

ABSTRACT
Extracellular vesicles (EVs) hold great potential as novel systems for nucleic acid delivery due to their natural composition. Our goal was to load EVs with microRNA that are synthesized by the cells that produce the EVs. HEK293T cells were engineered to produce EVs expressing a lysosomal associated membrane, Lamp2a fusion protein. The gene encoding pre-miR-199a was inserted into an artificial intron of the Lamp2a fusion protein. The TAT peptide/HIV-1 transactivation response (TAR) RNA interacting peptide was exploited to enhance the EV loading of the pre-miR-199a containing a modified TAR RNA loop. Computational modeling demonstrated a stable interaction between the modified pre-miR-199a loop and TAT peptide. EMSA gel shift, recombinant Dicer processing and luciferase binding assays confirmed the binding, processing and functionality of the modified pre-miR-199a. The TAT-TAR interaction enhanced the loading of the miR-199a into EVs by 65-fold. Endogenously loaded EVs were ineffective at delivering active miR-199a-3p therapeutic to recipient SK-Hep1 cells. While the low degree of miRNA loading into EVs through this approach resulted in inefficient distribution of RNA cargo into recipient cells, the TAT TAR strategy to load miRNA into EVs may be valuable in other drug delivery approaches involving miRNA mimics or other hairpin containing RNAs.
KEYWORDS: Exosomes, microRNA mimic, HIV-1 TAR RNA, TAT peptide, hepatocellular cancer, small hairpin RNA, shRNA
Introduction
Extracellular vesicles (EVs) are nano-sized structures that may be subdivided into exosomes, ectosomes, microvesicles and apoptotic bodies [1]. EVs are produced by many cell types including cells of the immune system [2,3], mesenchymal stem cells [4], endothelial [5], epithelial [6] and cancer cells [7–9]. EVs naturally contain a variety of cargo, including protein [8], DNA [10,11], mRNA [12], miRNA [12,13] and lncRNAs [14,15], and have been identified in many bodily fluids including plasma [16], serum [17], saliva [18], cerebral spinal fluid [19], breast milk [20], urine [21] and semen [22]. EVs are shed directly into bodily fluids and play important roles in cell–cell communication [23,24].
Research in this field has recently developed into many practical applications of EVs, including biomarkers of disease [25], predictors of therapeutic response [26] and as drug delivery carriers (reviewed in [27,28]). Unlike synthetic liposome or nanoparticle drug delivery systems, EVs are composed completely of natural ingredients. Their natural composition may allow EVs to avoid the toxicity and rapid clearance associated with liposomes or nanoparticles in the bloodstream [29,30]. EVs deliver their cargo to recipient cells through cell attachment via surface adhesion proteins [31] or by directly fusing to the plasma membrane [32]. EVs have been used as drug delivery carriers of siRNA [33], miRNA mimics [34,35], miRNA inhibitors [34], mRNA [36] and proteins expressed from plasmid DNA [37].
Work by us and others over the past decade or so has shown that a large number of miRNAs are differentially expressed during tumorigenesis [38]. miRNAs are attractive targets for therapy because (i) they regulate entire pathways rather than individual proteins, (ii) they may be easily inhibited with anti-miRs if overexpressed in the tumour or replenished by miRNA mimics if underexpressed and (iii) therapeutic oligonucleotides such as anti-miRNAs or miRNA mimics accumulate in highly perfused organs like liver. We previously reported that miR-199a-3p, a miRNA shown in several studies to be reduced in hepatocellular carcinoma (HCC), targets CD44 and that the proliferation was reduced in CD44 positive but not CD44 negative HCC cells lines [39]. The goal of this study was to deliver miRNA mimics to HCC cells using miR-199a-3p as our proof of concept mimic.
HEK293/HEK293T cells have been used extensively for protein engineering. Because these cells produce high quantities of EVs [40] grow quickly and exhibit high transfection efficiency, they have been widely used as EV delivery systems [41,42]. The cargo of HEK293T is not enriched by cancer or disease related pathways [43]. Furthermore, HEK293T EVs did not display any toxicity or immunogenicity in vitro [44] or in vivo [45].
Two methods exist to deliver cargo RNAs to EVs (recently reviewed in [46]). These include exogenous (also known as chemical) loading, in which the oligo is introduced into the EVs [33,37,47,48] and endogenous (i.e. active) loading where the EV producing cells synthesizes both the delivery system and the RNA cargo [36,49]. We developed here an active delivery modality that exploits the HIV-1 TAR RNA-TAT peptide interaction by swapping the wild type pre-miR-199a loop with the TAR RNA loop. The modified pre-miR-199a is designed to recognise the TAT peptide that was introduced into the EVs using a Lamp2a fusion protein. The loading of the miR-199a into EVs was enhanced using this TAT–TAR interaction.
Material and methods
Cell lines and tissue culture
HEK293T cell line (CRL-11268) was purchased from American Type Culture Collection (Manassas, VA). HEK293T cells were cultured as adherent cells in T-flasks or 150 mm dishes using Dulbecco’s Modified Eagle Medium (Life Technologies, Grand Island, NY) supplemented with 10% fetal bovine serum (FBS) (Sigma, St. Louis, MO). The collection of EVs from cells cultured used FBS that was first depleted of exogenous EVs by an 18 h centrifugation at 100,000 rcf, 4°C. Cells were cultured in a 5% CO2/humidified air incubator. HEK293T cells were passaged using trypsin/EDTA and only passages less than 35 were used throughout the study. Cultures were scaled up to 12 or 24, 150 mm dishes prior to isolating the EVs.
Cloning of vector containing the Lamp2a fusion protein and modified miR-199a
An overview of the approach to introduce miR-199a-3p into HEK293T EVs is shown in Figure 1(a). The pEGFP-1 vector expressing mouse Lamp2b was generously donated by Dr Matthew Wood. We substituted the mouse Lamp2b with the human Lamp2a sequence as Lamp2a is the most abundant of the three isoforms (Lamp2a, Lamp2b and Lamp2c). Lamp2a was cloned into the vector backbone using NheI and BamHI restriction sites. Four peptides sequences were introduced into the Lamp2a sequence: 3x Flag peptide for EV purification, PC94 peptide for HCC targeting [50], TAT peptide to bind to the modified TAR resembling miRNA sequence and His tag to verify that the protein was translated in frame. As HEK293T cells are neomycin resistant, the hygromycin cassette was introduced downstream of the SV40 promoter. The map of the vector is shown in Supplementary Figure 1(a). miR-199a hairpin loop was modified to resemble TAR RNA, as shown in Figure 1(b). The primary sequence of human miR-199a was inserted into an artificial intron of the Lamp2a gene. miR-199a sequence was introduced within the exonic junction of Lamp2a gene by cloning it within the two unique restriction sites: HindIII and SalI (Figure 1(c)). Table 1 highlights all the vector backbones that were generated for this study to develop engineered EVs.
Figure 1.
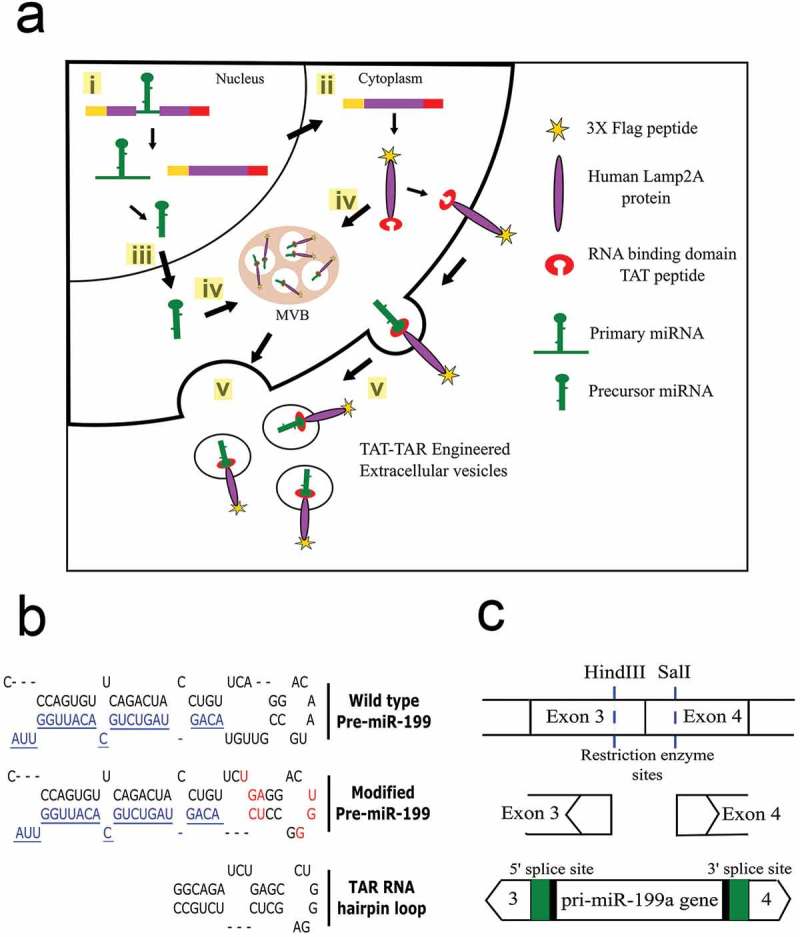
Overview of strategy to load cargo RNAs into EVs. (a) Sequence of events leading to loading of cargo RNA into the EVs. (i) The modified miR-199a gene engineered into an intron of the targeting fusion protein gene is spliced from the mRNA in the nucleus of the HEK293T cells. (ii) The fusion protein is translated from the spliced mRNA (iii) while the modified pri-miR-199a is processed to mature miR-199a-3p. (iv) Both the targeting, fusion protein and pre-miR-199a are directed into exosomes or microvesicles; the targeting protein through alignment of the Lamp2a transmembrane domain and the pre-miRNA-199a through binding of the modified precursor to the TAT peptide on the luminal C-terminus of the targeting protein. (v) EVs are shed into the extracellular space and collected by ultracentrifugation. (b) Unique restriction sites were located in exons 3 and 4 of the Lamp2a cDNA. The 370 bp chimeric intron containing the pri-miR-199a gene, 5′ and 3′ flanking sequences of exons 3 and 4, respectively and the 5′ and 3′ splice sites were cloned into the restriction sites of the targeting vector. Green colour depicts the consensus sequences that were engineered to increase the splicing efficiency. (c) Sequence of the wild type pri-miR-199a sequence (top) with the mature miR-199a-3p coloured blue. Eight nt contained within the loop and stem portions (red) were modified in the pri-miR-199a sequence (middle) to resemble that portion of the TAR RNA that binds to the TAT protein (bottom).
Table 1.
List of vector backbones that were used during the study to develop engineered HEK293T cells.
| Sr. No | Vectors generated to stably select HEK293T cells | Description |
|---|---|---|
| 1 | Empty | pEGFP-C1 backbone without Lamp2 fusion protein |
| 2 | FPLTH | 3X-Flag-PC94-Lamp2a-TAT-His fusion protein |
| 3 | FPLTH-Mod 199 | 3X-Flag-PC94-Lamp2a-TAT-His fusion protein containing modified precursor miR-199a TAR hairpin loop |
| 4 | FPL-Mod 199 | 3X-Flag-PC94-Lamp2a fusion protein containing modified precursor miR-199a TAR hairpin loop |
| 5 | FPLTH-WT 199 | 3X-Flag-PC94-Lamp2a-TAT-His fusion protein containing wild type precursor miR-199a hairpin loop |
Generation of stable cells
The pEGFP-C1 vector with and without the engineered Lamp2a protein was used to generate stable HEK293T cells. The cells were plated overnight in a six-well plate at a seeding density of 200,000 cells per well and 2.5 µg of the plasmid was transfected using Lipofectamine 3000. Forty-eight hours post-transfection, the cells were trypsinised, diluted 10 times and plated in another six-well plate. DMEM media was replaced with DMEM media containing 150 µg/ml of hygromycin. Cells were selected over a period of 1–1.5 months until visible colonies were formed. The colonies were subsequently harvested to verify for protein expression and cells were expanded to collect EVs from their supernatant.
Collection of EVs
EVs were isolated using a standard ultracentrifugation protocol [51]. In brief, 0.4 l of cell culture supernatant was centrifuged at 300g for 10 min, transferred to clean tubes and centrifuged at 2000g for 20 min to remove cell debris. The supernatant was transferred to clean tubes and centrifuged at 10,000g for 30 min, followed by vacuum filtration using a 0.22 μm filter (EMD Millipore, Billerica, MA). Filtered supernatant was transferred to ultracentrifuge tubes (#355622, Beckman Coulter, Brea, CA) and centrifuged at 110,000g for 4 h in a Beckman Coulter Optima™ XE ultracentrifuge with a Type 45 Ti rotor. The EV pellet was washed once with chilled, sterile phosphate-buffered saline (PBS, pH 7.4), followed by resuspension in 0.3–0.6 ml PBS with 1% DMSO. EV protein yield was measured using Pierce™ BCA Protein Assay (Life Technologies).
In silico computational analysis
In silico interaction between the modified pre-miR-199a sequence and TAT peptide was carried out by computational modelling through Rosetta and manual manipulation of NMR structures. Molecular dynamics simulation was also conducted on the final model to determine the stability of the structure and analyse the key interactions between the TAT peptide and the modified RNA. A detailed description of the molecular dynamics simulation is provided in the supplemental data section (Supplementary Figures 2–4).
Figure 2.
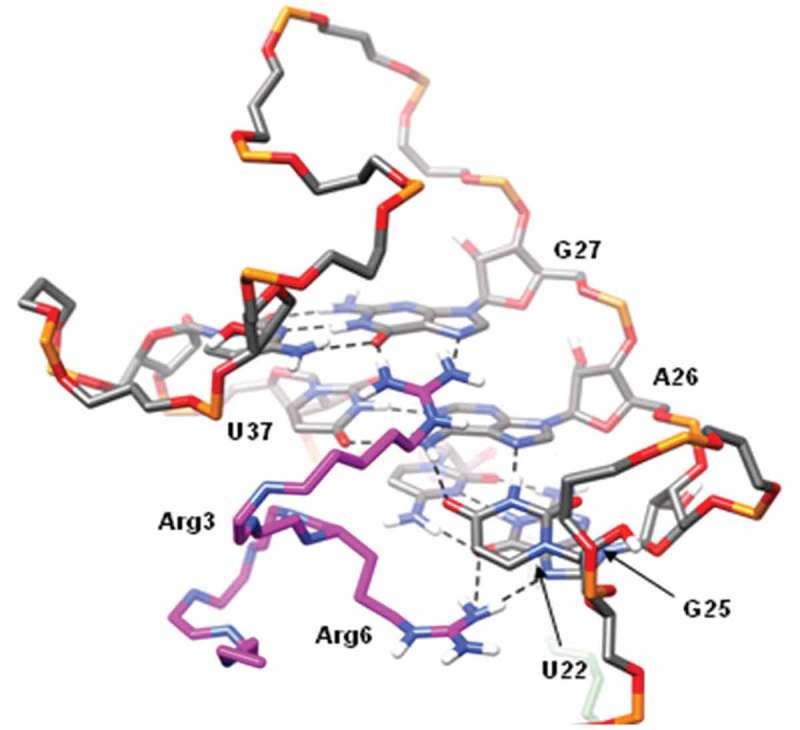
Interaction between TAT peptide and TAR RNA loop. The peptide is depicted in magenta with only the key arginine (Arg3 and Arg6) side chains are shown. Arg3 forms stable hydrogen bonds (dashed black lines) with G27, while Arg6 forms hydrogen bonds with G25. U22 and A26 of the U22-A26-U37 base triple are sandwiched by the two arginine residues.
Figure 4.
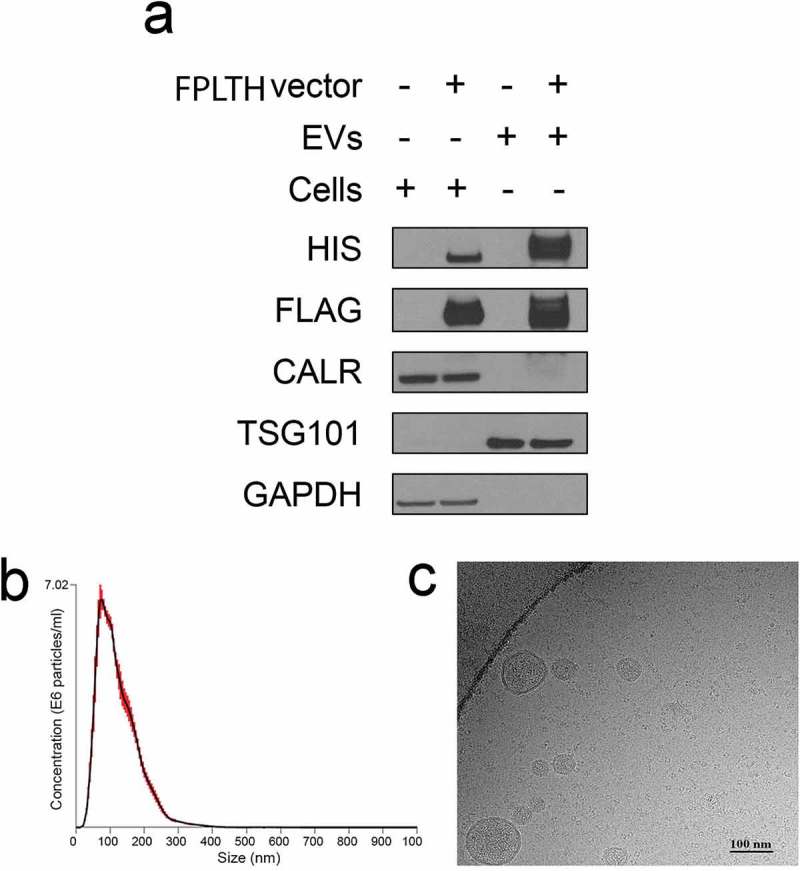
Characterisation of engineered EVs. (a) Protein isolated from the engineered EVs or the EV producing cells were detected by immunoblotting using antibodies against the various proteins shown. Cells were stably transfected with the FPLTH-Mod 199 vector (+) or empty vector (−). (b) Particle size distribution of engineered EVs as determined by nanoparticle tracking analysis. (c) Cryo-TEM image of engineered EVs collected by ultracentrifugation.
EMSA gel shift assay
Tar RNA loop sequence (UCUGAGCCUGGGAGCUC) was used to replace the wild type human miR-199a-1 sequence. Primers were designed to amplify the engineered pre-miRNA-199a-1 with T7 promoter at the 5′ end. The forward primer containing T7 promoter sequence (underlined) and reverse primers, are 5′- TAATACGACTCACTATAGGCAGTGTTCAGACTACCTGTTCTCTGAGCCTGG-3′ and reverse 5′-TAAGCAATGTGCAGACTACTGTTCGAGCTCCCAGGCTCAGAGAACAGGTAGTC-3′. DNA templates were amplified by PCR under a standard protocol which consisted of 94°C for 2 min, 40 cycles of 94°C for 15 s, 60°C for 20 s, 72°C for 20 s and 72°C for 10 min as a final extension step. Amplicons were purified with Qiagen Nucleotide Removal Kit (Cat#28034, Qiagen, Valencia CA USA). Template concentration was measured with NanoDrop Spectrophotometer. In-vitro T7 transcription was performed with 200 ng of DNA templates with T7 promoter in a 30 µl reaction using AmpliScribe™ T7-Flash™ Transcription Kit (Cat# ASF3507, EpiCentre USA). The reaction contained 3 µl of AmpliScribe T7-Flash 10X Reaction Buffer, 2.7 µl of ATP, CTP, GTP and UTP at 100 mM, 3 µl of DTT at 100 mM, and 3 μl of AmpliScribe T7-Flash Enzyme. One microlitre of [α-32P] ATP solution at 40 mCi/ml was added into the reaction to label the transcripts. The reaction was incubated 42°C for 4 h. Fifty nanograms of low molecular weight markers (10–100 nt) (Affymetrix # 76410) were 5′ end labelled with [γ-32P]-ATP (6000 Ci/mmol) at 37°C for 5 min in a 20 µl reaction. 32P-labelled RNA transcripts and markers were purified with Centri Spin-10 Column (Cat# CS-101, Princeton Separations) following the manufacture protocol. T7 transcripts for the modified miR-199a were incubated without or with increasing amounts of TAT peptide (RPRGTRGKGRRIRR) custom ordered from Lifetein, USA. The TAT peptide was added at increasing molar ratios of peptide to RNA: 1, 25, 50, 75, 100, 125 and 150. The samples were then resolved on a 4% native polyacrylamide gel, 4°C at 200 V for 3 h before the autoradiographic development.
In vitro DICER processing
One microgram of 32P-labelled T7 transcripts for modified miR-199a in the presence and absence of 10-fold molar ratio of TAT binding peptide were incubated at 37°C overnight in a 20 µl of reaction using recombinant human Dicer enzyme (Can# T510002, Genlantis USA). The samples were resolved on a native polyacrylamide gel as mentioned above.
Luciferase reporter assay
HEK293T cells were seeded at 80,000 cells per well in 24-well plates. Cells were allowed to attach overnight and were transfected with lipofectamine 2000 according to the manufacturer’s instructions. One hundred nanograms of psiCHECK-2 vector expressing entire 3′ UTR of CD44 gene [39] was cotransfected with 500 ng of plemiR vector expressing the modified precursor miR-199a. Thirty-six hours post transfection, cells were harvested by the addition of 500 μl passive lysis buffer. Renilla and firefly luciferase activities in the cell lysate were measured with the Dual Luciferase assay system (Promega, Madison, WI).
Immunoblotting
Immunoblotting was performed using manufacturer protocols and standard conditions. The EV pellet was lysed using RIPA buffer with 1X Protease and Phosphatase Inhibitor Cocktail. Protein was assayed using the BCA protein assay. Protein lysates were mixed with 4X non-reducing lithium dodecyl sulfate sample loading buffer containing 3% 2-mercaptoethanol. Twenty micrograms total protein were loaded per lane of a gradient 4–12% precast SDS-PAGE gel (NuPAGE®, Life Technologies). Following separation, proteins were transferred to a nitrocellulose membrane then blocked with 5% bovine serum albumin (BSA) before being incubated with primary antibodies. The primary antibodies used included: anti-Flag (F1804, Sigma-Aldrich), anti-His (D3I1O, Cell Signaling), anti-TSG101 (SC7964, Santa Cruz) anti-calreticulin (CALR, 2891S, Cell Signaling) and anti-GAPDH (SC-32233, Santa Cruz Biotechnology, Dallas, TX). The membrane was washed using 0.05% Tween-20 in 1X TBS-T wash buffer then incubated with 1% BSA in TBS-T and secondary antibody (goat anti-mouse or goat-anti-rabbit) for 1 h. Presence of proteins was visualised using an enhanced chemiluminescence detection system (GE Healthcare Bio-Sciences, Pittsburgh, PA). GAPDH and TSG101 protein were used as loading controls for cells and EVs, respectively.
RNA isolation and qRT-PCR
Total RNA was isolated from the cells or EVs using Trizol reagent (Invitrogen, USA) with standard conditions. RNA was converted to cDNA using random primers and SuperScript II reverse transcriptase enzyme or using TaqMan microRNA assays (Invitrogen, USA) as previously described [52]. qRT-PCR was performed in an Applied Biosystems 7900HT qPCR instrument. A custom TaqMan probe (LNA sequence TG+AGCC+T+GG+GA) was designed to the modified pre-miR-199a loop amplified using forward primer 5′- CCCAGTGTTCAGACTACCTGTTC-3′ and reverse primer 5′- TAACCAATGTGCAGACTACTGT-3′. Data were normalised to U6 or 18S rRNA and the relative expression was determined using the 2−ΔCT method.
Cryo transmission electron microscopy
Isolated EVs were re-suspended in 1X PBS buffer solution. To disperse any aggregated EVs, the sample was sonicated for 30 s immediately prior to freezing. Sample preparation for Cryo-TEM was performed in the EM Core of the University of Florida’s Interdisciplinary Center for Biotechnology Research. Three microlitre aliquots of suspended EVs sample were applied to C-flat holey carbon grids (Protochips, Inc.) and vitrified using a Vitrobot™ Mark IV (FEI Co.) operated at room temperature and with 100% humidity in the control chamber. The vitrified sample was stored under liquid nitrogen to avoid ice crystals formation and transferred into a Gatan cryo holder (Model 626/70) for imaging. The sample was examined using a 16-megapixel CCD camera (Gatan, Inc.) on a Tecnai (FEI Co.) G2 F20-TWIN Transmission Electron Microscope operated at a voltage of 200 kV using low dose conditions (~20 e/Å2). Images were recorded with a defocus of approximately 3 µm to improve contrast.
Small RNA sequencing
The small RNA of the EVs or the producing cells was sequenced using the Illumina sequence kit according to manufacturer’s recommendations by the gene expression core at the Ohio State University. EVs were collected as described above. RNA was isolated from the cells or EVs using the Trizol method. A detailed description of the methodology and data analysis may be found in the supplemental data section. Data were uploaded to NCBI gene omnibus GSE99430.
Nanoparticle tracking analysis
The size and concentration of purified EVs was determined using nanoparticle tracking analysis (NTA) on a Nanosight NS300 instrument (Malvern Instruments, Westborough, MA).
Uptake of EV cargo into recipient cells
HCC cell lines SK-Hep1 and Huh7 were maintained in DMEM medium supplemented with 10% heat-inactivated FBS culture in T75 culture flasks. Before plating the cells, both the cell-lines were re-suspended in DMEM-EV free media and 40,000 cells were plated in 24-well plate (Falcon, Corning Life Sciences). EVs were isolated from Empty and FPLTH-Mod 199 engineered cells and using NTA, approximately 2 × 1011 particles of EVs were added to the recipient cells and incubated. Twenty-four hours post-incubation cells were harvested for RNA isolation and both precursor and mature miR-199a-3p was measured by qRT-PCR. The relative expression of miRNA-199a-3p validated mRNA targets was also evaluated by qRT-PCR. Primer sequences are available upon request.
Flag enriched EVs column purification
Engineered EVs were purified using modifications to an established protocol (J. Leonard, unpublished data). Two hundred millilitres of cell culture supernatant isolated from the engineered HEK293T cells were centrifuged at 2000g and 10,000g to eliminate cell debris and apoptotic vesicles, respectively. The supernatant was filtered through 0.22 µm filter and was subsequently degassed to remove air bubbles which could cause the gel bed to crack. Flag column purification was setup using Anti-Flag M2 affinity gel from Sigma (Catalog #A2220) and the procedure was carried out using manufacture’s guidelines. The supernatant was passed three times through the column in order to ensure complete binding of the Flag expressing EVs prior to elution with 3X Flag peptide from Sigma (Catalog# F4799). The EV fractions collected post-elution were then concentrated to smaller volume using 3 kDa cut-off centrifugal filters from EMD Millipore (Catalog# UFC900396). Western blotting and qRT-PCR were performed on the Flag enriched EV fraction to determine its protein expression and miR-199 expression levels. To determine EV uptake into recipient cells, EVs isolated by ultracentrifugation or column purification (0.5 × 1010), or the column flow through were added to recipient cells SK-Hep1 and Huh7 cells. RNA was isolated after 3, 6 and 12 h and the amount of pre-miR-199a within the recipient cells were measured by qRT-PCR.
Transwell insert assay
SK-Hep1 or Huh7 cells were co-cultured along with engineered HEK293T cells to study the uptake of engineered EVs. Biological triplicates of HCC cells were plated at a density of 1.5 × 105 cells in DMEM/10% FBS and left to attach overnight in the lower chamber of 6 well plates separated with a 0.4 µm polyester membrane Transwell filters (Sigma #CLS3450). The following day, media was then replaced with DMEM without FBS. Engineered, donor HEK293T cells were plated on the culture insert at a density of 2.5 × 105 in exosome free DMEM/10% FBS. Following 24 h of co-culture, cells were harvested for RNA isolation.
Results
In silico prediction and in vitro confirmation of TAT peptide and modified pre-miR-199a interaction
In silico computation was performed to determine if the modified pre-miR-199a would affect binding to the TAT peptide. A molecular dynamics simulation was conducted to determine the stability of the structure and to analyse the key interactions between the TAT peptide and the modified pre-miR-199a. The key interactions between TAT peptide and the TAR RNA loop that is present on the modified pre-miR-199a involve Arg3 and Arg6 sandwiching U22 of the UCU bulge (Figure 2). Additionally, Arg3 forms hydrogen bonds with G27, while Arg6 forms hydrogen bonds with G25. All of these interactions were stable throughout the entire molecular dynamics simulation and the peptide remained bound to the major groove of the RNA hairpin. Importantly, the stem portion of the RNA is not bound to peptide allowing the option to insert any hairpin sequence into the targeting vector without effecting peptide binding.
Modified precursor is processed to functionally active mature miR-199a-3p
The interaction between the TAT peptide and the modified pre-miR-199a was further validated by EMSA gel shift assays. TAT peptide produced a shift in the mobility of the modified pre-miR-199a, demonstrating binding (Figure 3(a)). rDicer digestion, qRT-PCR for the mature miRNA and luciferase reporter experiments were conducted to ensure that the modified pre-miR-199a was processed by Dicer to the active, mature miR-199a. We hypothesised that Dicer would cleave the TAT peptide bound pre-miR-199a since the current model of Dicer processing postulates that Dicer anchors on the base of the hairpin’s stem portion and the cleavage site is selected by measuring from the 5′ end of the dsRNA [53]. To test this hypothesis, we performed rDicer digests of peptide bound and unbound T7 transcribed modified pre-miRNA-199a. The ~22 nt mature miRNA was processed from the precursor following a 12 h incubation with rDicer even in the presence of 10-fold TAT peptide (Figure 3(b)). This demonstrates that peptide bound to the loop of the modified pre-miRNA does not affect its processing by Dicer.
Figure 3.
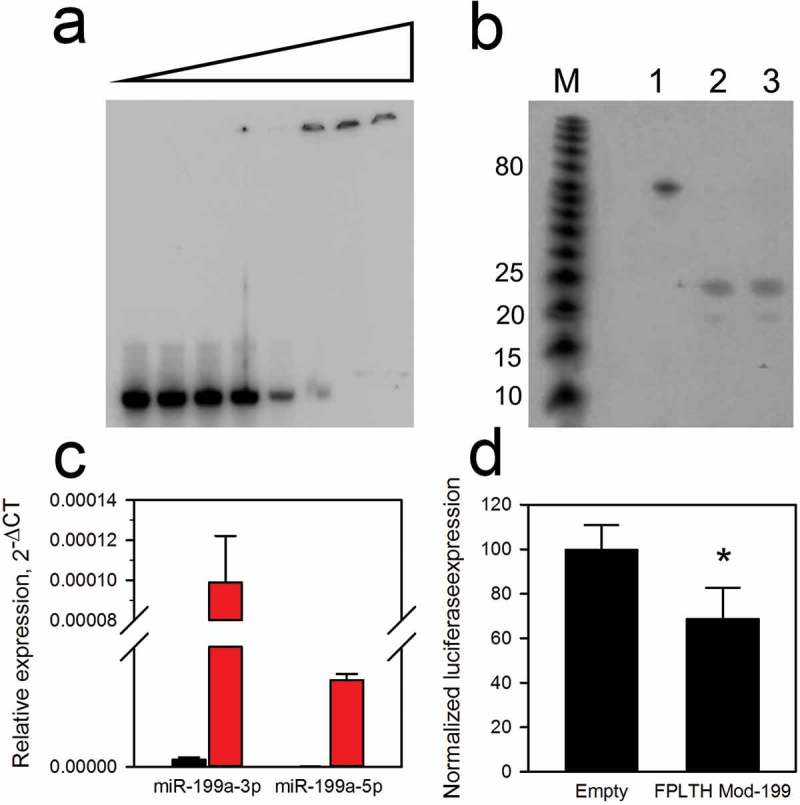
Modified pre-miR-199a binding, processing and functionality. (a) EMSA gel shift of modified pre-miR-199a. The 32P-labelled transcript of the modified pre-miR-199a was incubated in the absence (first lane) or with increasing amounts of TAT peptide. (b) rDicer processing of peptide bound modified pre-miR-199a. A 10-fold excess of TAT peptide to RNA was bound to the 62 nt modified pre-miR-199a transcript (lane 3). Lane two represents no TAT peptide added. Transcripts were exposed to rDicer for 18 h (lanes 2–3) or mock treatment (lane 1). The 23 nt mature miR-199a was generated by rDicer in the absence (lane 2) or presence (lane 3) of the TAT peptide. (c) Generation of mature miR-199a from modified precursor as determined by TaqMan qPCR. The black and red bars represent the untransfected cells and FPLTH vector transfected cells, respectively. (d) CD44 3′ UTR was inserted downstream of the luciferase reporter gene in psiCheck-2 vector and was co-transfected into HEK293T cells along with a pLemiR vector containing FPLTH-Mod 199 or empty vector control. *p < 0.05.
To establish accurate splicing and expression of the pre-miR-199a from the intron of the Lamp2a gene, we transiently transfected the vector containing the FPLTH-Mod 199 fusion protein into HEK293T cells. PCR was performed to demonstrate correct splicing of the Lamp2a gene. RNA was isolated from the stable cells and cDNA was generated using primers spanning the miR-199a intronic portion (Supplementary Figure 5(a)). The plasmid vector backbones with and without the modified miR-199a portion were used as positive controls. The cDNA generated from the stable cells containing miR-199a shows a band of ~100 bp compared to the control vector band of ~400 bp (Supplementary Figure 5(a)). The difference in the band sizes relates to the correct splicing of the intronic portion.
Figure 5.
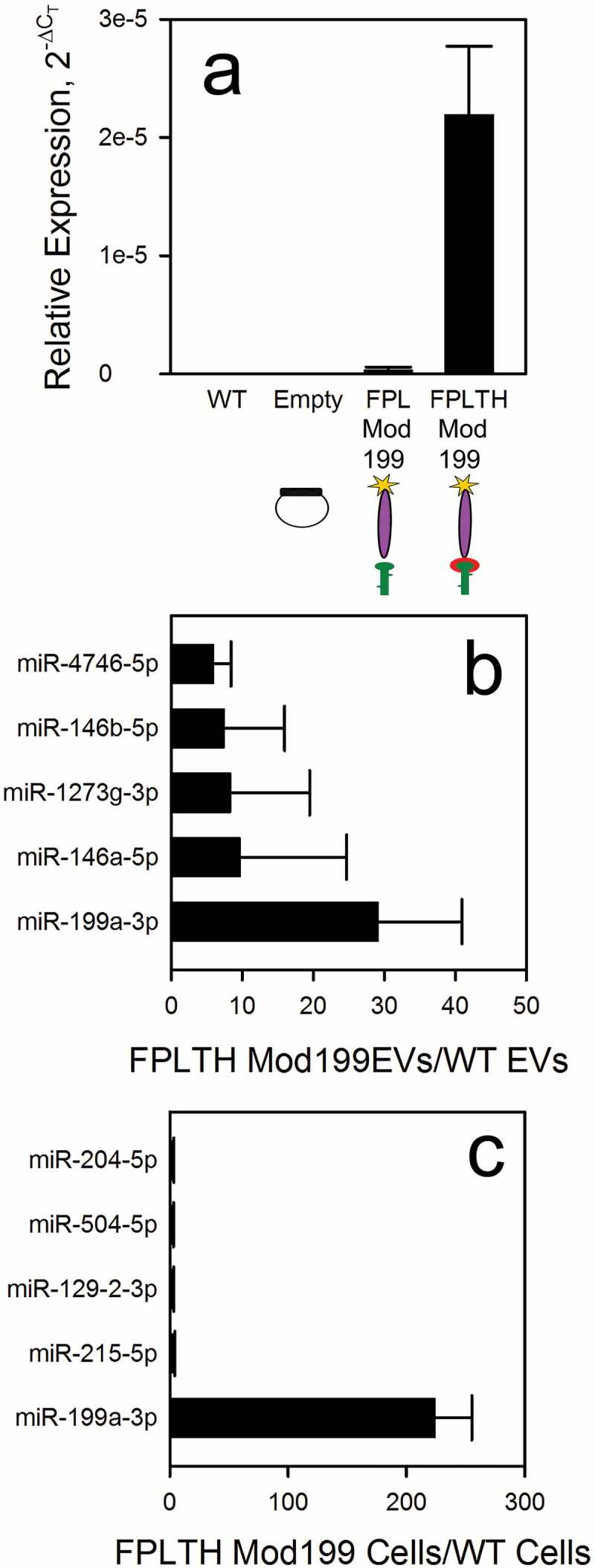
Modified pre-miR-199a is preferentially loaded into TAT expressing cells. (a) Modified pre-miR-199a was measured by qRT-PCR on RNA isolated from wild type HEK293T cells (WT), empty vector expressing cells (Empty), Flag-PC94-Lamp2a vector containing modified miR-199 expressing cells (FPL Mod 199) and Flag-PC94-Lamp2a-TAT-His vector containing modified miR-199 expressing cells (FPLTH-Mod 199). Small RNA sequencing was performed on RNA isolated from FPLTH-Mod 199 or wild type EVs (b) or the producing cells (c). Data are presented as the ratios of the amount of mature miRNA in the FPLTH-Mod 199 to wild type.
RNA isolated from the HEK293T cells stably transfected with the FPLTH-Mod 199 vector was assayed by TaqMan qRT-PCR to verify the expression levels of precursor and mature miR-199a. This assay has two levels of sequence specificity for the mature miRNA, 3′ priming of the cDNA and the TaqMan probe for detection [54]. Precursor miR-199 levels were confirmed as high expression of the miRNA was obtained in the cells (CT = 23.5) (Supplementary Figure 5(b)). Both mature miR-199a-3p and miR-199a-5p were detected, further demonstrating effective processing of the primary and precursor transcript (Figure 3(c)). Similar to the endogenously expressed miRNAs [55], the expression of the miR-199a-3p was greater than that of the miR-199a-5p. Mature miR-199a-3p transcribed and processed from the FPLTH-Mod 199 vector was functional as transfection of the vector into HEK293T cells along with a luciferase reporter construct containing the 3′ UTR of CD44, a validated target of miR-199a-3p [39], was reduced (Figure 3(d)).
Characterisation of engineered EVs
Engineered HEK293T cells and EVs collected by ultracentrifugation of the cell culture supernatant were characterised by immunoblotting to assess the presence of the N- and C-terminus Flag tag and 6X His tags, respectively. Hung and Leonard report that addition of an N-linked glycosylation site to the fusion protein reduces the protease cleavage of expressed Lamp2b fusion peptides on the surface of EVs [49]. Similarly, we introduced N-linked glycosylation site to our fusion protein for the detection of the N-terminus Flag tag [49]. Immunoblotting of the purified EV protein lysate demonstrated that the purified EVs contained Flag and His antibody, demonstrating translation occurs in frame following splicing of the pre-miR-199a containing intron from the Lamp2b fusion protein gene and ensures there was no cleavage or truncation of the fusion protein (Figure 4(a)). To further evaluate the purity of the EVs, CALR (endoplasmic reticulum and apoptotic body marker) and TSG101 (EV marker) was blotted in both cells and EV lysates. CALR absence in EV lysates indicates the purity of pelleted EVs from endoplasmic reticulum and apoptotic bodies. Enrichment of EV marker TSG101 was present in the EV lysates but not the cell lysate (Figure 4(a)).
EVs collected by ultracentrifugation of the cell culture supernatant were characterised by NTA. EVs purified from wild type HEK293T cells had a median diameter of 175.9 nm while the FPLTH-Mod 199 EVs had a mean diameter of 178 nm (Table 2). The data presented in Table 2 shows that we were able to collect 8.2 × 1011 and 9.9 × 1011 EVs per batch from the wild type HEK293T and FPLTH-Mod 199 cells, respectively. Mean total protein per batch and protein to EV particle ratios are also included in Table 2. To further validate the size and morphology of the EVs, purified EVs were imaged using cryo-TEM (Figure 4(c)).
Table 2.
Production yields and batch assessment of EVs collected from supernatant of 24, 150 mm dishes.
| EV type | Batches | Particle size (nm) | Number of particles | Total protein (µg) | Protein/particle |
|---|---|---|---|---|---|
| Wild type 293T | 4 | 175.9 ± 15.3 | 8.2 × 1011 | 1251 ± 104 | 1 µg/6.53 × 108 |
| FPLTH-Mod 199 | 6 | 178.0 ± 18.3 | 9.9 × 1011 | 751 ± 649 | 1 µg/1.31 × 109 |
Modified pre-miR-199a is preferentially loaded into TAT peptide expressing EVs
In order to validate proper loading of the miR-199a into the EVs, we used TaqMan qRT-PCR to measure the presence of pre-miR-199a. To demonstrate proof of concept for our TAT TAR loading strategy, the loading of the precursor miR-199a in the vector expressing FPLTH was compared to those cells stably expressing the fusion protein that lacked the TAT peptide. There was a 65-fold enrichment of the miR-199a-3p in the EVs isolated from those cells stably expressing FPLTH-Mod 199 vector compared to those cells expressing the FPL-Mod 199 fusion protein demonstrating significant enrichment occurs due to the TAT peptide TAR RNA loop interaction (Figure 5(a)).
We further performed small RNA seq to determine the relative ranking of miR-199a-3p compared to other endogenous miRNAs that are present in the producing cells and EVs. Small RNA was isolated on triplicate samples of EV producing cells and EVs isolated from those cells. The miRNA expression was ranked in both the cells and EVs. Using the ratio of miRNAs loaded in the EVs from the engineered to wild type cells, miR-199a-3p ranked first in both the EVs (Figure 5(b)) and engineered cells (Figure 5(c)).
Limited activity of endogenously loaded EVs
Sk-Hep-1 and Huh7 cells were cultured in the presence of FPLTH-Mod 199 and Empty EVs and both precursor miR-199 and mature miR-199a-3p levels were measured in the recipient cells by qRT-PCR. Pre-miR-199a levels were significantly increased in both SK-Hep1 and Huh7 cells by 8- and 10-fold, respectively, indicating successful uptake of the EVs into the recipient cell lines (Figure 6(a)). We also measured the active mature miR-199a-3p which significantly increased only in the Huh7 cells (Figure 6(b)). Despite the significant increase of the modified pre-miR-199a levels in the recipient cells, the limited amount of mature miR-199a-3p in the cells was unable to alter the mRNA levels of several miRNA-199a-3p targets (Figure 6(c)).
Figure 6.
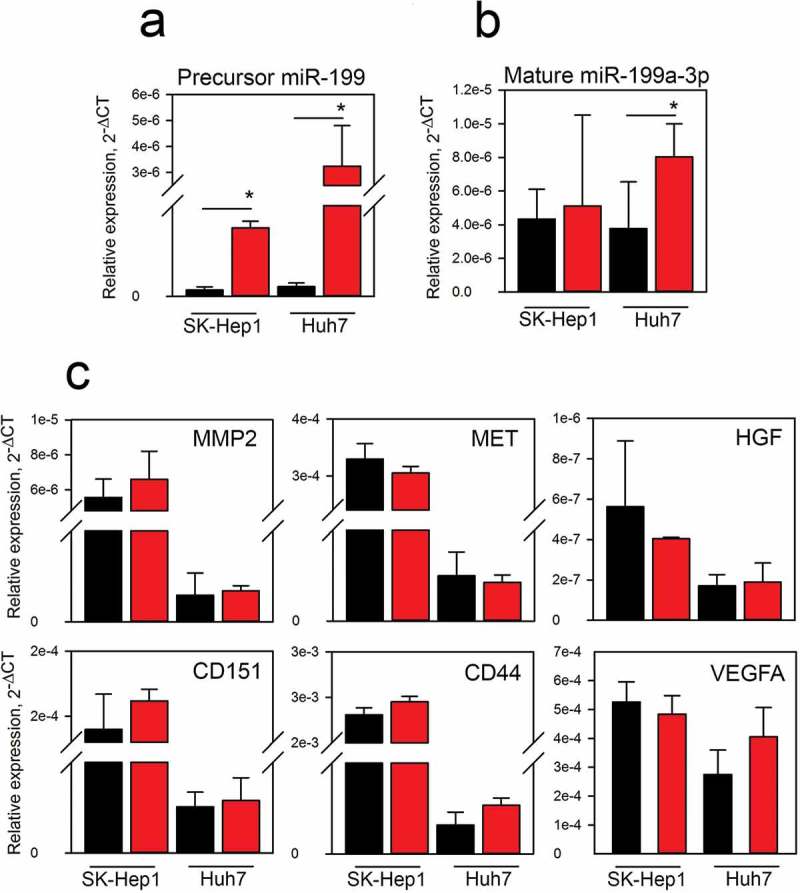
Uptake and activity of EVs in hepatocellular carcinoma (HCC) recipient cells. qRT-PCR analysis was performed to measure modified pre-miR-199a (a) and mature miR-199a-3p (b) in Sk-Hep1 and Huh7 HCC recipient cells. Cells were exposed to 2 × 1010 EVs for 24 h. (c) The expression levels of miR-199a-3p target genes were measured by qRT-PCR in the SK-Hep1 and Huh7 cells treated with EVs. Black and red bars represent cells treated with Empty and FPLTH-Mod 199 EVs, respectively.
EVs containing the FPLTH fusion peptide are loaded more abundantly with pre-miR-199a through the TAT TAR interaction (Figure 5(a)). Anti-Flag protein affinity pulldown of the loaded EVs could result in even higher yields. Cell culture supernatant was purified on a column containing Flag antibody coated agarose beads. Several fractions of the elution were collected and immunoblotting and NTA was carried out to confirm the expression of the Flag protein and particle size/number of the isolated EVs (Figure 7(a,b)). Moreover, EVs were collected from the various fractions using ultracentrifugation. Particle counts for column purified EVs was lower compared to the EVs isolated from column flow-through or ultracentrifuged cell culture supernatant (Figure 7(c)). RNA was subsequently isolated from the EVs and the amount of pre-miR-199a was determined using qRT-PCR. A higher amount of pre-miR-199a was detected in the column purified EVs compared to the EVs isolated from the ultracentrifuged flow through or supernatant (Figure 7(d)). Column purification of Flag containing EVs, though less in number, had a higher amount of pre-miR-199a loaded per EV.
Figure 7.
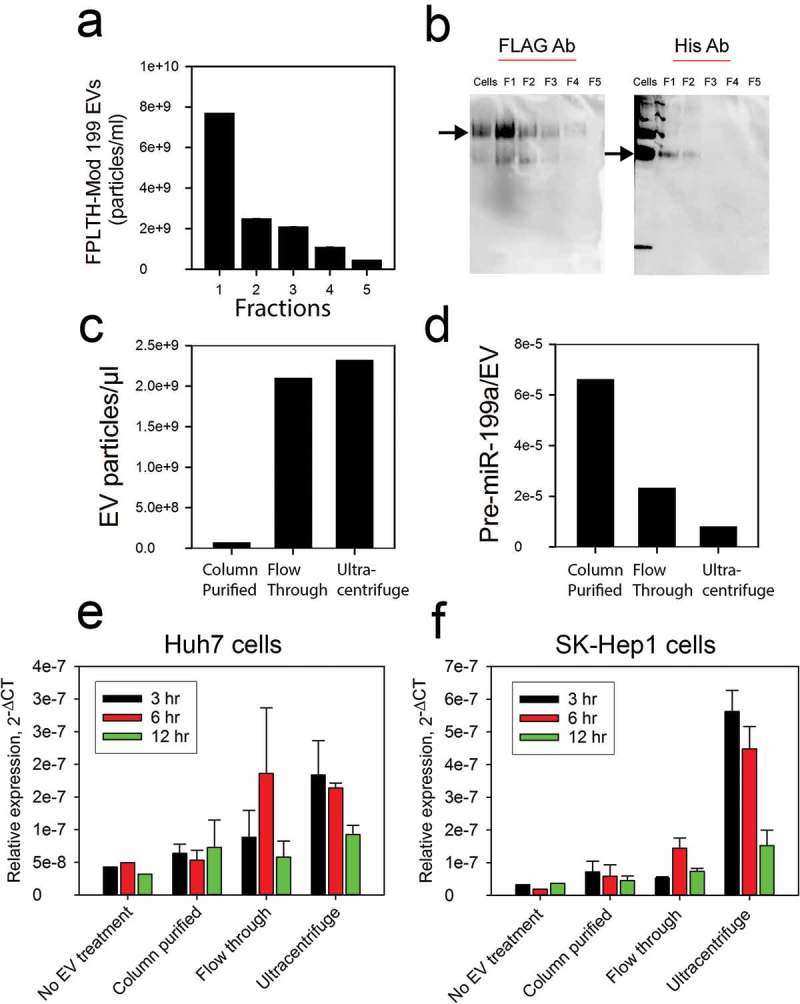
Characterisation and cellular uptake of EVs isolated by ultracentrifugation and column chromatography affinity pulldown. (a) Fractions were collected from the anti-FLAG affinity column purification; EV particle count was determined by NTA. (b) Immunoblotting was performed using anti-FLAG and anti-HIS antibodies on the fractions to confirm the presence of the engineered EVs. (c) Particle count on EVs present in the ultracentrifuged flow through, ultracentrifuged supernatant or isolated using column purification. (d) qRT-PCR analysis showing the pre-miR-199a expression per EV particle on EVs present in the ultracentrifuged flow through, ultracentrifuged supernatant or isolated using column purification. qRT-PCR measured the modified pre-miR-199a levels in Huh7 (e) or SK-Hep1 (f) cells treated with EVs present in the ultracentrifuged flow through, ultracentrifuged supernatant or isolated using column purification.
The ability of the EVs to deliver active cargo was determined by seeding EVs isolated using the techniques mentioned above on SK-Hep1 and Huh7 recipient cells. Since the column purified EVs were enriched for modified pre-miR-199a (Figure 7(d)), we expected to observe an increased uptake of the miRNA in recipient cells treated with column purified EVs. However, EVs collected by ultracentrifugation delivered the largest amount of modified pre-miR-199a (Figure 7(e,f)). The maximum amount of pre-miR-199a from ultracentrifuge purification was delivered at 3 h with the levels decreasing at the 6 and 12 h time points (Figure 7(e,f)).
We then considered used a transwell insert approach similar to Cha, et al. [56] to study the cellular uptake of the EVs. Using this approach, the EVs released by the producing cells in the upper chamber are free to interact with and be internalised into the recipient cells in the lower chamber (Figure 8(a)), thus the activity of EVs may be studied without purification. The cellular uptake of mature miR-199a-3p was increased in the recipient Sk-Hep1 and Huh7 cells by approximately 2-fold (Figure 8(b,c)). As before, we measured the mRNA of several miR-199a-3p targets by qRT-PCR in both co-culture experiments, however no change in expression was noted (data not shown).
Figure 8.
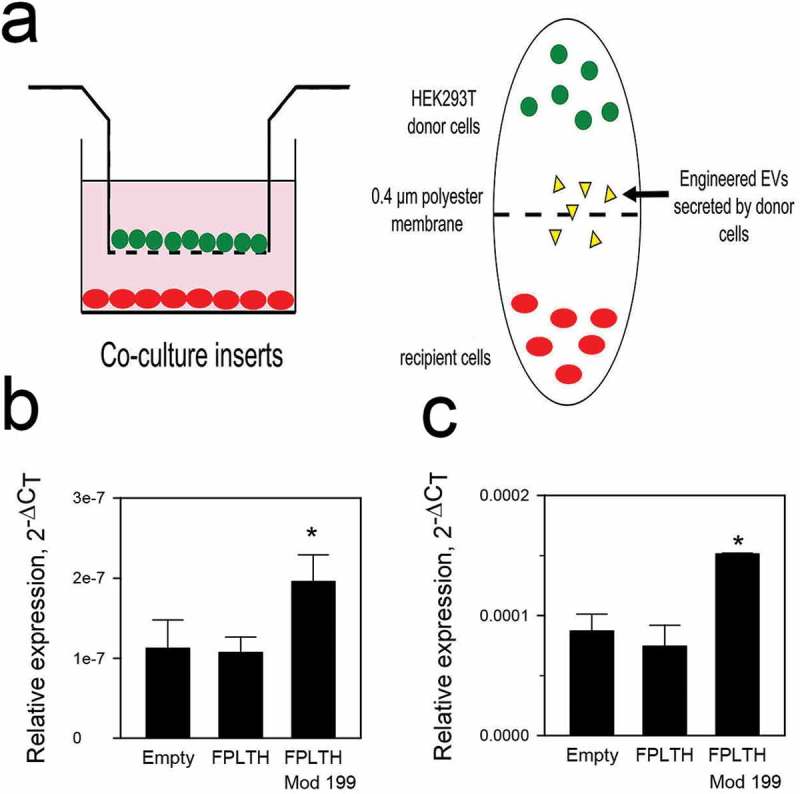
EV uptake study using transwell insert. (a) Schematic of transwell insert approach to study the transfer of EVs from producing cells to recipient cells. qRT-PCR analysis showing mature miR-199 expression levels when Empty, FPLTH and FPLTH-Mod 199 expressing stable cells were co-cultured with recipient (b) SK-Hep1 or (b) Huh7 cells.
Discussion
EVs have been proposed as carriers for a variety of nucleic acid therapeutics including siRNA, plasmid DNA, miRNA mimics, miRNA inhibitors and mRNA [33–37]. For many of these applications, the nucleic acid cargo was introduced into the EV producing cells or EVs using transfection reagents or electroporation [33–35,37]. Others used an endogenous approach by applying targeting moieties within the cargo RNA so that it could be directed into the EVs [36]. Similar to Hung and Leonard [36], we considered endogenous loading of cargo RNA into EVs by designing a system in which the HEK293T cells would generate both the EV delivery system and the RNA cargo (Figure 1). To enhance the loading of the RNA cargo into the EVs, the sequence of the precursor miR-199a was modified. Introduction of the TAR RNA/TAT peptide interaction between the Lamp2a fusion protein and the modified pre-miR-199a loop enhanced pre-miR-199a loading into the EVs by 65-fold (Figure 5(a)). Not only does this demonstrate proof of concept for the TAT TAR interaction to enhance pre-miR-199a loading into EVs but also shows that the TAT peptide is in the correct conformation with the cells and EVs.
The binding affinity between the TAT peptide and TAR RNA is high (Kd = 10 nM) [57]. In order for the miR-199a that is expressed from the HEK293T cells to be functional, the modified pri-miR-199a must be processed by Drosha and then Dicer. Introduction of the modified miR-199a gene into an artificial intron encoding Lamp2a did not affect its processing as the mature miR-199a was generated (Figure 3(c)). Moreover, insertion of the artificial intron did not alter the translation of the FPLTH fusion protein. Functionally active, mature miRNA was produced from the protein bound pre-miRNA following Dicer processing. Since the fusion peptide was bound predominately to the bulge of the pre-miRNA (Figure 2), Dicer cleavage released the miRNA from the targeting protein and allowed it to be processed to mature miRNA via miRISC. Dicer anchors on the base of the hairpin’s stem and the cleavage site is measured from a terminal phosphate group on the 5′ end of the dsRNA [53]. Therefore, the modifications made to the pre-miR-199a did not affect Dicer processing, as the stem portions are essentially unchanged.
Since the stem portion of the pre-miRNA hairpin of our modified pre-miR-199a does not come into play during the binding to the TAT peptide (Figure 2), the stem portion of the hairpin could be replaced with other pre-miRNA sequences, shRNA or anti-miRNAs. mRNA could conceivably be loaded into EVs by including a TAT RNA stem portion to the mRNA. Despite these advances, there are several shortcomings to our work. Since the amount of the miR-199a loaded into the EVs is quite low (0.003 copies/EV or 340 EVs/copy), it was necessary to add approximately 19 billion EV particles to SK-Hep1 and Huh7 to show that the engineered cargo was successfully taken up by the recipient cells (Figure 6(a)). In spite of a significant increase of the modified pre-miR-199a delivered, the amount of the active, mature miR-199a is likely too low to modify the miRNA’s target genes. Hung and Leonard observed a 40-fold enrichment of cargo RNA for EVs expressing the vesicular stomatitis virus glycoprotein, however even with this impressive enhancement of loading, the overall delivery of the engineered EVs was quite low, as the EVs were rapidly degraded in the recipient cells [36].
The protein and RNA yields during EV isolation using ultracentrifugation are dependent upon the rotor type and the centrifugation time [58]. With a focus to overcome the challenges associated with ultracentrifugation method and to further enrich the EVs with our engineered cargo, we carried out EV isolation using column chromatography. We showed that the miRNA copy per EV increases when EVs are isolated using a column affinity approach compared with the ultracentrifugation method (Figure 7(d)). However, for unknown reasons, this increase in miR-199a copy per EV using affinity purification did not equate to increased uptake of mature miR-199a in the recipient cells (Figure 7(e,f)). We also explored a co-culture system to facilitate the exchange of EVs without purification between the two cell lines. While the recipient cells show a significant uptake of mature miR-199, these levels were unable to reduce the miRNA’s target gene expression.
Both our work and that of Hung and Leonard [36] demonstrate some of the challenges of endogenous loading of RNA cargo into EVs. An alternative to endogenous delivery of RNA cargo into EVs is the exogenous loading. Li, et al, recently demonstrated activity of miR-195 exogenously loaded into liver stellate cells EV in a cholangiocarcinoma in vivo tumour model [59]. Didiot et al. report that conjugating siRNA with cholesterol significantly improves the siRNA loading and in vivo activity of the EVs [47]. The reported loading of cholesterol conjugating shRNA (1000–3000 copies/EV) [47] compared to our endogenous loading (0.003 copies/EV) represents a potential means of overcoming the lack of activity observed with endogenously loaded EVs.
Another possibility for our inability to modulate miR-199a-3p targets in the recipient cells could be due to the inability of the EVs to escape the endosomal pathway or due to the rapid degradation of EV cargo upon internalisation. EVs are internalised into recipient cells through binding to the cell surface and depending upon the membrane proteins present on the EV surface, they either release their cargo into the cytoplasm or become internalised by the endosomal proteins [32]. It is also possible that the membrane proteins present on the surface of EV’s isolated from HEK293T cells cause them to be trafficked into the lysosomal pathway and thus no therapeutic efficacy is observed. Moreover, a recent study demonstrates that EV loaded miRNAs are rapidly degraded in vitro by the XRN1 exonuclease [60]. This mechanism could provide another explanation why we do not see any changes within the miR-199a mRNA targets. Thus, our work as well as the published work by Hung et al. [36] show ineffective delivery of therapeutic cargo when EVs were isolated from HEK293T and HEK293FT cells, respectively. Evaluating EVs produced from other cell types such as dendritic or mesenchymal stem cells that express different surface membrane proteins offer a comparison to determine how EV producing cell types might affect the successful delivery of therapeutic EVs.
We report an endogenous method to load miRNA-199a into EVs. While the endogenous delivery of EVs is an attractive option to load EVs by non-synthetic means, careful consideration should be taken. Stably selecting cells to overexpress a gene could have indirect consequences on the phenotype of the EV. Overexpressing a miRNA could result in gene expression changes in the donor cells by affecting its targets, causing regulatory changes in the cellular contents that become packaged into the EVs. Our technique has low overall cargo loading and was ineffective in delivering active RNA cargo to recipient cells. Still the TAT TAR strategy described here to load miRNA into EVs may be valuable in drug delivery approaches involving other hairpin containing RNAs.
Supplementary Material
Acknowledgement
We acknowledge Dr Matthew Wood for providing the pEGFPC-1 vector and Dr Joshua Leonard for his protocol for the affinity column purification and for his assistance with the glycosylation engineering.
Responsible Editor Raymond Michel Schiffelers, Universitair Medisch Centrum, Utrecht, The Netherlands
Funding Statement
This work was supported by the National Institutes of Health grant 1UH2TR000914 to T.D.S. and M.A.P.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplemental data
Supplemental data for this article can be accessed here.
References
- [1].Lotvall J, Hill AF, Hochberg F, et al. Minimal experimental requirements for definition of extracellular vesicles and their functions: a position statement from the International Society for Extracellular Vesicles. J Extracell Vesicles. 2014;3:26913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Théry C, Regnault A, Garin J, et al. Molecular characterization of dendritic cell-derived exosomes. Selective accumulation of the heat shock protein hsc73. J Cell Biol. 1999;147(3):599–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Zitvogel L, Regnault A, Lozier A, et al. Eradication of established murine tumors using a novel cell-free vaccine: dendritic cell-derived exosomes. Nat Med. 1998;4(5):594–600. [DOI] [PubMed] [Google Scholar]
- [4].Lai RC, Arslan F, Lee MM, et al. Exosome secreted by MSC reduces myocardial ischemia/reperfusion injury. Stem Cell Res. 2010;4(3):214–222. [DOI] [PubMed] [Google Scholar]
- [5].Walker JD, Maier CL, Pober JS.. Cytomegalovirus-infected human endothelial cells can stimulate allogeneic CD4+ memory T cells by releasing antigenic exosomes. J Immunol. 2009;182(3):1548–1559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].van Niel G, Raposo G, Candalh C, et al. Intestinal epithelial cells secrete exosome-like vesicles. Gastroenterology. 2001;121(2):337–349. [DOI] [PubMed] [Google Scholar]
- [7].André F, Schartz NE, Chaput N, et al. Tumor-derived exosomes: a new source of tumor rejection antigens. Vaccine. 2002;20(Suppl 4):A28–31. [DOI] [PubMed] [Google Scholar]
- [8].Hegmans JP, Bard MP, Hemmes A, et al. Proteomic analysis of exosomes secreted by human mesothelioma cells. Am J Pathol. 2004;164(5):1807–1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Wolfers J, Lozier A, Raposo G, et al. Tumor-derived exosomes are a source of shared tumor rejection antigens for CTL cross-priming. Nat Med. 2001;7(3):297–303. [DOI] [PubMed] [Google Scholar]
- [10].Kahlert C, Kalluri R. Exosomes in tumor microenvironment influence cancer progression and metastasis. J Mol Med. 2013;91(4):431–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Kahlert C, Melo SA, Protopopov A, et al. Identification of double-stranded genomic DNA spanning all chromosomes with mutated KRAS and p53 DNA in the serum exosomes of patients with pancreatic cancer. J Biol Chem. 2014;289(7):3869–3875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Valadi H, Ekström K, Bossios A, et al. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat Cell Biol. 2007;9(6):654–659. [DOI] [PubMed] [Google Scholar]
- [13].Hunter MP, Ismail N, Zhang X, et al. Detection of microRNA expression in human peripheral blood microvesicles. Plos ONE. 2008;3(11):e3694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Gezer U, Özgür E, Cetinkaya M, et al. Long non-coding RNAs with low expression levels in cells are enriched in secreted exosomes. Cell Biol Int. 2014;38(9):1076–1079. [DOI] [PubMed] [Google Scholar]
- [15].Kogure T, Yan IK, Lin WL, et al. Extracellular vesicle-mediated transfer of a novel long noncoding RNA TUC339: a mechanism of intercellular signaling in human hepatocellular cancer. Genes Cancer. 2013;4(7–8):261–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Caby MP, Lankar D, Vincendeau-Scherrer C, et al. Exosomal-like vesicles are present in human blood plasma. Int Immunol. 2005;17(7):879–887. [DOI] [PubMed] [Google Scholar]
- [17].Ochieng J, Pratap S, Khatua AK, et al. Anchorage-independent growth of breast carcinoma cells is mediated by serum exosomes. Exp Cell Res. 2009;315(11):1875–1888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Palanisamy V, Sharma S, Deshpande A, et al. Nanostructural and transcriptomic analyses of human saliva derived exosomes. Plos One. 2010;5(1):e8577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Street JM, Barran PE, Mackay CL, et al. Identification and proteomic profiling of exosomes in human cerebrospinal fluid. J Transl Med. 2012;10:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Admyre C, Johansson SM, Qazi KR, et al. Exosomes with immune modulatory features are present in human breast milk. J Immunol. 2007;179(3):1969–1978. [DOI] [PubMed] [Google Scholar]
- [21].Pisitkun T, Shen RF, Knepper MA. Identification and proteomic profiling of exosomes in human urine. Proc Natl Acad Sci U S A. 2004;101(36):13368–13373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Vojtech L, Woo S, Hughes S, et al. Exosomes in human semen carry a distinctive repertoire of small non-coding RNAs with potential regulatory functions. Nucleic Acids Res. 2014;42(11):7290–7304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Bang C, Batkai S, Dangwal S, et al. Cardiac fibroblast-derived microRNA passenger strand-enriched exosomes mediate cardiomyocyte hypertrophy. J Clin Invest. 2014;124(5):2136–2146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Hoshino A, Costa-Silva B, Shen TL, et al. Tumour exosome integrins determine organotropic metastasis. Nature. 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Properzi F, Logozzi M, Fais S. Exosomes: the future of biomarkers in medicine. Biomark Med. 2013;7(5):769–778. [DOI] [PubMed] [Google Scholar]
- [26].Hong C-S, Muller L, Whiteside TL, et al. Plasma exosomes as markers of therapeutic response in patients with acute myeloid leukemia. Front Immunol. 2014;5:160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Batrakova EV, Kim MS. Using exosomes, naturally-equipped nanocarriers, for drug delivery. J Control Release. 2015;219:396–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Marcus ME, Leonard JN. FedExosomes: engineering therapeutic biological nanoparticles that truly deliver. Pharmaceuticals (Basel). 2013;6(5):659–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Lundqvist M, Stigler J, Elia G, et al. Nanoparticle size and surface properties determine the protein corona with possible implications for biological impacts. Proc Natl Acad Sci U S A. 2008;105(38):14265–14270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Lynch Iad KA. Protein-nanoparticle interactions. Nanotoday. 2008;3(1–2):40–47. [Google Scholar]
- [31].Théry C, Ostrowski M, Segura E. Membrane vesicles as conveyors of immune responses. Nat Rev Immunol. 2009;9(8):581–593. [DOI] [PubMed] [Google Scholar]
- [32].Mulcahy LA, Pink RC, Carter DR. Routes and mechanisms of extracellular vesicle uptake. J Extracell Vesicles. 2014;3:10.3402/jev.v3.24641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Alvarez-Erviti L, Seow Y, Yin H, et al. Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat Biotechnol. 2011;29(4):341–345. [DOI] [PubMed] [Google Scholar]
- [34].Momen-Heravi F, Bala S, Bukong T, et al. Exosome-mediated delivery of functionally active miRNA-155 inhibitor to macrophages. Nanomedicine. 2014;10(7):1517–1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Ohno SI, Takanashi M, Sudo K, et al. Systemically injected exosomes targeted to EGFR deliver antitumor microRNA to breast cancer cells. Mol Ther. 2013;21(1):185–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Hung ME, Leonard JN. A platform for actively loading cargo RNA to elucidate limiting steps in EV-mediated delivery. J Extracell Vesicles. 2016;5:31027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Kanada M, Bachmann MH, Hardy JW, et al. Differential fates of biomolecules delivered to target cells via extracellular vesicles. Proc Natl Acad Sci U S A. 2015;112(12):E1433–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Calin GA, Croce CM. MicroRNA signatures in human cancers. Nat Rev Cancer. 2006;6(11):857–866. [DOI] [PubMed] [Google Scholar]
- [39].Henry JC, Park JK, Jiang J, et al. miR-199a-3p targets CD44 and reduces proliferation of CD44 positive hepatocellular carcinoma cell lines. Biochem Biophys Res Commun. 2010;403(1):120–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Yeo RW, Lai RC, Zhang B, et al. Mesenchymal stem cell: an efficient mass producer of exosomes for drug delivery. Adv Drug Deliv Rev. 2013;65(3):336–341. [DOI] [PubMed] [Google Scholar]
- [41].Ohno S, Takanashi M, Sudo K, et al. Systemically injected exosomes targeted to EGFR deliver antitumor microRNA to breast cancer cells. Mol Ther. 2013;21(1):185–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Mizrak A, Bolukbasi MF, Ozdener GB, et al. Genetically engineered microvesicles carrying suicide mRNA/protein inhibit schwannoma tumor growth. Mol Ther. 2013;21(1):101–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Li J, Chen X, Yi J, et al. Identification and characterization of 293T cell-derived exosomes by profiling the protein, mRNA and microRNA components. Plos One. 2016;11(9):e0163043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Rosas LE, Elgamal OA, Mo X, et al. In vitro immunotoxicity assessment of culture-derived extracellular vesicles in human monocytes. J Immunotoxicol. 2016;13(5):652–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Zhu X, Badawi M, Pomeroy S, et al. Comprehensive toxicity and immunogenicity studies reveal minimal effects in mice following sustained dosing of extracellular vesicles derived from HEK293T cells. J Extracell Vesicles. 2017;6:1324730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Sutaria DS, Badawi M, Phelps MA, et al. Achieving the promise of therapeutic extracellular vesicles: the devil is in details of therapeutic loading. Pharm Res. 2017;34:1053–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Didiot MC, Hall LM, Coles AH, et al. Exosome-mediated delivery of hydrophobically modified siRNA for huntingtin mRNA silencing. Mol Ther. 2016;24:1836–1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Kooijmans SA, Stremersch S, Braeckmans K, et al. Electroporation-induced siRNA precipitation obscures the efficiency of siRNA loading into extracellular vesicles. J Control Release. 2013;172(1):229–238. [DOI] [PubMed] [Google Scholar]
- [49].Hung ME, Leonard JN. Stabilization of exosome-targeting peptides via engineered glycosylation. J Biol Chem. 2015;290(13):8166–8172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Lo A, Lin CT, Wu HC. Hepatocellular carcinoma cell-specific peptide ligand for targeted drug delivery. Mol Cancer Ther. 2008;7(3):579–589. [DOI] [PubMed] [Google Scholar]
- [51].Thery C, Amigorena S, Raposo G, et al. Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Curr Protoc Cell Biol. 2006;Chapter 3:Unit 3.22; doi: 10.1002/0471143030.cb0322s30 [DOI] [PubMed] [Google Scholar]
- [52].Schmittgen TD, Lee EJ, Jiang J, et al. Real-time PCR quantification of precursor and mature microRNA. Methods. 2008;44(1):31–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Park JE, Heo I, Tian Y, et al. Dicer recognizes the 5′ end of RNA for efficient and accurate processing. Nature. 2011;475(7355):201–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Chen C, Ridzon DA, Broomer AJ, et al. Real-time quantification of microRNAs by stem-loop RT-PCR. Nucleic Acids Res. 2005;33(20):e179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Kozomara A, Griffiths-Jones S. miRBase: annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 2014;42(Database issue):D68–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Cha DJ, Franklin JL, Dou Y, et al. KRAS-dependent sorting of miRNA to exosomes. Elife. 2015;4:e07197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Slice LW, Codner E, Antelman D, et al. Characterization of recombinant HIV-1 Tat and its interaction with TAR RNA. Biochemistry. 1992;31(48):12062–12068. [DOI] [PubMed] [Google Scholar]
- [58].Cvjetkovic A, Lötvall J, Lässer C. The influence of rotor type and centrifugation time on the yield and purity of extracellular vesicles. J Extracell Vesicles. 2014;3:10.3402/jev.v3.23111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Li L, Piontek K, Ishida M, et al. Extracellular vesicles carry microRNA-195 to intrahepatic cholangiocarcinoma and improve survival in a rat model. Hepatology. 2017;65(2):501-514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Zangari J, Ilie M, Rouaud F, et al. Rapid decay of engulfed extracellular miRNA by XRN1 exonuclease promotes transient epithelial-mesenchymal transition. Nucleic Acids Research.2017;45(7):4131–4141. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.