6 ± 0.1 | 50.7 ± 0.1 | 35.4 ± 0.1 | 4.4 ± 0.1 | 30.8 ± 0.1 | 32.4 ± 0.1 | 32.4 ± 0.1 |
| 202 | – | – | – | – | 0.9 ± 0.1 | 42.9 ± 0.7 | 33.0 ± 0.4 | 23.2 ± 0.8 |
| 203 | 0.1 ± 0.1 | 13.6 ± 1.6 | 40.6 ± 4.1 | 37.5 ± 5.0 | 0.0 ± 0.0 | 15.7 ± 1.9 | 39.2 ± 3.7 | 39.0 ± 4.9 |
| 204‡ | 0.5 ± 0.1 | 5.5 ± 0.8 | 43.5 ± 3.6 | 46.8 ± 4.1 | 0.9 ± 0.2 | 6.6 ± 0.8 | 40.1 ± 3.2 | 48.0 ± 3.8 |
| 207 | – | – | – | 8.4 ± 0.7 | 11.9 ± 0.6 | 51.2 ± 1.0 | 28.5 ± 0.8 | |
Circular dichroism-derived values using different sample preparations.
MD values starting from the CG DMD structures of Ref. 203 and using OPLS-AA and SPC/E. Similar values are obtained using OPLS-AA and TIP3P.
Figure 5.
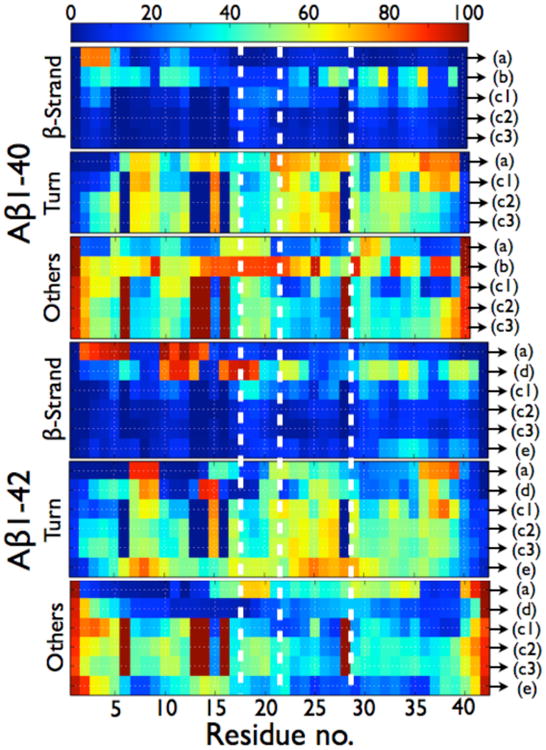
Per residue probabilities of β-strand, turn and others (coil and α-helix) for the Aβ1-40 and Aβ1-42 dimers as computed by different simulation protocols. As seen in Table 2, the total α-helix content amounts to less than 1.3% in Ref. 195, 202, 203 and 204 so others represent coil. In ref. 195, there is a total of 4.4% of α-helix in the dimers of Aβ1-42 and there is a probability of 27% for residues 23-28 to form α-helix. In Ref. 204, there is a total of 8.4% of α-helix in the dimers of Aβ1-42 and there is a probability of 45% for residues 13-17 to form a α-helix. (A) HT-REMD with the CG OPEP model and implicit solvent.(195) (B) united-atom CHARMM19 REMD with the SASA implicit solvent.(199) (C1) CG DMD with implicit solvent.(203) (C2) all-atom MD with OPLS-AA/SPCE.(204) (C3) all-atom MD with OPLS-AA/TIP3P.(204) (D) All-atom MC with implicit solvent.(181a) (E) all-atom REMD with OPLS-AA/TIP3P.(207) All secondary structure probabilities were computed using STRIDE; except for (b) for which the secondary structures are determined using information on the φ and Ψ dihedral angles only, without consideration of the H-bond network. As such, the β-sheet probabilities for (b) must be considered as extended conformations and no turn probability can be determined. The vertical dotted white lines delimit the four regions: N-terminal (residues 1-16), CHC (residues 17-21), loop region in the fibril (residues 22-28), and C-terminal (residues 29-40/42).
4.4. Aggregation of High Order Assemblies
Simulations of higher order assemblies from random states are very challenging as the number of minima scales exponentially with the number of particles. CG and all-atom models coupled to implicit solvent schemes enable long timescales that are not reachable by all-atom explicit solvent MD.
The structural ensemble of the Aβ17-42 trimer was investigated using REMD and the six-bead CG OPEPv3, with 1.2 μs for each replica.(208) This fragment was selected because it covers the β-strand–loop–β-strand in the Aβ1-42 fibril. At equilibrium and 300 K, the trimer adopts globular conformations with 46% of turn, 35% of random coils, 8.7% of helix and 7% of β-strand. Using a RMSD cut-off of 3 Ǻ, 35% of all sampled conformations can be described by two clusters. The first cluster with a population of 19% displays one chain with a β-hairpin spanning residues F17-L34, and the other two chains with a disordered β-α-β-turn-β motif. In this motif, the α-helix spans residues E22-K28, the turn spans G37-G38, and the β-strand signal is rather weak elsewhere. The second cluster (15.4%) is more disordered with an inter-peptide antiparallel β-sheet spanning the CHC region and residues I31-I34, a α-helix spanning A21-N26, and turns at positions G37-G38. The third (13.3%) and fifth (8.2%) clusters are random coil in character, but display inter-molecular antiparallel β-sheets between V36-G38 and V39-V41 (cluster 3) or between I31-G33 and G38-V40 (cluster 5). Overall, the preference for parallel β-sheet is not encoded in Aβ17-42 trimer. This picture is fully different from the REMD results of Aβ10-40 tetramer using CHARMM19/SASA, showing rather amorphous states that are structurally similar to the dimers.(200)
A total of five all-atom MD simulations of 200 ns each with OPLS-AA(164) and GB/SA(209) on a system of 20 Aβ1-42 at a concentration of 0.8 mM starting from various structures and dispersed peptides(210) reveals that the early aggregation pathways at 300 K are very diverse and are dominated by unstructured oligomers characterized by 82% coil, 7.6% β-strand and 10% α-helix, consistent with atomistic REMD of Aβ1-42 dimer in explicit solvent. (207) The conformations are characterized by strong intermolecular interactions involving residues 31-42 and 17-21 and several differences between Aβ1-42 and Aβ1-40 aggregation are observed from the intermolecular contact maps. The oligomer mass distribution, though out of equilibrium within 200 ns, displays a higher population for dimers, tetramers, hexamers, octamers (globular shape), 12-mers and 18-mers (elongated shape), in agreement with experimental results.(135) A maximum flow transition network analysis unveils a complex aggregation process, although key preferential pathways are found: for instance the trimer serves as building block for the hexamer, while the dimer preferentially aggregates into tetramer and octamer.(210)
Finally, CG DMD simulations have been used to study the aggregation of 32 peptides of both Aβ alloforms.(184b,203) The simulations reproduce qualitatively the main features of the oligomer size distribution of Aβ1-40 and Aβ1-42 as measured experimentally. Overall, these oligomers, as for the dimer, form rather amorphous aggregates with a low propensity of β-sheet that are stabilized by intermolecular contacts between the CHC and C-terminal regions. The main differences between Aβ1-42 and Aβ1-40 oligomers are the larger β-strand propensity at the C-terminal region and turn propensity at G37-G38, and the larger flexibility and solvent-exposure of the N-terminal of Aβ1-42. Interestingly, there is no substantial increase of β-strand content from dimers to hexamers and larger oligomers, with all β contents varying between 14% and 22%. We recall that the β-sheet content amounts to 50% in fibrils. DMD further indicates that Aβ1-40 dimers and hexamers have indistinguishable intramolecular contact maps and tertiary structures, whereas the Aβ1-42 transition from dimers to hexamers is accompanied by a partial loss of intramolecular contacts within the CHC.
In summary, higher-resolution experimental data going beyond cross-collision sections (IM-MS), secondary structure content averaged over different oligomer sizes (CD) and hydrodynamic radii (diffusion NMR) as well as standardized simulations will help converge on the most relevant dimer and oligomer structures. While the simulations agree on a few structural aspects, they wildly differ on the equilibrium ensemble due to the difficulties associated with correct sampling and force field accuracy. Comparisons between simulations with different force fields are required as well as multiscale approaches that couple cheap potential for fast sampling and more reliable force fields for refinement of selected poses.(204,208) Another possibility is to run CG simulations for a short time and then switch to atomistic simulations for a few ps, and so on. It may be at this cost that we will obtain a convergent and reliable free energy landscape of dimers and high order oligomers.
5. Aβ Nucleus in Aqueous Solution
5.1. Nucleation and Protein Aggregation
The nucleation of amyloid fibrils is a process associated with the generation of nanoscale fibrils or protofilaments that have the property of irreversible growth.(114) Unless the nanofibril size exceeds the size Nc of the so-called critical nucleus, the nanofibril is more likely to dissolve rather than grow. Only if the number of monomers becomes larger than Nc, the system can grow irreversibly into a macro-scale amyloid fibril. From a thermodynamic point of view, the size of critical nucleus may be defined as a turnover point of the free energy plotted as a function of the number of chains (Fig. 6A).(211)
Figure 6.
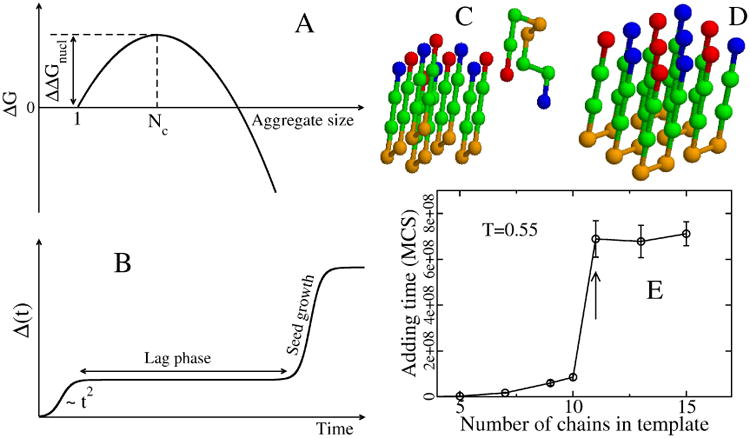
(A) Schematic plot of free energy of the aggregate, relative to the monomer, as a function of the aggregate size. The critical nucleus size corresponds to the peak of ΔG, while ΔΔGnucl is the barrier to nucleation. (B) Time dependence of the fibril mass M(t). Within homogeneous nucleation theory M(t) ∼ t2 at short time scales. The plateau corresponds to the lag phase, whose duration is proportional to exp(ΔΔGnucl/kBT). (C) A typical initial conformation for the (5+1) system in the lattice models with 8-bead sequence +HHPPHH-, where + and − refer to charged residues, while H and P denote hydrophobic and polar residues.(229) (D) The final fibril conformation with the lowest energy. (E) Dependence of the adding time τadd on the number of monomers that belong to the preformed template. Results are averaged over 50 Monte Carlo trajectories. At a concentration of 290 μM, τadd becomes independent of N for Ntemplate larger than 11. Arrow refers to Nc=11.(230)
Protein aggregation might occur through three possible pathways. In homogeneous nucleation,(212) new aggregates are generated at a rate that depends on the concentration of monomers alone and is independent of the concentration of existing fibrils. In the fragmentation process, the rate of generation of new aggregates depends only on the concentration of existing fibrils.(213) Finally, within the secondary nucleation process, the rate depends both on the concentration of the monomer and of existing fibrils.(214) The six-bead CG OPEPv3 simulations revealed that the fibril formation of short linear peptides occurs via the homogeneous nucleation mechanism.(215a) OPEP has been optimized by discriminating native from non-native structures of proteins and successfully folding peptides to their NMR structures.(179,180a) Using an off-lattice model coupled to enhanced sampling, the rate-limiting step has been suggested prior to nucleation to be associated with a change in the width of the fibrillar aggregate of 3.5.(216) Aβ1-42 aggregation was shown to proceed through the secondary nucleation pathway rather than through a classical mechanism of homogeneous primary nucleation using a combination of kinetic studies, and selective radiolabeling experiments.(14)
In homogeneous nucleation, the lag phase (Fig.6B) is weak and at short times the fibril mass M(t) scales with CNc+2 t2, where C is the monomer concentration. Thus, from the concentration dependence of the slope of logM-logt2 one can extract the size of critical nucleus.(217) Nc can also be estimated from the dependence of the lag phase time on protein concentration, as approximately C-Nc+1/2. (218)
Using a simple two-state model and Langevin dynamics, the lag phase and the nucleus size Nc has been shown to vary from 4 to 35 linear peptides depending on the energy difference between the amyloid-competent and amyloid-protected minima and one can generate fibril topologies resembling those observed experimentally, e.g., twist and multifilament composition.(102) Using a lattice model and Monte Carlo (MC) simulations, it was evidenced that the balance between electrostatic and hydrophobic interactions not only modulates the populations of the amyloid-competent monomeric state and the lag phase, but also the topology of the fibrils.(219) Using a more complex onlattice model and dynamic MC, 10 linear peptides of seven amino acids with an alternative hydrophobic and hydrophilic pattern remain stable at low temperature. When the short fibrils are subsequently simulated in a grand canonical ensemble, further growth of the structure is observed, indicating that Nc is at least equal to 9.(220) These Nc values are much higher than that derived by atomistic simulations followed by ab initio calculations where Nc = 3 was found to be sufficient to trigger fibril growth of the GNNQQNY peptide.(221) This low Nc value is likely due to the neglect of conformational entropy. Indeed, based on multi-scale simulations, De Simone showed that a comprehensive description of the flexibility of the all states must also be considered for self-assembly.(222)
By using a mesoscopic model similar to that defined by Caflisch, one can establish a connection between the early nucleation events and the kinetic information available in the later stages of the aggregation process. Using an energy difference between amyloid-competent and non-amyloid-competent states from all-atom simulations and translational and rotational diffusion constants from experiment, the nucleus was estimated as Nc=4 for Aβ1-42 using dynamic MC.(223) This estimate does not agree with quasi-elastic light scattering (QLS) experiment at a Aβ concentration of 1.16 mM or 0.47 mM in 0.1 M HCl where the experimental kinetic data at low pH are reproduced correctly when the number of peptides involved in the critical nucleus of Aβ1-40 is 10.(224) The kinetic data vary, however, with the experimental conditions used, as described below. In contrast, calculating ΔG as a function of the number of monomers with the help of a CG model, Fawzi et al. obtained Nc=10 for Aβ1-40.(225) Based on the experimental(226) and theoretical(227-228) observations that the binding of monomers to a preformed fibril obeys the dock-lock mechanism in which a monomer first docks and then undergoes the structural arrangement necessary to lock onto the template, Li et al. proposed that the time for adding a new monomer, τadd, is expected to become independent of the template size when it exceeds Nc.(219,229) By using a lattice model with 8 beads for Aβ1-40 (Fig. 6C-D), Nc was found to be 11 (Fig. 6E). However the population of the amyloid-competent monomer is found to be on the order of 9% at the folding temperature, a value that is possibly overestimated. Nevertheless, one can show that this approach(230) provides an estimate of Nc consistent with the dependence of the free energy variation on the number of monomers.(211)
By using classical nucleation theory to describe amyloid nucleation, Cabriolu et al. predict the nucleus size and the fibril nucleation rate as a function of the super-saturation of the protein solution. It was found that Nc is 15 for Aβ1-40 at a protein concentration of 120 μM, but variation in the supersaturation of the phase can cause Nc to increase to 50.(231a) This is rather consistent with the experimental estimate of the size of the critical nucleus (Nc > 29) using fluorescence correlation spectroscopy at a supersaturation of 100 μM Aβ1-40 solution.(231b) Note that Auer et al. argued that, in some cases, the dependence of the fibril nucleation rate on the concentration of monomer protein is stepwise and not power-law.(232) If this were the case also for Aβ1-40 and Aβ1-42, a treatment of the nucleation process based only on CNT would be only approximately correct.
5.2. Nucleus of C-terminal Aβ Fragments by Atomistic Simulations
Recent advances in sampling techniques allowed studying by an all atom description the early stages of the aggregation process of the octa-valine-peptide (Val8) and the Aβ35-40 peptide.(233,234) Several studies reported the importance of the residues 35-40 in triggering the aggregation process of the whole Aβ peptide.(235-237) The micro-crystalline structure of this peptide in a amyloid-like configuration reveals antiparallel (AP) β-strands within the sheets and parallel (P) β-sheets.(83a) Aβ35-40 was modeled with AMBER99sb/TIP3P at a concentration of 120 mM and 350 K.(234) At this concentration, the peptide spontaneously forms a compact disordered aggregate and the “rare event” is the formation of an ordered nucleus. The process was studied by bias-exchange metadynamics(238) allowing reconstructing multidimensional free energy landscapes with large barriers and in which a reliable reaction coordinate is unknown.
The free energy landscape of 18 Aβ35-40 peptides displays a funnel with two local minima at its bottom and a third local minimum at a free energy approximately 40 kcal/mol higher (Fig. 7).(234) Basin 1 includes structures that are mainly disordered. Basin 2 contains a much larger fraction of anti-parallel β-sheets (up to 10-12 β-strands). In this basin the β-strands, although common, are not organized in a stable configuration and contacts between different layers form only transiently. Basins 1 and 2 are separated by a relatively low barrier that can be crossed on the time scale of a few tens of ns. Basin 3 includes structures with a high content of anti-parallel β-sheets, which are closely packed on top of each other in a steric zipper. This basin might represent a viable seed for the formation of an amyloid-like ordered aggregate. The structure is similar but not identical to those reported by Sawaya et al.(83a) In particular the layer of β-sheets (top) is shifted by two residues with respect to the layer (below), as compared to the experimental structure. Consequently, inter-sheet contacts involve different side chains, providing a better screening from the solvent. The barrier associated with the disruption of the structure of basin 3 is 16 kcal/mol at 300 K, indicating that it is remarkably stable, at variance with basin 2, and at variance with the experimental structure, that, if used for constructing a model of an aggregate with less than 30 chains, is stable only for a few ns. These results led Baftizadeh et al. to hypothesize that the rate-limiting step for the nucleation of Aβ35-40 is not associated with the formation of AP β-sheets, but with the formation of specific inter-digitation of the side chains observed in basin 3. Indeed, structures with a content of β-sheets comparable to the one observed in basin 3 will become disordered, while structures arranged in a correct steric zipper, are orders of magnitudes more stable. This scenario is qualitatively consistent with the dock-lock mechanism.(230)
Figure 7.
Free energy landscape of 18 Aβ35-40 peptides estimated by atomistic metadynamics simulations with explicit solvent as a function of the number of antiparallel β-sheets and the number of number of antiparallel frontal packing (different β-sheets facing each other).(178a) Basin 1 includes structures that are mainly disordered, with secondary structure elements formed only transiently. Basin 2 contains a much larger fraction of antiparallel β-sheets (up to 10–12 β-strands). The main characteristic of this basin is that the β-strands, although common, are not organized in a stable nucleus. These structures are only metastable, and can convert to the disordered melt in a few tenths of ns. Basin 3 includes instead structures with a specific inter-digitation of the side chains, which are stable at least on the ms time scale.
An unbiased REMD simulation of 16 Aβ37-42 peptides with CHARMM27/TIP3P was performed using 48 replicas, each for 500 ns.(239) Aβ37-42 with opposite charges at the termini is particularly intriguing because it forms amyloid fibrils with AP sheets and P β-strands. Despite frequent β-sheet formation/fragmentation events and 20% of free monomers, the population of 4-5 fully P β-strands, consistent with the fibril structure, is 1-2%, while the population of 4-5 fully AP β-strands is 3-8%. The global free energy minimum consists of structures with 2-3 β-sheets, each of 2-3 mixed AP/P β-strands and a variety of sheet-to-sheet pairing angles surrounded by random coil peptides.(239) The aggregates of low-to-medium free energies consist of mixed P/AP β-strands in agreement with integrative temperature sampling simulations of the same peptide with 16 copies using AMBER99 and GB/SA,(240) CG-REMD of 20 NNQQ and GNNQQNY peptides, and other amyloid-forming peptides.(241-245) This free energy picture is also consistent with atomistic metadynamics of 18 Val8 peptides in explicit solvent,(233) where the maximum free energy involves a transition from P/AP to P orientations when a sufficient number of parallel sheets is formed so that the free energy starts to decrease with fully P β-sheets. The REMD simulation for Aβ37-42 indicates that Nc is > 8, but whether Nc is around 12-16 as estimated for the Val8 peptide(220,233) cannot be determined due to finite-size effects.
5.3. Structures of the Nuclei for Aβ1-40 and Aβ1-42 Peptides
Simulating the formation of the critical nucleus of full length Aβ starting from a disordered aggregate and by describing the system with an atomistic Hamiltonian in explicit water is still not possible with current computational resources. However, the results obtained from the nucleation process of smaller fragments, the MD structural ensemble of the Aβ1-40/42 monomers, and experimental data on small oligomers, allow drawing some hypotheses on the structure of the nucleus of Aβ1-40/42.
What is clear from various experimental studies is that small Aβ1-40/1-42 oligomers are rich in antiparallel β-sheets.(22,63,111,123,124) A solution NMR structure of Aβ1-40 monomer with a dimer protein is also available,(114) where the CHC and C-terminal forms a β-hairpin spanning residues 17-36, with the loop region resistant to proteolysis and the rest of Aβ residues disordered. This specific structure is also observed in three atomistic simulations of Aβ1-40 monomer using three different force fields,(154b,174,178) CG OPEP-REMD of the monomer of Aβ17-36(144) and the trimer of Aβ17-42,(208) albeit with low probabilities. Based on these observations, one can speculate that during aggregation, soluble oligomers may form by stacking β-hairpin-like structures with a loop formed, and loose β-strands at positions 30-35 and 17-20. The next step towards the formation of the fibril, once a critical nucleus is formed, would be the crossing of a high energy barrier associated with a concerted conformational transition in which the β-sheets become parallel and pass from out-of-register to in-register arrangements via chains reptation.(246-250)
This scenario explains why the Aβ peptide with a lactam bridge between residues 23 and 28 does not display any lag phase,(251,252) and the FAD mutations and Pro replacement at positions 21-23 change the time for aggregation.(52,53,253) This scenario is also supported by IM-MS on Aβ1-42 where the first region to aggregate spans residues 20-35, followed by residues 36-42 and then residues 1-19,(56) and the fact that a turn-nucleating D-ProGly motif in the V24-N27 region largely accelerates fibril formation of Aβ1-40.(146) Finally, there is strong theoretical evidence on several amyloid peptides that β-hairpins formed in the monomer provide a perfect seed for further growth of the aggregates and reduce lag phases for fibril formation. This is supported by simulations on Aβ25-35 peptides,(254) prion fragment PrP106-122,(255) β2-microglobulin 20-41 and 83-99 peptides,(256,257) and human islet amyloid polypeptide hIAPP1-37.(258,259) For instance, simulations on Aβ25-35 showed that, although the monomer preferentially forms a β-hairpin, a transition from compact β-hairpin conformations to extended β-strand structures occurs between dimer and trimer.(254)
An alternative Aβ nucleus is based on the atomistic metadynamics simulations of Val8,(233) OPEP REMD simulations of GNNQQNY and Aβ16-22,(61) PROFASI MC simulations of Aβ16-22 and Aβ25-35,(260) and enhanced sampling simulations of Aβ37-42 peptides(239,240) where a mixing of AP/P β-strands dominate in the early low-order oligomers. In this case, the maximum free energy involves a transition from mixed P/AP to fully P orientations, which occurs when a sufficient number of P β-strands is formed so that the free energy starts to decrease to a minimum. Clearly the transition time varies with the frequency of fragmentation events dependent on the concentration. Aggregation could also start in the C-terminal region or at the CHC. Initiation in the C-terminal region is supported by the two IM-MS experiments,(56,137b) and the propensity of Aβ35-42 fibrils to display either P or AP β-strands within the sheets.(261a) Initial aggregation at CHC is supported by mutagenesis where Pro replacement in any residue of the region 17-21 leads to the loss of Aβ fibril formation,(253) and replacement of residues F19 and F20 by Leu or Ile does not prevent aggregation, but enhances amyloid formation.(261b)
Lastly, it is possible that fully extended metastable states with a population of 7% as described in metadynamics of Aβ1-40 monomer(178) and REMD of Aβ1-28 monomer (see Section 10) in explicit solvent represent seeds for polymerization. Overall, several nuclei could co-exist with populations depending on the experimental conditions such as T, pH, agitation, etc.
6. Interactions of Aβ Peptides with Membranes
AD pathology is linked to interactions between various types of assemblies of Aβ peptides (e.g., oligomers, channels and fibrils) and neural cell membranes, the membrane integrity being directly affected. Several recent experimental and theoretical studies have been aimed at unveiling the details of the specific molecular interactions between Aβ peptides and lipid membranes, providing a wealth of information. Key findings and related hypotheses are:
Membranes become more permeable to ions in the presence of Aβ peptides. In contrast, monomers or fully developed fibrils have little or no effect on membrane permeability.(262,263) Lipid vesicles may also become more permeable in the presence of attached growing (i.e., not mature) fibrils.(264) Mature fibrils can also affect the structure of membranes to some extent,(265) but the effect is thought to be much less dramatic. Moreover, if the Aβ peptides are modified such that amyloid fibril formation is accelerated and the formation of small soluble oligomers is decreased, both their toxicity and their propensity for binding to lipid membranes are attenuated.(266)
Several different mechanisms that could lead to membrane leakage(267,268)have been proposed and discussed: (i) The simple mechanical “carpeting” byfibrillar peptide aggregates on one leaflet of the membrane surface, whichdestabilizes the membrane by creating an asymmetric pressure between theleaflets. (ii) The detergent effect - a result of the surfactant-like propertiesassociated with the amphiphilic nature of Aβ, which causes the removal oflipids from the membrane, leading subsequently to thinning or evenoccurrence of holes in membranes. (iii) The formation of toroid-like Aβ poresand membrane channels. It is well accepted that neuronal death in AD isrelated to disturbances in Ca2+ homeostasis. The formation of Ca2+ channelsin lipid bilayers was directly observed in experiments where Aβ1-40 peptideswere incorporated into planar phosphatidylserine bilayers. A linear current–voltage relationship in symmetrical solutions was recorded and, using AFM,an 8 to 12 nm doughnut-shaped structure with a 1 to 2 nm internal pore cavitythat protrudes approximately 1 nm above the embedded bilayer surface wasrevealed.(263) These channels are composed of 3, 4, 5 or 6 subunits with themost common structures being those with four or five subunits (Fig. 8).(269)
Aβ aggregation can be significantly accelerated by the presence ofmembranes.(270-272) An important factor favouring membrane binding is the presence of electrostatic attractions between negatively charged lipid head-groups and peptides, which even persists in solutions with high ionic strength, where electrostatic interactions are almost fully screened.(270) It has been suggested that electrostatics drives the initial binding of Aβ peptides while preventing a deeper insertion into the membrane.(273) Using two novel mouse models expressing membrane-anchored or non-anchored versions of the human Aβ1-42 peptide, membrane-anchored Aβ accelerates amyloid formation. This strongly suggests that Aβ-membrane interactions play a pivotal role in early-onset AD and exacerbate toxicity in mice.(274)
Another factor proposed to favour membrane binding is the propensity of Aβ to form weakly stable α-helical configurations that anchor the peptide to the membrane.(275,276) Several effects have been discussed that might promote the aggregation of membrane-bound peptides.(277,278) Peptides that bind to membranes are oriented and accumulated, and they have a reduced diffusion constant. Furthermore, membranes may induce conformational changes in the binding peptides themselves (i.e., may lower their unfolding activation barriers), and they may even serve as a template for fibril formation.(270)
Figure 8.
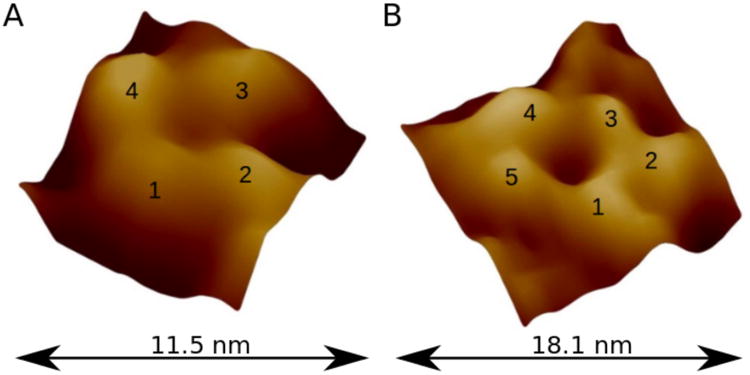
High-resolution AFM images of individual Aβ1-42 channels. They are most often observed as (A) tetrameric, or (B) pentameric subunits assemblies. Other types (e.g., hexameric) of pore-like Aβ structures were also reported.(269)
Many techniques are used to demonstrate the membrane-mediated effect on Aβ aggregation, including imaging methods such as AFM and TEM,(263,279) binding to amyloid-specific dyes, such as Thioflavin T, and techniques monitoring changes in protein size, i.e., gel electrophoresis, SEC, and DLS. AFM is the main technique to demonstrate amyloid pore formation in membranes.(262,267) CD and attenuated total reflection Fourier transform infrared (ATR-FTIR) spectroscopy monitors secondary structure changes upon Aβ-membrane interactions. Electrophysiological techniques allow studying amyloid-enabled membrane leakage.(263,270) Total internal reflection fluorescence microscopy (TIRFM) enables the visualization of individual Aβ species on membranes and the characterization of their oligomeric states, all at biologically relevant nanomolar concentrations.(58b,280,281)
The numerous recent experimental studies examining Aβ-membrane interactions have led to key, central questions that remain open, such as: (i) What is the main pathway for the α-to-β molecular transition accompanying the aggregation of membrane-bound Aβ? Though some studies have suggested an alpha helical conformation for the membrane-attached Aβ, there is also evidence that the oligomers that attach the membrane are beta-sheet rich.(281) (ii) What is the primary molecular reason behind the toxicity of aggregated Aβ peptides, and (iii) Why are amyloid oligomers the most toxic species?(149b,282) Molecular simulations may help answer these questions at greater detail as they allow the investigation of Aβ-membrane interactions at atomistic level.
The direct simulation of entire peptide aggregation processes using atomistic models remains a challenge by speed limitations of today's computers.(210) All-atom MD simulations can give insight into the early stages of peptide adsorption and peptide-membrane interactions. Davis and Berkowitz have used REMD simulations and umbrella sampling to study the adsorption of Aβ1-42 on bilayers(283,284) and possible mechanisms of dimerization, focusing in particular on the role of electrostatic interactions.(285) They found that lipid-protein interactions dominate the behaviour of Aβ on dipalmitoyl-phosphatidylcholine (DPPC) bilayers, whereas protein-protein interactions prevail on negatively charged 2-dioleoyl-sn-glycero-3-phospho-l-serine (DOPS) bilayers. Independent simulation studies arrived at the conclusion that the adsorption of Aβ on membranes follows a two-step mechanism. First, electrostatic interactions between charged residues and phosphate lipid head-groups drive the initial binding of Aβ to the membrane surface. Once Aβ is anchored to the membrane, hydrophobic interactions involving residues 17-21 and the C-terminal region from residue 30 onwards gain in importance, stabilizing membrane-bound Aβ.(272,283,284,286,287) Upon adsorption on the lipid bilayer, the Aβ peptides appear to preferentially adopt a structure involving two helices: a more flexible α-helix in the N-terminal half of Aβ, and a second one, with a higher conformational stability, involving residues 30-36 (Fig. 9A). In addition, binding to the membrane seems to induce the formation of the intrapeptide D23-K28 salt bridge in Aβ. This conformational propensity was determined from all-atom REMD with both implicit(288) and explicit(286) membranes, and is in agreement with NMR studies.(275,276) The Aβ peptides are localized at the interface between membrane and water, with the C-terminal helix penetrating into the membrane core while the polar N-terminal region interacts mainly with the bilayer surface.
Figure 9.
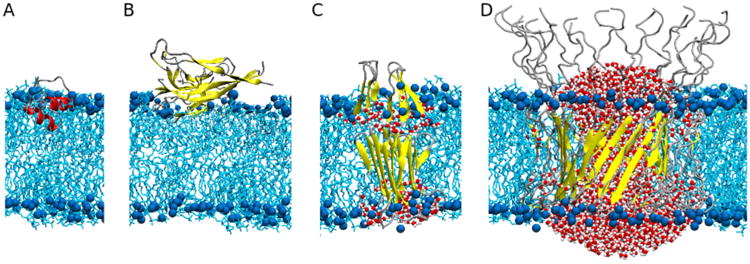
Various models of membrane-bound Aβ studied by MD simulations. (A) Monomeric Aβ1-40 is localized as a helical structure at the interface between membrane and solvent with the C-terminal helix inserting into the membrane.(288) (B) A fibrillar Aβ1-40 oligomer interacting with the membrane on the surface causes both a loss of β structure and a thinning of the membrane.(273) (C) The membrane-inserted β -sheet tetramer composed of Aβ1-42 allows water to permeate through the membrane(297) and can further assemble into a pore structure.(296) (D) Aβ1-42 barrel structure leading to pore formation in the membrane, which allows water and ion transport across the membrane.(293,294) Note that structures shown here are simulation-based models, as experimental high-resolution structures of membrane-bound Aβ peptides have yet to be resolved. The structural and mechanistic relationships between the simulation models are unknown. Aβ peptides are represented according to their local secondary structure: helices (red), β-sheets (yellow), and turns (silver). The bilayer phosphorus atoms (blue) are shown as van der Waals spheres and the lipid tails as licorice (cyan). For clarity, only water molecules inside the membrane are shown (van der Waals spheres).
Tofoleanu and Buchete probed the molecular interactions between preformed fibrillar Aβ oligomers and lipid bilayers, in the presence of explicit water molecules, using atomistic MD.(273) They studied the adsorption of models of Aβ1-40 dimer fibrillar oligomers on phosphatidylethanolamine (POPE) lipid membranes (composing about a quarter of all phospholipids in living cells) under different relative orientations between membrane and fibrils. They investigated the relative contributions of different structural elements and interaction factors to the dynamics and stability of Aβ protofilament segments near membranes, and simulated the first steps in the mechanism of fibril– membrane interaction. The Aβ1-40 fibril structures used here were constructed based on atomistic constraints from ss-NMR(28,67,79) and refined by MD simulations.(289,290) They identified the electrostatic interactions between Aβ charged side chains and lipid head-groups to be the main force driving conformational transitions, together with hydrogen bonds formed between specific residues in the Aβ protofilaments and the lipid head-groups. These interactions facilitate synergistically the insertion of the hydrophobic C-terminal segment of Aβ peptides through the lipid head-groups, leading both to a loss of the β-sheet-rich fibril structure, and to local membrane-thinning effects (Fig.9B).(273) Additional computational studies showed that the chemical composition of the lipid head-groups can control in a specific manner both the type and magnitude of interactions between Aβ protofilaments and membranes.(291) These findings suggest a polymorphic structural character of amyloid ion channels embedded in lipid bilayers.(292) Atomistic computational models suggest that putative amyloid channel structures could also be stabilized by inter-peptide hydrogen bonds (leading to the formation of long-range ordered β-strands) though Aβ channels may also present a significant helical content in peptide regions (e.g., the N- and C-peptide termini) that are subject to direct interactions with lipids rather than with neighbouring Aβ peptides. Before experimental high-resolution structures of Aβ amyloid channels become available, various models of Aβ pore-like structures traversing lipid bilayers were constructed, including helical, β-sheet and combinations of these two secondary structures.(292)
Nussinov et al. developed a model for Aβ channel structures, which break into mobile β-sheet subunits and enable toxic ionic flux (Fig.9D).(293-295) The subunits are preferentially tetramers or hexamers, which could serve as building blocks for the transmembrane pores reported from AFM studies (Fig. 8).(295) Strodel et al. also proposed Aβ pore models composed of tetramer to hexamer β-sheet subunits, which emerged from a global optimization study of transmembrane Aβ (Fig.9C).(296) Large-scale MD of these β-sheet subunits revealed that the tetramers themselves are sufficient to cause membrane damage while transmembrane Aβ monomers do not perturb the membrane sufficiently to make them permeable.(297) The membrane damaging effect of the β-sheet tetramers is further enhanced by Aβ mutations (e.g., the “Arctic” E22G mutation), and may explain the higher toxicity of these mutants compared to wild-type Aβ peptides.(297,298) Also, for helical Aβ peptides in their monomeric states, the membrane-inserted stability was tested for several pure and mixed model membranes.(297,299-301) Different insertion depths were considered, with either K28, V24, D23 or K16 located at the membrane-water interface. The most stable transmembrane α-helix was observed for Aβ peptides positioned at residue D23 at the interface of a DPPC membrane (297) while unsaturated lipids or smaller insertion depths cause a loss of α-helix, in some cases in favour of β-strands. The first stages of Aβ self-assembly inside mixed bilayers were recently tested by MD, revealing the formation of a β-sheet between two peptides in the presence of cholesterol(302) or ganglioside GM1.(303,304a-b) In the absence of GM1, no β-sheet formation is observed as GM1 mediates the initial interactions between Aβ peptides leading to oligomerization. These computational findings are in agreement with the observation that lipid rafts (i.e., cholesterol and sphingolipids enriched highly ordered membrane micro-domains) are potential modulators of Aβ production, aggregation and toxicity.(305) Finally, one MD simulation with umbrella sampling has recently focused on the effect of attached Aβ1-42 monomers on the free energy of membrane pores modelled with DPPC lipids. They found that the attached Aβ1-42 monomers reduce the free energy of membranes pores by 2kcal/mol, increase the lifetime of pores and enlarge the pore density.(306) Most of the simulations studying membrane interactions with Aβ are summarized in Table 3.
Table 3.
Summary of molecular simulation studies focused on interactions of Aβ peptides with lipid membranes. DMPC stands for dimyristoylphosphatidyl-choline, POPC for palmitoyloleoylphosphatidylcholine, POPS for 1- palmitoyl, 2-oleoyl-sn-glycero-3-phosphoserine, and POPG for palmitoyloleoyl-phosphatidylglycerol.
| References | Aβ system | Type of membrane | Simulation | Key notes |
|---|---|---|---|---|
| 288 | Aβ40 monomer | Implicit IMM1 | All-atom REMD | Helical Aβ structure at membrane-water interface. |
| 299 | Aβ40 monomer | DPPC | All-atom MD | Helical Aβ structure in bilayer with tendency to exit the membrane and localize at membrane-water interface. |
| 286 | Aβ(10-40) monomer | DMPC | All-atom REMD | Helical Aβ structure at membrane-water interface. |
| 283-285 | Aβ42 monomer and dimer | DPPC and DOPS | All-atom REMD and umbrella sampling MD | Binding of Aβ to membranes. Dimerization of membrane-bound Aβ. |
| 273,291,292 | Aβ40 protofilament | POPE and POPC | All-atom MD | Interactions between Aβ fibrillar oligomers and membranes influence of lipid composition, structural effects. |
| 300,301 | Aβ40 monomer | DPPC, POPC, POPS, POPC/POPS, rafts | All-atom MD | Stability of membrane-inserted Aβ. Effect of insertion depth and membrane composition |
| 302-304 | Aβ42 dimer | Mixed bilayers with cholesterol and/or GM1 mimicking rafts | All-atom MD | Effect of rafts on membrane-binding and dimerization of Aβ. |
| 296-298 | Aβ42 monomer and oligomers | Implicit IMM1 POPC, POPG, DPPC | All-atom Monte Carlo and MD | Structure prediction for transmembrane Aβ. Testing the stability and membrane effects of the resulting β-sheet structures. Effect of mutations. |
| 293-295 | Aβ barrels | DOPC, POPC, POPG | All-atom MD | Models for Aβ channel structures. Water and ion flux across membranes through Aβ pores. Effect of Aβ mutations. |
| 311 | Aβ40 & Aβ42 monomers | 3 bead model for Lipids | Coarse-grained MD | Aβ adsorption and aggregation on small vesicles |
For studying the membrane-mediated aggregation of Aβ peptides on larger time scale, one must resort to coarse-grained simulations. A number of models have been proposed.(307-311) One may distinguish between specific models, where amyloids are represented by a specific sequence,(308a) and phenomenological models, which are designed to reproduce the aggregation process in general.(309) One of the latter, the two-state mesoscopic model for proteins(310) has been combined with a simple solvent-free three-bead model for lipids by Friedman et al to study peptide adsorption and aggregation on small vesicles and peptide-induced membrane damage.(311) In agreement with the experimental picture, this Langevin dynamics study found that vesicle leakage occurs primarily due to transient defects during filament growth; mature fibrils did not damage the vesicles. Studying the fibril degradation in the presence of vesicles, the simulation showed that it results in protofibrillar intermediates whose structure differs from those formed upon aggregation or upon disaggregation without lipid vesicles.(311)
All these results suggest that, in spite of the high complexity of the systems including lipid membranes, computational studies are becoming increasingly more feasible due both to accelerated hardware and methodological developments and may guide new experiments that could test more efficiently the assembly and structural features of membrane-formed amyloid channels. Future simulations should be able to unravel how membranes can facilitate the aggregation of Aβ peptides and modulate the formation of oligomers, fibrils and channels, while novel experiments are still needed to provide high-resolution structures of membrane-bound Aβ aggregates.
7. Interactions of Aβ Peptides with Metal Ions
7.1. Relevance of the Interaction of Metal Ions (Cu, Zn and Fe) with Aβ
The accumulation of zinc, copper and iron ions in amyloid plaques, a hallmark of AD, is well documented.(312)) Interestingly, human plaques showed higher metal content than plaques in AD model mice.(313) Raman studies further suggested that Zn(II) and Cu(II) are bound directly to Aβ, the main constituent of amyloid plaques. Amyloid plaques are also enriched in iron, mainly present as particles containing Fe(III) supposed to originate from ferritin. Whether ionic, mononuclear iron is bound to Aβ is not clear.(314)
Although there is clear evidence for Zn and Cu interactions with Aβ in amyloid plaques, it is not known at which time point or which aggregation state these metal ions bind to Aβ in vivo. However, it seems that under normal physiological conditions Zn(II) and Cu(I/II) do not bind to monomeric Aβ, and the hypothesis is that only upon metal and/or Aβ deregulation the formation of metal-Aβ complexes can occur.(315) This is in agreement with the finding that amyloid plaques are formed around synapses in which high concentrations of Zn and Cu are released in the synaptic cleft. These released metal ions are a peculiar pool. Their ligands are not known, but these metals are readily accessible for chelation by ligands with moderate affinity. This is in contrast to classical metalloproteins, where metal ions are strongly bound and often buried in the proteins. This suggests that these synaptic metal pools are kinetically labile and thermodynamically moderate stable.(316)
Numerous other studies, from in vitro to in vivo, report evidence of a connection between metal metabolism, Aβ metabolism and AD. This includes, for instance, reports on a Cu pool in the blood as AD marker, the mutual regulating effects of APP and metal ions, the association of a single nucleotide polymorphism of the Cu transporter ATPase (ATP7B) with sporadic AD and the decrease of amyloid plaque load in AD mice after disruption of the Zn transporter ZnT-3.(317-318)
Taken together, these data suggest that metal ions like Zn and Cu can bind to Aβ under AD conditions and could have two important impacts directly linked to AD: modulation of the aggregation behavior and, for Cu, catalysis of toxic reactive oxygen species (ROS) production.
7.2. Structure of the Cu, Zn and Fe-binding Sites of Aβ
The metal-binding sites of Cu(I/II), Zn(II) and Fe(II) within the Aβ sequence have been studied during the last two decades. Complexes of Fe(III) with Aβ at neutral pH are not stable enough to inhibit precipitation of Fe(III) as iron oxide.(314) This suggests that the interaction of Fe(III) with Aβ is only relevant in a ternary complex with another biomolecule or with particles containing Fe(III).There is a general agreement that the main binding sites for Cu(I/II), Zn(II) and Fe(II) are located in the first 16 amino acids of the peptide, at least for the monomeric form.(314) The truncated Aβ1-16 peptide containing the metal-binding domain, which is more soluble and hence more appropriate for studies in solution and does not form amyloids, is often used instead of the full-length peptide. Indeed, several spectroscopic studies showed that the metal binding of Aβ1-16 is very similar to the full-length Aβ1-40/42. However, as small changes can have a large impact on aggregation, further experimental and theoretical studies well connected to experiments are needed to elucidate the detailed structures of truncated and full-length Aβ.
The current knowledge on the metal-binding sites of the most relevant metal ions is summarized in Fig.10.(314,319-322) They have been identified for the soluble, monomeric complexes, but might be different in the aggregated Aβ. A general feature for all these metal-binding complexes is that they are very flexible and dynamic. Monomeric Aβ remains an intrinsically disordered peptide upon metal binding, as fast ligand exchange reactions and equilibrium between different binding sites exist leading to a polymorphism in the coordination environment.
Figure 10.
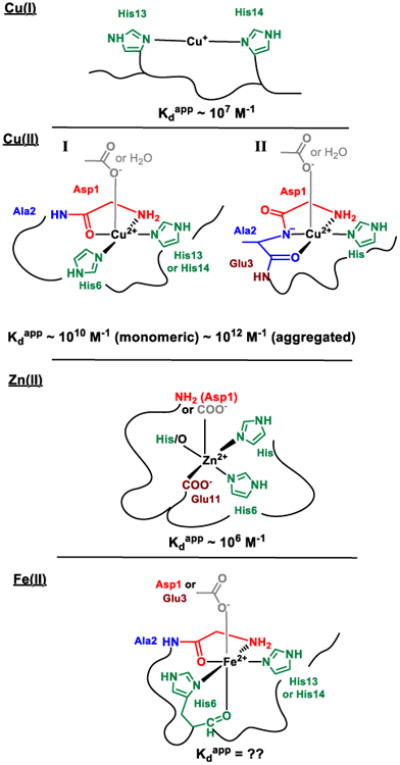
Models of the coordination sphere of different monomeric metal-Aβ complexes. Kdapp stands for apparent dissociation constant (i.e. dissociation constant at pH 7.4 in 0.1M salt and in the absence of buffer)
There is consensus about the Cu(I)-binding site within Aβ at pH 6-8, that consist of a linear site with two His as ligands. The main site is with H13 and H14 as ligands (Fig.10 top), but this site is in fast exchange (< sec) with linear Cu(I) bound to H6 and H13/H14.(314) Cu(II)-Aβ around neutral pH exists with two different types of coordination spheres, called component I and II (Fig.10) which are in fast equilibrium (< sec). Moreover, in each of these two components, further exchange between the same type of ligand occurs (e.g. between H13 and H14). Recent MD simulations confirmed this polymorphism of Cu(II)-Aβ.(323) There was a long debate about the coordination sphere of component I, but a consensus has been reached following the application of site specific isotope labelling and advanced EPR and other experimental and theoretical methods.(314,319,322) Regarding component II there are still different models proposed, but most results support the structure shown in Fig.10. Drew described however an alternative structure.(319)
Mutations of amino acids in Aβ (like A2V, H6R, D7N), even when they concern residues not directly involved in metal-binding, can have a large impact on the coordination site via second sphere interactions.(322) This is exemplified by the comparison of Cu(II)-binding to human with murine Aβ, in which the mutation responsible for a dramatic change of the major coordination is R5G and leads to the following major coordination sphere: D1 (via NH2, O=C) and H6 (N imidazole and N amide). The impact of mutants outside the metal-binding domain remains to be established.(325)
Less is known about the binding sites in aggregated Aβ (oligomers, amyloids etc). Most studies suggest the same type of residues coordinating Cu(II) for the soluble Aβ, but these might come from two different Aβ molecules. A very recent study based on advanced EPR methods of fibrillar Cu(II)-Aβ1-40, confirmed the same equatorial coordination sphere of component I as in monomeric Cu(II)-Aβ1-16.(326) The results propose that the Cu(II) sites along the fibrils alternate between the two sub-components Ia (D1, H6, H13) and Ib (D1, H6, H14). This would be in contrast to the soluble form, in which a fast (< sec) equilibrium between subcomponents Ia and Ib exists. Cu(II)-binding to amyloid Aβ1-40 fibrils was also studied by ss-NMR coupled to MD.(327) In general, the results agreed with the EPR measurements, because H13 and H14 resonances were broadened upon addition of Cu(II) (H6 and D1 were not addressed). Other residues were also affected in particular the C-terminal COO- and Glu side chains. This might be explained by axial binding to Cu(II), as it was also suggested for COO- groups in monomeric Cu(II)-Aβ1-16.(328)
In contrast to Cu, the coordination spheres of Zn(II) and Fe(II) have been less investigated. Current favoured models are given in Fig.10. The models show the main ligands involved, but due to their flexibility a polymorphism in the coordination can be expected.(329) Indeed, recent MD simulations on Zn(II)-Aβ reported that the COO- from either D1 or E7 can bind, but in two different conformations and with a higher population for E7. The binding of either D1 or E7 had an impact on the preferred partner (i.e. D22 or E23) of the salt-bridge with K28.(330) Due to the insolubility of Fe(III) even in the presence of Aβ no well-defined species for structural studies was so far obtained.(320,331)
The apparent binding constants of metal ions to Aβ have been determined by several methods (Fig. 10 and Refs. 331,332). After some discussions in the literature, an apparent dissociation constant Kdapp (pH 7.4 in the absence of buffer) of Cu(II) from monomeric Aβ on the order of 10-10 M is now relatively consensual.(332) Interestingly, the affinity for aggregated Aβ is about 2 orders of magnitude higher.(333) A consensual value for Kdapp for Zn around 1-10 μM is reported, with an up to 10 fold higher affinity for aggregated Aβ.(321) No values are reported for Fe(II/III). The binding affinity of Cu(I) to Aβ is still under debate, with values for Kdapp from 10-7 to 10-10 M.(334,335) In general, the affinities obtained for Cu(I/II)- and Zn(II)-Aβ are several magnitudes below the affinities of Cu- and Zn-proteins with a defined 3D structure. This is in line with the entropic penalty of metal binding to a disordered peptide. Moreover, this suggests that metals might only be able to bind Aβ at the Zn- and Cu-rich synapses and under Alzheimer conditions where metal deregulation occurs.
7.3. Role of Metal Ions in the Aggregation of Aβ
The effects of metal ions on Aβ aggregation, i.e. in terms of kinetics, thermodynamics, structures formed and their populations, are not clear and are condition-dependent.(320,326,336a) There are two effects on which there is a wide agreement in the literature: i) metal ions (mainly Cu(II) and Zn(II) are studied) modulate the aggregation and ii) the effects are metal-specific, e.g. Cu(II) affects Aβ differently from Zn(II). We did recently a survey of the literature about the effects of Cu(II) and Zn(II) on the aggregation of Aβ.(320,325) Several tendencies could be identified: i) Zn(II) and Cu(II) at high μM concentrations and/or in large superstoichiometric ratios compared to Aβ promote amorphous type aggregates (precipitation) over the ordered formation of fibrillar amyloids. ii) Metal ions affect the kinetics of Aβ aggregation, with the most significant impact on the nucleation phase; and iii) Cu(II) and Zn(II) affect the population and/or the type of aggregation intermediates formed.
At least two parameters might be important in the influence of Zn(II) and Cu(II) binding on Aβ aggregation: changes in the 3D structure(s) and in the overall charge of the Aβ complexes. At neutral pH Aβ has an overall charge of about -3: divalent metal ion-binding hence yields a more neutral charge (about -2 with Cu(I/II)-Aβ and -1 to -2 with Zn(II)-Aβ at pH 7.4: note that one has not only to consider the charge of the metal ion, but also the replacement of protons by metal-binding) and a faster aggregation is expected. The fact, that the aggregation behavior is metal-dependent, shows clearly that the structural changes upon metal-binding (which are also metal specific, see Fig.10) play an important role as well.
In a more general way, metal ions can promote amorphous aggregates and amyloid-type aggregates in a condition dependent way. It seems that the system proneness to aggregation is crucial to determine which type of aggregates are formed (amorphous vs. amyloid). If conditions are such that aggregation of Aβ is already fast (as with high concentrations of Aβ, Aβ1-42, pH close to pI, etc), Cu(II) or Zn(II)-binding (in particular at high concentrations or ratios) accelerates aggregation and favors amorphous aggregates. Aggregation is too fast, however, to properly align the Aβ peptides into an ordered β-sheet structure like in amyloids. For a system with a low propensity to aggregate (as with higher pH, low concentrations of Aβ, Aβ1-40, etc.), metal ions (in particular at lower concentrations or ratios) favor formation of amyloid-type aggregates. Using MD simulations, Miller et al. showed that Zn ions promote Aβ aggregation via a population shift of polymorphic states.(336b)
It is not well known how much the structure of amyloid fibrils differs from metal-free Aβ peptides to metal-Aβ peptides for Cu(II, I) and Fe(II, III). For Cu(II), addition of this metal to preformed amyloid fibrils of Aβ does not change the peptide structure, as monitored by ss-NMR.(327) The conformation of the Zn(II)-attached fibrils has also been investigated by ss-NMR.336c The data show the absence of the D23-K28 salt-bridge, but the presence of the F19-L34 contact. Also, Zn(II) tends to accelerate the precipitation of the oligomers without changing the overall solubility of the peptide, which may help explain why Zn(II) at low concentrations lowers Aβ toxicity.336d
7.4. Cu-Aβ as a Catalyst for the Production of Reactive Oxygen Species
A large body of evidence suggests that oxidative stress is implicated in AD, but it is not clear if it is a primary cause or a consequence.(337) The production of reactive oxygen species (ROS) is a major contributor to oxidative stress, and indeed AD affected tissue shows signs of enhanced ROS production, in particular around the amyloid plaques. Cu is well known to be able to catalyze the production of ROS and hence it is possible that the complex Cu-Aβ is implicated in the production of ROS in AD.(331)
Indeed, in vitro experiments reported that Cu-Aβ is able to catalyze the production of H2O2 and HO° in the presence of a biologically relevant reducing agent (like ascorbate) and dioxygen (Fig.11 top). There is an ongoing discussion if Aβ is an antioxidant or prooxidant. Aβ is considered as an antioxidant based on the fact that Cu in buffer catalyses ROS production more efficiently than Cu-Aβ. However, “free” Cu concentrations are extremely low in biology as Cu metabolism is tightly controlled. Hence, an antioxidant activity of Aβ compared to “free” copper might be only relevant under particular conditions where “free” Cu reaches higher concentrations. It seems not evident that this ever occurs, when taking into account the presence of high concentrations of potential ligands (like glutathione, histidine, cysteine etc.).
Figure 11.
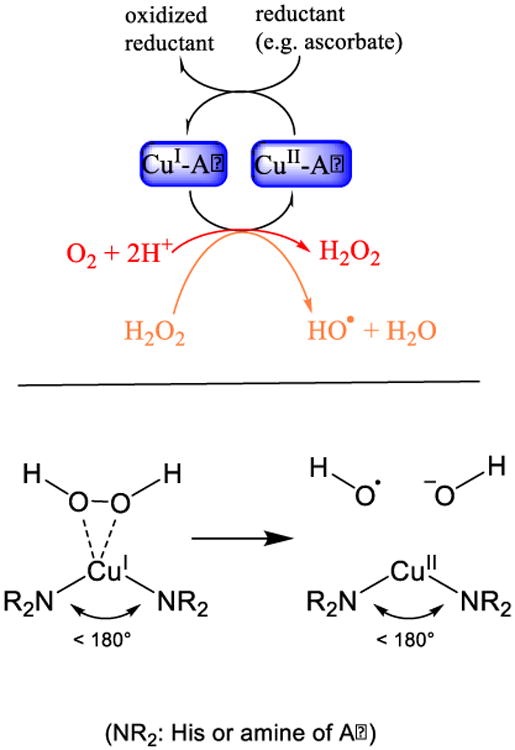
Catalytic role of Cu-Aβ in the production of reactive oxygen species. Top: Cu-Aβ is able to catalyze the production of H2O2 and HO° in the presence of a reducing agent and dioxygen. Bottom: The catalytic active state capable to catalyze the Fenton type reaction. Active are only the conformations with a diagonal ligation by the peptide remote from a angle of 180° and in which the H2O2 binds to Cu(I) side on.
So in the framework of an imbalance of Cu, it seems more relevant to compare the efficiency of Cu-Aβ with the Cu-pool from which Aβ obtains Cu in AD instead of “free” Cu. However the identity of this Cu-pool is not known. In order to address this, the ROS efficiency of Cu-Aβ was compared with several biological relevant Cu-peptide or Cu-protein complexes. Generally, Cu-Aβ was quite active, and hence one can conclude that that Cu-Aβ has the potential to contribute to oxidative stress in AD. This is supported by the finding that oligomeric Cu-Aβ aggregates have a higher ROS production activity than monomeric Cu-Aβ, in line with the higher toxicity of oligomeric Aβ.(331)
Electrochemistry of Cu-Aβ suggested that the reduction and oxidation does not occur directly between the two ground states (most populated), shown in Fig.10.(314) The reorganization energy is too important as can be seen from the very different structures of Cu(I) and Cu(II)-Aβ. Instead a low populated (0.1%), intermediate state exists in equilibrium with the ground states, but only this intermediate state undergoes a rapid redox reaction. Thus this state can be considered as a kind of transient entatic state. This suggests that this low populated, intermediate state is responsible for all the redox activity and hence such a type of state might also be responsible for the reactivity with dioxygen and a biological reducing agent to produce ROS. Recent advances were made in the understanding of this intermediate, “hot” state.(338) First, the ligands of this intermediate state were assessed and H13, H14 and D1 were identified. Interestingly H6 is not involved. This shows that the intermediate redox competent state is different from the two ground states.
Further computational studies using MD (Car–Parrinello) and DFT suggested that the highly reactive Cu(I)-Aβ state consists of N-Cu(I)-N coordination with an angle far from 180°, and high water crowding at the open side (Fig.11 bottom).(339) This allows side on entrance of H2O2 and its cleavage to form a hydroxyl radical. Interestingly, a reactive Cu(I)-Aβ state was more easily originated when starting with a dimer model (Cu(II)-Aβ2) compared to a monomer (Cu(II)-Aβ), likely due to structural constrains of the peptide. This in line with the higher ROS production reactivity of Cu(II)-Aβ oligomers.
7.5. Metal-based therapeutics
As above discussed, AD involves a mismetabolism of Cu and Zn. This mismetabolism is rather an imbalance than a general overload or lack. The imbalance tends toward an extracellular increase and an intracellular decrease of Zn and Cu. Moreover, the extracellular Cu(II) is prone to catalyze ROS.(340) Based on that, therapeutical strategies have been developed in order to use compounds that bind the misplaced Zn and/or Cu pool and diminish the Cu prooxidant activity (i.e. redox silence it that it does not catalyze ROS production).(312,341) This can be achieved by moderate affinity ligands. Such drugs should have the following properties i) not be toxic: ii) cross the blood brain barrier iii) have the right affinity, higher than Aβ but lower than essential metalloproteins, and iv) the complex of Cu-ligand should not itself produce ROS.(342)
Moreover, it seems also more advantageous not only to bind the misplaced pool and redox silence it, but to transport it back into the cell.(343) Thus a fifth property can be added: v) relocation of the metals, from extracellular to intracellular. The free ligand as well as its metal complex should be able to cross the membrane. A driving force is needed to release the metal intracellularly. In the case of Cu with two chemical ligands gtsm (glyoxal-bis(N(4)-methyl thiosemicarbazone) and PBT2 (hydroxyquinoline), the driving force is the reduction of Cu(II) to Cu(I) and subsequent decomplexation.(343) These properties are referred to as ionophores or chaperones. Several compounds have been synthesized along these lines and tested in vitro or in AD models as inhibitors of Aβ aggregation or toxicity (for some examples see Fig.12). The compound PBT2 went to a phase IIa clinical trial and showed improvements in two executive function component tests in a battery of neuropsychological tests.(312,315) These effects have been attributed the ability of PBT2 to facilitate intracellular copper uptake (point v).
Figure 12.
A selection of ligands studied in the context of metalmismetabolism in AD. A and B have ionophoric properties, a dimeric form(compound 15) is derived from clioquinol where the covalent attachment oftwo hydroxyquinoline increase Cu(II) affinity and selectivity, C are water-soluble Cu(II) chelators, C and D are brain-penetrating Cu(II) ligands and Eand F are bi-functional Cu(II) chelators with Aβ-targeting unit (for more detailssee Refs. 342 and 344).
8. Aβ interactions with Protein Receptors
Understanding the interactions that Aβ establishes with various cellular components is a key challenge to unveil the molecular mechanisms at the onset of AD. Aberrant interactions with membrane associated proteins and receptors can mediate the neurotoxic effects of Aβ1-42 oligomers, such as in the case of the highly specific binding to the cellular prion protein (PrPC). (34,35,345) This controversial interplay has been associated with impaired activity of N-methyl-D-aspartate (NMDA) receptors,(345,346) which mediate critical functions in the central nervous system, in conjunction with copper binding from both Aβ oligomers and prion protein. Other proposed receptors for toxic Aβ assemblies include mGluR5(347), EphB2(348) and GM1(349), for which the first simulations of GM1 complexes with Aβ1-42 in lipid membranes have been reported.(304,305)
In contrast, other proteins are functionally employed as a primary biological defence against the effects of Aβ aggregation. In this context, both intracellular and extracellular chaperones are able to bind and stabilize misfolded oligomer species in such a way to prevent further fibrilization or dissociation. In particular clusterin, highlighted in genome wide association studies, is thought to play role as an extracellular chaperone.(350,351) Furthermore serum albumin, the most abundant protein in blood plasma and cerebrospinal fluid (CSF), inhibits Aβ fiber formation.(352)
In addition to PrP, Aβ can bind to other amyloid peptides, in particular serum amyloid P (SAP),(353) islet amyloid polypeptide (IAPP. Ref. 354) and transthyretin.(355) Both SAP and PrP have been found within plaques of AD patients.(356,357) Finally we should not forget that Aβ1-42 interactions with other forms of Aβ, for example Aβ1-40, that may be crucial to the misassembly process.(358,359)
8.1 Aβ – Prion Protein
The membrane anchored cellular prion protein (PrPC) has been identified as a cell surface receptor of Aβ. Specifically a screen of more than 200,000 proteins, using an unbiased cDNA expression library, has identified PrPC as a principle candidate to bind to Aβ.(34) This study also showed that interaction between PrPC and Aβ1-42 oligomers leads to the inhibition of long-term potentiation (LTP) in the hippocampal slices from normal mice expressing PrPC. Crucially it was shown using a mouse model of AD with a knockout PrP that AD pathology was dependent on the expression of PrPC,(34) while PrP knockout mice can develop Aβ plaques but do not exhibit neurotoxicity.(360)
A nanomolar affinity between Aβ oligomers and PrPC has consistently been reported.(34,361-364) This interaction is generally accepted, what remains contested is the influence of PrPC on Aβ toxicity in vivo.(363-366) The conflicting observations might be explained by the multifactorial nature of AD, some of its pathology could be independent on PrPC while other aspects of Aβ toxicity could be PrPC dependent. The conflicting observations might simply reflect differences in the AD mouse model used, or in some instances the Aβ preparations used. There are however a growing number of reports in animal models and hippocampal primary culture, showing PrPC dependent Aβ toxic effects, which impair synaptic plasticity and cause special memory defects and axon degeneration.(34,345,360,367-372) Furthermore ex vivo AD brain extracts indicate the co-localisation of Aβ and PrP in amyloid plaques.(357,373,374)
Numerous lines of enquiry have consistently highlighted the natively unstructured N-terminal domain of PrPC as the recognition site for Aβ. For example, the α-helical folded domain of PrPC spanning residues 113-231 has no influence on Aβ fiber growth while the N-terminal half of PrPC spanning residues 23-126 inhibits amyloid fiber formation in favour of non-ThT binding Aβ oligomers.(35) These PrPC trapped Aβ oligomers bind the oligomer specific A11 antibody and SEC indicates they are 12 and 24 Aβ monomers in size. More recently larger protofibrils structures of Aβ have also been identified in the presence of PrPC.(372) Solution NMR indicates that the interaction between Aβ and PrPC is conformation-dependent. Aβ monomer has little affinity for PrPC and it is not until Aβ forms oligomers that it interacts with PrPC. It is also clear PrP profoundly inhibits fiber formation by trapping Aβ in an oligomer form and is capable of disassembling mature fibrils.(35) The ability to trap and concentrate Aβ into toxic oligomers suggests a mechanism by which PrPC might confer Aβ neurotoxicity in AD.
The structural bases of this interaction are currently unknown. A major limiting factor in this context is the inability of current techniques of structural investigation to characterise Aβ oligomers, which is largely due to the transient and heterogeneous nature of these aggregates. Several models of Aβ oligomers have been proposed based on direct and indirect experimental evidence. These range from highly structured assemblies (mainly composed of β-sheets scaffold) to poorly ordered oligomers. Yet, a consensus is still elusive on the size of the most toxic assemblies, showing a dynamic distribution of assemblies ranging from 2 to 14 monomers. An additional barrier in the study of the interaction between Aβ oligomers and PrPC is associated with the unstructured nature of the N-terminal domain of PrPC (residues 23-126), which poses significant challenges of studying intrinsically disordered proteins.(375,376) It was proposed that the N-terminus of PrPC is the locus of the interaction with poorly structured, highly toxic Aβ oligomers.(362) This interaction was recently studied using computational approaches based on extensive MD simulations of dodecameric Aβ assemblies featuring short antiparallel β-hairpins at the C-terminus of the protein monomers.(377) The resulting oligomer models were used to infer on the interaction with the unstructured N-terminal tail of residues 23-127 by using PrPC models from an experimental NMR ensemble (PDB code 1QLX) and by performing mutant deletions according to Ref. 362. While this study could only rely to a massive use of modelling and simulations, it evidenced a conceptual model for the interaction between PrPC and toxic Aβ oligomers that can be useful for seeding new experiments.
While the function and misfunction roles of the disordered N-terminal domain of PrPC remain largely elusive, a large number of studies have dissected the misfolding pathways of the C-terminal PrPC domain in the mechanisms leading to PrPSc, the scrapie fibrillar form of the protein that is associated with the prion disorders. The large number of NMR structures of the C-terminus domain provides an important starting point to sample misfolding pathways using computational(378-380) or experimental(381-382) approaches. One of the most accredited scenarios, which accounts for the role of a number pathological mutations, is the misfolding of the native interface between two subdomains of PrPC, the first spanning strands S1 and S2 and helix H1 and the second spanning helices H2 and H3.(383-385) This pathway, corroborated by a series of experimental evidences, possibly can interplay also in the mechanisms of interaction with Aβ oligomers by the exposure of hydrophobic surfaces that are natively hidden in the interior of the folded part of the protein.
It has been proposed that Aβ has an effect on CNS function mediated by NMDA receptors activity including strong inhibition of long-term potentiation and enhancement of long-term depression.(346) Interestingly, it has been shown that PrPC limits excessive NMDA receptors activity that might otherwise promote neuronal damage.(386) Significantly, PrPC only affects the NMDA receptor in a copper-loaded state.(345) A mechanism for the PrPC dependent Aβ toxicity has been proposed which indicates Aβ disrupts copper homeostasis at the synapse which is required for normal PrPC dependent inhibition of excessive NMDA receptors activity.(345) Aβ released at the synapse, with a picomolar affinity for Cu2+,(387) may disrupt Cu2+ binding to PrPC and so, in part, mediate neuronal and synaptic injury.(345) The mechanism by which PrP mediates Aβ toxicity and NMDA activity(346) may also involve the Fyn receptor.(369) Several evidences have been reported on the direct binding between Aβ and NMDA receptors both in vitro and in vivo(388-391) as well as on the activation of NMDA receptors by Aβ oligomers.(392) Furthermore Aβ promotes endocytosis of NMDA receptors and so reduces the surface of NMDA receptors.(346)
The structural details of the Aβ-PrP interaction are clearly of interest and yet to be fully elucidated, indeed if the PrP-Aβ interaction is responsible, at least in part, for Aβ toxicity then identifying a molecule that blocks this interaction represents a novel pharmaceutical target.(367)
8.2 Aβ - Clusterin
Genome wide association studies have highlighted link between the development of AD and an ATP-independent chaperone, clusterin.(393) Clusterin belongs to a family of extracellular protein-folding chaperones, including α2-macroglobulin, haptoglobin, and αS1- and β-casein, which have been shown in vitro to stabilize proteins and prevent their aggregation under condition that normally lead to the formation of amyloids.(350,351,394-396) Clusterin is able to intervene in amorphous aggregation of a broad range of proteins in such a way to redirect the aggregation process in the assembly process of soluble high-molecular-weight aggregates.(395,397) It has been shown by single-molecule fluorescence that clusterin interacts with small Aβ oligomers ranging from dimers to 50-mers in such a way to form long-lived, stable complexes.(351) In this way clusterin can interplay with both aggregation and disaggregation processes of Aβ sequestering small oligomers, which have been shown to be the most toxic forms of Aβ, thereby mitigating the toxic effects of Aβ aggregation. This Alzheimer's disease mechanism is corroborated by the recent discovery of co-localization of clusterin with extracellular amyloid deposits containing Aβ.(350) It has indeed been suggested that clusterin may interplay in a novel extracellular proteostasis system, in which a series of extracellular chaperones bind to misfolded proteins in vivo to keep them soluble and to inhibit the formation of toxic aggregates in order to facilitate their bulk uptake and degradation via receptor-mediated endocytosis.(398) The Aβ-clusterin interactions remain to be studied by computer means.
8.3 Aβ - Albumin
An extra-cellular binding partner identified for Aβ is human serum albumin (HSA). This interaction was first described when Aβ was isolated from blood plasma, with 90-95% of Aβ within blood plasma directly bound to albumin.(399, 400) It is suggested that this interaction might explain why, unlike systemic amyloid related diseases, although Aβ is found at a similar concentration in blood plasma and CSF (0.1–0.5 nM),(401,402) Aβ plaque deposits are typically only observed in the brain and not peripheral tissue.
Albumin is the most abundant protein found in blood plasma with a concentration of ca. 640 μM. Concentrations of albumin in the CSF are much lower (3 μM)(403). Although markedly less concentrated than in blood, this still constitutes the most abundant protein in the CSF. The affinity of monomeric Aβ for HSA has been determined and a dissociation constant (Kd) of 5-10 μM, for both Aβ1-40 and Aβ1-42 has been consistently reported.(400,404,405) Despite the relatively weak micro-molar affinity (Kd = 5-10 μM) of Aβ for albumin, a concentration in the CSF of 3 μM suggests the capacity of albumin to bind approximately half of Aβ in the brain CSF. Furthermore this will be quite sensitive to changes in HSA concentrations. It is generally presumed that Aβ will bind to the hydrophobic pockets within albumin, which are often occupied by fatty acids, although this is not confirmed. There are reports to indicate HSA binds monomeric(400,404) or oligomeric(406,407) Aβ.
Recently it has been shown in vitro that physiological micro-molar levels of albumin found in the CSF does indeed inhibit Aβ amyloid fiber formation, significantly increasing the time before fiber nucleation occurs and decreasing the total amount of fibrils produced,(352) as shown in Fig.13. Furthermore, it was shown that the amount of amyloid fibers generated directly correlates to the proportion of Aβ not competitively bound to HSA.(352) Indeed it is likely nearly half of Aβ in the CSF will be bound to HSA and inhibited from forming fibers. This suggests a role for HSA in regulating Aβ fibril growth in the brain interstitium and computer simulations should be able soon to provide insights into the Aβ-HAS energy landscape as reported for HAS interacting with other molecules.(408) Thus levels of albumin in CSF should represent a risk factor and therapeutic target in AD. It is therefore perhaps surprising that the correlation between albumin levels in CSF and risk of developing AD pathology is yet to be conclusively identified. It might suggest that typically Aβ has already formed fibers within intra-cellular vesicles before release into the synapse. Alternatively Alzheimer's is a multi-factorial disease and albumin might just be one of many risk factors associated with the disease. Small variations in albumin levels in middle age may not be easily recognised, masked by a multitude of other factors that protect against or exacerbated AD pathology.
Figure 13.
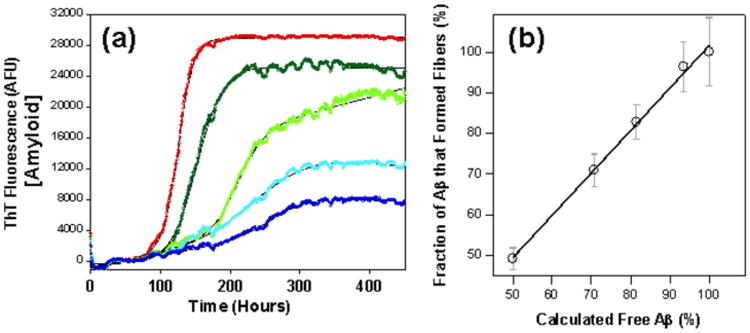
Physiological micro-molar concentrations of albumin inhibit fibre formation. (a) Kinetics of Aβ fibre formation in the presence of: no albumin (red); 1 μM (dark green); 3 μM (green); 5 μM (light blue); and 10 μM (dark blue) of albumin. (b) Competitive effects of albumin on total Aβ fibrils generated with a strong direct correlation between fraction of fibres generated and the calculated fraction of Aβ free to form fibres (not bound to increasing concentrations of albumin), based upon a Kd of 5 mM. Adapted from Stanyon et al.(352).
In summary we have characterised Aβ-protein interactions into three broad themes: cell surface interactions, for which we have focused on the prion protein (PrPC), and its connection with NMDA receptors activity. In addition, we have looked at protective defences against the effects of protein misfolding and aggregation in vivo, in particular by the extracellular chaperone clusterin and also human serum albumin. Finally, the interaction with other amyloidogenic proteins suggests possible interconnections between different protein misfolding diseases. For Aβ-Tau interactions, the reader can find recent data in Refs.186 and 408b. Overall, Aβ may exert its toxicity on neurons via more than one mechanism. Furthermore, different forms of Aβ, monomeric, oligomeric and fibrillar, can present different recognition sites to different binding partners. There is still much to be understood about the molecular interactions of Aβ, both intra- and extra-cellular and at the synaptic cleft, as it is these interactions that may constitute new pharmaceutical targets.
9. Interactions of Aβ with Inhibitors
The failures of recent phase III clinical trials on two Aβ-targeting monoclonal antibodies, bapineuzumab and solanezumab, in the summer of 2012 indicate that the timing of the intervention of AD needs to be reconsidered.(42,409) In the light of these reports, are we back to 2008 when news in Nature reported that the major conundrum in the field is: are we just treating people too late? The good news is that three studies are to be launched in 2014 on asymptomatic individuals identified as being at increased risk of developing AD on the basis of genetic predisposition or amyloid levels.(42)
First of all, several protein-like drugs are briefly discussed. A homodimeric protein of 58-residues, ZAβ3, was found to bind to Aβ1-40 monomer and inhibit the fibrilization process. The structure of the ZAβ3:Aβ1-40 complex by solution NMR revealed that the Aβ1-40 is locked into a β-hairpin conformation spanning residues 17-36 with the rest of the amino acids disordered, and the edges of Aβ1-40 β-sheets were capped by the 2 β-strands of ZAβ3, thus blocking the β-sheet extension of Aβ1-40.(114) Similarly, Reelin, an extracellular matrix protein, was reported to stabilize the Aβ1-42 oligomers, leading to reduced toxicity and delayed fibrillization.(410)
A series of N-methylated peptides or D-amino acid peptides were also found capable of inhibiting Aβ amyloid formation, targeting either Aβ residues 32-37(411) or 16-21 (SEN304, Ref. 412). For instance, SEN304 was found to bind to Aβ1-42 monomer and oligomers and to promote the formation of non-toxic aggregates. With a concentration as low as 100 nM, SEN304 was able to almost completely remove the inhibition of long-term potentiation (LTP) by 1μM of Aβ1-42 in a hippocampal slice.(412) Recently, two inhibitors with alternating D- and L-amino acids of lengths 21 and 23 designed by MD simulations to form α-sheet, α-sheet having all its peptide groups orientated in the same direction, were found to reduce Aβ1-42 aggregation and toxicity at molar ratio of at least 10:1.(413)
Small molecules as potential drug candidates against AD have been investigated intensively in recent years. Though differing in size, geometry and chemical properties, the compounds in general exhibit inhibitory effects by three modes. First, the compounds can bind to fibrils and reduce toxicity by limiting fibril fragmentation. In a recent study, several compounds, including BAF31 shown in Fig.14, were found to reduce Aβ1-42 cytotoxicity against mammalian cells by up to 90%.(9b) The compounds were identified through virtual screening of 18,000 purchasable molecules and ranked according to their calculated binding energies to fibrillar Aβ16-21 segments. What is interesting in this study is that the compound binding increases the fiber stability to limit fragmentation, rather than reducing fiber formation, and the compounds do not bind to oligomers.
Figure 14.
Chemical structures of small compound inhibitors of Aβ aggregation and toxicity.
Second, the compounds can accelerate the formation of fibrils and reduce the lifetime of toxic oligomers. An example is an orcein-related polyphenol, O4 (Fig.14). The molecule was found to bind directly to oligomers and promote the conversion into larger amyloid fibrils, with O4 interacting with hydrophobic residues of Aβ1-42.(14)
In the third mode, the small molecules interact with oligomers and prevent fibrillization. The resulting complexes are believed to be off-pathway and nontoxic. Several polyphenols were reported to rescue AD in this way. ε-Viniferin glucoside (EVG for short, Fig.14) is one of the polyphenols that inhibits fibril formation in Aβ25–35, Aβ1-40 and Aβ1-42 and protects against PC12 cell death induced by these peptides.(415) Electrospray ionization mass spectrometry showed a non-covalent complex between one Aβ1–40 peptide and two EVG molecules.(415) Solution NMR and molecular modelling were used to characterize the interaction between the compounds, using 1mM Aβ40 and 2mM EVG in DMSO.(416) EVG induces the formation of turns in the 10-12 and 28-30 regions of Aβ. Chemical shift perturbations and short-range intra-molecular NOEs confirmed that EVG predominantly interacts with clefts formed by Y10, V12, Q15 and F19 or by K28, G29, A30 and I31, although no intermolecular NOE's were observed.(416)
The epigallocatechin gallate (EGCG, Fig.14) is another polyphenol showing similar effects as EVG. EGCG is currently undergoing a phase 2-3 clinical test against early stages of Alzheimer's disease (NCT00951834), which is expected to complete in June 2015. EGCG was shown to redirect the Aβ aggregation pathway, and generate off-pathway non-toxic oligomers which are incapable of amyloid fibrillogenesis.(417) EGCG can also remodel mature Aβ fibrils into non-toxic oligomers,(418) suggesting its therapeutic potential for treatment of AD patients. Thermodynamic analysis using isothermal titration calorimetry for EGCG and Aβ fragments/full length peptides by Wang et al. reported that EGCG mainly interacts with Aβ residues 1-16 through hydrogen bonding and residues 17-42 through hydrophobic interactions.(419,420) Higher resolution structures of Aβ/EGCG complex are also available. Solution-state NMR measurements performed by Lopez del Amo et al. showed that the EGCG-induced Aβ1-40 oligomers adopt a well-defined structure, rather than disordered, in which residues 22-39 maintained a β-sheet conformation, and residues 1-20 were unstructured.(421) Atomistic REMD simulations for Aβ1-42 dimer with 10 EGCG found that the equilibrium structures of Aβ42 dimer in the presence of EGCG were characterized by the existence of 5% free Aβ42 monomers. Upon EGCG binding, the intermolecular contacts between the CHC and residues 29-42 were also greatly impacted.(207) The simulations also revealed EGCG was most likely to interact with F4, R5, H6, Y10, L17-F20, I31-I32, L34-V36, V39 and I41 (Fig.15A).
Figure 15.
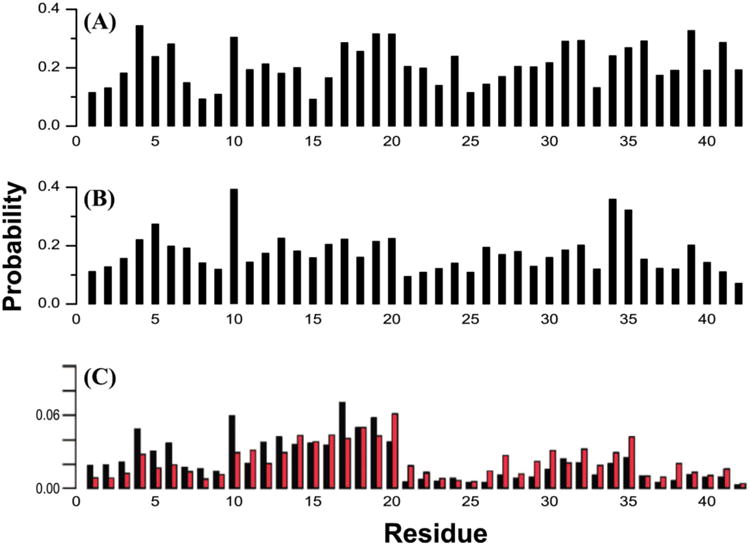
Contact probabilities of each Aβ1-42 each amino acid with compounds. (A) Between EGCG and Aβ1-42 dimer from atomistic REMD simulation.(207) (B) Between NQTrp and Aβ1-42 dimer from atomistic REMD simulation.(432) (C) Between 10 small organic molecular fragments (e.g., dimethyl ketone, furan and pyrazole) and Aβ1-42 monomer from REMD and FTMap.(175) Non-bonded (black bars) and hydrogen bonding (red bars) interactions are shown separately.
Apart from the polyphenols discussed above, non-polyphenol molecules were also found to inhibit Aβ fibrilization through binding to oligomers. Carnosine (β-alanyl-l-histidine, Fig.14), a naturally occurring dipeptide, was found to bind Aβ and inhibit fibril formation. With MD simulations and NMR experiments, again without any detection of intermolecular NOE's, carnosine was found to form transient salt bridges with charged residues in Aβ (R5, K16 and K28) and hydrophobic contacts with the CHC and flanking regions.(422) Arai et al. also designed a non-peptidic inhibitor targeting CHC under the assumption that intermolecular side chain and main chain interactions must be optimal and minimal, respectively.(423)
Recently, the auto-oxidation of polyphenols, (+)-taxifolin(424) and EGCG(425) into quinone-derivatives was reported to be essential. The experiments revealed the potential of quinone-derivatives as Aβ aggregation inhibitors. Indeed, the quinones and quinone derivatives were found to inhibit amyloid aggregation several years ago.(426-428) In 2010, Scherzer-Attali et al. observed that a quinone derivative, NQTrp (1,4-naphthoquinon-2-yl-L-tryptophan, Fig.14), was able to inhibit fibril formation by Aβ1-42 and completely recover the phenotype in a transgenic AD drosophila model.(429) An extensive REMD simulation using the coarse-grained OPEP force field, followed by all-atom docking calculations, picked the NQTrp molecule as the best ligand of Aβ17-42 trimeric structures among five small-molecule drugs, including 3 polyphenols.(208) The NMR study with the presence of a 0.25 molar ratio of NQTrp to Aβ12–28 monomer found that the structures of NQTrp-bound Aβ12-28 were characterized by 2 turns formed by residues 18-20, and 22-26, although no intermolecular NOEs were observed.(429) While several simulations focusing on the binding of NQTrp to Aβ fragments were performed,(430,431) an all-atom REMD simulation of Aβ1-42 dimer with the presence of 2 NQTrp molecules in explicit solvent provided a different binding picture.(432) The structure representing the first most populated cluster shown in Fig.16A is characterized by two helices spanning the CHC or part of CT (residues 30–35) region in each chain. The second cluster in Fig.16B is essentially random coil with two short helices spanning residues 3–6 in one chain and residues 24–28 in the other chain. A structure with 3 β-strands is found in Cluster 8 (Fig.16C) where a β-hairpin formed by residues 32–34 and 37–40 of one chain packed against a third strand formed by residues 39–41 of the other chain. Overall, 555 clusters were identified and the residues with high probabilities to interact with NQTrp are F4-D7, Y10, H13-H14, K16-L17, F19-F20, S26, K28, I31-I32, L34-M35 and V39 (Fig.15B).(432)
Figure 16.
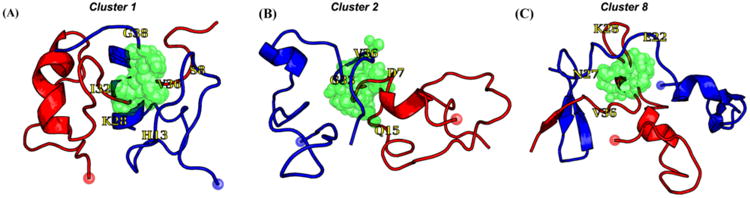
Representative structures of Aβ1-42 dimer/NQTrp from atomistic REMD simulations. (A) Cluster 1, (B) Cluster 2, and (C) Cluster 8. Chains A and B are colored red and blue, respectively. The red and blue spheres represent the Cα atoms of Asp1. The center of mass of NQTrp is shown as green spheres, and we show NQTrp if it forms a contact < 4 Å with any heavy atom of Aβ1-42. Residues colored yellow assist in the reading.
Complexes of EGCG or NQTrp to Aβ both involve several hydrophobic residues including Y10, L17, F19-F20, I31-I32, L34 and V39, though overall, the sites with the highest interaction probability are clearly different (Fig. 15A and 15B). R5, K16, K28 and the CHC region are found in the binding sites of both NQTrp and Carnosine. The proclivity of Aβ1-42 monomer to form pockets able to bind small molecules was investigated by Zhu et al. with a 100ns REMD simulation with AMBER99sb/TIP3P.(175) The 35 most populated Aβ1-42 monomer centroids were subjected to fragment-based calculations, and the most populated binding pockets identified using FTMap(433) and FRED.(435) The CHC residues were found to have the highest propensity to bind small molecular fragments, but F4, Y10, and M35 were also involved in many of the hotspots (Fig.15C).(175) In contrast, the central region (particularly 22-26) had a much lower tendency to form binding hot spots. This reduced probability agrees with the reduced experimental 1H-15N HSQC chemical shift perturbation of central region residues when certain small compounds are titrated into Aβ monomer sample.(435,436)
Obtaining high-resolution structures of Aβ monomer/inhibitor complexes from NMR with intermolecular NOEs remains a challenge. The strategy adopted in REMD simulations of Aβ/EGCG and Aβ/NQTrp complexes suggests a general first-order approach to screen Aβ/inhibitor interactions, but this remains a very difficult task because current inhibitors interacting with Aβ monomer, dimer and trimer show many binding sites with small occupancies and contact surfaces.(207,432,437) Apart from simulations, the cost-effective virtual screening favors the discovery of novel inhibitors with high-quality receptor structures. Recently, a ligand-based drug design provided a different approach from virtual screening to the discovery of novel inhibitors.(438) The method used a known inhibitor, a substituted Aβ1-42 peptide termed [Nle35, D-Pro37] Aβ1-42. The structure of residues 35-40 from the inhibitor was stripped of side-chain atoms except D-Pro, and used as query to screen for small molecules with similar spatial geometry and recapitulated hydrogen-bonding interactions. A compound was found to inhibit Aβ1-42 aggregation with an IC50 of 13 μM. The approach is of great interest as it provides a good demonstration for both theoretical and experimental work of how to cooperate and the great benefits.
10. Truncated Variants of Aβ and Pathogenic and Protective Aβ Mutations
Familial forms of Alzheimer's Disease (FAD) represent only a small fraction of all AD cases and show an autosomal dominant pattern of inheritance, which often results in early onset symptoms (in general between 40 and 65 years old). FAD mutations occur on the presenilin PSEN1 and PSEN2 genes as well as on the APP gene from which Aβ is processed.(439) We will therefore focus on APP mutations and particularly within Aβ spanning residues 672-714 for the 42 amino acid sequence.
10.1 Experimental Findings
Over 30 mutations in the APP gene are known today, 25 of which are pathogenic and autosomal dominant with an early-onset disease phenotype and two of which are reported to be protective mutations against AD.(440) In order to better understand the pathogenic and/or protective effects of AD mutations, it is important to genetically screen significantly large groups of AD and non-AD patients to obtain very good statistics SNPs.
Four types of FAD genetic aberrations have been observed to happen within the APP gene: complete gene duplications, single-point missense substitutions and deletion or insertion of a nucleotide. Gene duplications cause an overexpression of APP, which inevitably implies an overproduction of Aβ(441) and all the consequential toxicity known to accompany it. Some mutations occur near the APP-to- Aβ β- and γ-cleavage sites, which generally results in an overproduction of Aβ(442,443) or shifts the relative amounts of Aβ1-40 and Aβ1-42 towards a higher production of the more toxic Aβ1-42.
Many pathogenic FAD mutations increase Aβ propensity to aggregate in vitro.(442,443) In particular, the mutations located in and near the CHC, the Flemish (A21G), Dutch (E22Q), Italian (E22K), Arctic (E22G) and Iowa (D23N) mutations, are also known to increase the toxicity mediated by Aβ. Because of their close proximity to the α-cleavage site (K16-L17), some of these mutations have been reported to also decrease the production of non-amyloid products and increase Aβ levels while making the mutant Aβ resistant to the Aβ-degrading enzyme neprilysin.(444) Teplow et al. characterized the role of various residues and reported that Aβ1-40 is mostly sensitive to mutations at positions 22 and 23 such as E22G and D23N, while Aβ1-42 is most affected by A21G.(150) It has been shown that A21G decreases fibril elongation and promotes protofibril and toxic oligomer formation, while E22G is observed to increase the rate of protofibril formation. The effects of the FAD A21G and E22G mutations have also been studied using IM-MS showing that the early oligomer distributions differ for each mutant and the Aβ alloform.(53) Another pathogenic FAD mutation is the Osaka E22Δ mutation, which consists of a deletion of residue 22. E22Δ is known to induce a cholesterol-mediated toxicity as the mutation modulates levels of intracellular and extracellular Aβ, the secretion of which normally regulates cholesterol efflux.(445) The de novo D23Y mutant and proline substitutions in the CHC have also been shown to affect self-assembly and toxicity.(139,446)
Experiments have also highlighted the importance of the N-terminal residues 1 to 16,(50,53-55,444,447) whose role has been underestimated in the past owing to its highly disordered structure in synthetic Aβ fibrils. These include the pathogenic FAD H6R (English), D7H (Taiwanese) and D7N (Tottori) mutations. In particular, D7N accelerates the kinetics of transition to β-sheet-rich configurations and promotes the early formation of higher order oligomers with more α/β structures that are significantly more toxic compared to WT Aβ1-40 and Aβ1-42.(149a) An IM-MS study also showed that the FAD D7N mutation leads to early oligomer distributions that differ from Aβ1-40 to Aβ1-42.(53) The double substitution D1E/A2V also affects Aβ1-40 fibrillogenesis and predominantly forms neurotoxic aggregates.(448) Finally, a novel FAD mutation, K16N, found in one family was shown to increase Aβ production as it is a poorer substrate for α-secretase. The mutant K16N Aβ is itself not harmful, but becomes toxic when mixed upon equimolar ratio of WT Aβ, inhibiting WT Aβ1-42 fibril formation and producing more Aβ oligomers.(444) In many cases, key side chain interactions are reported to be at the origin of Aβ toxicity. Based on the Aβ1-42 fibril model,(29) the K16N mutation is reported to add a hydrogen bond between the side chains of K16 and N16 in hetero-tetramers, increasing therefore the stability of the aggregates.(444) The importance of the lysine residues in Aβ has been further highlighted by Sinha et al. who rationally designed K16A and K28A mutants.(151a) K16 is known to be important for driving fibrillogenesis while K28 is thought to stabilize a loop driving Aβ folding. What they found is that each mutation by alanine has profound effects on Aβ assembly and dramatically reduces Aβ toxicity, suggesting the design of inhibitors targeting K16 and K28. Another example is provided by the FAD D23N mutation, which by preventing the formation of a salt bridge with K28, also modifies toxicity. Finally, the English mutation by adding one charged residue and the Taiwanese and Tottori mutations, by deleting one charged residue, are also likely to change the network and populations of all salt bridges.
Surprisingly, there are no toxic FAD mutation reported in the C-terminal region (residues 30-42), though a high percentage of Aβ with a Met-sulfoxide at position 35 is present in the AD brain.(449) Yet the de novo Aβ G33A and GI33 variants have been shown to promote the aggregation process in vitro by increasing the population of large oligomers (16- to 20-mers) at the expense of small oligomers (2-to 4-mers); however, how they affect the structures of the early formed Aβ1-42 oligomers is an open question.(450) On the basis of in vitro and in vivo experiments, Aβ1-42 oligomers with substitution of G33 by alanine and isoleucine are much less toxic than the WT Aβ1-42, suggesting that G33 may represent the critical residue linking toxicity and oligomerization, adding therefore complexity to the origin of toxicity.(450) Enhanced aggregation propensity of Aβ1-40 was also confirmed in the double de novo mutants G33V/V40A and I31L/M35L(451) and, in a more extensive study Hecht et al. demonstrated that particular nonpolar side chains in the C-terminal half of Aβ1-42 are not required for aggregation and amyloidogenesis.(452)
Besides the Aβ1-40 and Aβ1-42, the truncated Aβ4-42 and Aβ5-42,(46) Aβ1-26 and Aβ1-30 and Aβ1-39 peptides are found in amyloid plaques.(40) The Aβ1-43 peptide, extended by a single threonine at the C-terminus relative to Aβ1-42, has a stronger neural toxicity and higher aggregation capacity than Aβ1-42,(453) and increases the rate of extent of protofibril aggregation and confers slow C-terminal motions in the monomeric and protofibril-bound forms of Aβ1-43.(454) In addition, many post-translational modifications of Aβ peptides are also observed in amyloid plaques. Among the modifications, proteolytic removal of D1 and A2 and the subsequent cyclizing of E3 and E11 to a pyroglumate (Aβ3(pE) and Aβ11(pE)) are particularly interesting.(47-49) Aβ(pE) is more cytotoxic and aggregates more rapidly than conventional Aβ. Only 5% of Aβ3(pE)-42 mixed with 95% of WT Aβ1-42 is enough to significantly enhance the cytotoxicity in vivo through the formation of hybrid Aβ3(pE)-42/Aβ1-42 oligomers. In light of these observations, it was postulated that Aβ3(pE) might trigger AD by propagating through a template-folding prion-like mechanism with Aβ1-42. This truncated variant is also known to be acting in a tau-dependent manner and to be particularly resistant to degradation.(50)
The most intriguing and interesting mutations are undoubtedly the AD-protective ones, but they have not been extensively studied experimentally and theorically. Two protective mutations at the position 2 of Aβ (position 673 in APP) have been reported. A rare genetic mutation observed in a single Italian kindred, A2V, causes an early-onset of AD when it is only inherited from both parents, while heterozygous carriers of A2V are unaffected. A2V enhances Aβ1-40 aggregation kinetics by a factor of four, but the mixture of the Aβ1-40 WT and A2V peptides protects against AD.(54) Using multiple low-resolutions methods, an equimolar solution of Aβ1-42 WT and A2V produces smaller aggregates with much slower kinetics than Aβ1-42 WT, suggesting instability of the mixed aggregates.(455)
The A2T mutation, on the other hand, is always a protective mutation, independently of its homozygous or heterozygous form. It was reported in 79% of a non-AD control group with better cognitive test results compared to an AD group by using a large-scale DNA screening of Icelanders.(55) Thus far, this mutation has not been observed in non-Nordic populations.(456,457) This mutation reduces Aβ production by 40%, at variance with A2V in its homozygous state which enhances Aβ production(54). Importantly, ThT fluorescence essays reveal that the mutations, while having little effect on Aβ 1-42 peptide aggregation, drastically modify the properties of the Aβ1-40 pool with A2V accelerating and A2T delaying aggregation of the peptides. In agreement with the results of Ref.455 on the mixed A2V/WT aggregates, A2T forms smaller aggregates than the WT peptides.(458) This finding on the kinetics and oligomer sizes should be however confirmed by Nile red binding and static light scattering experiments, respectively. Whether more unstable oligomers render them more available for degradation is sufficient to explain in vitro experiments showing that A2T attenuates the APP-mediated intracellular cell-death(459) remains to be determined. Taken together, the physical properties of the Aβ1-40/42 A2V/A2T and Aβ3(pE)-42 peptides raise many questions and opens new drug-design perspectives.
10.2 What we have learned from Mutational Computational Studies
Most of the simulations studying the effects of mutations are summarized in Table 4. For each mutant, we give its polymeric state, the method used and the main findings. Many simulations of the Arctic E22G mutation have been performed and highlight its destabilizing effect on the region 20-30(184b,202) and its increased β-strand effect on the N-terminal.(184b) The other mutations at the position 22 also have significant effects on the CHC structure and flexibility and either induce more(176,177,460) or less(176) α-helix structure, although all mutants remain essentially disordered. Lin et al., on the basis of thousands of MD trajectories simulated with AMBER99sb/TIP3P have therefore postulated that there might exist a link between α-helix propensity and aggregation kinetics.(177) As a result, an increased helix-helix interaction between dimers may result in altered kinetics of oligomerization.
Table 4.
Computational studies on the variants. n.a. stands for not applicable.
| Ref. | Mutations | Aβ alloforms | Oligomer size | Force field & solvent model | Method | Timescale | Theoretical findings upon mutation |
|---|---|---|---|---|---|---|---|
| 473 | A2V | 1-28 | 1 | CHARMM22* TIP3P | T-REMD | 9.8 μs |
|
| 465 | H6R | 1-40, 1-42 | 1, 2 | OPLS-AA TIP4P | MD | 3.7 μs |
|
| 466 | D7H | 1-40, 1-42 | 1 | OPLS-AA GB/SA | T-REMD | 24 μs |
|
| 464 | D7N | 1-40, 1-42 | 1, 2 | OPLS-AA TIP3P | MD | 7.4 μs |
|
| 462 | A21G | 1-40, 1-42 | 2 | GROMOS96 GB/SA | MD | 60 ns |
|
| 177 | E22K. E22Q E22G, D23N | 1-42 | 1 | AMBERFF99SBTIP3P | MD | > 1000 μs |
|
| 460 | E22Δ | 1-40, 1-42 | 1 | AMBERFF99SBImplicit GB/SA | T-REMD | 2.4 μs |
|
| 178 | E22K | 1-42 | l | AMI3ERFF99SB TIP3P | MD | >700 μs |
|
| 184b | E22G | 1-40, 1-42 | l | CGImplicit | DMD | 60*107 steps |
|
| 463 | D23N | 15-40 | 2 | AMBERFF99SB TIP4P-Ew | Minimization ab-initio FMO | n. a. |
|
| 202 | F20E E22G E22G 131E | 1-42 | 2 | PHOFAS1 Implicit | MC | 80*1010 steps |
|
| 474 | Pyroglutamate variants | 3-40, 3-42, 11-40, 11-42 | 32 | CG Implicit | DMD | 3.2*109 steps |
|
Other mutations in the region 20-23 have been shown experimentally to modulate the rate of aggregation.(442,443) This effect is observed in several computational studies showing an increased aggregation propensity for E22Q,(461) and a reduced aggregation propensity for F20E dimers.(202) In some cases, the CHC residues may also become solvent exposed and then serve as docking sites for Aβ deposition onto fibril (E22Q).(461) MD simulations on A21G report a decrease in β-strand propensity for the Aβ1-40 and Aβ1-42 dimers upon substitution,(462) and this was confirmed by IM-MS experiment.(53) The simulations also reveal that the per-residue β probability varies from Aβ1-40 and Aβ1-42 dimers upon A21G. The D3N mutant in both alloforms was also studied by all-atom MC simulations and OPEP CG REMD simulations both in implicit solvent and showed that, by perturbing the side-chain H-bond network, the peptide remains compact(202) or displays a rather independent N-terminus.(195) In particular, the latter simulation showed that the D23N mutation causes non-local perturbations of the WT conformational ensemble by increasing the β-sheet propensity at the C-terminal region. This result is in agreement with the ss-NMR structure of a highly synaptic Aβ1-40 toxic oligomer with a stable N-terminal β-strand.(126) Finally, the dimers of Aβ15-40 WT and D23N were studied using a selected numbers of parallel and antiparallel molecular mechanics-generated conformations that were refined by ab-initio FMO (fragment molecular orbital) calculations. It is found that in water, the parallel conformation is more stable than the antiparallel one, due to the larger hydration energy for the parallel conformation for both the WT-and the D23N- Aβ15–40 dimers.(463)
Simulations of the FAD D7N and H6R variants also proposed different mechanisms for the increased Aβ aggregation.(464-466) For instance, all-atom MD simulations showed that D7N enhances the aggregation rate by decreasing the turn propensity at residues 8-9, by perturbing salt-bridges half-lives and by reducing the bending free energy of the loop region.(464) Simulations on H6R, in contrast, the rate of fibril formation of Aβ1-42 increases due to increased β-structure at the C-terminal in both monomer and dimer and enhanced stability of salt bridge Asp23-Lys28 in monomer, while the enhancement of turn at residues 25-29 and reduction of coil in regions 10-13, 26-19, and 30-34 would play the key role for Aβ1-40.(465) The results of the FAD D7H simulations(466) are discussed in Table 4.
De novo mutations were also investigated such as the mutation D23Y, whose effect on the hexamer of Aβ1-40 was reported to favour the locking of Aβ monomer onto fibrils, thus promoting fibril growth. Simulations found that the interactions with the aromatic ring of Y23 are more fibril-compatible than with the negatively charged D23.(467) The de novo G33A and G33I mutations were also explored on the monomer and dimer of Aβ29-42, and the REMD simulations showed a significant reduction of the β-hairpin population upon both mutations and a destabilization of the dimer due to an increase in hydrophobicity.(468)
Atomistic MD simulations of monomeric Aβ1-40 Met-ox found that M35 oxidation decreases the β-strand content of the C-terminal (residues 29–40), with a specific effect on the secondary structure of residues 33–35, thus potentially impeding aggregation. Further, there is an important interplay between oxidation state and solution conditions, with pH and salt concentration augmenting the effects of oxidation.(469) REMD simulations of Aβ1-43 dimers followed by ab initio calculations revealed a ring-shaped conformation, in which T43 is hydrogen bonded to R5, that is absent in Aβ1-42 dimers.(470) Simulations of Aβ1-39 monomer and dimer with CHARMM/SASA revealed a very high percentage of α-helices, which is likely due to the very high bias for α-helix of this force field.(471,472)
Finally, the A2V variant and the Aβ3(pE)-42 peptides have started to be studied by computer simulations. First of all, Nguyen et al. have found via all-atom REMD simulations that the A2V mutant of Aβ1-28 is much less intrinsically disordered than the WT peptide, increases the propensity to form β-hairpins and enhances the α-helix in the region 17-24. Both peptides display a non-negligible population (7%) of extended metastable conformations differing however in their atomic details that represent ideal seeds for polymerization. More importantly, the two conformational ensembles are totally different, suggesting unstable dimers.(473) This result is in agreement with the increase in β-propensity of pure A2V aggregates by light scattering and provides a first answer for the reduced aggregation kinetics of the mixture of WT and A2V peptides.(54) Atomistic REMD simulations of WT-WT, WT-A2V and WT-A2T Aβ1-40 dimers are in progress.
A DMD-CG simulation in aqueous solution of 32 AβpE3 and AβpE11 peptides lacking pyroglumate at position 3 and 11 reported than truncation of the N-terminal residues in Aβ3-40, Aβ3-42, Aβ11-40 and Aβ11-42 shifts the oligomer size distribution towards larger oligomers as observed experimentally.(474) Moreover, the fact that the N-terminal of the AβpE3-40/42 and Aβ11-42 variants is more flexible than the Aβ1-40/42 WT peptides could be related to their increased toxicities relative to the WT peptides. The activity of Aβ3(pE)-42 pores has been studied using a planar bilayer recording and their architectures provided by all-atom MD simulations showing that the N-termini β-strands tend to reside in the hydrophobic lipid core, in contrast to WT Aβ1-42 peptides.(475) Using multiple experimental essays and MD simulations, Lee et al. compared the adsorbed and membrane-inserted oligomeric species of AβpE3-42 and Aβ1-42 peptides. They found lower concentrations and larger dimensions for both species of membrane-associated AβpE3-42 oligomers. The larger dimensions are attributed to the faster self-assembly kinetics of AβpE3-42. Membrane-inserted AβpE3-42 oligomers were also found to modify the mechanical properties of the membrane.(476)
11. Conclusions
We have reviewed what experiments and computer simulations can tell us about the amyloid-β protein and its link to Alzheimer's disease. Our knowledge of the structures of synthetic Aβ1-40/1-42 fibrils, protofibrils, and large oligomers has markedly increased in the recent years and it is clear that polymorphism is present from the monomer to fibrils. We know that fibrils with different molecular structures can result from environment-dependent self-assembly and kinetic rather than thermodynamic control. We also know that metastable states can be alleviated by using appropriate seeds or under shear flow and the structural models of the Aβ1-40 fibrils that build up take on different structures in the brain of diseased Alzheimer's patients with different AD symptoms. This high degree of polymorphism, which arises from many physical factors and persists in vitro and in brain tissues, is correlated to different phenotypes and is rather a bad news for drug design because one drug may be efficient for one patient but not for another.
Structural and dynamical characterization of the smallest oligomers, the most toxic species, and the monomers has been moving at a lower pace due to their transient character and intrinsic disorders, but with the help of new experimental methods and efficient sampling methods using multiple force fields and representations, our knowledge of these species in aqueous solution, in proximity or in the membranes, with and without ion metals should significantly increased, although polymorphism of the aggregates and high sensitivity of external conditions will not facilitate the reproducibility of the experimental readouts and the convergence of the simulations. One particular advantage of computer simulations, however, is that calculations can be repeated using different pH conditions and model membranes and the effects of site-specific mutations can be investigated.
Characterization of the primary nucleus/nuclei and the population of amyloid-competent monomeric state(477) prior to the lag phase remain difficult both experimentally and theoretically due to the sensitivity of the experimental conditions and the amino acid sequence. One amino acid substitution is sufficient to change the free energy landscape as evidenced from the kinetics and the oligomer sizes distribution of FAD Aβ variants and the recent isotope-edited and ss-NMR findings that Aβ16-22 with E22Q displays by unexpected antiparallel β-strand orientation intermediates that later transition completely into parallel β-strands, suggesting a new nucleation mechanism in a progressive assembly pathway.(478)
Understanding the interactions that the Aβ1-40/42, Aβ1-40/42 A2V and A2T variants and the Aβ3(pE)-42 peptides in both monomeric and oligomeric forms, establishes with metal ions and various cellular components is a top challenge to unveil the molecular mechanisms at the onset of AD. Again both biochemical and biophysical experiments along with simulations have just started to give a more precise picture, but important efforts towards this direction should be pursued.
How the structures of Aβ may relate to the mechanism of toxicity is still unknown since toxicity comes from all oligomers to the fibrils. One source of toxicity comes from membrane channel formation, and the cylindrin conformation has been suggested to be toxic. But other antiparallel beta-sheet conformations are also considered toxic. In addition a single amino acid change is able either to reduce (A2T, A2V) or increase (FAD, K16A) toxicity. Another source of toxicity comes from metal-ions and the interactions with the cellular partners, but our understanding is still limited. To this end, we are currently investigating the polymerization and depolymerization process of Aβ WT, A2V and A2T assembly in the absence and presence of PrP as well as the toxicity of the assemblies.
Despite extensive studies, drug after drug aimed at targeting Aβ has failed to slow the progression of AD in clinical trials. If it is true that we are treating people too late, there are however two other hurdles for drug improvement. While many groups are working on developing drugs that bind to Aβ fibrils (reducing therefore the fragmentation process) or bind to Aβ oligomers to slow down or accelerate fibrillation, and in all cases reduce Aβ cytotoxicity, how any interact with Aβ1-42 and Aβ3(pE)-42 oligomers is unknown at an atomic resolution. Yet, obtaining high-resolution structures of the Aβ oligomers/drug complexes is a prerequisite to optimize the kinetic and thermodynamic binding properties of promising compounds (and thus their specificity), prior to cell viability essays, animal models for AD and clinical trials. The second hurdle is that repeated identification of the same types of molecules as promising hits against different proteins is polluting the chemical literature. For instance, quinones are redox cycler, metal complexer and covalent modifier.(479) It has also been found that curcumin from turmeric, EGCG from green tea, and resveratrol from grapes, which reduce Aβ aggregation in vitro, also alter lipid bilayer properties and the function of diverse membrane proteins.(480) So the effect of one drug might not be what we expect, and in this context we recently showed using expressed and then produced Aβ1-28, Aβ1-40 and Aβ1-42 peptides with multiple essays and different readouts that the NQTrp inhibitor exerts its inhibitory effect via other mechanisms than direct interactions with Aβ peptides (O. Berthoumieu et al., unpublished).
Along with the next steps already described at the end of each section, some challenges should be considered. The first challenge is that the population of dimers, trimers and dodecamers (Aβ*56) in brain tissues vary with aging, indicating that the species to be targeted at early or late-onset AD are not the same. By using 75 cognitively intact individuals, ranging from young children to the elderly, and 58 impaired subjects with mild cognitive impairment or probable Alzheimer's disease, It was found that Aβ*56 may play a pathogenic role very early in the pathogenesis of AD.(481)
The second challenge is that experiments have reported drastic acceleration of fibril formation for the Aβ1-40 peptide in shear flow.(482-484) The origin of such a kinetic speed up is still debated. In addition, while atomistic simulations in explicit solvent of the full aggregation of the Aβ1-40/42 peptides and causing- or protecting-variants are still out of reach, coarse-grained simulations in implicit solvent require the treatment of hydrodynamics effects.(61,485) In Fig.17 we report preliminary results of the early steps of the aggregation of 18 Aβ16-22 blocked by acetyl and amine under shear flow as obtained from simulations using the CG model OPEP with hydrodynamic interactions (S. Melchionna, P. Derreumaux and F. Sterpone, unpublished). For sake of exemplarity the simulations were performed at rather high concentration (109 mM) and in the absence of shear rates or in the presence of high shear rates (108, 109 and 1010 s-1). With hydrodynamics effects, the early steps of aggregation are already very different from Langevin dynamics as seen from the size of the oligomers. With high shear rates, we observe formation of a unique elongated aggregate within 10 ns. For this highly concentrated system the presence of the laminar shear flow seems not affecting the overall kinetics of the collapse, but however does influence the way the aggregation proceeds. Namely increasing the rate an oscillatory behaviour in the aggregation process emerges (see left panel reporting the time evolution of the system's gyration radius). The highest shear rate also breaks down the hydrogen bonds and side-chain contacts in the aggregates, thus a non-monotonic behaviour as a function of the hydrodynamic perturbation is expected for similar aggregation processes of proteins. We are currently investigating the dynamics over a much longer time scale.
Figure 17.
Aβ16-22 aggregation simulations under several conditions. Top panels. Pictorial representation of monomer aggregation for several simulation schemes: i) Langevin dynamics, ii) Langevin dynamics with Hydrodynamics Interactions (HI), and iii) Langevin dynamics with HI and longitudinal shear, shear rate Δv/Δz=108 s-1. The time-evolutions show the effect of HI on the aggregation kinetics. When shear-flow is activated also the details of the aggregation change: i.e. first two separate aggregates form and their relative motion in the shear flow (translation and rotation) guides their further encounter and fusion. Bottom panels. Time evolution of key parameters describing the aggregation process in shear-flow for different values of the shear rate Δv/Δz (0,108,109,1010 s-1): the gyration radius Rg (left panel), the number of inter-peptide hydrogen bonds formed between backbone NH and O groups (mid panel) and number of inter-peptide side-chain contacts (right panel). The system is composed of 18 peptides placed in a cubic box of L=65 Å and the shear gradient is generated along the Z direction.
A third challenge is to have a direct observation of Aβ protein self-assembly in live cells as a result of crowding effects. Using noninvasive fluorescence lifetime recordings and super-resolution fluorescence, the formation of Aβ1– 40 and Aβ1–42 aggregates in live cells was dissected. Both peptides are retained in lysosomes, where their accumulation leads to aggregation, but the kinetics of Aβ1–42 aggregation are considerably faster than those of Aβ1–40 and, unlike Aβ1–40, show no detectable lag phase. Compact amyloid aggregates were observed for both alloforms.(486) While, these experiments represent one step ahead towards understanding aggregation in the cells, higher spatial resolution methods and how the cellular environment affects the dynamics of Aβ1-40/42 and their variants are major concerns. To this end, in cell-NMR of Aβ protein is an ideal tool for gaining information at the atomic level, but several obstacles remain.(487-488)
It is also a challenge to determine the impact of PrP and HAS on Aβ oligomerization. We are addressing both aspects experimentally by polymerization and de-polymerization kinetic experiments and theoretically by performing exascale simulations. In this respect, we have recently shown that it is possible to get the dynamics of 18 000 HAS proteins comprising 80 millions of particles with hydrodynamics interactions consistent with the experimental translational and rotational diffusion constants as a function of the density of the system.(61)
Finally, pathogenic events involve an imbalance between the production and the clearance of the Aβ peptide. It is important to understand at atomic detail how the Aβ is cleaved by γ-secretase and how this process is affected by A2V and A2T mutations. Clearly, the recent release of the 3D structure of the human γ-secretase complex at 4.5 A(489) combined with simulations going beyond the Aβ1-55 dimer(490) should help clarify this issue.
While many inhibitors have been designed to target a specific region of Aβ, it would be interesting to study in cells the cumulative effect of inhibitors designed to recognize different regions of Aβ. It will be also of great interest to combine different drugs targeting Aβ processing and rendering Aβ aggregates very unstable and more prone to degradation. Today, we are just seeing the top of the iceberg in understanding phenotype-related toxicity and aggregation propensity of WT Aβ and their familial-disease and protective variants. But continuous and synergetic efforts between in vitro and in vivo studies (including basic verifications such as purity and reproducibility of the results using various readouts or transgenic animals with different sex and times of AD incubation) and theoretical studies (using multiple approaches) should get us closer to finding a cure for AD.
Acknowledgments
Jessica Nassica-Labouze thanks funding from the European Social Fund (ESF), Op. Programme2007-2013, Ob. 2, Axis 5, TALENTS FVG Programme - Activity 2 - OutgoingScheme - FP code: 1418521006, managed by AREA Science Park. Phuong Nguyen, Bogdan Tarus and Philippe Derreumaux thank DARI (grant x2014077198) for computer resources. Fabio Sterpone and Simone Melchionna acknowledge funding from ERC under the European Community's seventh framework program (FP7/2007-2013) grant agreement no. 258748. Simulations with shear forces used HPC resources from GENCI (CINES and TGCC, grants 2012 c2012086818 and 2013 x201376818. Olivia Berthoumieu and Peter Faller would like to thank their collaborators and in particular Drs. Christelle Hureau, Emmanuel Gras, Fabrice Collin, Giovanni La Penna, for helpful discussions. Viorel Buchette and Birgit Strodel would like to thank Ratnesh Lal and Ruth Nussinov for providing the AFM images shown in Fig.8, and John E. Straub and Ruth Nussinov for providing the structure files of the membrane-bound Aβ models illustrated in Fig.9. Li Mai thanks Narodowe Centrum Nauki in Poland (Grant 2011/01/B/NZ1/01622). Yuguang Mu acknowledges Tier 1 grant RG 23/11 from Nanyang Technological University, Singapore.
Finally Philippe Derreumaux thanks support of University of Paris Diderot, ANR SIMI7 GRAL 12-BS07-0017, ANR LABEX Grant “DYNAMO” (ANR-11-LABX-0011), 6th European PRCD (Immunoprion, FP6-Food023144), IUF, French/Singapore Merlion PhD program (Grant 5.08.10), Pierre de Gilles de Gennes Foundation and its international PhD grant program, Fudan University in China, CNRS – Polish Academy of Sciences (Grant 168836) and the CNRS Institute of Chemistry (INC).
Biographies
Jessica Nasica-Labouze: Institut de Biologie Physico-Chimique - CNRS UPR 9080 - 13, rue Pierre et Marie Curie - F-75005 Paris, France, and Statistical and biological physics, SISSA, Via Bonomea, 265 - 34136 Trieste, Italy.
Jessica Nasica-Labouze is a Postdoctoral Fellow on the Talents FVG outgoing mobility Fellowship (Consorzio per l'AREA di ricerca di Trieste) and is involved in a collaborative project between UPR9080 CNRS, Paris and SISSA, Italy. She got her Ph.D. in biophysics at the Universite de Montreal (Canada) where she worked on characterizing the self-assembly mechanism of amyloid peptides and then did a postdoc at SISSA, investigating the dynamics of chromatin via coarsegrained simulations. Her current research interests focus mainly on the effects of protective and pathogenic mutations on the folding and interactions of the amyloid-beta protein, involved in Alzheimer's disease, via a wide spectrum of computational methods.

Olivia Berthoumieu: CNRS, Laboratoire de Chimie de Coordination, 205 Route de Narbonne, 31077 Toulouse, France.
Olivia Berthoumieu is a post-doctoral researcher at the Coordination Chemistry Department in Toulouse, France. She graduated from Oxford University, where she received a PhD in Biochemistry. Her research interests include (a) membrane proteins as tools for bionanotechnological applications; (b) ligand-receptor interactions study and high resolution imaging using atomic force microscopy (AFM); (c) metal ions interactions with the amyloid-beta peptide and inhibition of Aβ aggregation.

Sébastien Côté: Department of Physics, Université de Montréal, C.P. 6128, succ. Centre-ville, Montréal (Quebec), Canada H3C 3J7.
Sébastien Côté is currently a Ph.D. student under the supervision of Professor Normand Mousseau in the Department of Physics at Université de Montréal in Montréal, Canada. He received his Honours B.Sc. in Physics at McGill University, Montréal, Canada in 2009. His research interests are non-amyloid/amyloid proteins folding/oligomerization, peptide–membrane interactions, and the development of new coarse-grained methodologies.

Normand Mousseau: Normand Mousseau is Professor of Physics and Canada Research Chair in Computational Physics of Complex Materials at Université de Montréal. He obtained his Ph.D. from Michigan State University and pursued post-doctoral studies at Oxford University, UK, and Université de Montréal. His research interests focus on the kinetics of complex systems at the atomic scale studied with the help of various computational methods often developed in his group. Over the last few years, in biophysics, he has been working on the first steps of amyloid aggregation as well as protein flexibility. He is currently working on the development of new coarse-grained potentials in addition to pursuing a number of avenues for accelerated sampling techniques.

Tong Zhang: School of Biological Sciences, Nanyang Technological University (NTU), 60 Nanyang Drive, Singapore, 637551, and UPR9080 CNRS, Paris.
Tong Zhang is a Ph.D. student under supervision of Prof. Derreumaux and Associate Professor Yuguang Mu in the School of Biological Sciences at NTU Singapore. He received his B.Sc. in Biology in the Nanyang Technological University, Singapore in 2010. His research interests are to use computer aided approaches to understand the folding and aggregation of Alzheimer's Aβ, Aβ/small molecule interactions. tzhang2@e.ntu.edu.sg

Yuguang Mu: School of Biological Sciences, Nanyang Technological University, 60 Nanyang Drive, Singapore, 637551.
Yuguang Mu is an Associate Professor in the School of Biological Sciences of Nanyang Technological University in Singapore. He got his Ph.D. in Shandong University, China and postdoc training under the Alexander von Humboldt foundation in Germany. His research interests are (a) MD simulation method and data analysis method development; (b) Peptide, protein folding, unfolding study, specially aimed at folding, misfolding mechanism which could lead to amyloid fibril; (c) DNA dynamics, DNA –protein, DNA-counterions interaction study.

Andrew Doig: Manchester Institute of Biotechnology, 131 Princess Street, University of Manchester, Manchester M1 7DN, UK.
Andrew Doig is a Professor of Biochemistry at the University of Manchester. He got his Ph.D. at the University of Cambridge, UK and postdoc training in the Department of Biochemistry at Stanford University, USA. His research interests are (a) drug discovery for Alzheimer's Disease; (b) bioinformatics studies on drug target proteins, genes in mouse development, amyloidoses and protein vibrational spectroscopy; (c) effects of β-amyloid on the proteome, metabolome and cytoskeleton.

Birgit Strodel: Forschungszentrum Jülich GmbH, Institute of Complex Systems: Structural Biochemistry (ICS-6), 52425 Jülich, Germany.
Birgit Strodel is the Head of the Multiscale Modeling Group at the Jülich Research Centre and is an Assistant Professor at the Institute of Theoretical and Computational Chemistry at the Heinrich Heine University Düsseldorf. Her main research interests are in the thermodynamics and kinetics of protein aggregation, and protein-protein interactions. A large part of the simulations performed in her lab focus on the aggregation of amyloidogenic peptides, aiming to understand the molecular basis of Alzheimer's disease.

Nicolae-Viorel Buchete: School of Physics, University College Dublin, Belfield, Dublin 4, Ireland.
Nicolae-Viorel (Vio) Buchete is a Lecturer of Theoretical & Computational Nano-Bio Physics in the School of Physics, and a PI in the Complex & Adaptive Systems Laboratory, a research institute in the University College Dublin (UCD), Ireland. He got his PhD from Boston University, and he held a postdoctoral research fellowship at the National Institutes of Health in Bethesda, MD, USA. Ongoing research projects in his group at UCD are concerned with statistical mechanics and conformational dynamics of biomolecular systems, protein folding, amyloid aggregation, structural aspects of systems biology and bioinformatics, and with multiscale modeling of biomolecules and complex fluids.

Alfonso De Simone: Department of Life Sciences, Imperial College London, South Kensington, London SW7 2AZ.
Alfonso De Simone is Lecturer and Group Leader at the Department of Life Sciences of the Imperial College London. He obtained the Ph.D. in 2007 in structural biology at the University of Padova. He was then postdoctoral research associate at the University of Cambridge in the laboratory of Chris Dobson, working at new combinations of biomolecular NMR and molecular simulations to characterize structure and dynamics of transient protein states along biochemical processes, including protein aggregation into amyloids, enzymatic reactions and biomaterial assembly. In 2011 he established his group at Imperial College London where he is working at the definition of advanced methods at the interface of solution/solid-state NMR and molecular simulations. Current research topics span intrinsically disordered proteins, membrane proteins, biotechnologically relevant enzymes and biomaterials.

Peter Faller: CNRS, Laboratoire de Chimie de Coordination, and University Paul Sabatier of Toulouse205, Route de Narbonne, 31077 Toulouse, France.
Professor in Chemistry at the University of Toulouse Paul Sabatier (F) and group leader at the Laboratoire de Chimie de Coordination du CNRS. Born in St. Gallen (CH); trained teacher for elementary school (Kreuzlingen, CH); Study of (Bio)Chemistry (Univ.Zürich, CH); PhD in (Bio)Chemistry on metallothioneins (Univ. Zürich); post-doc on photosystem II. Ongoing research projects of the group are on the interaction of amyloidogenic peptides with metal ions, inhibitors and markers.

Angel E. Garcia: Department of Physics, Applied Physics and Astronomy, and Center for Biotechnology and Interdisciplinary Studies, Rensselaer Polytechnic Institute, Troy, New York. USA
Angel E. Garcia is a Professor of Physics at Rensselaer Polytechnic Institute. He obtained a PhD in Theoretical Physics from Cornell University. His main research interest is the understanding of the physico-chemical principles that determine protein folding, stability and dynamics, and the application of these principles to built computer models that describe these processes. A main effort in his laboratory has been the characterization of the ensemble of conformations adopted by the Aβ-peptides in the monomeric state. He uses state of the art enhanced sampling methods and parallel computers to perform atomistic simulations of various forms of the Aβ peptides in explicit solvent, and over multiple microseconds timescales. These simulations are carefully validated against J-couplings, RDC and other data.

Alessandro Laio: Statistical and biological physics, SISSA, Via Bonomea 265, 34136, Trieste, Italy. Alessandro Laio is Associate Professor at the Physics department of SISSA. He got a Ph.D. in solid-state physics in SISSA. Then he moved for a postdoc to ETH, Switzerland, where he worked first on a QM/MM approach to molecular biochemistry, then on the development of techniques for computer simulations on rare events. His current research interests are: (a) the development of techniques aimed at computing the free energy by atomistic simulations. (b) Protein folding, protein aggregation and protein-protein interaction. (c) structural bioiformatics and algorithm development for data mining.

Mai Suan Li: Institute of Physics Polish Academy of Sciences, Al. Lotnikow 32/46, 02-668 Warsaw, Poland.
Mai Suan Li is a Professor of Physics at the Institute of Physics Polish Academy of Sciences. He got his Ph.D. at the Kishinev State University, Moldova and habilitation degree at the Institute of Physics Polish Academy of Sciences. His research interests are (a) development of coarse-grained models for studying protein folding and misfolding; (b) protein unfolding under external mechanical force; (c) mechanisms of protein and peptide aggregation; (d) computer-aided drug design for Alzheimer's disease, influenza A virus and breast cancer.

Simone Melchionna: IPCF, Consiglio Nazionale delle Ricerche, P.le A. Moro 2, 00185, Rome, Italy.
Simone Melchionna is a researcher at the Institute for the Chemico-Physical Processes, of the Consiglio Nazionale delle Ricerche. He has a Ph.D. in chemistry from the University of Rome – La Sapienza, during which we developed techniques for Molecular Dynamics of biological systems, such as constrained mechanics, enhanced sampling and isothermal-isobaric dynamical approaches. Then he moved for three years to Cambridge where he worked on confined fluids and water via density functional and other theoretical approaches. Subsequently, he worked on Lattice Boltzmann and multiscale simulation numerical methods, with applications to DNA translocation. His research focuses on high-performance computing applied to proteins and other biological systems.

Phuong Nguyen: Laboratoire de Biochimie Theorique, IBPC, 13 rue Pierre et Marie Curie, 75005, Paris, France.
Phuong Nguyen is CR1 permanent researcher at CNRS. He got a Ph.D in Condensed Matter Theory at the Physics Department of Bielefeld University, Germany. His postdoc at the Chemistry Department of Frankfurt University, Germany focused on nonequilibrium molecular dynamics simulations aimed at understanding energy flows, photoinduced conformational dynamics in biomolecules. His current research focuses on the development and application of efficient, physically rigorous theoretical methods for studying equilibrium and nonequilibrium structure, dynamics and thermodynamics of single and amyloid proteins at all-atom and coarse-grained levels.

Anant Paravastu: Department of Chemical and Biomedical Engineering, Florida State University and Florida A&M University and the National High Magnetic Field Laboratory, 2525 Pottsdamer St., Tallahassee, Florida, 32310 USA
Anant Paravastu is an Associate Professor of Chemical and Biomedical Engineering at Florida State University. He received his Ph.D. in chemical engineering at the University of California, Berkeley, USA and his postdoctoral training at the National Institutes of Health. His primary research interests are in solid state NMR structural analysis of peptide assembles, including Aβ oligomers and designer peptide nanofibers with applications in regenerative medicine.

Samuela Pasquali: Laboratoire de Biochimie Théorique, CNRS UPR 9080 – Université Paris Diderot. Samuela Pasquali is Associate professor in the Biology/Biochemistry department of the university of Paris Diderot, where she teaches math and bioinformatics to biology students. She got a Ph.D. in statistical physics in 2005 at New York University, having worked on theoretical modeling of RNA folding and polymer folding under confinement. She then moved to Paris at ESPCI for a 3-years postdoc on numerical approaches to compute Casimir forces, both classical (Van der Waals) and quantum. In 2008 she was hired as an associate professor at Paris Diderot and since then she devoted her research to the problem of RNA folding for which she developed efficient coarse-grained model and the improvement of the coarse-grained protein model for amyloid and non-amyloid proteins.

David J Rosenman: Department of Biology, Rensselaer Polytechnic Institute, 110 8th Street, Troy, NY 12180, USA
David J. Rosenman got his B.S. with Honors in Biology from the California Institute of Technology in 2009. He became a PhD student in the Biology Department at Rensselaer Polytechnic Institute in 2010, under the mentorship of Dr. Angel E.García and Dr. Chunyu Wang. His research focuses on the combined use of molecular dynamics and nuclear magnetic resonance techniques to characterize the conformational ensembles of the amyloid beta (Aβ) peptides implicated in Alzheimer's disease.

Fabio Sterpone: Laboratoire de Biochimie Théorique, IBPC, 13 Rue Pierre et Marie Curie, Paris, France.
Fabio Sterpone is currently researcher at the CNRS, France. He graduated from the University of Paris UPMC (Biophysics) and occupied several post-doc positions later on; he dealt with quantum classical simulations of materials and the effect of solvent on biomolecular structure and dynamics. Presently, he is mainly interested in the study of protein stability and aggregation in extreme-environments by applying and developing multi-scale simulation methodologies.

Bogdan Tarus: Laboratory of Theoretical Biochemistry - UPR 9080, IBPC, 13 rue Pierre et Marie Curie, 75005 Paris, France.
Bogdan Tarus is postdoc at the Laboratory of Theoretical Biochemistry, Institut de Biologie Physico-Chimique. He got a PhD in physical chemistry at Boston University, Massachusetts. Then he moved for a postdoc to Heidelberg University, Germany, where he continued to work on amyloid protein aggregation. Moving to INRA, France, he worked on designing of small chemical compounds that interact with proteins of biological interest. His work at IBPC involves studying of amyloid protein aggregation and developing of chemical compounds to inhibit the aggregation. His current research interests are: protein dynamics and aggregation using computational methods at atomistic and coarsegrained resolution, drug design, protein-drug interaction, and inhibition of protein aggregation.

John H Viles: School of Biological and Chemical Sc., Queen Mary, University of London, London E1 4AS Dr John Viles has an active research program studying protein misfolding associated with amyloid formation in neurodegenerative diseases. Biophysical approaches include: NMR; CD; IR; EPR and transmission electron microscopy. His main research areas include: Copper and zinc in Alzheimer's disease; the structure and misfolding of the prion protein in TSE's ‘mad-cow’ disease; Amyloid-beta and its protein binding partners. He received his Ph.D. from the University of London in 1994 and continued his research with Prof P J Sadler (FRS) studying metallo-proteins using NMR. In 1997 he took up a post-doctoral position with Prof P Wright at the Scripps Research Institute, California. In collaboration, with the Nobel Laureate, Prof S Prusiner, he has published a number of significant papers on the structure of the prion protein. He returned to the UK in 2000 to take up a lectureship position at Queen Mary and is currently an Associate Professor (Reader) in Biochemistry at Queen Mary, University of London.

Chunyu Wang: Department of Biological Sciences, and Center for Biotechnology and Interdisciplinary Studies, Rensselaer Polytechnic Institute, Troy, New York. USA Chunyu Wang is an Associate Professor of Biology at Rensselaer Polytechnic Institute. He obtained his PhD in Biochemistry from Cornell University and MD from Peking Union Medical College. He mainly uses solution NMR technique to study the structure, dynamics and interactions of Amyloid-β peptide (Aβ), in combination with molecular dynamics simulation. His other interests include intein-mediated protein splicing and related phenomena, such as Hedgehog autoprocessing.

Philippe Derreumaux: Laboratoire de Biochimie Théorique, UPR9090 CNRS, IBPC, 13 Rue Pierre et Marie Curie, Paris, France
Philippe has been director of UPR9080 since 2007 and is senior member of IUF. He got his HDR in Physics at Pierre and Marie Curie University. He has worked with Gerard Vergoten, Warner Peticolas, Martin Karplus and Tamar Schlick during 1993-2004. Presently, he is interested in the development of enhanced conformational techniques, and coarse-grained protein (OPEP) and ARN (Hire-RNA) models. The theoretical advances are used to understand the aggregation process of amyloid proteins, and notably the Aβ protein, in aqueous solution, and comprehend the equilibrium conformation ensemble and dynamics of Aβ in a crowding environment with the prion protein and serum albumin or in a real cell environment.

References
- 1.Alzheimer US Association. http://www.alz.org/
- 2.Hardy J, Selkoe DJ. The Amyloid Hypothesis of Alzheimer's Disease: Progress and Problems on The Road To Therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 3.Hernández-Rodríguez M, Correa-Basurto J, Martínez-Ramos F, Padilla-Martínez II, Benítez-Cardoza CG, Mera-Jiménez E, Rosales-Hernández MC. Design of Multi-target Compounds as AChE, BACE1, and amyloid-β(1-42) Oligomerization Inhibitors: in silico and in vitro Studies. J Alzheimers Dis. 2014;41:1073–1085. doi: 10.3233/JAD-140471. [DOI] [PubMed] [Google Scholar]
- 4.Selkoe DJ. Alzheimer's Disease: Genes, Proteins, and Therapy. Physiol Rev. 2001;81:741–766. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- 5.Tan CC, Yu JT, Wang HF, Tan MS, Meng XF, Wang C, Jiang T, Zhu XC, Tan L. Efficacy and Safety of Donepezil, Galantamine, Rivastigmine, and Memantine for the Treatment of Alzheimer's Disease: A Systematic Review and Meta-analysis. J Alzheimers Dis. 2014;41:615–631. doi: 10.3233/JAD-132690. [DOI] [PubMed] [Google Scholar]
- 6.Valverde E, Sureda FX, Vázquez S. Novel Benzopolycyclic Amines with NMDA Receptor Antagonist Activity. Bioorg Med Chem. 2014;22:2678–2683. doi: 10.1016/j.bmc.2014.03.025. [DOI] [PubMed] [Google Scholar]
- 7.Yan R, Vasser R. Targeting the β secretase BACE1 for Alzheimer's Disease Therapy. Lancet Neurol. 2014;13:319–329. doi: 10.1016/S1474-4422(13)70276-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wischik CM, Harrington CR, Storey JM. Tau-aggregation Inhibitor Therapy for Alzheimer's Disease. Biochem Pharmacol. 2014;88:529–539. doi: 10.1016/j.bcp.2013.12.008. [DOI] [PubMed] [Google Scholar]
- 9.(a) Doig AJ, Derreumaux P. Inhibition of Protein Aggregation and Amyloid Formation by Small Molecules. Curr Opin Struct Biol. 2015;30C:50–56. doi: 10.1016/j.sbi.2014.12.004. [DOI] [PubMed] [Google Scholar]; (b) Jiang L, Liu C, Leibly D, Landau M, Zhao M, Hughes MP, Eisenberg DS. Structure-based Discovery of Fiber-binding Compounds that Reduce the Cytotoxicity of Amyloid beta. Elife. 2013;2:e00857. doi: 10.7554/eLife.00857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, Brett FM, Farrell MA, Rowan MJ, Lemere CA, Regan CM, Walsh DM, Sabatini BL, Selkoe DJ. Amyloid-beta Protein Dimers Isolated Directly from Alzheimer's Brains Impair Synaptic Plasticity and Memory. Nat Med. 2008;14:837–842. doi: 10.1038/nm1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.(a) Bernstein SL, Dupuis NF, Lazo ND, Wyttenbach T, Condron NN, Bitan G, Teplow DB, Shea JE, Ruotolo BT, Robinson CV, Bowers MT. Amyloid-β Protein Oligomerization and the Importance of Tetramers and Dodecamers in the Aetiology of Alzheimer's Disease. Nat Chem. 2009;1:326–331. doi: 10.1038/nchem.247. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Sherman MA, Lesné SE. Detecting Aβ*56 Oligomers in Brain Tissues. Methods Mol Biol. 2011;670:45–56. doi: 10.1007/978-1-60761-744-0_4. [DOI] [PubMed] [Google Scholar]
- 12.Jan A, Adolfsson O, Allaman I, Buccarello AL, Magistretti PJ, Pfeifer A, Muhs A, Lashuel HA. Abeta42 Neurotoxicity is Mediated by Ongoing Nucleated Polymerization Process Rather Than by Discrete Abeta42 Species. J Biol Chem. 2011;286:8585–8596. doi: 10.1074/jbc.M110.172411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Knowles TP, Waudby CA, Devlin GL, Cohen SI, Aguzzi A, Vendruscolo M, Terentjev EM, Welland ME, Dobson CM. An Analytical Solution to the Kinetics of Breakable Filament Assembly. Science. 2009;326:1533–1537. doi: 10.1126/science.1178250. [DOI] [PubMed] [Google Scholar]
- 14.Cohen SI, Linse S, Luheshi LM, Hellstrand E, White DA, Rajah L, Otzen DE, Vendruscolo M, Dobson CM, Knowles TP. Proliferation of Amyloid-β42 Aggregates Occurs Through a Secondary Nucleation Mechanism. Proc Natl Acad Sci USA. 2013;110:9758–9763. doi: 10.1073/pnas.1218402110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schmit JD, Gosh K, Dill K. What Drives Amyloid Molecules to Assemble into Oligomers and Fibrils? Biophys J. 2011;100:450–458. doi: 10.1016/j.bpj.2010.11.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jeong JS, Ansaloni A, Mezzenga R, Lashuel HA, Dietler G. Novel Mechanistic Insight into the Molecular Basis of Amyloid Polymorphism and Secondary Nucleation During Amyloid Formation. J Mol Biol. 2013;425:1765–1781. doi: 10.1016/j.jmb.2013.02.005. [DOI] [PubMed] [Google Scholar]
- 17.Qiang W, Kelley K, Tycko R. Polymorph-specific Kinetics and Thermodynamics of β-amyloid Fibril Growth. J Am Chem Soc. 2013;135:6860–6871. doi: 10.1021/ja311963f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tycko R, Wickner RB. Molecular Structures of Amyloid and Prion Fibrils: Consensus versus Controversy. Acc Chem Res. 2013;46:1487–1496. doi: 10.1021/ar300282r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Meisl G, Yang X, Hellstrand E, Frohm B, Kirkegaard JB, Cohen SI, Dobson CM, Linse S, Knowles TP. Differences in Nucleation Behavior Underlie the Contrasting Aggregation Kinetics of the Aβ40 and Aβ42 peptides. Proc Natl Acad Sci USA. 2014;111:9384–9389. doi: 10.1073/pnas.1401564111. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Assarsson A, Hellstrand E, Cabaleiro-Lago C, Linse S. Charge Dependent Retardation of Amyloid β Aggregation by Hydrophilic Proteins. ACS Chem Neurosci. 2014;4:266–274. doi: 10.1021/cn400124r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kirkitadze MD, Condron MM, Teplow DB. Identification andCharacterization of Key Kinetic Intermediates in Amyloid beta-protein Fibrillogenesis. J Mol Biol. 2001;312:1103–1119. doi: 10.1006/jmbi.2001.4970. [DOI] [PubMed] [Google Scholar]
- 21.Hou L, Shao H, Zhang Y, Li H, Menon NK, Neuhaus EB, Brewer JM, Byeon IJL, Ray DG, Vitek MP, Iwashita T, Makula RA, Przybyla AB, Zagorski MG. Solution NMR studies of the Abeta(1-40) and Abeta(1-42) Peptides Establish that the Met35 Oxidation State Affects the Mechanism of Amyloid Formation. J Am Chem Soc. 2004;126:1992–2005. doi: 10.1021/ja036813f. [DOI] [PubMed] [Google Scholar]
- 22.Sarroukh R, Cerf E, Derclaye S, Dufrêne YF, Goormaghtigh E, Ruysschaert JM, Raussens V. Transformation of Amyloid beta(1-40) Oligomersinto Fibrils is Characterized by a Major Change in Secondary Structure. Cell Mol Life Sci. 2011;68:1429–1438. doi: 10.1007/s00018-010-0529-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bertini I, Gallo G, Korsak M, Luchinat C, Mao J, Ravera E. Formation Kinetics and Structural Features of beta-amyloid Aggregates by Sedimented solute NMR. Chembiochem. 2013;14:1891–1897. doi: 10.1002/cbic.201300141. [DOI] [PubMed] [Google Scholar]
- 24.Kim BH, Lyubchenko YL. Nanoprobing of Misfolding and Interactions of Amyloid β42 Protein. Nanomedicine. 2014;4:871–878. doi: 10.1016/j.nano.2013.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.García S, Cuscó C, Brissos RF, Torrents E, Caubet A, Gamez P. Dual Role of Cu2+ Ions on the Aggregation and Degradation of soluble Aβ Oligomers and Protofibrils Investigated by Fluorescence Spectroscopy and AFM. J Inorg Biochem. 2012;116:26–36. doi: 10.1016/j.jinorgbio.2012.07.007. [DOI] [PubMed] [Google Scholar]
- 26.Lv Z, Roychaudhuri R, Condron MM, Teplow DB, Lyubchenko YL. Mechanism of Amyloid β–protein dimerization determined using single–molecule AFM force spectroscopy. Sci Rep. 2013;3:2880–2888. doi: 10.1038/srep02880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Petkova AT, Leapman RD, Guo ZH, Yau WM, Mattson MP, Tycko R. Self-Propagating, Molecular-Level Polymorphism in Alzheimer's beta-Amyloid Fibrils. Science. 2005;307:262–265. doi: 10.1126/science.1105850. [DOI] [PubMed] [Google Scholar]
- 28.Petkova AT, Ishii Y, Balbach JJ, Antzutkinm ON, Leapman RD, Delaglio F, Tycko R. A Structural Model for Alzheimer's β-amyloid Fibrils based on Experimental Constraints from Solid State NMR. Proc Natl Acad Sci USA. 2002;99:16742–16747. doi: 10.1073/pnas.262663499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Luhrs T, Ritter C, Adrian M, Riek-Loher D, Bohrmann B, Doeli H, Schubert D, Riek R. 3D Structure of Alzheimer's Amyloid-β(1–42) Fibrils. Proc Natl Acad Sci USA. 2005;102:17342–17347. doi: 10.1073/pnas.0506723102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lu JX, Qiang W, Yau WM, Schwieters CD, Meredith SC, Tycko R. Molecular Structure of β-amyloid Fibrils in Alzheimer's Disease Brain tissue. Cell. 2013;154:1257–1268. doi: 10.1016/j.cell.2013.08.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gunderson W, Hernández-Guzmán J, Karr J, Sun L, Szalai V, Warncke K. Local Structure and Global Patterning of Cu2+ Binding in Fibrillar Amyloid-β [Aβ(1–40)] Protein. J Am Chem Soc. 2012;134:18330–18337. doi: 10.1021/ja306946q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Eury H, Bijani C, Faller P, Hureau C. Copper(II) Coordination to Amyloid β : Murine versus Human Peptide. Angew Chem Int Ed. 2011;50:901–905. doi: 10.1002/anie.201005838. [DOI] [PubMed] [Google Scholar]
- 33.Stanyon HF, Viles JH. Human Serum Albumin Can Regulate Amyloid-β Peptide Fiber Growth in the Brain Interstitium. J Biol Chem. 2012;287:28163–28168. doi: 10.1074/jbc.C112.360800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Laurén J, Gimbel DA, Nygaard HB, Gilbert JW, Strittmatter SM. Cellular Prion Protein Mediates Impairment of Synaptic Plasticity by Amyloid-beta Oligomers. Nature. 2009;457:1128–1132. doi: 10.1038/nature07761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Younan ND, Sarell CJ, Davies P, Brown DR, Viles JH. The Cellular Prion Protein Traps Alzheimer's Aβ in an Oligomeric Form and Disassembles Amyloid Fibers. FASEB J. 2013;27:1847–1858. doi: 10.1096/fj.12-222588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.(a) Zou K, Gong JS, Yanagisawa K, Michikawa M. A Novel Function of Monomeric Amyloid beta-protein Serving as an Antioxidant Molecule against Metal-induced Oxidative Damage. J Neurosci. 2002;22:4833–4841. doi: 10.1523/JNEUROSCI.22-12-04833.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) State of aggregation. Nat Neurosci | Editorial Nat Neurosci. 2011;14:399. doi: 10.1038/nn0411-399. [DOI] [PubMed] [Google Scholar]
- 37.Götz J, Matamales M, Götz NN, Ittner LM, Eckert A. Alzheimer's Disease Models and Functional Genomics—How Many Needles are there in the Haystack? Front Physiol. 2012;3:320. doi: 10.3389/fphys.2012.00320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kim T, Vidal GS, Djurisic M, William CM, Birnbaum ME, Garcia KC, Hyman BT, Shatz CJ. Human LilrB2 Is a β-Amyloid Receptor and Its Murine Homolog PirB Regulates Synaptic Plasticity in an Alzheimer's Model. Science. 2013;341:1399–1404. doi: 10.1126/science.1242077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zempel H, Luedtke J, Kumar Y, Biernat J, Dawson H, Mandelkow E, Mandelkow EM. Amyloid-β Oligomers Induce Synaptic Damage via Tau-dependent Microtubule Severing by TTLL6 and Spastin. EMBO J. 2013;32:2920–2937. doi: 10.1038/emboj.2013.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Um JW, Kaufman AC, Kostylev M, Heiss JK, Stagi M, Takahashi H, Kerrisk ME, Vortmeyer A, Wisniewski T, Koleske AJ, Gunther EC, Nygaard HB, Strittmatter SM. Metabotropic Glutamate Receptor 5 Is a Coreceptor for Alzheimer Aβ Oligomer Bound to Cellular Prion Protein. Neuron. 2013;79:887–902. doi: 10.1016/j.neuron.2013.06.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kanekiyo T, Xu B, Bu G. ApoE and Aβ in Alzheimer's Disease: Accidental Encounters or Partners? Neuron. 2014;81:740–754. doi: 10.1016/j.neuron.2014.01.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Aisen PS, Vellas B, Hampel H. Moving towards early clinical trials for amyloid-targeted therapy in Alzheimer's disease. Nat Rev Drug Discov. 2013;4:324. doi: 10.1038/nrd3842-c1. [DOI] [PubMed] [Google Scholar]
- 43.Abbott A. The plaque plan. Nature. 2008;458:161–164. doi: 10.1038/456161a. [DOI] [PubMed] [Google Scholar]
- 44.Nguyen P, Derreumaux P. Understanding Amyloid Fibril Nucleation and Aβ Oligomer/Drug Interactions from Computer Simulations. Acc Chem Res. 2014;47:603–611. doi: 10.1021/ar4002075. [DOI] [PubMed] [Google Scholar]
- 45.Moore BD, Chakrabarty P, Levites Y, Kukar TL, Baine AM, Moroni T, Ladd TB, Das P, Dickson DW, Golde TE. Overlapping Profiles of Aβ Peptides in the Alzheimer's Disease and Pathological Aging Brains. Alzheimers Res Ther. 2012;4:18. doi: 10.1186/alzrt121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rembach A, Faux NG, Watt AD, Pertile KK, Rumble RL, Trounson BO, Fowler CJ, Roberts BR, Perez KA, Li QX, Laws SM, Taddei K, Rainey-Smith S, Robertson JS, Vandijck M, Vanderstichele H, Barnham KJ, Ellis KA, Szoeke C, Macaulay L, Rowe CC, Villemagne VL, Ames D, Martins RN, Bush AI, Masters CL AIBL research group. Changes in Plasma Amyloid beta in a Longitudinal Study of Aging and Alzheimer's Disease. Alzheimers Dement. 2014;10:53–61. doi: 10.1016/j.jalz.2012.12.006. [DOI] [PubMed] [Google Scholar]
- 47.Rezaei-Ghaleh N, Amininasab M, Giller K, Kumar S, Stündl A, Schneider A, Becker S, Walter J, Zweckstetter M. Turn Plasticity Distinguishes Different Modes of Amyloid-β Aggregation. J Am Chem Soc. 2014;136:4913–4919. doi: 10.1021/ja411707y. [DOI] [PubMed] [Google Scholar]
- 48.Atwood CS, Perry G, Zeng H, Kato Y, Jones WD, Ling KQ, Huang X, Moir RD, Wang D, Sayre LM, Smith MA, Chen SG, Bush AI. Copper Mediates Dityrosine Cross-Linking of Alzheimer's Amyloid-beta. Biochemistry. 2004;43:560–568. doi: 10.1021/bi0358824. [DOI] [PubMed] [Google Scholar]
- 49.Masters CL, Selkoe DJ. Biochemistry of Amyloid β-Protein and Amyloid Deposits in Alzheimer Disease. Cold Spring Harb Perspect Med. 2012;2:a006262. doi: 10.1101/cshperspect.a006262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nussbaum JM, Schilling S, Cynis H, Silva A, Swanson E, Wangsanut T, Tayler K, Wiltgen B, Hatami A, Rönicke R, Reymann K, Hutter-Paier B, Alexandru A, Jagla W, Graubner S, Glabe CG, Demuth HU, Bloom GS. Prion-like Behaviour and Tau-dependent Cytotoxicity of Pyroglutamylated Amyloid-β. Nature. 2012;485:651–655. doi: 10.1038/nature11060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Deng B, Lian Y, Wang X, Zeng F, Jiao B, Wang YR, Liang CR, Liu YH, Bu XL, Yao XQ, Zhu C, Shen L, Zhou HD, Zhang T, Wang YJ. Identification of a Novel Mutation in the Presenilin 1 Gene in a Chinese Alzheimer's Disease Family. Neurotox Res. 2014;26:211–215. doi: 10.1007/s12640-014-9462-3. [DOI] [PubMed] [Google Scholar]
- 52.Tomiyama T, Nagata T, Shimada H, Teraoka R, Fukushima A, Kanemitsu H, Takuma H, Kuwano R, Imagawa M, Ataka S, Wada Y, Yoshioka E, Nishizaki T, Watanabe Y, Mori H. A New Amyloid beta Variant Favoring Oligomerization in Alzheimer's-type Dementia. Ann Neurol. 2008;63:377–387. doi: 10.1002/ana.21321. [DOI] [PubMed] [Google Scholar]
- 53.Gessel MM, Bernstein S, Kemper M, Teplow DB, Bowers MT. Familial Alzheimer's Disease Mutations Differentially Alter Amyloid β-Protein Oligomerization. ACS Chem Neurosci. 2012;3:909–918. doi: 10.1021/cn300050d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Di Fede G, Catania M, Morbin M, Rossi G, Suardi S, Mazzoleni G, Merlin M, Giovagnoli AR, Prioni S, Erbetta A, Falcone C, Gobbi M, Colombo L, Bastone A, Beeg M, Manzoni C, Francescucci B, Spagnoli A, Cantù L, Del Favero E, Levy E, Salmona M, Tagliavini FA. Recessive Mutation in the APP Gene with Dominant-negative Effect on Amyloidogenesis. Science. 2009;323:1473–1477. doi: 10.1126/science.1168979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jonsson T, Atwal JK, Steinberg S, Snaedal J, Jonsson PV, Bjornsson S, Stefansson H, Sulem P, Gudbjartsson D, Maloney J, Hoyte K, Gustafson A, Liu Y, Lu Y, Bhangale T, Graham RR, Huttenlocher J, Bjornsdottir G, Andreassen OA, Jönsson EG, Palotie A, Behrens TW, Magnusson OT, Kong A, Thorsteinsdottir U, Watts RJ, Stefansson K. A Mutation in APP Protects against Alzheimer's Disease and Age-related Cognitive Decline. Nature. 2012;488:96–99. doi: 10.1038/nature11283. [DOI] [PubMed] [Google Scholar]
- 56.Zhang Y, Rempel DL, Zhang J, Sharma AK, Mirica LM, Gross ML. Pulsed Hydrogen–deuterium Exchange Mass Spectrometry Probes Conformational Changes in Amyloid beta (Aβ) Peptide Aggregation. Proc Natl Acad Sci USA. 2013;110:14604–14609. doi: 10.1073/pnas.1309175110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Santi S, Musi V, Descrovi E, Paeder V, Di Francesco J, Hvozdara L, van der Wal P, Lashuel HA, Pastore A, Neier R, Herzig HP. Real-time Amyloid Aggregation Monitoring with a Photonic Crystal-based Approach. Chemphyschem. 2013;14:3476–3482. doi: 10.1002/cphc.201300633. [DOI] [PubMed] [Google Scholar]
- 58.Johnson RD, Schauerte JA, Chang CC, Wisser KC, Althaus JC, Carruthers CJ, Sutton MA, Steel DG, Gafni A. Single-molecule ImagingReveals aβ42:aβ40 Ratio-dependent Oligomer Growth on Neuronal Processes. Biophys J. 2013;104:894–903. doi: 10.1016/j.bpj.2012.12.051. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Narayan P, Ganzinger KA, McColl J, Weimann L, Meehan S, Qamar S, Carver JA, Wilson MR, St George-Hyslop P, Dobson CM, Klenerman D. Single Molecule Characterization of the Interactions between Amyloid-β Peptides and the Membranes of Hippocampal Cells. J Am Chem Soc. 2013;135:1491–1498. doi: 10.1021/ja3103567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gu L, Liu C, Guo Z. Structural Insights into Aβ42 Oligomers using Site-directed Spin Labeling. J Biol Chem. 2013;288:18673–18683. doi: 10.1074/jbc.M113.457739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Drew SC, Noble CJ, Masters CL, Hanson GR, Barnham KJ. Pleomorphic Copper Coordination by Alzheimer's Disease Amyloid-β Peptide. J Am Chem Soc. 2009;131:1195–1207. doi: 10.1021/ja808073b. [DOI] [PubMed] [Google Scholar]
- 61.Sterpone F, Melchionna S, Tuffery P, Pasquali S, Mousseau N, Cragnolini T, Chebaro Y, St-Pierre J, Kalimeri M, Barducci A, Laurin Y, Tek A, Baaden M, Nguyen PH, Derreumaux P. The OPEP Protein Model: From Single Molecules, Amyloid Formation, Crowding and Hydrodynamics to DNA/RNA Systems. Chem Soc Rev. 2014;43:4871–4893. doi: 10.1039/c4cs00048j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tycko R. Solid-State NMR Studies of Amyloid Fibril Structure. Annu Rev Phys Chem. 2011;62:279–299. doi: 10.1146/annurev-physchem-032210-103539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sarroukh R, Goormaghtigh E, Ruysschaert J, Raussens V. ATR-FTIR: A “Rejuvenated” Tool to Investigate Amyloid Proteins. Biochim Biophys Acta. 2013;1828:2328–2338. doi: 10.1016/j.bbamem.2013.04.012. [DOI] [PubMed] [Google Scholar]
- 64.Miller Y, Ma B, Nussinov R. Polymorphism in Alzheimer Aβ Amyloid Organization Reflects Conformational Selection in a Rugged Energy Landscape. Chem Rev. 2010;110:4820–4838. doi: 10.1021/cr900377t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Faendrich M, Schmidt M, Grigorieff N. Recent Progress in Understanding Alzheimer's β-Amyloid Structures. Trends Biochem Sci. 2011;36:338–345. doi: 10.1016/j.tibs.2011.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Meinhardt J, Sachse C, Hortschansky P, Grigorieff N, Faendrich M. Aβ(1-40) Fibril Polymorphism Implies Diverse Interaction Patterns in Amyloid Fibrils. J Mol Biol. 2009;386:869–877. doi: 10.1016/j.jmb.2008.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Paravastu AK, Petkova AT, Tycko R. Polymorphic Fibril Formation by Residues 10-40 of the Alzheimer's β-Amyloid Peptide. Biophys J. 2006;90:4618–4629. doi: 10.1529/biophysj.105.076927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Paravastu AK, Leapman RD, Yau WM, Tycko R. Molecular Structural Basis for Polymorphism in Alzheimer's β-Amyloid Fibrils. Proc Natl Acad Sci USA. 2008;105:18349–18354. doi: 10.1073/pnas.0806270105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chen B, Thurber KR, Shewmaker F, Wickner RB, Tycko R. Measurement of Amyloid Fibril Mass-Per-Length by Tilted-Beam Transmission Electron Microscopy. Proc Natl Acad Sci USA. 2009;106:14339–14344. doi: 10.1073/pnas.0907821106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Nilsson MR. Techniques to Study Amyloid Fibril Formation In Vitro. Methods. 2004;34:151–160. doi: 10.1016/j.ymeth.2004.03.012. [DOI] [PubMed] [Google Scholar]
- 71.Fraser P, Nguyen J, Surewicz W, Kirschner D. pH-Dependent Structural Transitions of Alzheimer Amyloid Peptides. Biophys J. 1991;60:1190–1201. doi: 10.1016/S0006-3495(91)82154-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sunde M, Serpell LC, Bartlam M, Fraser PE, Pepys MB, Blake CCF. Common Core Structure of Amyloid Fibrils by Synchrotron X-ray Diffraction. J Mol Biol. 1997;273:729–739. doi: 10.1006/jmbi.1997.1348. [DOI] [PubMed] [Google Scholar]
- 73.Eanes ED, Glenner GG. X-ray Diffraction Studies on Amyloid Filaments. J Histochem Cytochem. 1968;16:673–677. doi: 10.1177/16.11.673. [DOI] [PubMed] [Google Scholar]
- 74.Serpell LC, Fraser PE, Sunde M. X-ray Fiber Diffraction of Amyloid Fibrils. Methods Enzymol. 1999;309:526–536. doi: 10.1016/s0076-6879(99)09036-9. [DOI] [PubMed] [Google Scholar]
- 75.Morris KL, Serpell LC. X-Ray Fibre Diffraction Studies of Amyloid Fibrils Amyloid Proteins: Methods and Protocols. (Second) 2012;849:121–135. doi: 10.1007/978-1-61779-551-0_9. [DOI] [PubMed] [Google Scholar]
- 76.Kisilevsky R. Review: Amyloidogenesis - Unquestioned Answers and Unanswered Questions. J Struct Biol. 2000;130:99–108. doi: 10.1006/jsbi.2000.4222. [DOI] [PubMed] [Google Scholar]
- 77.Kheterpal I, Williams A, Murphy C, Bledsoe B, Wetzel R. Structural Features of the Aβ Amyloid Fibril Elucidated by Limited Proteolysis. Biochemistry. 2001;40:11757–11767. doi: 10.1021/bi010805z. [DOI] [PubMed] [Google Scholar]
- 78.Torok M, Milton S, Kayed R, Wu P, McIntire T, Glabe CG, Langen R. Structural and Dynamic Features of Alzheimer's Aβ Peptide in Amyloid Fibrils Studied by Site-Directed Spin Labeling. J Biol Chem. 2002;277:40810–40815. doi: 10.1074/jbc.M205659200. [DOI] [PubMed] [Google Scholar]
- 79.Petkova AT, Yau WM, Tycko R. Experimental Constraints on Quaternary Structure in Alzheimer's β-Amyloid Fibrils. Biochemistry. 2006;45:498–512. doi: 10.1021/bi051952q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Makin OS, Sikorski P, Serpell LC. Diffraction to Study Protein and Peptide Assemblies. Curr Opin Chem Biol. 2006;10:417–422. doi: 10.1016/j.cbpa.2006.08.009. [DOI] [PubMed] [Google Scholar]
- 81.Jahn TR, Makin OS, Morris KL, Marshall KE, Tian P, Sikorski P, Serpell LC. The Common Architecture of Cross-β Amyloid. J Mol Biol. 2010;395:717–727. doi: 10.1016/j.jmb.2009.09.039. [DOI] [PubMed] [Google Scholar]
- 82.Sipe JD, Cohen AS. Review: History of the Amyloid Fibril. J Struct Biol. 2000;130:88–98. doi: 10.1006/jsbi.2000.4221. [DOI] [PubMed] [Google Scholar]
- 83.(a) Sawaya MR, Sambashivan S, Nelson R, Ivanova MI, Sievers SA, Apostol MI, Thompson MJ, Balbirnie M, Wiltzius JJW, McFarlane HT, Madsen AO, Riekel C, Eisenberg D. Atomic Structures of Amyloid Cross-β Spines Reveal Varied Steric Zippers. Nature. 2007;447:453–457. doi: 10.1038/nature05695. [DOI] [PubMed] [Google Scholar]; (b) Eisenberg D, Jucker M. The Amyloid State of Proteins in Human Diseases. Cell. 2012;148:1188–1203. doi: 10.1016/j.cell.2012.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Park J, Kahng B, Hwang W. Thermodynamic Selection of Steric Zipper Patterns in the Amyloid Cross-β Spine. PLoS Comput Biol. 2009;5:e1000492. doi: 10.1371/journal.pcbi.1000492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Tycko R. Biomolecular Solid State NMR: Advances in Structural Methodology and Applications to Peptide and Protein Fibrils. Annu Rev Phys Chem. 2001;52:575–606. doi: 10.1146/annurev.physchem.52.1.575. [DOI] [PubMed] [Google Scholar]
- 86.Benzinger TLS, Gregory DM, Burkoth TS, Miller-Auer H, Lynn DG, Botto RE, Meredith SC. Propagating Structure of Alzheimer's β-amyloid(10-35) is Parallel β-sheet with Residues in Exact Register. Proc Natl Acad Sci U S A. 1998;95:13407–13412. doi: 10.1073/pnas.95.23.13407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lansbury PT, Costa PR, Griffiths JM, Simon EJ, Auger M, Halverson KJ, Kocisko DA, Hendsch ZS, Ashburn TT, Spencer RGS, Tidor B, Griffin RG. Structural Model for the β-Amyloid Fibril Based on Interstrand Alignment of an Antiparallel-Sheet Comprising a C-Terminal Peptide. Nat Struct Biol. 1995;2:990–998. doi: 10.1038/nsb1195-990. [DOI] [PubMed] [Google Scholar]
- 88.Lazo ND, Downing DT. Amyloid Fibrils May be Assembled from β-Helical Protofibrils. Biochemistry. 1998;37:1731–1735. doi: 10.1021/bi971016d. [DOI] [PubMed] [Google Scholar]
- 89.Fabian H, Nauman D, Otvos L, Schultz C, Backmann J. Impact of Point Mutations and Amino Acid Modifications on the Structure and Stability of Peptides and Proteins Probed by FTIR Spectroscopy. J Mol Struct. 1995;348:5–8. [Google Scholar]
- 90.Halverson K, Fraser PE, Kirschner DA, Lansbury PT., Jr Molecular Determinants of Amyloid Deposition in Alzheimer's Disease: Conformational Studies of Synthetic β-Protein Fragments. Biochemistry. 1990;29:2639–2644. doi: 10.1021/bi00463a003. [DOI] [PubMed] [Google Scholar]
- 91.Ishii Y. C-13-C-13 Dipolar Recoupling Under Very Fast Magic Angle Spinning in Solid-State Nuclear Magnetic Resonance: Applications to Distance Measurements, Spectral Assignments, and High-Throughput Secondary-Structure Determination. J Chem Phys. 2001;114:8473–8483. [Google Scholar]
- 92.Ishii Y, Balbach JJ, Tycko R. Measurement of Dipole-Coupled Lineshapes in a Many-Spin System by Constant-Time Two-Dimensional Solid State NMR with High-Speed Magic-Angle Spinning. Chem Phys. 2001;266:231–236. [Google Scholar]
- 93.Tycko R. Symmetry-Based Constant-Time Homonuclear Dipolar Recoupling in Solid State NMR. J Chem Phys. 2007;126:064506–064506. doi: 10.1063/1.2437194. [DOI] [PubMed] [Google Scholar]
- 94.(a) Balbach JJ, Ishii Y, Antzutkin ON, Leapman RD, Rizzo NW, Dyda F, Reed J, Tycko R. Amyloid Fibril Formation by Aβ(16-22), a Seven-Residue Fragment of the Alzheimer's β-Amyloid Peptide, and Structural Characterization by Solid State NMR. Biochemistry. 2000;39:13748–13759. doi: 10.1021/bi0011330. [DOI] [PubMed] [Google Scholar]; (b) Ma B, Nussinov R. Stabilities and Conformations of Alzheimer's β-Amyloid Peptide Oligomers (Aβ(16-22), Aβ(16-35), and Aβ(10-35)): Sequence Effects. Proc Natl Acad Sci USA. 2002;99:14126–14131. doi: 10.1073/pnas.212206899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Mikros E, Benaki D, Humpfer E, Spraul M, Loukas S, Stassinopoulou C, Pelecanou M. High-resolution NMR Spectroscopy of the beta-amyloid(1-28) Fibril Typical for Alzheimer's Disease. Angew Chem Int Edit. 2001;40:3603–3065. doi: 10.1002/1521-3773(20011001)40:19<3603::AID-ANIE3603>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 96.Antzutkin ON, Balbach JJ, Leapman RD, Rizzo NW, Reed J, Tycko R. Multiple Quantum Solid-State NMR Indicates a Parallel, not Antiparallel, Organization of β-Sheets in Alzheimer's β-Amyloid Fibrils. Proc Natl Acad Sci U S A. 2000;97:13045–13050. doi: 10.1073/pnas.230315097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Chi EY, Frey SL, Winans A, Lam KL, Kjaer K, Majewski J, Lee KY. Amyloid-β Fibrillogenesis Seeded by Interface-Induced Peptide Misfolding and Self-Assembly. Biophys J. 2010;98:2299–2308. doi: 10.1016/j.bpj.2010.01.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Bertini I, Gonnelli L, Luchinat C, Mao JF, Nesi A. A New Structural Model of Aβ(40) Fibrils. J Am Chem Soc. 2011;133:16013–16022. doi: 10.1021/ja2035859. [DOI] [PubMed] [Google Scholar]
- 99.Lopez del Amo JM, Schmidt M, Fink U, Dasari M, Fandrich M, Reif B. An Asymmetric Dimer as the Basic Subunit in Alzheimer's Disease Amyloid β Fibrils. Angew Chem Int Ed Engl. 2012;51:6136–6139. doi: 10.1002/anie.201200965. [DOI] [PubMed] [Google Scholar]
- 100.Wu C, Bowers MT, Shea JE. Molecular Structures of Quiescently Grown and Brain-Derived Polymorphic Fibrils of the Alzheimer Amyloid Aβ9-40 Peptide: A Comparison to Agitated Fibrils. PLoS Comput Biol. 2010;6:e1000693. doi: 10.1371/journal.pcbi.1000693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Qiang W, Yau W, Luo Y, Mattson MP, Tycko R. Antiparallel β-sheet Architecture in Iowa-Mutant β-amyloid Fibrils. Proc Natl Acad Sci USA. 2012;109:4443–4448. doi: 10.1073/pnas.1111305109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Pellarin R, Schuetz P, Guarnera E, Caflisch A. Amyloid FibrilPolymorphism is Under Kinetic Control. J Am Chem Soc. 2010;132:14960–14970. doi: 10.1021/ja106044u. [DOI] [PubMed] [Google Scholar]
- 103.Paravastu AK, Qahwash I, Leapman RD, Meredith SC, Tycko R. Seeded Growth of β-Amyloid Fibrils from Alzheimer's Brain-Derived Fibrils Produces a Distinct Fibril Structure. Proc Natl Acad Sci USA. 2009;106:7443–7448. doi: 10.1073/pnas.0812033106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Snyder SW, Ladror US, Wade WS, Wang GT, Barrett LW, Matayoshi ED, Huffaker HJ, Krafft GA, Holzman TF. Amyloid-β Aggregation: Selective Inhibition of Aggregation in Mixtures of Amyloid with Different Chain Lengths. Biophys J. 1994;67:1216–1228. doi: 10.1016/S0006-3495(94)80591-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Liosatos M, Morgan TE, Rozovsky I, Trommer B, Viola KL, Wals P, Zhang C, Finch CE, Drafft GA, Klein WL. Diffusible, Nonfibrillar Ligands Derived from Aβ1-42 are Potent Central Nervous System Neurotoxins. Proc Natl Acad Sci USA. 1998;95:6448–6453. doi: 10.1073/pnas.95.11.6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Kodali R, Wetzel R. Polymorphism in the Intermediates and Products of Amyloid Assembly. Curr Opin Struct Biol. 2007;17:48–57. doi: 10.1016/j.sbi.2007.01.007. [DOI] [PubMed] [Google Scholar]
- 107.Walsh DM, Selkoe DJ. Aβ Oligomers - A Decade of Discovery. J Neurochem. 2007;101:1172–1184. doi: 10.1111/j.1471-4159.2006.04426.x. [DOI] [PubMed] [Google Scholar]
- 108.Roychaudhuri R, Yang M, Hoshi MM, Teplow DB. Amyloid β-Protein Assembly and Alzheimer disease. J Biol Chem. 2009;284:4749–4753. doi: 10.1074/jbc.R800036200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Rangachari V, Moore BD, Reed DK, Sonoda LK, Bridges AW, Conboy E, Hartigan D, Rosenberry TL. Amyloid-β(1-42) Rapidly Forms Protofibrils and Oligomers by Distinct Pathways in Low Concentrations of Sodium Dodecylsulfate. Biochemistry. 2007;46:12451–12462. doi: 10.1021/bi701213s. [DOI] [PubMed] [Google Scholar]
- 110.Nichols MR, Moss MA, Reed DK, Lin W, Mukhopadhyay R, Hoh JH, Rosenberry TL. Growth of β-Amyloid(1-40) Protofibrils by Monomer Elongation and Lateral Association. Characterization of Distinct Products by Light Scattering and Atomic Force Microscopy. Biochemistry. 2002;41:6115–6127. doi: 10.1021/bi015985r. [DOI] [PubMed] [Google Scholar]
- 111.Tay WM, Huang D, Rosenberry TL, Paravastu AK. The Alzheimer's Amyloid-β(1-42) Peptide Forms Off-Pathway Oligomers and Fibrils that are Distinguished Structurally by Intermolecular Organization. J Mol Biol. 2013;425:2494–2508. doi: 10.1016/j.jmb.2013.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Sandberg A, Luheshi LM, Sollvander S, Pereira de Barros T, Macao B, Knowles TP, Biverstal H, Lendel C, Ekholm-Petterson F, Dubnovitsky A, Lannfelt L, Dobson CM, Hard T. Stabilization of Neurotoxic Alzheimer Amyloid-β Oligomers by Protein Engineering. Proc Natl Acad Sci U S A. 2010;107:15595–15600. doi: 10.1073/pnas.1001740107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Hard T. Protein Engineering to Stabilize Soluble Amyloid β-Protein Aggregates for Structural and Functional Studies. FEBS J. 2011;278:3884–3892. doi: 10.1111/j.1742-4658.2011.08295.x. [DOI] [PubMed] [Google Scholar]
- 114.Hoyer W, Gronwall C, Jonsson A, Stahl S, Hard T. Stabilization of a β-hairpin in Monomeric Alzheimer's Amyloid-β Peptide Inhibits Amyloid Formation. Proc Natl Acad Sci USA. 2008;105:5099, 5104. doi: 10.1073/pnas.0711731105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Klein WL. Aβ Toxicity in Alzheimer's Disease: Globular Oligomers(ADDLs) as New Vaccine and Drug Targets. Neurochem Int. 2002;41:345–352. doi: 10.1016/s0197-0186(02)00050-5. [DOI] [PubMed] [Google Scholar]
- 116.Barghorn S, Nimmrich V, Striebinger A, Krantz C, Keller P, Janson B, Bahr M, Schmidt M, Bitner RS, Harlan J, Barlow E, Ebert U, Hillen H. Globular Amyloid β-Peptide(1-42) Oligomer - A Homogenous and Stable Neuropathological Protein in Alzheimer's Disease. J Neurochem. 2005;95:834–847. doi: 10.1111/j.1471-4159.2005.03407.x. [DOI] [PubMed] [Google Scholar]
- 117.Moore BD, Rangachari V, Tay WM, Milkovic NM, Rosenberry TL. Biophysical Analyses of Synthetic Amyloid-β(1-42) Aggregates Before and After Covalent Cross-Linking. Implications for Deducing the Structure of Endogenous Amyloid-β Oligomers. Biochemistry. 2009;48:11796–11806. doi: 10.1021/bi901571t. [DOI] [PubMed] [Google Scholar]
- 118.Chimon S, Shaibat MA, Jones CR, Calero DC, Aizezi B, Ishii Y. Evidence of Fibril-Like β-sheet Structures in a Neurotoxic Amyloid Intermediate of Alzheimer's β-Amyloid. Nat Struct Mol Biol. 2007;14:1157–1164. doi: 10.1038/nsmb1345. [DOI] [PubMed] [Google Scholar]
- 119.Matsumura S, Shinoda K, Yamada M, Yokojima S, Inoue M, Ohnishi T, Shimada T, Kikuchi K, Masui D, Hashimoto S, Sato M, Ito A, Akioka M, Takagi S, Nakamura Y, Nemoto K, Hasegawa Y, Takamoto H, Inoue H, Nakamura S, Nabeshima Y, Teplow DB, Kinjo M, Hoshi M. Two Distinct Amyloid β-protein (Aβ) Assembly Pathways Leading to Oligomers and Fibrils Identified by Combined Fluorescence Correlation Spectroscopy, Morphology, and Toxicity Analyses. J Biol Chem. 2011;286:11555–11562. doi: 10.1074/jbc.M110.181313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Scheidt HA, Morgado I, Huster D. Solid-state NMR Reveals a Close Structural Relationship between Amyloid-β Protofibrils and Oligomers. J Biol Chem. 2012;287:22822–22826. doi: 10.1074/jbc.M112.367474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Scheidt HA, Morgado I, Rothemund S, Huster D, Fandrich M. Solid-State NMR Spectroscopic Investigation of Aβ Protofibrils: Implication of a β-Sheet Remodeling Upon Maturation into Terminal Amyloid Fibrils. Angew Chem Int Ed Engl. 2011;50:2837–2840. doi: 10.1002/anie.201007265. [DOI] [PubMed] [Google Scholar]
- 122.Doi T, Masuda Y, Irie K, Akagi K, Monobe Y, Imazawa T, Takegoshi K. Solid-State NMR Analysis of the β-Strand Orientation of the Protofibrils of Amyloid β-Protein. Biochem Biophys Res Commun. 2012;428:458–462. doi: 10.1016/j.bbrc.2012.10.096. [DOI] [PubMed] [Google Scholar]
- 123.Yu L, Edalji R, Harlan JE, Holzman TF, Lopez AP, Labkovsky B, Hillen H, Barghorn S, Ebert U, Richardson PL, Miesbauer L, Solomon L, Bartley D, Walter K, Johnson RW, Hajduk PJ, Olejniczak ET. Structural Characterization of a Soluble Amyloid β-peptide Oligomer. Biochemistry. 2009;48:1870–1877. doi: 10.1021/bi802046n. [DOI] [PubMed] [Google Scholar]
- 124.Ahmed M, Davis J, Aucoin D, Sato T, Ahuja S, Aimoto S, Elliott JI, Nostrand WEV, Smith SO. Structural Conversion of Neurotoxic Amyloid-β1-42 Oligomers to Fibrils. Nat Struc Mol Biol. 2010;17:561–568. doi: 10.1038/nsmb.1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Spencer RK, Li H, Nowick JS. X-ray Crystallographic Structures of Trimers and Higher-Order Oligomeric Assemblies of a Peptide Derived from Aβ. J Am Chem Soc. 2014;136:5595–5598. doi: 10.1021/ja5017409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Haupt C, Leppert J, Ronicke R, Meinhardt J, Yadav JK, Ramachandran R, Ohlenschlager O, Reymann KG, Gorlach M, Fandrich M. Structural Basis of β-Amyloid-Dependent Synaptic Dysfunctions. Angew Chem Int Ed Engl. 2012;51:1576–1579. doi: 10.1002/anie.201105638. [DOI] [PubMed] [Google Scholar]
- 127.(a) Sarkar B, Mithu VS, Chandra B, Mandal A, Chandrakesan M, Bhowmik D, Madhu PK, Maiti S. Significant Structural Differences Between Transient Amyloid-β Oligomers and Less-Toxic Fibrils in Regions Known To Harbor Familial Alzheimer's Mutations. Angew Chem Int Ed. 2014;53:6888–6892. doi: 10.1002/anie.201402636. [DOI] [PubMed] [Google Scholar]; (b) Laganowsky A, Liu C, Sawaya MR, Whitelegge JP, Park J, Zhao M, Pensalfini A, Soriaga AB, Landau M, Teng PK, Cascio D, Glabe C, Eisenberg D. Atomic View of a Toxic Amyloid Small Oligomer. Science. 2012;335:1228–31. doi: 10.1126/science.1213151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.(a) Wei G, Mousseau N, Derreumaux P. Sampling the Self-Assembly Pathways of KFFE Hexamers. Biophys J. 2004;87:3648–3656. doi: 10.1529/biophysj.104.047688. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Mousseau N, Derreumaux P. Exploring the Early Steps of Amyloid Peptide Aggregation by Computers. Acc Chem Res. 2005;38:885–891. doi: 10.1021/ar050045a. [DOI] [PubMed] [Google Scholar]
- 129.Melquiond A, Mousseau N, Derreumaux P. Structures of Soluble Amyloid Oligomers from Computer Simulations. Proteins. 2006;65:180–191. doi: 10.1002/prot.21100. [DOI] [PubMed] [Google Scholar]
- 130.Song W, Wei G, Mousseau N, Derreumaux P. Self-Assembly of the β2-microglobulin NHVTLSQ Peptide Using a Coarse-Grained Protein Model Reveals a β-barrel Species. J Phys Chem B. 2008;112:4410–4418. doi: 10.1021/jp710592v. [DOI] [PubMed] [Google Scholar]
- 131.Irback A, Mitternacht S. Spontaneous β-barrel Formation: an All-atom Monte Carlo Study of Aβ16-22 Oligomerization. Proteins. 2008;71:207–214. doi: 10.1002/prot.21682. [DOI] [PubMed] [Google Scholar]
- 132.Bellesia G, Shea JE. Effect of β-sheet Propensity on Peptide Aggregation. J Chem Phys. 2009;130:145103. doi: 10.1063/1.3108461. [DOI] [PubMed] [Google Scholar]
- 133.(a) De Simone A, Derreumaux P. Low Molecular Weight Oligomers of Amyloid Peptides Display β-barrel Conformations: a Replica Exchange Molecular Dynamics Study in Explicit Solvent. J Chem Phys. 2010;132:165103. doi: 10.1063/1.3385470. [DOI] [PubMed] [Google Scholar]; (b) Hatami A, Albay R, 3rd, Monjazeb S, Milton S, Glabe C. Monoclonal Antibodies against Aβ42 Fibrils Distinguish Multiple Aggregation State Polymorphisms in vitro and in Alzheimer Disease Brain. J Biol Chem. 2014;289:32131–43. doi: 10.1074/jbc.M114.594846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Bitan G, Lomakin A, Teplow D. Amyloid β-protein Oligomerization - Prenucleation Interactions Revealed by Photo-Induced Cross-Linking of Unmodified Proteins. J Biol Chem. 2001;276:35176–35184. doi: 10.1074/jbc.M102223200. [DOI] [PubMed] [Google Scholar]
- 135.Bitan G, Kirkitadze MD, Lomakin A, Vollers SS, Benedek GB, Teplow DB. Amyloid β-protein (Aβ) assembly: Aβ40 and Aβ42 oligomerize through distinct pathways. Proc Natl Acad Sci USA. 2003;100:330–335. doi: 10.1073/pnas.222681699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Roychaudhuri R, Yang M, Deshpande A, Cole GM, Frautschy S, Lomakin A, Benedek GB, Teplow DB. C-terminal Turn Stability Determines Assembly Differences Between Aβ40 and Aβ42. J Mol Biol. 2013;425:292–308. doi: 10.1016/j.jmb.2012.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.(a) Kloniecki M, Jablonowska A, Poznanski J, Langridge J, Hughes C, Campuzano I, Giles K, Dadlez M. Ion Mobility Separation Coupled with MS Detects Two Structural States of Alzheimer's Disease Aβ1-40 Peptide Oligomers. J Mol Biol. 2011;407:110–124. doi: 10.1016/j.jmb.2011.01.012. [DOI] [PubMed] [Google Scholar]; (b) Sitkiewicz E, Kłoniecki M, Poznański J, Bal W, Dadlez M. Factors Influencing Compact–Extended Structure Equilibrium in Oligomers of Aβ1–40 Peptide—An Ion Mobility Mass Spectrometry Study. J Mol Biol. 2014;426:2871–2885. doi: 10.1016/j.jmb.2014.05.015. [DOI] [PubMed] [Google Scholar]; (c) Chen YR, Glabe CG. Distinct Early Folding and Aggregation Properties of Alzheimer amyloid-β Peptides Aβ40 and Aβ42: Stable Trimer or Tetramer Formation by Aβ42. J Biol Chem. 2006;281:24414–24422. doi: 10.1074/jbc.M602363200. [DOI] [PubMed] [Google Scholar]
- 138.Liu R, McAllister C, Lyubchenko Y, Sierks MR. Residues 17-20 and 30-35 of β-amyloid Play Critical Roles in Aggregation. J Neurosci Res. 2004;75:162–171. doi: 10.1002/jnr.10859. [DOI] [PubMed] [Google Scholar]
- 139.(a) Williams AD, Portelius E, Kheterpal I, Guo JT, Cook KD, Xu Y, Wetzel R. Mapping Aβ Amyloid Fibril Secondary Structure Using Scanning Proline Mutagenesis. J Mol Biol. 2004;335:833–842. doi: 10.1016/j.jmb.2003.11.008. [DOI] [PubMed] [Google Scholar]; (b) Bernstein SL, Wyttenbach T, Baumketner A, Shea JE, Bitan G, Teplow DB, Bowers MT. Amyloid β-protein: Monomer Structure and Early Aggregation States of Aβ42 and its Pro19 Alloform. J Am Chem Soc. 2005;127:2075–2084. doi: 10.1021/ja044531p. [DOI] [PubMed] [Google Scholar]; (c) Masuda Y, Uemura S, Ohashi R, Nakanishi A, Takegoshi K, Shimizu T, Shirasawa T, Irie K. Identification of Physiological and Toxic Conformations in Aβ42 Aggregates. Chembiochem. 2009;10:287–295. doi: 10.1002/cbic.200800411. [DOI] [PubMed] [Google Scholar]
- 140.Gu L, Ngo S, Guo Z. Solid-support Electron Paramagnetic Resonance (EPR) Studies of Abeta40 Monomers Reveal a Structured State with Three Ordered Segments. J Biol Chem. 2012;287:9081–9089. doi: 10.1074/jbc.M111.292086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Bramucci E, Paiardini A, Bossa F, Pascarella S. PyMod: Sequence Similarity Searches, Multiple Sequence-Structure Alignments, and Homology Modeling within PyMOL. BMC Bioinformatics. 2012;13(4):S2. doi: 10.1186/1471-2105-13-S4-S2.S2-2105-13-S4-S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Lazo ND, Grant MA, Condron MC, Rigby AC, Teplow DB. On the Nucleation of Amyloid β-Protein Monomer Folding. Protein Sci. 2005;14:1581–1596. doi: 10.1110/ps.041292205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Chen W, Mousseau N, Derreumaux P. The Conformations of the Amyloid-β(21-30) Fragment can be Described by Three Families in Solution. J Chem Phys. 2006;125:084911. doi: 10.1063/1.2337628. [DOI] [PubMed] [Google Scholar]
- 144.Chebaro Y, Mousseau N, Derreumaux P. Structures and Thermodynamics of Alzheimer's Amyloid-β Aβ(16-35) Monomer and Dimer by Replica Exchange Molecular Dynamics Simulations: Implication for Full-Length Aβ Fibrillation. J Phys Chem B. 2009;113:7668–7675. doi: 10.1021/jp900425e. [DOI] [PubMed] [Google Scholar]
- 145.(a) Cruz L, Rao JS, Teplow DB, Urbanc B. Dynamics of Metastable β-hairpin Structures in the Folding Nucleus of Amyloid β-protein. J Phys Chem B. 2012;116:6311–6325. doi: 10.1021/jp301619v. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Smith MD, Cruz L. Effect of Ionic Aqueous Environments on the Structure and Dynamics of the Aβ(21-30) Fragment: A Molecular-Dynamics Study. J Phys Chem B. 2013;117:6614–6624. doi: 10.1021/jp312653h. [DOI] [PubMed] [Google Scholar]
- 146.Doran TM, Anderson EA, Latchney SE, Opanashuk LA, Nilsson BL. Turn Nucleation Perturbs Amyloid β Self-Assembly and Cytotoxicity. J Mol Biol. 2012;421:315–328. doi: 10.1016/j.jmb.2012.01.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.(a) Tsubuki S, Takaki Y, Saido TC. Dutch, Flemish, Italian, and Arctic Mutations of APP and Resistance of Aβ to Physiologically Relevant Proteolytic Degradation. Lancet. 2003;361:1957–1958. doi: 10.1016/s0140-6736(03)13555-6. [DOI] [PubMed] [Google Scholar]; (b) Smith MD, Cruz L. Changes to the Structure and Dynamics in Mutations of Aβ(21-30) Caused by Ions in Solution. J Phys Chem B. 2013;117:14907–14915. doi: 10.1021/jp408579v. [DOI] [PubMed] [Google Scholar]
- 148.Maji SK, Ogorzalek Loo RR, Inayathullah M, Spring SM, Vollers SS, Condron MM, Bitan G, Loo JA, Teplow DB. Amino Acid Position-Specific Contributions to Amyloid β-Protein Oligomerization. J Biol Chem. 2009;284:23580–23591. doi: 10.1074/jbc.M109.038133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.(a) Ono K, Condron MM, Teplow DB. Effects of the English (H6R) and Tottori (D7N) Familial Alzheimer Disease Mutations on Amyloid β-Protein Assembly and Toxicity. J Biol Chem. 2010;285:23186–23197. doi: 10.1074/jbc.M109.086496. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ono K, Condron MM, Teplow DB. Structure-Neurotoxicity Relationships of Amyloid β-Protein Oligomers. Proc Natl Acad Sci USA. 2009;106:14745–14750. doi: 10.1073/pnas.0905127106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Bitan G, Vollers SS, Teplow DB. Elucidation of Primary Structure Elements Controlling Early Amyloid β-Protein Oligomerization. J Biol Chem. 2003;278:34882–34889. doi: 10.1074/jbc.M300825200. [DOI] [PubMed] [Google Scholar]
- 151.(a) Sinha S, Lopes DH, Bitan GA. Key Role for Lysine Residues in Amyloid β-Protein Folding, Assembly and Toxicity. ACS Chem Neurosci. 2012;3:473–481. doi: 10.1021/cn3000247. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Xu W, Zhang C, Derreumaux P, Gräslund A, Morozova-Roche L, Mu Y. Intrinsic Determinants of Aβ(12-24) pH-dependent Self-assembly Revealed by Combined Computational and Experimental Studies. PLoS One. 2011;6:e24329. doi: 10.1371/journal.pone.0024329. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Chi EY, Frey SL, Winans A, Lam KL, Kjaer K, Majewski J, Lee KY. Amyloid-beta Fibrillogenesis Seeded by Interface-induced Peptide Misfolding and Self-assembly. Biophys J. 2010;98:2299–308. doi: 10.1016/j.bpj.2010.01.056. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Tycko R. Physical and Structural Basis for Polymorphism in Amyloid Fibrils. Protein Sci. 2014;23:1528–39. doi: 10.1002/pro.2544. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Adler J, Scheidt HA, Krüger M, Thomas L, Huster D. Local Interactions Influence the Fibrillation Kinetics, Structure and Dynamics of Aβ(1-40) but Leave the General Fibril Structure Unchanged. Phys Chem Chem Phys. 2014;16:7461–71. doi: 10.1039/c3cp54501f. [DOI] [PubMed] [Google Scholar]
- 152.(a) Zhang S, Iwata K, Lachenmann MJ, Peng JW, Li S, Stimson ER, Lu Y, Felix AM, Maggio JE, Lee JP. The Alzheimer's Peptide Aβ Adopts a Collapsed Coil Structure in Water. J Struct Biol. 2000;130:130–141. doi: 10.1006/jsbi.2000.4288. [DOI] [PubMed] [Google Scholar]; (b) Lim KH, Collver HH, Le YT, Nagchowdhuri P, Kenney JM. Characterizations of Distinct Amyloidogenic Conformations of the Aβ(1-40) and (1-42) Peptides. Biochem Biophys Res Commun. 2007;353:443–449. doi: 10.1016/j.bbrc.2006.12.043. [DOI] [PubMed] [Google Scholar]
- 153.Yan Y, McCallum SA, Wang C. M35 Oxidation Induces Aβ40-Like Structural and Dynamical Changes in Aβ42. J Am Chem Soc. 2008;130:5394–5395. doi: 10.1021/ja711189c. [DOI] [PubMed] [Google Scholar]
- 154.Rosenman DJ, Connors CR, Chen W, Wang C, Garcia AE. Aβ Monomers Transiently Sample Oligomer and Fibril-Like Configurations: Ensemble Characterization Using a Combined MD/NMR Approach. J Mol Biol. 2013:425, 3338–3359. doi: 10.1016/j.jmb.2013.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Yan Y, Wang C. Aβ42 is More Rigid than Aβ40 at the C Terminus: Implications for Aβ Aggregation and Toxicity. J Mol Biol. 2006;364:853–862. doi: 10.1016/j.jmb.2006.09.046. [DOI] [PubMed] [Google Scholar]
- 156.Yan Y, Liu J, McCallum SA, Yang D, Wang C. Methyl Dynamics of the Amyloid-β peptides Aβ40 and Aβ42. Biochem Biophys Res Commun. 2007;362:410–414. doi: 10.1016/j.bbrc.2007.07.198. [DOI] [PubMed] [Google Scholar]
- 157.(a) Vivekanandan S, Brender JR, Lee SY, Ramamoorthy A. A partially Folded Sructure of Amyloid-beta(1-40) in an Aqueous Environment. Biochem Biophys Res Commun. 2011;411:312–316. doi: 10.1016/j.bbrc.2011.06.133. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Bhowmik D, MacLaughlin CM, Chandrakesan M, Ramesh P, Venkatramani R, Walker GC, Maiti S. pH Changes the Aggregation Propensity of Amyloid-β without Altering the Monomer Conformation. Phys Chem Chem Phys. 2014;16:885–9. doi: 10.1039/c3cp54151g. [DOI] [PubMed] [Google Scholar]
- 158.Hwang TL, van Zijl PC, Mori S. Accurate Quantitation of Water-amide Proton Exchange Rates using the Phase-modulated CLEAN Chemical Exchange (CLEANEX-PM) Approach with a fast-HSQC (FHSQC) Detection Scheme. J Biomol NMR. 1998;11:221–226. doi: 10.1023/a:1008276004875. [DOI] [PubMed] [Google Scholar]
- 159.Rezaei-Ghaleh N, Andreetto E, Yan LM, Kapurniotu A, Zweckstetter M. Interaction between Amyloid beta Peptide and an Aggregation Blocker Peptide Mimicking Islet Amyloid Polypeptide. PLoS One. 2011;6:e20289. doi: 10.1371/journal.pone.0020289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Bitan G, Teplow DB. Rapid Photochemical Cross-linking – A New Tool for Studies of Metastable, Amyloidogenic Protein Assemblies. Acc Chem Res. 2004;37:357–364. doi: 10.1021/ar000214l. [DOI] [PubMed] [Google Scholar]
- 161.(a) Hukushima K, Nemoto K. Exchange Monte Carlo Method and Application to Spin glass Simulations. J Phys Soc Jpn. 1996;65:1604–1608. [Google Scholar]; (b) Sugita Y, Okamoto Y. Replica-exchange Molecular Dynamics Method for Protein Folding. Chem Phys Lett. 1999;314:141–151. [Google Scholar]
- 162.Nguyen PH, Okamoto Y, Derreumaux P. Communication: Simulated Tempering with Fast On-the-fly Weight Determination. J Chem Phys. 2013;138:061102. doi: 10.1063/1.4792046. [DOI] [PubMed] [Google Scholar]
- 163.(a) Laio A, Parrinello M. Escaping Free-energy Minima. Proc Natl Acad Sci USA. 2002;99:12562–12566. doi: 10.1073/pnas.202427399. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Laio A, Gervasio FL. Metadynamics: A Method to Simulate Rare Events and Reconstruct the Free Energy in Biophysics, Chemisty and Material Science. Rep Prog Phys. 2008;71:126601. doi: 10.1088/0034-4885/71/12/126601. [DOI] [Google Scholar]; (c) Barducci A, Bonomi M, Derreumaux P. Assessing the Quality of the OPEP Coarse-grained Force Field. J Chem Theory Comput. 2011;7:1928–1934. doi: 10.1021/ct100646f. [DOI] [PubMed] [Google Scholar]
- 164.Kaminski GA, Friesner RA, Tirado-Rives J, Jorgensen WL. Evaluation and Reparametrization of the OPLS-AA Force field for Proteins via Comparison with Accurate Quantum Chemical Calculations on Peptides. J Phys Chem B. 2001;105:6474–6487. [Google Scholar]
- 165.(a) Hornak V, Abel R, Okur A, Strockbine B, Roitberg A, Simmerling C. Comparison of Multiple Amber Force Fields and Development of Improved Protein Backbone Parameters. Proteins Struct Funct Bioinf. 2006;65:712–725. doi: 10.1002/prot.21123. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Best RB, Hummer G. Optimized Molecular Dynamics Force Fields applied to the Helix-Coil Transition of Polypeptides. J Phys Chem B. 2009;113:9004–9015. doi: 10.1021/jp901540t. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Lindorff-Larsen K, Piana S, Palmo K, Maragakis P, Klepeis JL, Dror RO, Shaw DE. Improved Side-chain Torsion Potentials for the Amber ff99SB Protein Force Field. Proteins Struct Funct Bioinf. 2010;78:1950–1958. doi: 10.1002/prot.22711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Piana S, Lindorff-Larsen K, Shaw DE. How Robust are Protein Folding Simulations with respect to Force Field Parametrization? Biophys J. 2011;100:L47–L49. doi: 10.1016/j.bpj.2011.03.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of Simple Potential Functions for Simulating Liquid Water. J Chem Phys. 1983;79:926. [Google Scholar]
- 168.Berendsen HJC, Grigera JR, Straatsma TP. The Missing Term in Effective Pair Potentials. J Phys Chem. 1987;91:6269–6271. [Google Scholar]
- 169.Horn HW, Swope WC, Pitera JW, Madura JD, Dick TJ, Hura GL, Head-Gordon T. Development of an Improved Four-Site Water Model for Biomolecular Simulations: TIP4P-Ew. J Chem Phys. 2004;120:9665–9678. doi: 10.1063/1.1683075. [DOI] [PubMed] [Google Scholar]
- 170.Shaw DE, Maragakis P, Lindorff-Larsen K, Piana S, Dror RO, Eastwood MP, Bank JA, Jumper JM, Salmon JK, Shan Y, Wriggers W. Atomic-level Characterization of the Structural Dynamics of Proteins. Science. 2010;330:341–346. doi: 10.1126/science.1187409. [DOI] [PubMed] [Google Scholar]; (b) Lindorff-Larsen K, Piana S, Dror RO, Shaw DE. How Fast-folding Proteins Fold. Science. 2011;334:517–520. doi: 10.1126/science.1208351. [DOI] [PubMed] [Google Scholar]; (c) Lindorff-Larsen K, Maragakis P, Piana S, Eastwood MP, Dror RO, Shaw DE. SystematicValidation of Protein Force Fields against Experimental Data. PLoS One. 2012;7:e32131. doi: 10.1371/journal.pone.0032131. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Piana S, Lindorff-Larsen K, Shaw DE. Protein Folding Kinetics andThermodynamics from Atomistic Simulation. Proc Natl Acad Sci USA. 2012;109:17845–17850. doi: 10.1073/pnas.1201811109. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Lindorff-Larsen K, Trbovic N, Maragakis P, Piana S, Shaw DE. Structure and Dynamics of an Unfolded Protein Examined by Molecular Dynamics Simulation. J Am Chem Soc. 2012;134:3787–3791. doi: 10.1021/ja209931w. [DOI] [PubMed] [Google Scholar]; (f) Wang W, Ye W, Jiang C, Luo R, Chen HF. New Force Field on Modeling Intrinsically Disordered Proteins. Chem Biol Drug Des. 2014;84:253–269. doi: 10.1111/cbdd.12314. [DOI] [PubMed] [Google Scholar]
- 171.(a) Sgourakis NG, Yan Y, McCallum SA, Wang C, Garcia AE. TheAlzheimer's Peptides Adopt Distinct Conformations in Water: a Combined MD/NMR study. J Mol Biol. 2007;368:1448–1457. doi: 10.1016/j.jmb.2007.02.093. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Sgourakis NG, Merced-Serrano M, Boutsidis C, Drineas P, Du Z, Wang C, Garcia AE. Atomic-level Characterization of the Ensemble of the Aβ(1-42) Monomer in Water using Unbiased Molecular Dynamics Simulations and Spectral Algorithms. J Mol Biol. 2011;405:570–583. doi: 10.1016/j.jmb.2010.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Ball KA, Phillips AH, Nerenberg PS, Fawzi NL, Wemmer DE, Head-Gordon T. Homogeneous and Heterogeneous Tertiary Structure Ensembles of amyloid-beta Peptides. Biochemistry. 2011;50:7612–7628. doi: 10.1021/bi200732x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.(a) Choy WY, Forman-Kay JD. Calculation of Ensembles of Structures Representing the Unfolded State of an SH3 Domain. J Mol Biol. 2001;308:1011–1032. doi: 10.1006/jmbi.2001.4750. [DOI] [PubMed] [Google Scholar]; (b) Marsh JA, Forman-Kay JD. Structure and Disorder in an Unfolded State under Nondenaturing Conditions from Ensemble Models Consistent with a Large Number of Experimental Restraints. J Mol Biol. 2009;391:359–374. doi: 10.1016/j.jmb.2009.06.001. [DOI] [PubMed] [Google Scholar]; (c) Krzeminski M, Marsh JA, Neale C, Choy WY, Forman-Kay JD. Characterization of Disordered Proteins with ENSEMBLE. Bioinformatics. 2013;29:398–399. doi: 10.1093/bioinformatics/bts701. [DOI] [PubMed] [Google Scholar]
- 174.Ball KA, Phillips AH, Wemmer DE, Head-Gordon T. Differences in β-Strand Populations of Monomeric Aβ40 and Aβ42. Biophys J. 2013;104:2714–2724. doi: 10.1016/j.bpj.2013.04.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Zhu M, De Simone A, Schenk D, Toth G, Dobson CM, Vendruscolo M. Identification of Small-molecule Binding Pockets in the Soluble Monomeric Form of the Aβ42 Peptide. J Chem Phys. 2013;139:035101. doi: 10.1063/1.4811831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Lin YS, Bowman GR, Beauchamp KA, Pande VS. Investigating how Peptide Length and a Pathogenic Mutation Modify the Structural Ensemble of Amyloid beta Monomer. Biophys J. 2012;102:315–324. doi: 10.1016/j.bpj.2011.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Lin YS, Pande VS. Effect of Familial Mutations on the Monomer Structure of Aβ42. Biophys J. 2012;103:L47–L49. doi: 10.1016/j.bpj.2012.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.(a) Granata D. Ph D Thesis. SISSA, Italy: Octobre. 2013. Characterizing Structure and Free Energy Landscape of Proteins by NMR-guided Metadynamics. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Granata D, Camilloni C, Vendruscolo M, Laio A. Characterization of the free-energy landscapes of proteins by NMR-guided metadynamics. Proc Natl Acad Sci USA. 2013;110:6817–6822. doi: 10.1073/pnas.1218350110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Chebaro Y, Dong X, Laghaei R, Derreumaux P, Mousseau N. Replica Exchange Molecular Dynamics Simulations of Coarse-grained Proteins in Implicit Solvent. J Phys Chem B. 2009;113:267–274. doi: 10.1021/jp805309e. [DOI] [PubMed] [Google Scholar]
- 180.(a) Maupetit J, Tuffery P, Derreumaux P. A Coarse-grained Protein Force Field for Folding and Structure Prediction. Proteins. 2007;69:394–408. doi: 10.1002/prot.21505. [DOI] [PubMed] [Google Scholar]; (b) Derreumaux P. Folding a 20 amino acid alpha beta Peptide with the Diffusion process-controlled Monte Carlo Method. J Chem Phys. 1997;107:1941–1947. [Google Scholar]; (c) Derreumaux P. A Diffusion Process-controlled Monte Carlo Method for Finding the Global Energy Minimum of a Polypeptide Chain. 1. Formulation and Test on a Hexadecapeptide. J Chem Phys. 1997;106:5260–5270. [Google Scholar]; (d) Derreumaux P. Generating Ensemble Averages for Small Proteins from Extended Conformations by Monte Carlo Simulations. Phys Rev Lett. 2000;85:206–209. doi: 10.1103/PhysRevLett.85.206. [DOI] [PubMed] [Google Scholar]; (e) Derreumaux P, Mousseau N. Coarse-grained Protein Molecular Dynamics Simulations. J Chem Phys. 2007;126:025101. doi: 10.1063/1.2408414. [DOI] [PubMed] [Google Scholar]; (f) Derreumaux P, Vergoten G, Lagant P. A Vibrational Molecular-force field of Model Compounds with Biological Interest. 1. Harmonic Dynamics of Crystalline Urea at 123-K. J Comput Chem. 1990;11:560–568. [Google Scholar]; (g) Derreumaux P, Dauchez M, Vergoten G. The Structures and Vibrational Frequencies of a series of Alkanes using the SPASIBA Force Field. J Mol Struct. 1993;295:203–221. [Google Scholar]; (h) Thévenet P, Shen Y, Maupetit J, Guyon F, Derreumaux P, Tufféry P. PEP-FOLD: An Updated de novo Structure Prediction Server for both Linear and Disulfide bonded Cyclic Peptides. Nucleic Acids Res. 2012;40(Web Server issue):W288–93. doi: 10.1093/nar/gks419. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Shen Y, Maupetit J, Derreumaux P, Tufféry P. Improved PEP-FOLD Approach for Peptide and Miniprotein Structure Prediction. J Chem Theory Comput. 2014;10:4745–4758. doi: 10.1021/ct500592m. [DOI] [PubMed] [Google Scholar]
- 181.(a) Irbäck A, Mitternacht S, Mohanty S. An Effective All-Atom Potential for Proteins. PMC Biophys. 2009;2:2. doi: 10.1186/1757-5036-2-2. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Mitternacht S, Staneva I, Härd T, Irbäck A. Comparing the Folding Free-Energy Landscapes of Aβ42 Variants with Different Aggregation Properties. Proteins Struct Funct Bioinf. 2010;78:2600–2608. doi: 10.1002/prot.22775. [DOI] [PubMed] [Google Scholar]
- 182.Côté S, Derreumaux P, Mousseau N. Distinct Morphologies for Amyloid beta Protein Monomer : Aβ1-40, Aβ1-42, Aβ1-40(D23N) J Chem Theory Comput. 2011;7:2584–2592. doi: 10.1021/ct1006967. [DOI] [PubMed] [Google Scholar]
- 183.Sterpone F, Nguyen PH, Kalimeri M, Derreumaux P. Importance of the Ion-Pair Interactions in the OPEP Coarse-grained Force Field: Parametrization and Validation. J Chem Theory Comput. 2013;9:4574–4584. doi: 10.1021/ct4003493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.(a) Yang M, Teplow DB. Amyloid β-Protein Monomer Folding: Free Energy Surfaces Reveal Alloform Specific Differences. J Mol Biol. 2008;384:450–464. doi: 10.1016/j.jmb.2008.09.039. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Lam AR, Teplow DB, Stanley HE, Urbanc B. Effects of the Artic (E22 –> G) Mutation on Amyloid beta-protein Folding: Discrete Molecular Dynamics Study. J Am Chem Soc. 2008;130:17413–17422. doi: 10.1021/ja804984h. [DOI] [PubMed] [Google Scholar]
- 185.Rauscher S, Gapsys V, Volkhardt A, Blau C, de Groot BL, Grubmüller H. Structural Ensemble of Intrinsically Disordered Proteins Depend Strongly on Force Field. Biophys J. 2014;106:271a. [Google Scholar]
- 186.Jin M, Shepardson N, Yang T, Chen G, Walsh D, Selkoe DJ. Soluble Amyloid beta-protein Dimers Isolated from Alzheimer Cortex directly Induce Tau Hyperphosphorylation and Neuritic Degeneration. Proc Natl Acad Sci USA. 2011;108:5819–5824. doi: 10.1073/pnas.1017033108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Autiero I, Saviano M, Langella E. In silico Investigation and Targeting of amyloid β Oligomers of Different Size. Mol Biosyst. 2013;9:2118–2124. doi: 10.1039/c3mb70086k. [DOI] [PubMed] [Google Scholar]
- 188.Berhanu WM, Hansmann UH. Structure and Dynamics of amyloid-β Segmental Polymorphisms. PLoS One. 2012;7:e41479. doi: 10.1371/journal.pone.0041479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Yu X, Zheng J. Polymorphic Structures of Alzheimer's β-amyloid Globulomers. PloS One. 2011;6:e20575. doi: 10.1371/journal.pone.0020575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Ma B, Nussinov R. Polymorphic Triple beta-sheet Structures Contribute to Amide Hydrogen/deuterium (H/D) Exchange Protection in the Alzheimer amyloid-beta 42 Peptide. J Biol Chem. 2011;286:34244–34253. doi: 10.1074/jbc.M111.241141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Derreumaux P. From Polypeptide Sequences to Structures using Monte Carlo Simulations and an Optimized Potential. J Chem Phys. 1999;111:2301–2310. [Google Scholar]
- 192.Forcellino F, Derreumaux P. Computer Simulations Aimed at Structure Prediction of Supersecondary Motifs in Proteins. Proteins. 2001;45:159–166. doi: 10.1002/prot.1135. [DOI] [PubMed] [Google Scholar]
- 193.Chebaro Y, Pasquali S, Derreumaux P. The Coarse-grained OPEP Force Field for Non-amyloid and amyloid Proteins. J Phys Chem B. 2012;116:8741–8752. doi: 10.1021/jp301665f. [DOI] [PubMed] [Google Scholar]
- 194.Kalimari M, Derreumaux P, Sterpone F. Are Coarse-grained Models Apt to Detect Protein Thermal Stability? The Case of OPEP Force Field. J Non-Cryst Solids. 2015;407:494–501. doi: 10.1016/j.jnoncrysol.2014.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Côté S, Laghaei R, Derreumaux P, Mousseau N. Distinct Dimerization for Various Alloforms of the amyloid-beta Protein : Abeta(1-40), Abeta(1-42), and Abeta(1-40)(D23N) J Phys Chem B. 2012;116:4043–4055. doi: 10.1021/jp2126366. [DOI] [PubMed] [Google Scholar]
- 196.Laghaei R, Mousseau N, Wei G. Effect of the Disulfide Bond on the Monomeric Structure of Human Amylin Studied by Combined Hamiltonian and Temperature Replica Exchange Molecular Dynamics Simulations. J Phys Chem B. 2010;114:7071–7077. doi: 10.1021/jp100205w. [DOI] [PubMed] [Google Scholar]
- 197.MacKerell AD, Bashford D, Bellott M, Dunbrack RL, Evanseck JD, Field MJ, Fischer S, Gao J, Guo H, Ha S, Joseph-McCarthy D, Kuchnir L, Kuczera K, Lau FT, Mattos C, Michnick S, Ngo T, Nguyen DT, Prodhom B, Reiher WE, Roux B, Schlenkrich M, Smith JC, Stote R, Straub J, Watanabe M, Wiórkiewicz-Kuczera J, Yin D, Karplus M. All-atom Empirical Potential for Molecular Modeling and Dynamics Studies of Proteins. J Phys Chem B. 1998;102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 198.Ferrara P, Apostolakis J, Caflisch A. Evaluation of a Fast Implicit Solvent Model for Molecular Dynamics Simulations. Proteins. 2002;46:24–33. doi: 10.1002/prot.10001. [DOI] [PubMed] [Google Scholar]
- 199.Takeda T, Klimov DK. Probing the Effect of Amino-terminal Truncation for Abeta1-40 Peptides. J Phys Chem B. 2009;113:6692–6702. doi: 10.1021/jp9016773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Kim S, Takeda T, Klimov DK. Mapping Conformational Ensembles of Abeta Oligomers in Molecular Dynamics Simulations. Biophys J. 2010;99:1949–1958. doi: 10.1016/j.bpj.2010.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Lockhart C, Kim S, Kumar R, Klimov DK. Does Aminoacid Sequence Determine the Properties of Abeta Dimer? J Chem Phys. 2011;135:035103. doi: 10.1063/1.3610427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Mitternacht S, Staneva I, Hard T, Irback A. Monte Carlo Study of the Formation and Conformational Properties of Dimers of Abeta42 Variants. J Mol Biol. 2011;410:357–367. doi: 10.1016/j.jmb.2011.05.014. [DOI] [PubMed] [Google Scholar]
- 203.Urbanc B, Betnel M, Cruz L, Bitan G, Teplow DB. Elucidation of Amyloid β-protein Oligomerization Mechanisms: Discrete Molecular Dynamics Study. J Am Chem Soc. 2010;132:4266–4280. doi: 10.1021/ja9096303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Barz B, Urbanc B. Dimer Formation Enhances Structural Differences between Amyloid β-protein (1-40) and (1-42): an Explicit-solvent Molecular Dynamics Study. PloS One. 2012;7:e34345. doi: 10.1371/journal.pone.0034345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Chong SH, Ham S. Atomic-level Investigations on the Amyloid-β Dimerization Process and its Driving Forces in Water. Phys Chem Chem Phys. 2012;14:1573–1575. doi: 10.1039/c2cp23326f. [DOI] [PubMed] [Google Scholar]
- 206.(a) Yano A, Okamoto A, Nomura K, Higai S, Kurita N. Difference in Dimer Conformation between Amyloid-β(1-42) and (1-43) Proteins: Replica Exchange Molecular Dynamics Simulations in Water. Chem Phys Lett. 2014:595–596. 242–249. [Google Scholar]; (b) Nguyen PH, Mai LS, Derreumaux P. Amyloid Oligomer Structure Characterization from Simulations : A General Method. J Chem Phys. 2014;140:094105. doi: 10.1063/1.4866902. [DOI] [PubMed] [Google Scholar]
- 207.Zhang T, Zhang J, Derreumaux P, Mu Y. Molecular Mechanism of the Inhibition of EGCG on the Alzheimer Aβ(1-42) Dimer. J Phys Chem B. 2013;117:3993–4002. doi: 10.1021/jp312573y. [DOI] [PubMed] [Google Scholar]
- 208.Chebaro Y, Jiang P, Zang T, Mu Y, Nguyen PH, Mousseau N, Derreumaux P. Structures of Aβ17-42 Trimers in Isolation and with Five Small-molecule Drugs using a Hierarchical Computational Procedure. J Phys Chem B. 2012;116:8412–8422. doi: 10.1021/jp2118778. [DOI] [PubMed] [Google Scholar]
- 209.Qiu D, Shenkin PS, Hollinger FP, Still WC. The GB/SA Continuum Model for solvation. A Fast Analytical Method for the Calculation of Approximate Born Radii. J Phys Chem A. 1997;101:3005–3014. [Google Scholar]
- 210.Barz B, Olubiyi OO, Strodel B. Early Amyloid β-protein Aggregation Precedes Conformational Change. Chem Commun. 2014;50:5373–5375. doi: 10.1039/c3cc48704k. [DOI] [PubMed] [Google Scholar]
- 211.Ferrone F. Analysis of Protein Aggregation Kinetics. Methods in Enzymology. 1999;309:256–274. doi: 10.1016/s0076-6879(99)09019-9. [DOI] [PubMed] [Google Scholar]
- 212.Oosawa F, Asakura S. Thermodynamics of the Polymerization of Protein. Academic; Waltham, MA: 1975. [Google Scholar]
- 213.Wegner A. Spontaneous Fragmentation of Actin Filaments in Physiological Conditions. Nature. 1982;296:266–267. doi: 10.1038/296266a0. [DOI] [PubMed] [Google Scholar]
- 214.Ferrone FA, Hofrichter J, Eaton WA. Kinetics of Sickle Hemoglobin Polymerization: II. A Double Nucleation Mechanism. J Mol Biol. 1985;183:611–631. doi: 10.1016/0022-2836(85)90175-5. [DOI] [PubMed] [Google Scholar]
- 215.Nasica-Labouze J, Mousseau N. Kinetics of amyloid Aggregation: a Study of the GNNQQNY Prion Sequence. PLoS Comput Biol. 2012;8:1002782. doi: 10.1371/journal.pcbi.1002782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Irbäck A, Jonsson SÆ, Linnemann N, Linse B, Wallin S. Aggregate Geometry in Amyloid Fibril Nucleation. Phys Rev Lett. 2013;110:058101. doi: 10.1103/PhysRevLett.110.058101. [DOI] [PubMed] [Google Scholar]
- 217.Bhattacharyya AM, A K Thakur AK, Wetzel R. Polyglutamine Aggregation Nucleation: Thermodynamics of a Highly Unfavorable Protein Folding Reaction. Proc Natl Acad Sci USA. 2005;102:15400–15405. doi: 10.1073/pnas.0501651102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Fay N, Inoue YJ, Bousset L, Taguchi H, Melki R. Assembly of the Yeast Prion Ure2p into Protein Fibrils. Thermodynamic and Kinetic Characterization. J Biol Chem. 2003;278:30199–30205. doi: 10.1074/jbc.M303000200. [DOI] [PubMed] [Google Scholar]
- 219.Li MS, Co NT, Reddy G, Hu CK, Straub JE, Thirumalai D. Factors Governing Fibrillogenesis of Polypeptide Chains Revealed by Lattice Models. Phys Rev Lett. 2010;105:218101. doi: 10.1103/PhysRevLett.105.218101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Abeln S, Vendruscolo M, Dobson CM, Frenkel D. A Simple Lattice Model that Captures Protein Folding, Aggregation and Amyloid Formation. PLoS One. 2014;9:e85185. doi: 10.1371/journal.pone.0085185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Mompeán M, González C, Lomba E, Laurents DV. Combining Classical MD and QM Calculations to Elucidate Complex System Nucleation: A Twisted, Three-Stranded, Parallel β-Sheet Seeds Amyloid Fibril Conception. J Phys Chem B. 2014;118:7312–7316. doi: 10.1021/jp503742p. [DOI] [PubMed] [Google Scholar]
- 222.De Simone A, Kitchen C, Kwan AH, Sunde M, Dobson CM, Frenkel D. Intrinsic Disorder Modulates Protein Self-Assembly and Aggregation. Proc Natl Acad Sci USA. 2012;109:6951–6956. doi: 10.1073/pnas.1118048109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Bieler NS, Knowles TPJ, Frenkel D, Vacha R. Connecting Macroscopic Observables and Microscopic Assembly Events in Amyloid Formation Using Coarse Grained Simulations. PLoS Comput Biol. 2012;8:e1002692. doi: 10.1371/journal.pcbi.1002692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Lomakin A, Teplow DB, Kirschner DA, Benedek GB. Kinetic Theory of Fibrillogenesis of Amyloid β-Protein. Proc Natl Acad Sci USA. 1997;94:7942–7947. doi: 10.1073/pnas.94.15.7942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Fawzi NL, Okabe Y, Yap EH, Head-Gordon T. Determining the Critical Nucleus and Mechanism of Fibril Elongation of the Alzheimer's Aβ(1-40) Peptide. J Mol Biol. 2007;365:535–550. doi: 10.1016/j.jmb.2006.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Collins SR, Douglass A, Vale RD, Weissman JS. Mechanism of Prion Propagation: Amyloid Growth Occurs by Monomer Addition. PLoS Biol. 2004;2:1582–1590. doi: 10.1371/journal.pbio.0020321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Nguyen PH, Li MS, Stock G, Straub JE, Thirumalai D. Monomer Adds to Preformed Structured Oligomers of Aβ-Peptides by a Two-Stage Dock-Lock Mechanism. Proc Natl Acad Sci USA. 2007;104:111–116. doi: 10.1073/pnas.0607440104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Takeda T, Klimov DK. Dissociation of Aβ(16-22) Amyloid Fibrils Probed by Molecular Dynamics. J Mol Biol. 2007;368:1202–1213. doi: 10.1016/j.jmb.2007.02.066. [DOI] [PubMed] [Google Scholar]
- 229.Li MS, Klimov DK, Straub JE, Thirumalai D. Probing the Mechanisms of Fibril Formation Using Lattice Models. J Chem Phys. 2008;129:175101. doi: 10.1063/1.2989981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Co NT, Li MS. New Method for Determining Size of Critical Nucleus of Fibril Formation of Polypeptide Chains. J Chem Phys. 2012;137:095101. doi: 10.1063/1.4749257. [DOI] [PubMed] [Google Scholar]
- 231.(a) Cabriolu R, Auer S. Amyloid Fibrillation Kinetics: Insight from Atomistic Nucleation Theory. J Mol Biol. 2011;411:275–285. doi: 10.1016/j.jmb.2011.05.032. [DOI] [PubMed] [Google Scholar]; (b) Garai K, Sahoo B, Sengupta P, Maiti S. Quasihomogeneous Nucleation of Amyloid beta Yields Numerical Bounds for the Critical Radius, the Surface Tension, and the Free Energy Barrier for Nucleus Formation. J Chem Phys. 2008;128:045102. doi: 10.1063/1.2822322. [DOI] [PubMed] [Google Scholar]
- 232.Kashchiev D, Cabriolu R, Auer S. Confounding the Paradigm: Peculiarities of Amyloid Fibril Nucleation. J Am Chem Soc. 2013;135:1531–1539. doi: 10.1021/ja311228d. [DOI] [PubMed] [Google Scholar]
- 233.Baftizadeh F, Biarnes X, Pietrucci F, Affinito F, Laio A. Multidimensional View of Amyloid Fibril Nucleation in Atomistic Detail. J Am Chem Soc. 2012;134:3886–3894. doi: 10.1021/ja210826a. [DOI] [PubMed] [Google Scholar]
- 234.Baftizadeh F, Pietrucci F, Biarnes X, Laio A. Nucleation Process of a Fibril Precursor in the C-Terminal Segment of Amyloid-β. Phys Rev Lett. 2013;110:168103. doi: 10.1103/PhysRevLett.110.168103. [DOI] [PubMed] [Google Scholar]
- 235.Hilbich C, Kisters-Woike B, Reed J, Masters C, Beyreuther K. Substitutions of Hydrophobic Amino Acids Reduce the Amyloidogenicity of Alzheimer's Disease βA4 Peptides. J Mol Biol. 1992;228:460–473. doi: 10.1016/0022-2836(92)90835-8. [DOI] [PubMed] [Google Scholar]
- 236.Tjernberg L, Naslund J, Lindqvist F, Johansson J, Karlstrom A, Thyberg J, Terenius L, Nordstedt C. Arrest of β-Amyloid Fibril Formation by a Pentapeptide Ligand. J Biol Chem. 1996;271:8545–8548. doi: 10.1074/jbc.271.15.8545. [DOI] [PubMed] [Google Scholar]
- 237.Kheterpal I, Williams A, Murphy C, Bledsoe B, Wetzel R. Structural Features of the β Amyloid Fibril Elucidated by Limited Proteolysis. Biochemistry. 2001;40:11757–11767. doi: 10.1021/bi010805z. [DOI] [PubMed] [Google Scholar]
- 238.Piana S, Laio A. A Bias-Exchange Approach to Protein Folding. J Phys Chem B. 2007;111:4553–4559. doi: 10.1021/jp067873l. [DOI] [PubMed] [Google Scholar]
- 239.Nguyen PH, Derreumaux P. Conformational Ensemble and Polymorphism of the All-Atom Alzheimer's Aβ(37-42) Amyloid Peptide Oligomers. J Phys Chem B. 2013;117:5831–5840. doi: 10.1021/jp401563n. [DOI] [PubMed] [Google Scholar]
- 240.Yang YI, Gao YQ. Computer Simulation Studies of Aβ37-42 Aggregation Thermodynamics and Kinetics in Water and Salt Solution. J Phys Chem B. 2015;119:662–670. doi: 10.1021/jp502169b. [DOI] [PubMed] [Google Scholar]
- 241.Lu Y, Wei G, Derreumaux P. Structural, Thermodynamical, and Dynamical Properties of Oligomers Formed by the Amyloid NNQQ Peptide: Insights from Coarse-Grained Simulations. J Chem Phys. 2012;137:025101. doi: 10.1063/1.4732761. [DOI] [PubMed] [Google Scholar]
- 242.Wei G, Mousseau N, Derreumaux P. Computational Simulations of the Early Steps of Protein Aggregation. Prion. 2007;1:3–8. doi: 10.4161/pri.1.1.3969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Mousseau N, Derreumaux P. Exploring Energy Landscapes of Protein Folding and Aggregation. Front Biosci. 2008;13:4495–516. doi: 10.2741/3019. [DOI] [PubMed] [Google Scholar]
- 244.Wei G, Song W, Derreumaux P, Mousseau N. Self-assembly of Amyloid-forming Peptides by Molecular Dynamics Simulations. Front Biosci. 2008;13:5681–92. doi: 10.2741/3109. [DOI] [PubMed] [Google Scholar]
- 245.Nasica-Labouze J, Meli M, Derreumaux P, Colombo G, Mousseau N. A Multiscale Approach to Characterize the Early Aggregation Steps of the Amyloid-Forming Peptide GNNQQNY from the Yeast Prion Sup-35. PLoS Comput Biol. 2011;5:e1002051. doi: 10.1371/journal.pcbi.1002051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Santini S, Wei G, Mousseau N, Derreumaux P. Pathway Complexity of Alzheimer's beta-amyloid Abeta16-22 Peptide Assembly. Structure. 2004;12:1245–55. doi: 10.1016/j.str.2004.04.018. [DOI] [PubMed] [Google Scholar]
- 247.Santini S, Mousseau N, Derreumaux P. In Silico Assembly of Alzheimer's Aβ16-22 Peptide into β-Sheets. J Am Chem Soc. 2004;126:11509–11516. doi: 10.1021/ja047286i. [DOI] [PubMed] [Google Scholar]
- 248.Petty SA, Decatur SM. Intersheet Rearrangement of Polypeptides During Nucleation of β-Sheet Aggregates. Proc Natl Acad Sci USA. 2005;102:14272–14277. doi: 10.1073/pnas.0502804102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Kittner M, Knecht V. Disordered Versus Fibril-Like Amyloid β(25-35) Dimers in Water: Structure and Thermodynamics. J Phys Chem B. 2010;114:15288–15295. doi: 10.1021/jp1065264. [DOI] [PubMed] [Google Scholar]
- 250.Whittleston CS, Wales DJ. Thermodynamics and Kinetics of Aggregation for the GNNQQNY Peptide. J Am Chem Soc. 2007;129:16005–16014. doi: 10.1021/ja075346p. [DOI] [PubMed] [Google Scholar]
- 251.Sciarretta KL, Gordon DJ, Petkova AT, Tycko R, Meredith SC. Aβ40-Lactam(D23/K28) Models a Conformation Highly Favorable for Nucleation of Amyloid. Biochemistry. 2005;44:6003–6014. doi: 10.1021/bi0474867. [DOI] [PubMed] [Google Scholar]
- 252.Reddy G, Straub JE, Thirumalai D. Influence of Preformed Asp23-Lys28 Salt Bridge on the Conformational Fluctuations of Monomers and Dimers of Aβ Peptides with Implications for Rates of Fibril Formation. J Phys Chem B. 2009;113:1162–1172. doi: 10.1021/jp808914c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Wood SJ, Wetzel R, Martin JD, Hurle MR. Prolines and Amyloidogenicity in Fragments of the Alzheimer's Peptide β/A4. Biochemistry. 1995:34, 724–730. doi: 10.1021/bi00003a003. [DOI] [PubMed] [Google Scholar]
- 254.Larini L, Shea JE. Role of β-Hairpin Formation in Aggregation: The Self-Assembly of the Amyloid-β(25-35) Peptide. Biophys J. 2012;103:576–586. doi: 10.1016/j.bpj.2012.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Grabenauer M, Wu C, Soto P, Shea JE, Bowers MT. Oligomers of the Prion Protein Fragment 106-126 are Likely Assembled from β-Hairpins in Solution, and Methionine Oxidation Inhibits Assembly Without Altering the Peptide's Monomeric Conformation. J Am Chem Soc. 2010;132:532–539. doi: 10.1021/ja905595k. [DOI] [PubMed] [Google Scholar]
- 256.Liang C, Derreumaux P, Wei G. Structure and Aggregation Mechanism of β2-Microglobulin (83-99) Peptides Studied by Molecular Dynamics Simulations. Biophys J. 2007;93:3353–3362. doi: 10.1529/biophysj.107.105585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Liang C, Derreumaux P, Mousseau N, Wei G. The β-Strand-Loop-β-Strand Conformation Is Marginally Populated in β2-Microglobulin (20-41) Peptide in Solution as Revealed by Replica Exchange Molecular Dynamics Simulations. Biophys J. 2008;95:510–517. doi: 10.1529/biophysj.107.125054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Qiao Q, Bowman GR, Huang X. Dynamics of an Intrinsically Disordered Protein Reveal Metastable Conformations that Potentially Seed Aggregation. J Am Chem Soc. 2013;135:16092–16101. doi: 10.1021/ja403147m. [DOI] [PubMed] [Google Scholar]
- 259.Wu C, Shea JE. Structural Similarities and Differences Between Amyloidogenic and Non-Amyloidogenic Islet Amyloid Polypeptide (IAPP) Sequences and Implications for the Dual Physiological and Pathological Activities of These Peptides. PLoS Comput Biol. 2013;9:e1003211. doi: 10.1371/journal.pcbi.1003211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Cheon M, Chang I, Mohanty S, Luheshi LM, Dobson CM, Vendruscolo M, Favrin G. Structural Reorganisation and Potential Toxicity of Oligomeric Species Formed During the Assembly of Amyloid Fibrils. PLoS Comput Biol. 2007;3:1727–1738. doi: 10.1371/journal.pcbi.0030173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.(a) Colletier JP, Laganowsky A, Landau M, Zhao M, Soriaga AB, Goldschmidt L, Flot D, Cascio D, Sawaya MR, Eisenberg D. Molecular Basis for Amyloid-β Polymorphism. Proc Natl Acad Sci USA. 2011;108:16938–16943. doi: 10.1073/pnas.1112600108. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Armstrong AH, Chen J, McKoy AF, Hecht MH. Mutations That Replace Aromatic Side Chains Promote Aggregation of the Alzheimer's Aβ Peptide. Biochemistry. 2011;50:4058–4067. doi: 10.1021/bi200268w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Kayed R, Sokolov Y, Edmonds B, McIntire TM, Milton SC, Hall JE, Glabe CG. Permeabilization of Lipid Bilayers is a Common Conformation-Dependent Activity of Soluble Amyloid Oligomers in Protein Misfolding Diseases. J Biol Chem. 2004;279:46363–46366. doi: 10.1074/jbc.C400260200. [DOI] [PubMed] [Google Scholar]
- 263.Quist A, Doudevski L, Lin H, Azimova R, Ng D, Frangione B, Kagan B, Ghiso J, Lal R. Amyloid Ion Channels: A Common Structural Link for Protein-Misfolding Disease. Proc Natl Acad Sci USA. 2005;102:10427–10432. doi: 10.1073/pnas.0502066102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Engel MFM, Khemtemourian L, Kleijer CC, Meeldijk HJD, Jacobs J, Verkleij AJ, de Kruijff B, Killian JA, Hoppener JWM. Membrane Damage by Human Islet Amyloid Polypeptide Through Fibril Growth at the Membrane. Proc Natl Acad Sci USA. 2008;105:6033–6038. doi: 10.1073/pnas.0708354105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Kastorna A, Trusova V, Gorbenko G, Kinnunen P. Membrane effects of Lysozyme Amyloid Fibrils. Chem Phys Lipids. 2012;165:331–337. doi: 10.1016/j.chemphyslip.2012.02.002. [DOI] [PubMed] [Google Scholar]
- 266.Hung LW, Ciccotosto GD, Giannakis E, Tew DJ, Perez K, Masters CL, Cappai R, Wade JD. Amyloid-β Peptide (Aβ) Neurotoxicity Is Modulated by the Rate of Peptide Aggregation: AβDimers Trimers Correlate with Neurotoxicity. J Neurosci. 2008;28:11950–11958. doi: 10.1523/JNEUROSCI.3916-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Butterfield SM, Lashuel HA. Amyloidogenic Protein-Membrane Interactions: Mechanistic Insight from Model Systems. Angew Chem Int Ed Engl. 2010;49:5628–5654. doi: 10.1002/anie.200906670. [DOI] [PubMed] [Google Scholar]
- 268.Williams TL, Serpell LC. Membrane and Surface Interactions of Alzheimer's Aβ Peptide – Insights into the Mechanism of Cytotoxicity. FEBS J. 2011;278:3905–3917. doi: 10.1111/j.1742-4658.2011.08228.x. [DOI] [PubMed] [Google Scholar]
- 269.Connelly L, Jang H, Arce FT, Capone R, Kotler SA, Ramachandran S, Kagan BL, Nussinov R, Lal R. Atomic Force Microscopy and MD Simulations Reveal Pore-Like Structures of All-D-Enantiomer of Alzheimer's β-Amyloid Peptide: Relevance to the Ion Channel Mechanism of AD Pathology. J Phys Chem B. 2012;116:1728–1735. doi: 10.1021/jp2108126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Bokvist M, Lindstrom F, Watts A, Grobner G. Two Types of Alzheimer's β-Amyloid (1-40) Peptide Membrane Interactions: Aggregation Preventing Transmembrane Anchoring Versus Accelerated Surface Fibril Formation. J Mol Biol. 2004;335:1039–1049. doi: 10.1016/j.jmb.2003.11.046. [DOI] [PubMed] [Google Scholar]
- 271.Zhao H, Tuominen EK, Kinnunen PK. Formation of Amyloid Fibers Triggered by Phosphatidylserine-Containing Membranes. Biochemistry. 2004;43:10302–10307. doi: 10.1021/bi049002c. [DOI] [PubMed] [Google Scholar]
- 272.Yates EA, Owens SL, Lynch MF, Cucco EM, Umbaugh CS, Legleiter J. Specific Domains of Aβ Facilitate Aggregation on and Association with Lipid Bilayers. J Mol Biol. 2013;425:1915–1933. doi: 10.1016/j.jmb.2013.03.022. [DOI] [PubMed] [Google Scholar]
- 273.Tofoleanu F, Buchete NV. Molecular Interactions of Alzheimer's Aβ Protofilaments with Lipid Membranes. J Mol Biol. 2012;421:572–586. doi: 10.1016/j.jmb.2011.12.063. [DOI] [PubMed] [Google Scholar]
- 274.Nagarathinam A, Höflinger P, Bühler A, Schäfer C, McGovern G, Jeffrey M, Staufenbiel M, Jucker M, Baumann F. Membrane-anchored Aβ Accelerates Amyloid Formation and Exacerbates Amyloid-associated Toxicity in Mice. J Neurosci. 2013;33:19284–19294. doi: 10.1523/JNEUROSCI.2542-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Shao H, Jao S, Ma K, Zagorski MG. Solution Structures of Micelle-bound Amyloid beta-(1-40) and beta-(1-42) Peptides of Alzheimer's Disease. J Mol Biol. 1999;285:755–773. doi: 10.1006/jmbi.1998.2348. [DOI] [PubMed] [Google Scholar]
- 276.Coles M, Bicknell W, Watson AA, Fairlie DP, Craik DJ. Solution Structure of Amyloid beta-peptide(1-40) in a Water-micelle Environment. Is the Membrane-spanning Domain Where We Think It Is? Biochemistry. 1998;37:11064–11077. doi: 10.1021/bi972979f. [DOI] [PubMed] [Google Scholar]
- 277.Gorbenko GP, Trusova V. Protein Aggregation in a Membrane Environment. Advances in Protein Chemistry and Structural Biology. 2011;84:113–142. doi: 10.1016/B978-0-12-386483-3.00002-1. [DOI] [PubMed] [Google Scholar]
- 278.Gorbenko GP, Kinnunen PK. The Role of Lipid-protein Interactions in Amyloid-type Protein Fibril Formation. Chem Phys Lipids. 2006;142:72–82. doi: 10.1016/j.chemphyslip.2006.02.006. [DOI] [PubMed] [Google Scholar]
- 279.Lin H, Bhatia R, Lal R. Amyloid beta Protein Forms Ion Channels: Implications for Alzheimer's Disease Pathophysiology. FASEB J. 2001;15:2433–2444. doi: 10.1096/fj.01-0377com. [DOI] [PubMed] [Google Scholar]
- 280.Morita M, Hamada T, Tendo Y, Hata T, Vestergaard MC, Takagi M. Selective Localization of Alzheimer's Amyloid beta in Membrane Lateral Compartments. Soft Matter. 2012;8:2816–2819. [Google Scholar]
- 281.Nag S, Sarkar B, Chandrakesan M, Abhyanakar R, Bhowmik D, Kombrabail M, Dandekar S, Lerner E, Haas E, Maiti A. Folding Transition Underlies the Emergence of Membrane Affinity in amyloid-β. S. Phys Chem Chem Phys. 2013;15:19129–19133. doi: 10.1039/c3cp52732h. [DOI] [PubMed] [Google Scholar]
- 282.Benilova I, Karran E, De Strooper B. The Toxic Aβ Oligomer and Alzheimer's Disease: An Emperor in Need of Clothes. Nat Neurosci. 2012;15:349–357. doi: 10.1038/nn.3028. [DOI] [PubMed] [Google Scholar]
- 283.Davis CH, Berkowitz ML. Structure of the amyloid-beta (1-42) Monomer Absorbed to Model Phospholipid Bilayers: a Molecular Dynamics Study. J Phys Chem B. 2009;113:14480–14486. doi: 10.1021/jp905889z. [DOI] [PubMed] [Google Scholar]
- 284.Davis CH, Berkowitz ML. Interaction between Amyloid-beta (1-42) Peptide and Phospholipid Bilayers: a Molecular Dynamics Study. Biophys J. 2009;96:785–797. doi: 10.1016/j.bpj.2008.09.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Davis CH, Berkowitz ML. A Molecular Dynamics Study of the Early Stages of Amyloid-beta(1-42) Oligomerization: The Role of Lipid Membranes. Proteins. 2010;78:2533–2545. doi: 10.1002/prot.22763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Lockhart C, Klimov DK. Alzheimer's Aβ10-40 Peptide Binds and Penetrates DMPC Bilayer: An Isobaric-isothermal Replica Exchange Molecular Dynamics Study. J Phys Chem B. 2014;118:2638–2648. doi: 10.1021/jp412153s. [DOI] [PubMed] [Google Scholar]
- 287.Yu X, Zheng J. Cholesterol Promotes the Interaction of Alzheimer β-Amyloid Monomer with Lipid Bilayer. J Mol Biol. 2012;421:561–571. doi: 10.1016/j.jmb.2011.11.006. [DOI] [PubMed] [Google Scholar]
- 288.Miyashita N, Straub JE, Thirumalai D. Structures of beta-amyloid Peptide 1-40, 1-42, and 1-55 - The 672-726 Fragment of APP- in a Membrane Environment with Implications for Interactions with gamma-secretase. J Am Chem Soc. 2009;131:17843–17852. doi: 10.1021/ja905457d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Buchete NV, Tycko R, Hummer G. Molecular Dynamics Simulations of Alzheimer's beta-amyloid Protofilaments. J Mol Biol. 2005;353:804–821. doi: 10.1016/j.jmb.2005.08.066. [DOI] [PubMed] [Google Scholar]
- 290.Buchete NV, Hummer G. Structure and Dynamics of Parallel beta-sheets, Hydrophobic core, and Loops in Alzheimer's Abeta Fibrils. Biophys J. 2007;92:3032–3039. doi: 10.1529/biophysj.106.100404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Tofoleanu F, Brooks BR, Buchete NV. Modulation of Alzheimer's Aβ Protofilament-Membrane Interactions by Lipid Headgroups. ACS Chem Neurosci. 2015 doi: 10.1021/cn500277f. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Tofoleanu F, Buchete NV. Alzheimer Aβ Peptide Interactions with Lipid Membranes: Fibrils, Oligomers and Polymorphic Amyloid Channels. Prion. 2012;6:339–345. doi: 10.4161/pri.21022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Jang H, Ma B, Lal R, Nussinov R. Models of Toxic beta-sheet Channels of Protegrin-1 Suggest a Common Subunit Organization Motif Shared with Toxic Alzheimer beta-amyloid Ion Channels. Biophys J. 2008;95:4631–4642. doi: 10.1529/biophysj.108.134551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Jang H, Arce FT, Capone R, Ramachandran S, Lal R, Nussinov R. Misfolded Amyloid Ion Channels Present Mobile beta-sheet Subunits in contrast to Conventional Ion Channels. Biophys J. 2009;97:3029–3037. doi: 10.1016/j.bpj.2009.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Jang H, Arce FT, Ramachandran S, Kagan BL, Lal R, Nussinov R. Familial Alzheimer's Disease Osaka Mutant (ΔE22) β-barrels Suggest an Explanation for the Different Aβ1-40/42 Preferred Conformational States Observed by Experiment. J Phys Chem B. 2013;117:11518–11529. doi: 10.1021/jp405389n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Strodel B, Lee JW, Whittleston CS, Whales DJ. Transmembrane Structures for Alzheimer's Aβ(1-42) Oligomers. J Am Chem Soc. 2010;132:13300–13312. doi: 10.1021/ja103725c. [DOI] [PubMed] [Google Scholar]
- 297.Poojari C, Kukol A, Strodel B. How the Amyloid-β Peptide and Membranes Affect Each Other: An Extensive Simulation Study. Biochim Biophys Acta. 2013;1828:327–339. doi: 10.1016/j.bbamem.2012.09.001. [DOI] [PubMed] [Google Scholar]
- 298.Poojari C, Strodel B. Stability of Transmembrane Amyloid β-peptide and Membrane Integrity Tested by Molecular Modeling of Site-specific Aβ42 Mutations. PLoS One. 2013;8:e78399. doi: 10.1371/journal.pone.0078399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Xu Y, Shen J, Luo X, Zhu W, Chen K, Ma J, Jiang H. Conformational Transition of Amyloid beta-peptide. Proc Natl Acad Sci USA. 2005;102:5403–5407. doi: 10.1073/pnas.0501218102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Lemkul JA, Bevan DR. A Comparative Molecular Dynamics Analysis of the Amyloid beta-peptide in a Lipid Bilayer. Arch Biochem Biophys. 2008;470:54–63. doi: 10.1016/j.abb.2007.11.004. [DOI] [PubMed] [Google Scholar]
- 301.Lemkul JA, Bevan DR. Lipid Composition Influences the Release of Alzheimer's Amyloid β-peptide from Membranes. Protein Sci. 2011;20:1530–1545. doi: 10.1002/pro.678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.Zhao LN, Chiu SW, Benoit J, Chew LY, Mu Y. Amyloid β Peptides Aggregation in a Mixed Membrane Bilayer: a Molecular Dynamics Study. J Phys Chem B. 2011;115:12247–12256. doi: 10.1021/jp2065985. [DOI] [PubMed] [Google Scholar]
- 303.Hoshino T, Mahmood MI, Mori K, Matsuzaki K. Binding and Aggregation Mechanism of Amyloid β-peptides onto the GM1 Ganglioside-Containing Lipid Membrane. J Phys Chem B. 2013;117:8085–8094. doi: 10.1021/jp4029062. [DOI] [PubMed] [Google Scholar]
- 304.(a) Manna M, Mukhopadhyay C. Binding, Conformational Transition and Dimerization of Amyloid-β Peptide on GM1-containing Ternary Membrane: Insights from Molecular Dynamics Simulation. PLoS One. 2013;8:e71308. doi: 10.1371/journal.pone.0071308. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Devarajan S, Sharmika JE. Molecular Dynamics Study of GM1 Ganglioside Complex with Amyloid β peptide (Aβ42) in Lipid Membrane. J Mol Liquids. 2014;195:59–64. [Google Scholar]
- 305.Rushworth JV, Hooper NM. Lipid Rafts: Linking Alzheimer's Amyloid-β Production, Aggregation, and Toxicity at Neuronal Membranes. Int J Alzheimers Dis. 2010;2011:603052. doi: 10.4061/2011/603052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.Pobandt T, Knecht V. Free Energy of Lipid Bilayer Defects Affected by Alzheimer's Disease-associated Amyloid-β42 Monomers. J Phys Chem B. 2014;118:3507–3516. doi: 10.1021/jp410477x. [DOI] [PubMed] [Google Scholar]
- 307.(a) Wu C, Shea JE. Coarse-grained Models for Protein Aggregation. Curr Opin Struct Biol. 2011;21:209–220. doi: 10.1016/j.sbi.2011.02.002. [DOI] [PubMed] [Google Scholar]; (b) Shea JE, Urbanc B. Insights into Aβ Aggregation: a Molecular Dynamics Perspective. Curr Top Med Chem. 2012;12:2596–2610. doi: 10.2174/1568026611212220012. [DOI] [PubMed] [Google Scholar]
- 308.(a) Derreumaux, Coarse-grained models for protein folding and aggregation. Methods Mol Biol. 2013;924:585–600. doi: 10.1007/978-1-62703-017-5_22. [DOI] [PubMed] [Google Scholar]; (b) Morriss-Andrews A, Brown FL, Shea JE. A Coarse-grained Model for Peptide Aggregation on a Membrane Surface. J Phys Chem B. 2014;118:8420–32. doi: 10.1021/jp502871m. [DOI] [PubMed] [Google Scholar]
- 309.Friedman R. Aggregation of Amyloids in a Cellular Context: Modelling and Experiment. Biochem J. 2011;438:415–426. doi: 10.1042/BJ20110369. [DOI] [PubMed] [Google Scholar]
- 310.Pellarin R, Caflisch A. Interpreting the Aggregation Kinetics of Amyloid Peptides. J Mol Biol. 2006;360:882–892. doi: 10.1016/j.jmb.2006.05.033. [DOI] [PubMed] [Google Scholar]
- 311.Friedman R, Pellarin R, Caflisch A. Amyloid Aggregation on Lipid Bilayers and its Impact on Membrane Permeability. J Mol Biol. 2009;387:407–415. doi: 10.1016/j.jmb.2008.12.036. [DOI] [PubMed] [Google Scholar]
- 312.Ayton S, Lei P, Bush AI. Metallostasis in Alzheimer's Disease. Free Radical Biol Med. 2013;62:76–89. doi: 10.1016/j.freeradbiomed.2012.10.558. [DOI] [PubMed] [Google Scholar]
- 313.Leskovjan AC, Lanzirotti A, Miller LM. Amyloid Plaques in PSAPP Mice Bind less Metal than Plaques in Human Alzheimer's Disease. Neuroimage. 2009;47:1215–1220. doi: 10.1016/j.neuroimage.2009.05.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314.Hureau C. Coordination of Redox Active Metal Ions to the APP and to the amyloid-b Peptides Involved in AD. Part 1: An Overview. Coord Chem Rev. 2012;256:2164–2174. [Google Scholar]
- 315.Roberts BR, Ryan TM, Bush AI, Masters CL, Duce JA. The Role of Metallobiology and Amyloid-β Peptides in Alzheimer's Disease. Neurochem. 2012;120:149–166. doi: 10.1111/j.1471-4159.2011.07500.x. [DOI] [PubMed] [Google Scholar]
- 316.Faller P, Hureau C. A Bioinorganic View of Alzheimer's Disease: When Misplaced Metal Ions (Re)direct the Electrons to the Wrong Target. Chemistry. 2012;18:15910–20. doi: 10.1002/chem.201202697. [DOI] [PubMed] [Google Scholar]
- 317.Squitti RJ. Copper Dysfunction in Alzheimer's Disease: from Meta-analysis of Biochemical Studies to New Insight into Genetics. Trace Elem Med Biol. 2012;26:93–6. doi: 10.1016/j.jtemb.2012.04.012. [DOI] [PubMed] [Google Scholar]
- 318.Lee JY, Cole TB, Palmiter RD, Suh SW, Koh JY. Contribution by Synaptic Zinc to the Gender-disparate Plaque Formation in Human Swedish Mutant APP Transgenic Mice. Proc Natl Acad Sci USA. 2002;99:7705–7710. doi: 10.1073/pnas.092034699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319.Drew SC, Barnham KJ. The Heterogeneous Nature of Cu(2+) Interactions with Alzheimer's Amyloid-β Peptide. Acc Chem Res. 2011;44:1146–1155. doi: 10.1021/ar200014u. [DOI] [PubMed] [Google Scholar]
- 320.Faller P, Hureau C, Berthoumieu O. Role of Metal Ions in the Self-assembly of the Alzheimer's Amyloid-beta Peptide. Inorg Chem. 2013;52:12193–206. doi: 10.1021/ic4003059. [DOI] [PubMed] [Google Scholar]
- 321.Zawisza I, Rozga M, Bal W. Affinity of peptides (Aβ, APP, α-synuclein, PrP) for Metal ions (Cu, Zn) Coord Chem Rev. 2012;256:2297–2307. [Google Scholar]
- 322.Hureau C, Dorlet P. Coordination of Redox Active Metal Ions to the APP Protein and to the Amyloid-β Peptides Involved in Alzheimer Disease. Part 2: How Cu(II) Binding Sites Depend on Changes in the Aβ Sequences. Coord Chem Rev. 2012;256:2175–2187. [Google Scholar]
- 323.Xu L, Wang X, Shan S. Characterization of the Polymorphic States of Copper(II)-bound Abeta(1-16) Peptides by Computational Simulations. J Comput Chem. 2013;34:2524–36. doi: 10.1002/jcc.23416. [DOI] [PubMed] [Google Scholar]
- 324.Azimi S, Rauk A. On the Involvement of Copper Binding to the N-terminus of the Amyloid Beta Peptide of Alzheimer's Disease: A Computational Study on Model Systems. Int J Alzheimers Dis. 2011;2011:539762. doi: 10.4061/2011/539762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 325.Eury H, Bijani C, Faller P, Hureau C. CuII Coordination amyloid-β: Murine versus Human Peptide. Angew Chem Int Ed. 2011;50:901–905. doi: 10.1002/anie.201005838. [DOI] [PubMed] [Google Scholar]
- 326.Gunderson WA, Hernandez-Guzman J, Karr JW, Sun L, Szalai VA, Warncke KJ. Local Structure and Global Patterning of Cu2+ Binding in Fibrillar amyloid-beta [Abeta(1-40)] Protein. J Am Chem Soc. 2012;134:18330–7. doi: 10.1021/ja306946q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 327.Parthasarathy S, Long F, Miller Y, Xiao Y, McElheny D, Thurber K, Ma B, Nussinov R, Ishii Y. Molecular-level Examination of Cu2+ Binding Structure for Amyloid Fibrils of 40-residue Alzheimer's beta by Solid-state NMR Spectroscopy. J Am Chem Soc. 2011;133:3390–400. doi: 10.1021/ja1072178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 328.Hureau C, Coppel Y, Dorlet P, Solari PL, Sayen S, Guillon E, Sabater L, Faller P. Deprotonation of the Asp1-Ala2 Peptide Bond Induces Modification of the Dynamic Copper(II) Environment in the Amyloid-b Peptide near Physiological pH. Angew Chem Int Ed. 2009;48:9522–9525. doi: 10.1002/anie.200904512. [DOI] [PubMed] [Google Scholar]
- 329.Miller Y, Ma BY, Nussinov R. Metal Binding Sites in Amyloid Oligomers: Complexes and Mechanisms. Coord Chem Rev. 2012;256:2245–2252. [Google Scholar]
- 330.Xu L, Wang XJ, Wang XC. Effects of Zn2+ Binding on the Structural and Dynamic Properties of Amyloid B Peptide Associated with Alzheimer's Disease: Asp(1) or Glu(11)? ACS Chem Neuro. 2013;4:1458–1468. doi: 10.1021/cn4001445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 331.Chassaing S, Collin F, Dorlet P, Gout J, Hureau C, Faller P. Copper and Heme-mediated Abeta Toxicity: Redox Chemistry, Abeta Oxidations and anti-ROS Compounds. Curr Top Med Chem. 2012;12:2573–2595. doi: 10.2174/1568026611212220011. [DOI] [PubMed] [Google Scholar]
- 332.Alies B, Renaglia E, Rozga M, Bal W, Faller P, Hureau C. Cu(II) Affinity for the Alzheimer's Peptide: Tyrosine Fluorescence Studies Revisited. Anal Chem. 2013;85:1501–1518. doi: 10.1021/ac302629u. [DOI] [PubMed] [Google Scholar]
- 333.Jiang D, Zhang L, Grant GP, Dudzik CG, Chen S, Patel S, Hao Y, Millhauser GL, Zhou F. The Elevated Copper Binding Strength of Amyloid-beta Aggregates Allows the Sequestration of Copper from Albumin: A Pathway to Accumulation of Copper in Senile Plaques. Biochemistry. 2013;52:547–56. doi: 10.1021/bi301053h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 334.Alies B, Badei B, Faller P, Hureau C. Reevaluation of Copper(I) Affinity for Amyloid-b Peptides by Competition with Ferrozine, an Unusual Copper(I) Indicator. Chemistry. 2012;18:1161–1167. doi: 10.1002/chem.201102746. [DOI] [PubMed] [Google Scholar]
- 335.Xiao Z, Gottschlich L, van der Meulen R, Udagedara SR, Wedd AG. Evaluation of Quantitative Probes for Weaker Cu(i) Binding Sites Completes a set of Four Capable of Detecting Cu(i) Affinities from Nanomolar to Attomolar. Metallomics. 2013;5:501–13. doi: 10.1039/c3mt00032j. [DOI] [PubMed] [Google Scholar]
- 336.(a) Viles JH. Metal ions and Amyloid Formation in Neurodegenerative Diseases. Coord Chem Rev. 2012;256:2271–2284. [Google Scholar]; (b) Miller Y, Ma B, Nussinov R. Zinc Ions Promote Alzheimer Aβ Aggregation via Population Shift of Polymorphic States. Proc Natl Acad Sci USA. 2010;107:9490–9495. doi: 10.1073/pnas.0913114107. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Mithu VS, Sarkar B, Bhowmik D, Chandrakesan M, Maiti S, Madhu PK. Zn(++) Binding Disrupts the Asp(23)-Lys(28) Salt Bridge without Altering the hairpin-Shaped Cross-β Structure of Aβ(42) Amyloid Aggregates. Biophys J. 2011;101:2825–32. doi: 10.1016/j.bpj.2011.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Garai K, Sahoo B, Kaushalya SK, Desai R, Maiti S. Zinc Lowers Amyloid-beta Toxicity by Selectively Precipitating Aggregation Intermediates. Biochemistry. 2007;46:10655–63. doi: 10.1021/bi700798b. [DOI] [PubMed] [Google Scholar]
- 337.Zhu X, Su B, Wang X, Smith MA, Perry G. Causes of Oxidative Stress in Alzheimer Disease. Cell Mol Life Sci. 2007;64:2202–10. doi: 10.1007/s00018-007-7218-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 338.Cassagnes LE, Herve V, Nepveu F, Hureau C, Faller P, Collin F. The Catalytically Active Copper-amyloid-Beta State: Coordination Site Responsible for Reactive Oxygen Species Production. Angew Chem Int Ed. 2013;52:11110–3. doi: 10.1002/anie.201305372. [DOI] [PubMed] [Google Scholar]
- 339.La Penna G, Hureau C, Andreussi O, Faller P. Identifying, by First-Principles Simulations, Cu[amyloid-beta] Species Making Fenton-type Reactions in Alzheimer's Disease. J Phys Chem B. 2013;117:16455–67. doi: 10.1021/jp410046w. [DOI] [PubMed] [Google Scholar]
- 340.James SA, Volitakis I, Adlard PA, Duce JA, Masters CL, Cherny RA, Bush AI. Elevated Labile Cu Is Associated with Oxidative Pathology in Alzheimer Disease. Free Radical Biol Med. 2012;52:298–302. doi: 10.1016/j.freeradbiomed.2011.10.446. [DOI] [PubMed] [Google Scholar]
- 341.Ceccom J, Cosledan F, Halley H, Frances B, Lassalle JM, Meunier B. Copper Chelator Induced Efficient Episodic Memory Recovery in a non-Transgenic Alzheimer's Mouse Model. PLoS One. 2012;7:e43105. doi: 10.1371/journal.pone.0043105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 342.Rodriguez-Rodriguez C, Telpoukhovskaia M, Orvig C. The art of Building Multifunctional Metal-binding Agents from Basic Molecular Scaffolds for the Potential Application in Neurodegenerative Diseases. Coord Chem Rev. 2012;256:2308–2332. [Google Scholar]
- 343.Crouch PJ, Barnham KJ. Therapeutic Redistribution of Metal Ions to Treat Alzheimer's Disease. Acc Chem Res. 2012;45:1604–11. doi: 10.1021/ar300074t. [DOI] [PubMed] [Google Scholar]
- 344.Pithadia AS, Lim MH. Metal-associated Amyloid-beta Species in Alzheimer's Disease. Curr Opin Chem Biol. 2012;16:67–73. doi: 10.1016/j.cbpa.2012.01.016. [DOI] [PubMed] [Google Scholar]
- 345.You H, Tsutsui S, Hameed S, Kannanayakal TJ, Chen L, Xia P, Engbers JD, Lipton SA, Stys PK, Zamponi GW. Aβ Neurotoxicity Depends on Interactions between Copper Ions, Prion Protein, and N-methyl-D-aspartate Receptors. Proc Natl Acad Sci USA. 2012;109:1737–1742. doi: 10.1073/pnas.1110789109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 346.Snyder EM, Nong Y, Almeida CG, Paul S, Moran T, Choi EY, Nairn AC, Salter MW, Lombroso PJ, Gouras GK, Greengard P. Regulation of NMDA Receptor Trafficking by Amyloid-beta. Nat Neurosci. 2005;8:1051–1058. doi: 10.1038/nn1503. [DOI] [PubMed] [Google Scholar]
- 347.Renner M, Lacor PN, Velasco PT, Xu J, Contractor A, Klein WL, Triller A. Deleterious Effects of Amyloid beta Oligomers Acting as an Extracellular Scaffold for mGluR5. Neuron. 2010;66:739–754. doi: 10.1016/j.neuron.2010.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 348.Cisse M, Halabisky B, Harris J, Devidze N, Dubal DB, Sun B, Orr A, Lotz G, Kim DH, Hamto P, Ho K, Yu GQ, Mucke L. Reversing EphB2 Depletion Rescues Cognitive Functions in Alzheimer Model. Nature. 2011;469:47–52. doi: 10.1038/nature09635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 349.Yanagisawa K, Ihara Y. GM1 Ganglioside-bound Amyloid beta-protein in Alzheimer's Disease Brain. Neurobiol Aging. 1998;19(1 Suppl):S65–67. doi: 10.1016/s0197-4580(98)00032-3. [DOI] [PubMed] [Google Scholar]
- 350.Yerbury JJ, Poon S, Meehan S, Thompson B, Kumita JR, Dobson CM, Wilson MR. The Extracellular Chaperone Clusterin Influences Amyloid Formation and Toxicity by Interacting with Prefibrillar Structures. FASEB J. 2007;21:2312–2322. doi: 10.1096/fj.06-7986com. [DOI] [PubMed] [Google Scholar]
- 351.Narayan P, Orte A, Clarke RW, Bolognesi B, Hook S, Ganzinger KA, Meehan S, Wilson MR, Dobson CM, Klenerman D. The Extracellular Chaperone Clusterin Sequesters Oligomeric Forms of the Amyloid-β(1-40) Peptide. Nat Struct Mol Biol. 2011;19:79–83. doi: 10.1038/nsmb.2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 352.Stanyon HF, Viles JH. Human Serum Albumin can Regulate Amyloid-β Peptide Fiber Growth in the Brain Interstitium: Implications for Alzheimer Disease. J Biol Chem. 2012;287:28163–28168. doi: 10.1074/jbc.C112.360800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 353.Likó I, Mák M, Klement É, Hunyadi-Gulyás É, Pázmány T, Medzihradszky KF, Urbányi Z. Evidence for an Extended Interacting Surface between beta-amyloid Serum Amyloid P Component. Neurosci Lett. 2007;412:51–55. doi: 10.1016/j.neulet.2006.10.052. [DOI] [PubMed] [Google Scholar]
- 354.Andreetto E, Yan LM, Tatarek-Nossol M, Velkova A, Frank R, Kapurniotu A. Identification of Hot Regions of the Abeta-IAPP Interaction Interface as High-affinity Binding Sites in both Cross- and Self-association. Angew Chem Int Ed. 2010;49:3081–3085. doi: 10.1002/anie.200904902. [DOI] [PubMed] [Google Scholar]
- 355.Buxbaum JN, Ye Z, Reixach N, Friske L, Levy C, Das P, Golde T, Masliah E, Roberts AR, Bartfai T. Transthyretin Protects Alzheimer's Mice from the Behavioral and Biochemical Effects of Abeta Toxicity. Proc Nat Acad Sci USA. 2008;105:2681–2686. doi: 10.1073/pnas.0712197105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 356.Coria F, Castano E, Prelli F, Larrondo-Lillo M, Van Duinen S, Shelanski M, Frangione B. Isolation and Characterization of Amyloid PComponent from Alzheimer's Disease and Other Types of Cerebral Amyloidosis. Lab Invest. 1988;58:454–458. [PubMed] [Google Scholar]
- 357.Dohler F, Sepulveda-Falla D, Krasemann S, Altmeppen H, Schluter H, Hildebrand D, Zerr I, Matschke J, Glatzel M. High Molecular Mass Assembliesof Amyloid-β Oligomers Bind Prion Protein in Patients with Alzheimer's Disease. Brain. 2014;137:873–876. doi: 10.1093/brain/awt375. [DOI] [PubMed] [Google Scholar]
- 358.Kuperstein I, Broersen K, Benilova I, Rozenski J, Jonckheere W, Debulpaep M, Vandersteen A, Segers-Nolten I, Van Der Werf K, Subramaniam V, Braeken D, Callewaert G, Bartic C, D'Hooge R, Martins IC, Rousseau F, Schymkowitz J, De Strooper B. Neurotoxicity of Alzheimer's Disease Aβ Peptides is Induced by Small Changes in the Aβ42 to Aβ40 Ratio. EMBO J. 2010;29:3408–3420. doi: 10.1038/emboj.2010.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 359.Jan A, Gokce O, Luthi-Carter R, Lashuel HA. The Ratio of Monomeric to Aggregated Forms of Abeta40 and Abeta42 is an Important Determinant of Amyloid-beta Aggregation, Fibrillogenesis, and Toxicity. J Biol Chem. 2008;283:28176–89. doi: 10.1074/jbc.M803159200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 360.Gimbel DA, Nygaard HB, Coffey EE, Gunther EC, Lauren J, Gimbel ZA, Strittmatter SM. Memory Impairment in Transgenic Alzheimer Mice Requires Cellular Prion Protein. J Neurosci. 2010;30:6367–6374. doi: 10.1523/JNEUROSCI.0395-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 361.Gunther EC, Strittmatter SM. Beta-amyloid Oligomers and Cellular Prion Protein in Alzheimer's Disease. J Mol Med. 2010;88:331–338. doi: 10.1007/s00109-009-0568-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 362.Chen S, Yadav SP, Surewicz WK. Interaction between Human Prion Protein and Amyloid-beta (Abeta) Oligomers: Role of N-terminal Residues. J Biol Chem. 2010;285:26377–26383. doi: 10.1074/jbc.M110.145516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 363.Balducci C, Beeg M, Stravalaci M, Bastone A, Sclip A, Biasini E, Tapella L, Colombo L, Manzoni C, Borsello T, Chiesa R, Gobbi M, Salmona M, Forloni G. Synthetic Amyloid-beta Oligomers Impair Long-term Memory Idependently of Cellular Prion Protein. Proc Natl Acad Sci USA. 2010;107:2295–2300. doi: 10.1073/pnas.0911829107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 364.Calella AM, Farinelli M, Nuvolone M, Mirante O, Moos R, Falsig J, Mansuy IM, Aguzzi A. Prion protein and Abeta-related Synaptic Toxicity Impairment. EMBO Mol Med. 2010;2:306–310. doi: 10.1002/emmm.201000082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 365.Kessels HW, Nguyen LN, Nabavi S, Malinow R. The Prion Protein as a Receptor for Amyloid-beta. Nature. 2010;466:E3–4. doi: 10.1038/nature09217. discussion E4-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 366.Cisse M, Sanchez PE, Kim DH, Ho K, Yu GQ, Mucke L. Ablation of Cellular Prion Protein Does not Ameliorate Abnormal Neural Network Activity or Cognitive Dysfunction in the J20 Line of Human Amyloid Precursor Protein Transgenic Mice. J Neurosci. 2011;31:10427–10431. doi: 10.1523/JNEUROSCI.1459-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 367.Freir DB, Nicoll AJ, Klyubin I, Panico S, Mc Donald JM, Risse E, Asante EA, Farrow MA, Sessions RB, Saibil HR, Clarke AR, Rowan MJ, Walsh DM, Collinge J. Interaction between Prion Protein and Toxic Amyloid β Assemblies can be Therapeutically Targeted at Multiple Sites. Nat Commun. 2011;2:336. doi: 10.1038/ncomms1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 368.Barry AE, Klyubin I, Mc Donald JM, Mably AJ, Farrell MA, Scott M, Walsh DM, Rowan MJ. Alzheimer's Disease Brain-derived Amyloid-β-Mediated Inhibition of LTP in vivo is Prevented by Immunotargeting Cellular Prion Protein. J; Neurosci. 2011;31:7259–7263. doi: 10.1523/JNEUROSCI.6500-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 369.Um JW, Nygaard HB, Heiss JK, Kostylev MA, Stagi M, Vortmeyer A, Wisniewski T, Gunther EC, Strittmatter SM. Alzheimer Amyloid-β Oligomer Bound to Postsynaptic Prion Protein Activates Fyn to Impair Neurons. Nat Neurosci. 2012;15:1227–1235. doi: 10.1038/nn.3178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 370.Bate C, Williams A. Amyloid-β-induced Synapse Samage is Mediated via Cross-linkage of Cellular Prion Proteins. J Biol Chem. 2011;286:37955–37963. doi: 10.1074/jbc.M111.248724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 371.Kudo W, Lee HP, Zou WQ, Wang X, Perry G, Zhu X, Smith MA, Petersen RB, Lee HG. Cellular Prion Protein is Essential for Oligomeric Amyloid-β-induced Neuronal Cell Death. Hum Mol Genet. 2012;21:1138–44. doi: 10.1093/hmg/ddr542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 372.Nicoll AJ, Panico S, Freir DB, Wright D, Terry C, Risse E, Herron CE, O'Malley T, Wadsworth JD, Farrow MA, Walsh DM, Saibil HR, Collinge J. Amyloid-β Nanotubes are Associated with Prion Protein-dependent Synaptotoxicity. Nat Commun. 2013;4:2416. doi: 10.1038/ncomms3416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 373.Zou WQ, Xiao X, Yuan J, Puoti G, Fujioka H, Wang X, Richardson S, Zhou X, Zou R, Li S. Amyloid-beta42 Interacts Mainly with Insoluble Prion Protein in the Alzheimer Brain. J Biol Chem. 2011;286:15095–15105. doi: 10.1074/jbc.M110.199356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 374.Ferrer I, Blanco R, Carmona M, Puig B, Ribera R, Rey M, Ribalta T. Prion Protein Expression in Senile Plaques in Alzheimer's Disease. Acta Neuropathol. 2001;101:49–56. doi: 10.1007/s004010000271. [DOI] [PubMed] [Google Scholar]
- 375.Krieger J, Fusco G, Lewitzky M, Simister PC, Marchant J, Camilloni C, Feller SM, De Simone A. Conformational Recognition of an Intrinsically Disordered Protein. Biophys J. 2014;106:1771–1779. doi: 10.1016/j.bpj.2014.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 376.(a) Derreumaux P. Evidence that the 127-164 Region of Prion Proteins has Two Equi-energetic Conformations with beta or alpha Features. Biophys J. 2001;81:1657–65. doi: 10.1016/S0006-3495(01)75819-5. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Santini S, Claude JB, Audic S, Derreumaux P. Impact of the Tail and Mutations G131V and M129V on Prion Protein Flexibility. Proteins. 2003;51:258–65. doi: 10.1002/prot.10348. [DOI] [PubMed] [Google Scholar]; (c) Santini S, Derreumaux P. Helix H1 of the Prion Protein is Rather Stable against Environmental Perturbations: Molecular Dynamics of Mutation and Deletion Variants of PrP(90-231) Cell Mol Life Sci. 2004;61:951–60. doi: 10.1007/s00018-003-3455-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 377.Gallion SL. Modeling Amyloid-beta As HomogeneousDodecamers and in Complex with Cellular Prion Protein. Plos One. 2012;7:e49375. doi: 10.1371/journal.pone.0049375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 378.Chebaro Y, Derreumaux PJ. The Conversion of Helix H2 to Beta-sheet is Accelerated in the Monomer and Dimer of the Prion Protein upon T183A Mutation. Phys Chem B. 2009;113:6942–6948. doi: 10.1021/jp900334s. [DOI] [PubMed] [Google Scholar]
- 379.Barducci A, Chelli R, Procacci P, Schettino V, Gervasio FL, Parrinello M. Metadynamics Simulation of Prion Protein: Beta-structure Stability and the Early Stages of Misfolding. J Am Chem Soc. 2006;128:2705–2710. doi: 10.1021/ja057076l. [DOI] [PubMed] [Google Scholar]
- 380.De Simone A, Dodson GG, Fraternali F, Zagari A. Water Molecules as Structural Determinants among Prions of Low Sequence Identity. FEBS Lett. 2006;580:2488–2894. doi: 10.1016/j.febslet.2006.02.083. [DOI] [PubMed] [Google Scholar]
- 381.Cobb NJ, Apostol MI, Chen S, Smirnovas V, Surewicz WK. Conformational Stability of Mammalian Prion Protein Amyloid Fibrils is Dictated by a Packing Polymorphism within the Core Region. J Biol Chem. 2014;289:2643–2650. doi: 10.1074/jbc.M113.520718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 382.Cordeiro Y, Kraineva J, Ravindra R, Lima LM, Gomes MP, Foguel D, Winter R, Silva JL. Hydration and Packing Effects on Prion Folding and Beta-sheet Conversion. High Pressure Spectroscopy and Pressure Perturbation Calorimetry Studies. J Biol Chem. 2004;279:32354–32359. doi: 10.1074/jbc.M404295200. [DOI] [PubMed] [Google Scholar]
- 383.De Simone A, Zagari A, Derreumaux P. Structural and Hydration Properties of the Partially Unfolded States of the Prion Protein. Biophys J. 2007;93:1284–1292. doi: 10.1529/biophysj.107.108613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 384.Eghiaian F, Daubenfeld T, Quenet Y, van Audenhaege M, Bouin AP, van der Rest G, Grosclaude J, Rezaei H. Diversity in Prion Protein Oligomerization Pathways Results from Domain Expansion as Revealed by hydrogen/deuterium Exchange and Disulfide linkage. Proc Natl Acad Sci USA. 2007;104:7414–7419. doi: 10.1073/pnas.0607745104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 385.Schwarzinger S, Horn AH, Ziegler J, Sticht H. Rare Large Scale Subdomain Motions in Prion Protein can Initiate Aggregation. J Biomol Struct Dyn. 2006;23:581–590. doi: 10.1080/07391102.2006.10507083. [DOI] [PubMed] [Google Scholar]
- 386.Khosravani H, Zhang Y, Tsutsui S, Hameed S, Altier C, Hamid J, Chen L, Villemaire M, Ali Z, Jirik FR, Zamponi GW. Prion Protein Attenuates Excitotoxicity by Inhibiting NMDA Receptors. J Cell Biol. 2008;181:551–565. doi: 10.1083/jcb.200711002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 387.Sarell CJ, Syme CD, Rigby SE, Viles JH. Copper(II) Binding to Amyloid-beta Fibrils of Alzheimer's Disease Reveals a Picomolar Affinity: Stoichiometry and Coordination Geometry are Independent of Abeta Oligomeric forms. Biochemistry. 2009;48:4388–4402. doi: 10.1021/bi900254n. [DOI] [PubMed] [Google Scholar]
- 388.Hu NW, Nicoll AJ, Zhang D, Mably AJ, O'Malley T, Purro SA, Terry C, Collinge J, Walsh DM, Rowan MJ. MGlu5 Receptors and Cellular Prion Protein Mediate Amyloid-β-facilitated Synaptic Long-term Depression in Vivo. Nat Commun. 2014;5:3374. doi: 10.1038/ncomms4374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 389.Chen X, Lin R, Chang L, Xu S, Wei X, Zhang J, Wang C, Anwyl R, Wang Q. Enhancement of Long-term Depression by Soluble Amyloid β Protein in Rat Hippocampus is Mediated by Metabotropic Glutamate Receptor and Involves Activation of p38MAPK, STEP and Caspase-3. Neuroscience. 2013;253:435–443. doi: 10.1016/j.neuroscience.2013.08.054. [DOI] [PubMed] [Google Scholar]
- 390.Kessels HW, Nabavi S, Malinow R. Metabotropic NMDA Receptor Function is Required for β-amyloid-induced Synaptic Depression. Proc Natl Acad Sci USA. 2013;110:4033–4038. doi: 10.1073/pnas.1219605110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 391.Mota SI, Ferreira IL, Pereira C, Oliveira CR, Rego AC. Amyloid-beta Peptide 1-42 Causes Microtubule Deregulation through N-methyl-D-aspartate Receptors in Mature Hippocampal Cultures. Curr Alzheimer Res. 2012;9:844–856. doi: 10.2174/156720512802455322. [DOI] [PubMed] [Google Scholar]
- 392.Brito-Moreira J, Paula-Lima AC, Bomfim TR, Oliveira FB, Sepulveda FJ, De Mello FG, Aguayo LG, Panizzutti R, Ferreira ST. Aβ Oligomers Induce Glutamate Release from Hippocampal Neurons. Curr Alzheimer Res. 2011;8:552–562. doi: 10.2174/156720511796391917. [DOI] [PubMed] [Google Scholar]
- 393.Sleegers K, Lambert JC, Bertram L, Cruts M, Amouyel P, Van Broeckhoven C. The Pursuit of Susceptibility Genes for Alzheimer's Disease: Progress and Prospects. Trends Genet. 2010;26:84–93. doi: 10.1016/j.tig.2009.12.004. [DOI] [PubMed] [Google Scholar]
- 394.Narayan P, Meehan S, Carver JA, Wilson MR, Dobson CM, Klenerman D. Amyloid-β Oligomers are Sequestered by both Intracellular and Extracellular Chaperones. Biochemistry. 2012;51:9270–9276. doi: 10.1021/bi301277k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 395.Humphreys DT, Carver JA, Easterbrook-Smith SB, Wilson MR. Clusterin has Chaperone-like Activity Similar to that of Small Heat Shock Proteins. J Biol Chem. 1999;274:6875–81. doi: 10.1074/jbc.274.11.6875. [DOI] [PubMed] [Google Scholar]
- 396.Wyatt AR, Yerbury JJ, Poon S, Wilson MR. Therapeutic Targets in Extracellular Protein Deposition Diseases. Curr Med Chem. 2009;16:2855–2866. doi: 10.2174/092986709788803187. [DOI] [PubMed] [Google Scholar]
- 397.Wyatt AR, Yerbury JJ, Wilson MR. Structural Characterization of Clusterin-chaperone Client Protein Complexes. J Biol Chem. 2009;284:21920–21927. doi: 10.1074/jbc.M109.033688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 398.Wyatt AR, Yerbury JJ, Berghofer P, Greguric I, Katsifis A, Dobson CM, Wilson MR. Clusterin Facilitates in Vivo Clearance of Extracellular Misfolded Proteins. Cell Mol Life Sci. 2011;68:3919–3931. doi: 10.1007/s00018-011-0684-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 399.Biere AL, Ostaszewski B, Stimson ER, Hyman BT, Maggio JE, Selkoe DJ. Amyloid beta-peptide is Transported on Lipoproteins and Albumin in Human Plasma. J Biol Chem. 1996;271:32916–32922. doi: 10.1074/jbc.271.51.32916. [DOI] [PubMed] [Google Scholar]
- 400.Kuo YM, Kokjohn TA, Kalback WM, Luehrs D, Galasko DR, Chevallier N, Koo EH, Emmerling MR, Roher AE. Amyloid-beta Peptides Interact with Plasma Proteins and Erythrocytes: Implications for their Quantitation in Plasma. Biochem Biophys Res Commun. 2000;268:750–756. doi: 10.1006/bbrc.2000.2222. [DOI] [PubMed] [Google Scholar]
- 401.Seubert P, Vigo-Pelfrey C, Esch F, Lee M, Dovey H, Davis D, Sinha S, Schiossmacher M, Whaley J, Swindlehurst C. Isolation and Quantification of Soluble Alzheimer's beta-peptide From Biological Fluids. Nature. 1992;359:325–327. doi: 10.1038/359325a0. [DOI] [PubMed] [Google Scholar]
- 402.Lame ME, Chambers EE, Blatnik M. Quantification of Amyloid beta Peptides Aβ(1-38), Aβ(1-40), and Aβ(1-42) in Human Cerebrospinal Fluid by Ultra-performance Liquid Chromatography-tandem Mass Spectrometry. Anal Biochem. 2011;419:133–139. doi: 10.1016/j.ab.2011.08.010. [DOI] [PubMed] [Google Scholar]
- 403.Stevens R, Elmendorf D, Gourlay M, Stroebel E, Gaafar H. Application of Fluoroimmunoassay to Cerebrospinal Fluid Immunoglobulin G and Albumin. J Clin Microbiol. 1979;10:346–50. doi: 10.1128/jcm.10.3.346-350.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 404.Rózga M, Kloniecki M, Jablonowska A, Dadlez M, Bal W. The binding Constant for Amyloid Abeta40 Peptide Interaction with Human Serum Albumin. Biochem Biophys Res Commun. 2007;364:714–718. doi: 10.1016/j.bbrc.2007.10.080. [DOI] [PubMed] [Google Scholar]
- 405.Bohrmann B, Tjernberg L, Kuner P, Poli S, Levet-Trafit B, Näslund J, Richards G, Huber W, Döbeli H, Nordstedt C. Endogenous Proteins Controlling Amyloid beta-peptide Polymerization. Possible Implications for beta-amyloid Formation in the Central Nervous System and in Peripheral Tissues. J Biol Chem. 1999;274:15990–15995. doi: 10.1074/jbc.274.23.15990. [DOI] [PubMed] [Google Scholar]
- 406.Milojevic J, Esposito V, Das R, Melacini G. Understanding the Molecular Basis for the Inhibition of the Alzheimer's Abeta-peptide Oligomerization by Human Serum Albumin using Saturation Transfer Difference and Off-resonance Relaxation NMR Spectroscopy. J Am Chem Soc. 2007;129:4282–4290. doi: 10.1021/ja067367+. [DOI] [PubMed] [Google Scholar]
- 407.Milojevic J, Melacini G. Stoichiometry and Affinity of the Human Serum Albumin-Alzheimer's Aβ Peptide Interactions. Biophys J. 2011;100:183–192. doi: 10.1016/j.bpj.2010.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 408.Garg A, Manidhar DM, Gokara M, Mlleda C, Reddy CS, Subramanyam R. Elucidation of the Binding Mechanism of CoumarinDerivatives with Human Serum Albumin. Plos One. 2013;8(5):e63805. doi: 10.1371/journal.pone.0063805. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Raz Y, Miller Y. Interactions between Aβ and Mutated Tau Lead to Polymorphismand Induce Aggregation of Aβ-mutated tau Oligomeric Complexes. Plos One. 2013;8:e73303. doi: 10.1371/journal.pone.0073303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 409.Mullard A. Sting of Alzheimer's Failures Offset by Upcoming Prevention Trials. Nat Rev Drug Discov. 2012;11:657–660. doi: 10.1038/nrd3842. [DOI] [PubMed] [Google Scholar]
- 410.Pujadas L, Rossi D, Andrés R, Teixeira CM, Serra-Vidal B, Parcerisas A, Maldonado R, Giralt E, Crulla N, Soriano E. Reelin Delays Amyloid-beta Fibril Formation and Rescues Cognitive Deficits in a Model of Alzheimer's Disease. Nat Commun. 2014;5:3443. doi: 10.1038/ncomms4443. [DOI] [PubMed] [Google Scholar]
- 411.Pratim Bose P, Chatterjee U, Nerelius C, Govender T, Norström T, Gogoll A, Sandegren A, Göthelid E, Johansson J, Arvidsson PI. Poly-N-Methylated Amyloid beta-peptide (Abeta) C-terminal Fragments Reduce Abeta Toxicity in Vitro and in Drosophila Melanogaster. J Med Chem. 2009;52:8002–8009. doi: 10.1021/jm901092h. [DOI] [PubMed] [Google Scholar]
- 412.Amijee H, Bate C, Williams A, Virdee J, Jeggo R, Spanswick D, Scopes DIC, Mazzitelli J, Doig A. The N-methylated peptide SEN304 Powerfully Inhibits Aβ(1-42) Toxicity by Perturbing Oligomer Formation. Biochemistry. 2012;51:8338–8352. doi: 10.1021/bi300415v. [DOI] [PubMed] [Google Scholar]
- 413.Hopping G, Kellock J, Barnwal RP, Law P, Bryers J, Varani G, Caughey B, Daggett V. Designed α-sheet Peptides Inhibit Amyloid Formation byTargeting Toxic Oligomers. Elife. 2014;3:e01681. doi: 10.7554/eLife.01681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 414.Bieschke J, Herbst M, Wiglenda T, Friedrich RP, Boeddrich A, Schiele F, Kleckers D, Lopez del Amo JM, Grüning BA, Wang Q, Schmidt MR, Lurz R, Anwyl R, Schnoegl S, Fändrich M, Frank RF, Reif B, Günter S, Walsh DM, Wanker EE. Small-molecule Conversion of Toxic Oligomers to Nontoxic β-sheet-rich Amyloid Fibrils. Nat Chem Biol. 2012;8:93–101. doi: 10.1038/nchembio.719. [DOI] [PubMed] [Google Scholar]
- 415.Richard T, Poupard P, Nassra M, Papastamoulis Y, Iglésias ML, Krisa S, Waffo-Teguo P, Mérillon JM, Monti JP. Protective Effect of ε-viniferin on β-amyloid Peptide Aggregation Investigated by Electrospray Ionization Mass Spectrometry. Bioorg Med Chem. 2011;19:3152–3155. doi: 10.1016/j.bmc.2011.04.001. [DOI] [PubMed] [Google Scholar]
- 416.Richard T, Papastamoulis Y, Waffo-Teguo P, Monti JP. 3D NMR Structure of a Complex between the Amyloid beta peptide(1-40) and thePolyphenol epsilon-viniferin Glucoside: Implications in Alzheimer's Disease. Biochim Biophys Acta. 2013;1830(11):5068–5074. doi: 10.1016/j.bbagen.2013.06.031. 2013. [DOI] [PubMed] [Google Scholar]
- 417.Ehrnhoefer DE, Bieschke J, Boeddrich A, Herbst M, Masino L, Lurz R, Engemann S, Pastore A, Wanker EE. EGCG Redirects AmyloidogenicPolypeptides into Unstructured, off-pathway Oligomers. Nat Struct Mol Biol. 2008:15, 558–566. doi: 10.1038/nsmb.1437. [DOI] [PubMed] [Google Scholar]
- 418.Bieschke J, Russ J, Friedrich RP, Ehrnhoefer DE, Wobst H, Neugebauer K, Wanker EE. EGCG Remodels Mature alpha-synuclein andamyloid-beta Fibrils and Reduces Cellular Toxicity. Proc Nat Acad Sci USA. 2010;107:7710–7715. doi: 10.1073/pnas.0910723107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 419.Wang SH, Liu FF, Dong XY, Sun Y. Thermodynamic Analysis of the Molecular Interactions between Amyloid β-Peptide 42 and (−)-Epigallocatechin-3-gallate. J Phys Chem B. 2010;114:11576–11583. doi: 10.1021/jp1001435. [DOI] [PubMed] [Google Scholar]
- 420.Liu FF, Dong XY, He L, Middelberg AP, Sun Y. Molecular Insight into Conformational Transition of Amyloid β-Peptide 42 Inhibited by (−)-Epigallocatechin-3-gallate Probed by Molecular Simulations. J Phys Chem B. 2011;115:11879–11887. doi: 10.1021/jp202640b. [DOI] [PubMed] [Google Scholar]
- 421.Lopez del Amo JM, Fink U, Dasari M, Grelle G, Wanker EE, Bieschke J, Reif B. Structural Properties of EGCG-induced, nontoxic Alzheimer's disease Aβ Oligomers. J Mol Biol. 2012;421(4-5):517–524. doi: 10.1016/j.jmb.2012.01.013. [DOI] [PubMed] [Google Scholar]
- 422.Attanasio F, Convertino M, Magno A, Caflisch A, Corazza A, Harridas H, Esposito G, Cataldo S, Pignataro B, Milardi D, Rizzarelli E. Carnosine Inhibits Aβ42 Aggregation by Perturbing the H-Bond Network in and around the Central Hydrophobic Cluster. ChemBioChem. 2013;14:583–592. doi: 10.1002/cbic.201200704. [DOI] [PubMed] [Google Scholar]
- 423.Arai T, Araya T, Sasaki D, Taniguchi A, Sato T, Sohma Y, Kanai M. Rational Design Identification of a Non-peptidic Aggregation Inhibitor of Amyloidβ based on a Pharmacophore Motif obtained from cyclo[-Lys-Leu-Val-Phe-Phe] Angew Chem Int Ed Engl. 2014;53:8236–8239. doi: 10.1002/anie.201405109. [DOI] [PubMed] [Google Scholar]
- 424.Sato M, Murakami K, Uno M, Nakagawa Y, Katayama S, Akagi K, Masuda Y, Takegoshi K, Irie K. Site-specific Inhibitory Mechanism for Amyloid β42 Aggregation by Catechol-type Flavonoids Targeting the Lys Residues. J Biol Chem. 2013;288:23212–23214. doi: 10.1074/jbc.M113.464222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 425.Palhano FL, Lee J, Grimster NP, Kelly JW. Toward the Molecular Mechanism(s) by which EGCG Treatment Remodels Mature Amyloid Fibrils. J Am Chem Soc. 2013;135:7503–7510. doi: 10.1021/ja3115696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 426.Tomiyama T, Kaneko H, Kataoka K, Asano S, Endo N. Rifampicin Inhibits the Toxicity of Pre-aggregated Amyloid Peptides by Binding to Peptide Fibrils and Preventing Amyloid-cell Interactio. Biochem J. 1997;322:859–865. doi: 10.1042/bj3220859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 427.Pickhardt M, Gazova Z, von Bergen M, Khlistunova I, Wang Y, Hascher A, Mandelkow EM, Biernat J, Mandelkow E. Anthraquinones Inhibit Tau Aggregation and Dissolve Alzheimer's Paired Helical Filaments in vitro and in Cells. J Biol Chem. 2005;280:3628–3635. doi: 10.1074/jbc.M410984200. [DOI] [PubMed] [Google Scholar]
- 428.Necula M, Kayed R, Milton S, Glabe CG. Small Molecule Inhibitors of Aggregation Indicate that Amyloid beta Oligomerization and Fibrillization Pathways are Independent and Distinct. J Biol Chem. 2007;282:10311–10324. doi: 10.1074/jbc.M608207200. [DOI] [PubMed] [Google Scholar]
- 429.Scherzer-Attali R, Pellarin R, Convertino M, Frydman-Marom A, Egoz-Matia N, Peled S, Levy-Sakin M, Shalev DE, Caflisch A, Gazit E, Segal D. Complete Phenotypic Recovery of an Alzheimer's disease Model by a Quinonetryptophan Hybrid Aggregation Inhibitor. PLoS One. 2010;5:e11101. doi: 10.1371/journal.pone.0011101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 430.Convertino M, Vitalis A, Caflisch A. Disordered binding of smallmolecules to Aβ(12-28) J Biol Chem. 2011;286:41578–41588. doi: 10.1074/jbc.M111.285957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 431.Scherzer-Attali R, Convertino M, Pellarin R, Gazit E, Segal D, Caflisch A. Methylations of Tryptophan-modified Naphthoquinone Affect its Inhibitory Potential toward Aβ Aggregation. J Phys Chem B. 2013;117:1780–1789. doi: 10.1021/jp309066p. [DOI] [PubMed] [Google Scholar]
- 432.Zhang T, Xu W, Mu Y, Derreumaux P. Atomic and Dynamic Insights into the Beneficial Effect of the 1,4-naphthoquinon-2-yl-L-tryptophan Inhibitor on Alzheimer's Aβ1-42 Dimer in terms of Aggregation and Toxicity. ACS Chem Neurosci. 2013;5:148–159. doi: 10.1021/cn400197x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 433.Brenke R, Kozakov D, Chuang GY, Beglov D, Hall D, Landon MR, Mattos C, Vajda S. Fragment-based Identification of Druggable ‘hot spots’ of Proteins using Fourier Domain Correlation Techniques. Bioinformatics. 2009;25:621–627. doi: 10.1093/bioinformatics/btp036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 434.McGann M. FRED Pose Prediction and Virtual Screening Accuracy. J Chem Inf Model. 2011;51:578–596. doi: 10.1021/ci100436p. [DOI] [PubMed] [Google Scholar]
- 435.Kroth H, Ansaloni A, Varisco Y, Jan A, Sreenivasachary N, Rezaei-Ghaleh N, Giriens V, Lohmann S, Lopez-Deber MP, Adolfsson O, Pihlgren M, Paganetti P, Froestl W, Nagel-Steger L, Willbold D, Schrader T, Zweckstetter M, Pfeifer A, Lashuel HA, Muhs A. Discovery and Structure Activity Relationship of Small Molecule Inhibitors of Toxic β-amyloid-42 Fibril Formation. J Biol Chem. 2012;287:34786–34800. doi: 10.1074/jbc.M112.357665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 436.Connors CR, Rosenman DJ, Lopes DH, Mittal S, Bitan G, Sorci M, Belfort G, Garcia A, Wang C. Tranilast Binds to Aβ Monomers and Promotes Aβ Fibrillation. Biochemistry. 2013;52:3995–4002. doi: 10.1021/bi400426t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 437.Tarus B, Nguyen PH, Berthoumieu O, Faller P, Doig AJ, Derreumaux P. Molecular Structure of the NQTrp Inhibitor with the AlzheimerAβ1-28 Monomer. Eur J Med Chem. 2015;91:43–50. doi: 10.1016/j.ejmech.2014.07.002. [DOI] [PubMed] [Google Scholar]
- 438.Novick PA, Lopes DH, Branson KM, Esteras-Chopo A, Graef IA, Bitan G, Pande VS. Design of β-amyloid Aggregation Inhibitors from a Predicted Structural Motif. J Med Chem. 2012;55:3002–3010. doi: 10.1021/jm201332p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 439.Stefanova NA, Muraleva NA, Korbolina EE, Kiseleva E, Maksimova KY, Kolosova NG. Amyloid Accumulation is a Late Event in Sporadic Alzheimer's Disease-like Pathology in Nontransgenic Rats. Oncotarget. 2015;6:1396–413. doi: 10.18632/oncotarget.2751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 440.Alzheimer Disease & Frontotemporal Dementia Mutation Database. http://www.molgen.ua.ac.be/admutations/
- 441.Rovelet-Lecrux A, Tuominen H, Majamaa K, Campion D, Remes AM. APP Locus Duplication in a Finnish Family with Dementia and Intracerebral Haemorrhage. J Neurol Neurosurg Psychiatry. 2007;78:1158–1159. doi: 10.1136/jnnp.2006.113514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 442.St George-Hyslop PH. Molecular Genetics of Alzheimer's Disease. Biol Psychiatry. 2000;47:183–199. doi: 10.1016/s0006-3223(99)00301-7. [DOI] [PubMed] [Google Scholar]
- 443.Levy E, Carman MD, Fernandez-Madrid IJ, Power MD, Lieberburg I, van Duinen SG, Bots GT, Luyendijk W, Frangione B. Mutation of the Alzheimer's Disease Amyloid Gene in Hereditary Cerebral Hemorrhage, Dutch Yype. Science. 1990;248:1124–1126. doi: 10.1126/science.2111584. [DOI] [PubMed] [Google Scholar]
- 444.Kaden D, Harmeier A, Weise C, Munter LM, Althoff V, Rost BR, Hildebrand PW, Schmitz D, Schaefer M, Lurz R, Skodda S, Yamamoto R, Arlt S, Finckh U, Multhaup G. Novel APP/Aβ Mutation K16N Produces Highly Toxic Heteromeric Aβ Oligomers. EMBO Mol Med. 2012;4:647–659. doi: 10.1002/emmm.201200239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 445.Nomura S, Umeda T, Tomiyama T, Mori H. The E693Δ (Osaka) Mutation in Amyloid Precursor Protein Potentiates Cholesterol-mediated Intracellular Amyloid β Toxicity via its Impaired Cholesterol Efflux. J Neuro Res. 2013;91:1541–1550. doi: 10.1002/jnr.23278. [DOI] [PubMed] [Google Scholar]
- 446.Shimizu T, Fukuda H, Murayama S, Izumiyama N, Shirasawa T. Isoaspartate Formation at Position 23 of Amyloid beta Peptide Enhanced Fibril Formation and Deposited onto Senile Plaques and Vascular Amyloids in Alzheimer's Disease. J Neurosci Res. 2002;70:451–461. doi: 10.1002/jnr.10350. [DOI] [PubMed] [Google Scholar]
- 447.Hashimoto Y, Matsuoka M. A mutation Protective Against Alzheimer's Disease Renders Amyloid β Precursor Protein Incapable of Mediating Neurotoxicity. J Neurochem. 2014;130(2):291–300. doi: 10.1111/jnc.12717. [DOI] [PubMed] [Google Scholar]
- 448.Qahwash I, Weiland KL, Lu Y, Sarver RW, Kletzien RF, Yan R. Identification of a Mutant Amyloid Peptide that Predominantly Forms Neurotoxic Protofibrillar Aggregates. J Biol Chem. 2003;278:23187–23195. doi: 10.1074/jbc.M213298200. [DOI] [PubMed] [Google Scholar]
- 449.Moskovitz J, Maiti P, Lopes DH, Oien DB, Attar A, Liu T, Mittal S, Hayes J, Bitan G. Induction of Methionine-sulfoxide Reductases Protects Neurons from Amyloid β-protein Insults in vitro and in vivo. Biochemistry. 2011;50:10687–10697. doi: 10.1021/bi201426b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 450.Harmeier A, Wozny C, Rost BR, Munter LM, Hua H, Georgiev O, Beyermann M, Hildebrand PW, Weise C, Schaffner W, Schmitz D, Multhaup G. Role of Amyloid-Beta Glycine 33 in Oligomerization, Toxicity, and Neuronal Plasticity. J Neurosci. 2009;29:7582–7590. doi: 10.1523/JNEUROSCI.1336-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 451.Kim W, Hecht MH. Mutations Enhance the Aggregation Propensity of the Alzheimer's A Beta Peptide. J Mol Biol. 2008;377:565–574. doi: 10.1016/j.jmb.2007.12.079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 452.Kim W, Hecht MH. Generic Hydrophobic Residues Are Sufficient to Promote Aggregation of the Alzheimer's Abeta 42 Peptide. Proc Natl Acad Sci USA. 2006;103:15824–15829. doi: 10.1073/pnas.0605629103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 453.Sandebring A, Welander H, Winblad B, Graff C, Tjernberg LO. The Pathogenic Abeta43 Is Enriched in Familial and Sporadic Alzheimer Disease. PLoS One. 2013;8:e55847. doi: 10.1371/journal.pone.0055847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 454.Conicella AE, Fawzi NL. The C-terminal Threonine of Aβ43 Nucleates Toxic Aggregation via Structural and Dynamical Changes in Monomers and Protofibrils. Biochemistry. 2014;53:3095–105. doi: 10.1021/bi500131a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 455.Messa M, Colombo L, del Favero E, Cantù L, Stoilova T, Cagnotto A, Rossi A, Morbin M, Di Fede G, Tagliavini F, Salmona M. The Peculiar Role of the A2V Mutation in Amyloid-beta 1-42 Molecular Assembly. J Biol Chem. 2014;289:24143–24152. doi: 10.1074/jbc.M114.576256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 456.Liu YW, He YH, Zhang YX, Cai WW, Yang LQ, Xu LY, Kong QP. Absence of A673T Variant in APP Gene Indicates an Alternative Protective Mechanism Contributing to Longevity in Chinese Individuals. Neurobiol Aging. 2014;35:935.e11–e12. doi: 10.1016/j.neurobiolaging.2013.09.023. [DOI] [PubMed] [Google Scholar]
- 457.Bamne MN, Demirci FY, Berman S, Snitz BE, Rosenthal SL, Wang X, Lopez OL, Kamboh LI. Investigation of an Amyloid Precursor Protein Protective Mutation (A673T) in a North American Case-control Sample of Late-onset Alzheimer's Disease. Neurobiol Aging. 2014;35:1779e15–e16. doi: 10.1016/j.neurobiolaging.2014.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 458.Benilova I, Gallardo R, Ungureanu AA, Castillo Cano V, Snellinx A, Ramakers M, Bartic C, Rousseau F, Schymkowitz J, De Strooper B. The Alzheimer Disease Protective Mutation A2T Modulates Kinetic and Thermodynamic Properties of Amyloid-beta (Abeta) Aggregation. J Biol Chem. 2014;289:30977–30989. doi: 10.1074/jbc.M114.599027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 459.Maloney JA, Bainbridge T, Gustafson A, Zhang S, Kyauk R, Steiner P, van der Brug M, Liu Y, Ernst JA, Watts RJ, Atwal JK. Molecular Mechanisms of Alzheimer Disease Protection by the A673T Allele of Amyloid Precursor Protein. J Biol Chem. 2014;289:30990–31000. doi: 10.1074/jbc.M114.589069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 460.Coskuner O, Wise-Scira O, Perry G, Kitahara T. The Structures of the E22delta Mutan-type Amyloid-beta Alloforms and the Impact of E22delta Mutation on the Structures of the Wild-type Amyloid-beta Alloforms. ACS Chem Neurosci. 2013;4:310–320. doi: 10.1021/cn300149j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 461.Baumketner A, Krone MG, Shea JE. Role of the Familial Dutch Mutation E22Q in the Folding and Aggregation of the 15-28 Fragment of the Alzheimer Amyloid-beta Protein. Proc Natl Acad Sci USA. 2008;105:6027–6032. doi: 10.1073/pnas.0708193105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 462.Huet A, Derreumaux P. Impact of the Mutation A21G (Flemish Variant) on Alzheimer's Beta-amyloid Dimers by Molecular Dynamics Simulations. Biophys J. 2006;91:3829–3840. doi: 10.1529/biophysj.106.090993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 463.Okamoto A, Yano A, Nomura K, Higai S, Kurita N. Effect of D23N Mutation on the Dimer Conformation of Amyloid-beta Proteins: Ab Initio Molecular Simulations in Water. J Mol Graph Model. 2014;50:113–124. doi: 10.1016/j.jmgm.2014.03.004. [DOI] [PubMed] [Google Scholar]
- 464.Viet MH, Nguyen PH, Ngo ST, Li MS, Derreumaux P. Effect of the Tottori Familial Disease Mutation (D7N) on the Monomers and Dimers of Abeta40 and Abeta42. ACS Chem Neurosci. 2013;4:1446–1457. doi: 10.1021/cn400110d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 465.Viet MH, Nguyen PH, Derreumaux P, Li MS. Effect of the English Familial Disease Mutation (H6R) on the Monomers and Dimers of Abeta40 and Abeta42. ACS Chem Neurosci. 2014;5:646–657. doi: 10.1021/cn500007j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 466.Truong PM, Viet MH, Nguyen PH, Hu CK, Li MS. Effect of Taiwan Mutation (D7H) on Structures of Amyloid-beta Peptides: Replica Exchange Molecular Dynamics Study. J Phys Chem B. 2014;118:8972–8981. doi: 10.1021/jp503652s. [DOI] [PubMed] [Google Scholar]
- 467.Takeda T, Klimov DK. Side Chain Interactions Can Impede Amyloid Fibril Growth: Replica Exchange Simulations of Abeta Peptide Mutant. J Phys Chem B. 2009;113:11848–11857. doi: 10.1021/jp904070w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 468.Lu Y, Wei G, Derreumaux P. Effects of G33A and G33I Mutations on the Structures of Monomer and Dimer of the Amyloid-beta Fragment 29-42 by Replica Exchange Molecular Dynamics Simulations. J Phys Chem B. 2011;115:1282–1288. doi: 10.1021/jp110269a. [DOI] [PubMed] [Google Scholar]
- 469.Brown AM, Lemkul JA, Schaum N, Bevan DR. Simulations of Monomeric Amyloid-beta Peptide (1-40) with Varying Solution Conditions and Oxidation State of Met35: Implications for Aggregation. Archives of Biochemistry and Biophysics. 2014;545:44–52. doi: 10.1016/j.abb.2014.01.002. [DOI] [PubMed] [Google Scholar]
- 470.Yano A, Okamoto A, Nomura K, Higai S, Kurita N. Difference in Dimer Conformation Between Amyloid-beta(1-42) and (1-43) Proteins: Replica Exchange Molecular Dynamics Simulations in Water. Chemical Physics Letters. 2014;595:242–249. [Google Scholar]
- 471.Anand P, Nandel FS, Hansmann UH. The Alzheimer's Beta Amyloid (Abeta1-39) Monomer in an Implicit Solvent. J Chem Phys. 2008;128:165102. doi: 10.1063/1.2907718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 472.Anand P, Nandel FS, Hansmann UH. The Alzheimer Beta-amyloid (Abeta(1-39) Dimer in an Implicit Solvent. J Chem Phys. 2008;129:195102. doi: 10.1063/1.3021062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 473.Nguyen PH, Tarus B, Derreumaux P. Familial Alzheimer A2V Mutation Reduces the Intrinsic Disorder and Completely Changes the Free Energy Landscape of the Abeta1-28 Monomer. J Phys Chem B. 2014;118:501–510. doi: 10.1021/jp4115404. [DOI] [PubMed] [Google Scholar]
- 474.Meral D, Urbanc B. Discrete Molecular Dynamics Study of Oligomer Formation by N-terminally Truncated Amyloid Beta-protein. J Mol Biol. 2013;425:2260–2275. doi: 10.1016/j.jmb.2013.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 475.Gillman AL, Jang H, Lee J, Ramachandran S, Kagan BL, Nussinov R, TeranArce F. Activity and Architecture of Pyroglutamate-modified Amyloid-beta (AbetapE3-42) pores. J Phys Chem B. 2014;118:7335–7344. doi: 10.1021/jp5040954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 476.Lee J, Gillman AL, Jang H, Ramachandran S, Kagan BL, Nussinov R, Teran Arce F. Role of the Fast Kinetics of Pyroglutamate-modified Amyloid-beta Oligomers in Membrane Binding and Membrane Permeability. Biochemistry. 2014;53:4704–4714. doi: 10.1021/bi500587p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 477.Zhuravlev PI, Reddy G, Straub JE, Thirumalai D. Propensity to Form Amyloid Fibrils Is Encoded as Excitations in the Free Energy Landscape of Monomeric Proteins. J Mol Biol. 2014;426:2653–2666. doi: 10.1016/j.jmb.2014.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 478.Liang C, Ni R, Smith JE, Childers WS, Mehta AK, Lynn DG. Kinetic Intermediates in Amyloid Assembly. J Am Chem Soc. 2014;136:15146–14149. doi: 10.1021/ja508621b. [DOI] [PubMed] [Google Scholar]
- 479.Baell J, Walters MA. Chemistry: Chemical con Artists First Drug Discovery. Nature. 2014;513:481–483. doi: 10.1038/513481a. [DOI] [PubMed] [Google Scholar]
- 480.Ingólfsson HI, Thakur P, Herold KF, Hobart EA, Ramsey NB, Periole X, de Jong DH, Zwama M, Yilmaz D, Hall K, Maretzky T, Hemmings HC, Jr, Blobel C, Marrink SJ, Koçer A, Sack JT, Andersen OS. Phytochemical Perturb Membranes and Promiscuously Alter Protein Function. ACS Chem Biol. 2014;9:1788–1798. doi: 10.1021/cb500086e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 481.Lesné SE, Sherman MA, Grant M, Kuskowski M, Schneider JA, Bennett DA, Ashe KH. Brain Amyloid-beta Oligomers in Ageing and Alzheimer's Disease. Brain. 2013;136:1383–1398. doi: 10.1093/brain/awt062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 482.Hamilton-Brown P, Bekard I, Ducker WA, Dunstan D, Dave E. How Does Shear Affect Abeta Fibrillogenesis? J Phys Chem B. 2008;112:16249–16251. doi: 10.1021/jp805257n. [DOI] [PubMed] [Google Scholar]
- 483.Dunstan D, Hamilton-Brown P, Asimakis P, Duckerand W, Bertolini J. Shear Flow Promotes Amyloid-beta Fibrillization. Protein Eng Des Sel. 2009;22:741–746. doi: 10.1093/protein/gzp059. [DOI] [PubMed] [Google Scholar]
- 484.Lee C, Bird S, Shaw M, Jean L, Vaux DJ. Combined Effects of Agitation, Macromolecular Crowding, and Interfaces on Amyloidogenesis. J Biol Chem. 2012;287:38006–38019. doi: 10.1074/jbc.M112.400580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 485.Gruebele M, Thirumalai D. Perspective: Reaches of Chemical Physics in Biology. J Chem Phys. 2013;139:121701. doi: 10.1063/1.4820139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 486.Esbjörner EK, Chan F, Rees E, Erdelyi M, Luheshi LM, Bertoncini CW, Kaminski CF, Dobson CM, Kaminski Schierle GS. Direct Observations of Amyloid Beta Self-assembly in Live Cells Provide Insights into Differences in the Kinetics of Abeta(1-40) and Abeta(1-42) Aggregation. Chem Biol. 2014;21:732742. doi: 10.1016/j.chembiol.2014.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 487.Li C, Charlton LM, Lakkavaram A, Seale C, Wang G, Young GB, MacDonald JM, Pielak GJ. Differential Dynamical Effects of Macromolecular Crowding on an Intrinsically Disordered Protein and a Globular Protein: Implications for In-cell NMR spectroscopy. J Am Chem Soc. 2008;130:6310–6311. doi: 10.1021/ja801020z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 488.Li C, Liu M. Protein Dynamics in Living Cells Studied by In-cell NMR Spectroscopy. FEBS Lett. 2013;587:1008–1011. doi: 10.1016/j.febslet.2012.12.023. [DOI] [PubMed] [Google Scholar]
- 489.Lu P, Bai XC, Ma D, Xie T, Yan C, Sun L, Yang G, Zhao Y, Zhou R, Scheres SH, Shi Y. Three-dimensional Structure of Human Gamma-secretase. Nature. 2014;512:166. doi: 10.1038/nature13567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 490.Dominguez L, Foster L, Meredith SC, Straub JE, Thirumalai D. Structural Heterogeneity in Transmembrane Amyloid Precursor Protein Homodimer Is a Consequence of Environmental Selection. J Am Chem Soc. 2014;136:9619–9626. doi: 10.1021/ja503150x. [DOI] [PMC free article] [PubMed] [Google Scholar]