

Abstract
Delayed cerebral ischemia (DCI) is a significant cause of morbidity and mortality for patients surviving the rupture of an intracranial aneurysm. Despite an association between vasospasm and DCI, thrombosis and thromboembolism may also contribute to DCI. In this study we investigate the time course of intravascular microclot formation after experimental subarachnoid hemorrhage (SAH) and assess the effects of the following two drugs on microclot burden: mutant thrombin-activated urokinase-type plasminogen activator (scFv/uPA-T), which is bound to red blood cells for use as a thromboprophylactic agent, and clazosentan, an endothelin antagonist. In the first study, adult male C57BL/6 mice were sacrificed at 24 (n=5), 48 (n=6), 72 (n=8), and 96 (n=3) hours after SAH induced by filament perforation of the anterior cerebral artery. Sham animals (n=5) underwent filament insertion without puncture. In the second study, animals received scFv/uPA-T (n=5) 3 hours after hemorrhage, clazosentan (n=5) by bolus and subcutaneous pump after SAH just prior to skin closure, or a combination of scFv/uPA-T and clazosentan (n=4). Control (n=6) and sham (n=5) animals received saline alone. All animals were sacrificed at 48 hours and underwent intra-cardiac perfusion with 4% paraformaldehyde. The brains were then extracted and sliced coronally on a cryostat and processed for immunohistochemistry. An antibody recognizing thrombin–anti-thrombin complexes was used to detect microclots on coronal slices. Microclot burden was calculated for each animal and compared among groups. Following SAH, positive anti-thrombin staining was detected bilaterally in the following brain regions, in order of decreasing frequency: cortex; hippocampus; hypothalamus; basal ganglia. Few microclots were found in the shams. Microclot burden peaked at 48 hours and then decreased gradually. Animals receiving scFv/uPA-T and scFv/uPA-T+clazosentan had a lower microclot burden than controls, whereas animals receiving clazosentan alone had a higher microclot burden (p<0.005). The overall mortality rate in the time course study was 40%; mortality was highest among control animals in the second study. Intravascular microclots form in a delayed fashion after experimental SAH. Microclots may be safely reduced using a novel form of thromboprophylaxis provided by RBC-targeted scFv/uPA-T and represent a potential target for therapeutic intervention in the treatment of DCI.
Keywords: Microclots, Microthrombi, Subarachnoid hemorrhage, Thromboembolism, Vasospasm, Experimental, Urokinase, Clazosentan
Introduction
Delayed cerebral ischemia (DCI) with subsequent infarction is a significant cause of morbidity and mortality in patients with subarachnoid hemorrhage (SAH) (Juvela et al., 2005; Rinkel et al., 2005; Roos, 2000; Woertgen et al., 2003). Since the discovery that aneurysm rupture is associated with spasm of major cerebral arteries (Ecker and Riemenschneider, 1951), cerebral vasospasm has been advanced as the cause of DCI. However, differences in the time course, severity, and distribution of cerebral vasospasm and DCI (Mesis et al., 2006; Minhas et al., 2003; Ohkuma et al., 2000; Rabinstein et al., 2004; Rabinstein et al., 2005; Weidauer et al., 2007) call into question a cause and effect relationship (Macdonald et al., 2007; Nolan and Macdonald, 2006; Stein et al., 2006a). Not all patients with DCI experience vasospasm, and not all patients with vasospasm develop DCI (Rowe et al., 1995; Treggiari et al., 2003). Furthermore, drugs such as nimodipine, which reduce the risk of DCI after SAH and improve clinical outcomes, have no effect on vasospasm (Allen et al., 1983; Petruk et al., 1988; Feigin et al., 1998), and effective treatment of vasospasm does not ensure amelioration of DCI (Treggiari et al., 2003; Macdonald et al., 2008). Thus, several lines of evidence suggest that DCI cannot always be attributed to vasospasm.
Thrombosis and thromboembolism have been proposed as an alternative or parallel mechanism of DCI. Markers of hypercoagulation, (Antovic et al., 2002; Peltonen et al., 1997) and platelet activation (Denton et al., 1971; Ishikawa et al., 2009; Juvela et al., 1991) rapidly increase following aneurysm rupture; values are higher in the CSF and jugular blood compared to systemic levels, suggesting a cerebral origin (Hirashima et al., 1997; Kasuya et al., 1998; Suzuki et al., 1999). In addition, several autopsy studies have detected the presence of small intra-vascular blood clots, termed microclots, and have demonstrated a correlation between microclot density and the location and severity of histological evidence of ischemia (Stein et al., 2006b) or radiographic evidence of infarction (Suzuki et al., 1990). Transcranial Doppler (TCD) studies detected microembolic signals in the cerebral vessels of as many as 70% of surveyed patients (Romano et al., 2002) and the presence of microembolic signals was independently associated with ischemia and infarction on head computed tomography (Giller et al., 1998; Romano et al., 2008). Although causation has yet to be determined, the presence of intravascular microclots after SAH represents a potential novel target for therapeutic intervention.
Fibrinolytic agents or drugs with anti-thrombotic effects may play a role in the prevention or treatment of DCI by reducing microclot burden, if their effect can be safely targeted to the nascent pathological clots that may cause vascular occlusion, but not to hemostatic plugs that prevent further bleeding aggravation of SAH. Urokinase-type plasminogen activator (uPA) is a serine protease that converts plasminogen to the active protease plasmin (Collen et al., 1991), and the fibrinolytic agent may be bound to red blood cells (RBCs) for long acting thromboprophylaxis (Zaitsev et al., 2010a). We have devised a new mutant form of the single chain low molecular weight (L144–L411, lacking kringle and growth factor like domains) urokinase that is inactive until cleaved by thrombin (uPA-T) and fused it with a single chain fragment of an antibody specific to mouse glycophorin A (scFv/uPA-T) that safely anchors uPA-T on circulating RBCs (Zaitsev et al., 2010a). Upon intravenous (IV) injection scFv/uPA-T binds to RBCs, which allows the drug to exert its fibrinolytic effect only on newly forming blood clots and without lysis of preexisting clots. In addition to modified urokinase, anticoagulant activity is observed among several drugs currently used to treat SAH (Roos et al., 2003). Clazosentan is a selective endothelin (ET)-A receptor antagonist that decreases angiographic vasospasm in experimental and clinical studies (Macdonald et al., 2008; Roux et al., 1997; Vajkoczy et al., 2005); however it is unclear whether clazosentan also possesses anti-thrombotic properties.
Prior animal studies have documented the existence of microclots following SAH (Wang et al., 2010), yet their temporal and spatial distribution remains unknown. The safe and effective reduction of microclots in the setting of SAH must first be demonstrated in order to assess the contribution of thromboembolism to the development of DCI and to develop fibrinolytic treatment approaches. In this study, we set out to characterize the time course of microclot formation and dissolution after experimental SAH and to examine the effect of scFv/uPA-T and clazosentan on intravascular microthrombosis.
Materials and methods
Mouse SAH model
Adult male C57BL/6 mice, weighing 18–25 g, were anesthetized with an intraperitoneal injection of pentobarbital (65 mg/kg). A subcutaneous injection of 0.05 ml 0.25% bupivacaine was administered over the surgical site. Body temperature was maintained at 37.0±0.5 °C using a heat lamp and a heat pad.
Through a midline neck incision, the right common carotid artery was exposed and the external carotid artery was isolated. A blunted 5-0 monofilament nylon suture was passed via the external and common carotid arteries into the internal carotid artery. The suture was advanced approximately 5 mm until resistance was felt, and it was then advanced 3–5 mm further to puncture the anterior cerebral artery. The filament was immediately withdrawn, allowing reperfusion and SAH. In sham mice, the suture was advanced until resistance was felt and then immediately withdrawn to avoid causing SAH. The suture was then removed, the external carotid artery was ligated, and the skin was sutured. Intraperitoneal buprenorphine (2 mg/kg) was administered immediately after skin closure and prior to recovery from anesthesia. The endovascular filament perforation model has been shown to cause widespread cerebral ischemic changes; furthermore, the model closely replicates an autologous arterial bleed into the subarachnoid space that occurs with anterior circulation aneurysm rupture in humans (McGirt et al., 2002; Parra et al., 2002). All procedures were approved by the University of Pennsylvania Institutional Animal Care and Use Committee and followed institutional guidelines.
Experimental groups
In the first of two separate studies, to establish a time course of micro-clot formation and dissolution, mice underwent experimental SAH and were sacrificed at 24, 48, 72, and 96 hours after SAH. Sacrifice times were randomly assigned at the start of surgery. Sham animals were sacrificed at 72 hours after sham surgery. In the second study, to examine the effect of a bound uPA and clazosentan (Actelion Pharmaceuticals Ltd, Allschwil/Basel, Switzerland) on microthrombosis, the following five groups were established: (a) sham; (b) control; (c) SAH+scFv/uPA-T; (d) SAH+clazosentan; (e) SAH+scFv/uPA-T+clazosentan. Control animals underwent experimental SAH; sham and control animals received normal saline by jugular vein injection. In the SAH+scFv/uPA-T group, scFv/uPA-T (4 mg/kg) was administered by tail vein injection 3 hours after SAH. In the SAH+clazosentan group, a bolus of clazosentan (1 mg/kg) was administered via the right jugular vein after SAH and ligation of the external carotid artery, and a clazosentan pump (Alzet Micro-Osmotic pump, Model 1003D, Durect Corporation, Cupertino, CA) was then placed subcutaneously in the abdomen at the time of skin closure to deliver clazosentan over time (72 mg/kg, 1.0 μl per hour, 100 μl solution loaded 24 hours prior to SAH). In the SAH+scFv/uPA-T+clazosentan group, animals received IV scFv/uPA-T, IV clazosentan, and a subcutaneous clazosentan pump as above. Animals received clazosentan at the end of surgery and received scFv/uPA-T 3 hours after SAH. Based on findings of the time course study, animals in the second study were sacrificed at 48 hours. Tail vein injections were not performed by the surgeon.
Perfusion-fixation and histological preparation
All animals were given an overdose of 200 mg/kg sodium pentobarbital and transcardially perfused with heparinized saline followed by 4% paraformaldehyde. The brains were removed, post-fixed 3–4 days in 4% paraformaldehyde, depending on the firmness of the brains, and then immersed in 30% sucrose until saturated (4–5 days). The ventral surface of each brain was photographed to determine the presence or absence of SAH. Animals were excluded if the parenchyma was visualized through the blood clot on the ventral surface of the gross specimen (30). Representative hemorrhages are shown in Fig. 1. Brains were then snap-frozen in isopentane at −30 °C to −40 °C and sectioned coronally on a cryostat from 3 mm behind frontal poles to cerebellum at a thickness of 20 microns. Coronal brain slices were mounted serially on six glass slides, yielding six slide sets; individual sets were then used for H&E or immunohistochemistry. All brains were assigned a random number to ensure blinded histological evaluation with respect to time of sacrifice and treatment group.
Fig. 1.
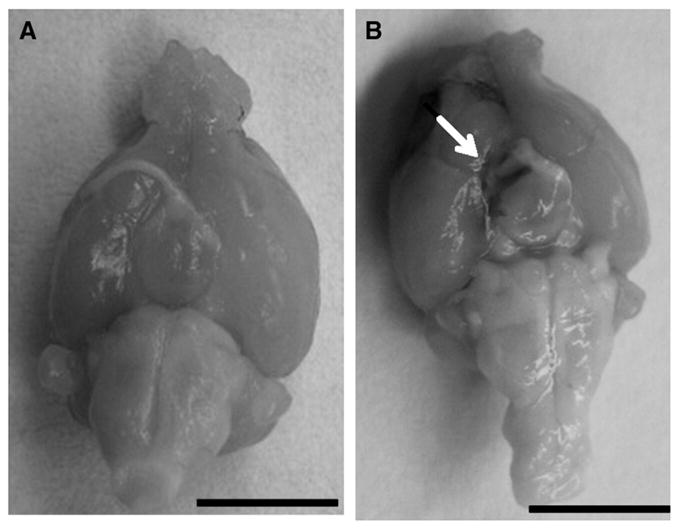
Ventral surface of the brain of a sham animal (A) and a control animal sacrificed 48 hours after hemorrhage. The arrow shows residual blood. Bar in lower left corner=1 cm.
Hematoxylin and eosin (H&E) staining and immunohistochemistry
In both studies, H&E staining was used to ensure that blood was restricted to the subarachnoid space and that the cortical surface had not been penetrated. For analysis of intravascular microthrombi, coronal brain slices were mounted on slides and incubated overnight with 1:400 dilution of a sheep polyclonal antibody used to recognize free antithrombin and antithrombin in antithrombin–protease complexes (Abcam, Cambridge, MA). Mounted tissue was then incubated with fluorochrome-conjugated anti-sheep IgG (H+L), 1:100 secondary antibody (Jackson Immuno Research Laboratories, West Grove, PA) for visualization. Samples were examined with a Nikon E600 microscope (Melville, NY) under 2, 10, and 20× magnifications.
Evaluation of intravascular microthrombi
For each brain specimen, the total number of microclots was counted for each coronal brain slice of one slide set, which involved an average of 60–70 brain slices. Criteria were consistently applied to identify clots, which included organized aggregates located within the intravascular compartment with positive antithrombin staining. H&E was used as an aid to assist in identifying the endothelium, assessing the organization of an aggregate, and to identify the anatomic region in which microclots were identified.
To estimate the density of microclots, the surface area of each microscopic specimen was computed from digital micrographs using SPOT Imaging software (SPOT Imaging Solutions, Sterling Heights, MI) and the number of microclots, regardless of size, per mm2 (microclot burden) was calculated for each section. Microclot density was then divided by slice thickness (20 microns) to obtain the number of microclots per mm3. Mice sacrificed at different time points and sham mice were compared with respect to microclot burden. Animals receiving saline, scFv/uPA-T, clazosentan, or scFv/uPA-T+clazosentan, and sham mice were also compared with respect to microclot burden. The counter was blinded to time of sacrifice in the first study and to experimental group in the second study.
Statistical analysis
Comparisons among different groups were performed using a one-way analysis of variance (ANOVA), followed by post-hoc Bonferroni tests for individual comparisons (Stata 9, StataCorp, College Station, TX). We considered differences for which the probability was <0.05 to be significant.
Results
General observations
For the time course study, 5 mice underwent sham surgery and 33 mice underwent experimental SAH with sacrifice at 24 hours (n=5), 48 hours (n=6), 72 hours (n=8), and 96 hours (n=3). The mortality rate was 11/33 (33%) among experimental mice; premature deaths most commonly occurred among animals to be sacrificed after 72 or 96 hours. For the second study, the number of mice per experimental group was as follows: shams (n=5); controls (n=6); SAH+scFv/uPA-T (n=5); SAH+clazosentan (n=5); SAH+scFv/uPA-T+clazosentan (n=4). The mortality rate for each experimental group was 3/9 (33%) in controls, 3/15 (20%) scFv/uPA-T, 1/6 (17%) clazosentan, 2/8 (25%) scFv/uPA-T+clazosentan. Increased numbers per group were required to compensate for exclusion due to parenchymal injury or histological artifact that precluded micro-clot counting (n=9). Three animals were excluded for lack of sufficient hemorrhage volume (Fig. 2).
Fig. 2.

H&E of coronal brain slice at 48 hours after SAH. Residual SAH is evident at the base (arrow) with extension into both Sylvian fissures. Original magnification 2×.
Microclot characterization
In both studies, microclots were visualized by positive anti-thrombin staining and seen throughout brain hemispheres both ipsilateral and contralateral to filament perforation. Microclots were seen in arterioles, veins, and venules of all sizes, ranging from 10 to 100 microns in diameter (Fig. 3A). Microclots generally appeared in clusters, although density varied considerably within each animal; some brain slices contained over 350 microclots. Single clots in isolation were noted in several specimens. There were almost no microclots in the sham animals. The anatomic distribution of microclots varied, but included the cortex, hippocampus, hypothalamus, and basal ganglia, in order of decreasing microclot counts. Microclots filled the lumens of several vessels (Fig. 3B).
Fig. 3.

Photomicrographs showing immunofluorescence staining of representative brain slices from time course study. Microclots of various sizes are shown in small cortical vessels (A) and larger penetrating arteries and veins (B). Original magnification 20×. Bar in lower right corner=50 microns.
Time course of microclot formation and dissolution
Among surviving animals, the density of microclots increased within 24 hours of SAH and increased drastically from 24 to 48 hours. Microclot density reached a peak of 27 microclots/mm3 at 48 hours and decreased more gradually thereafter (Fig. 4). The microclot burden was significantly higher (p<0.001) among animals sacrificed at 48 hours compared to all other groups. Two microclots/mm3 were found on average in sham animals. Microclots remained present at 96 hours after SAH.
Fig. 4.

Microclot burden (#/mm3) for each group. Microclot burden reached a maximum of 27 microclots/mm3 at 48 hours. Mean values for 48-hour mice are significantly higher (p<0.001) than for all other groups. Vertical bars represent standard errors for each group.
Effect of RBC-targeted scFv/uPA-T and clazosentan on microclot burden
Animals receiving scFv/uPA-T alone or in combination with clazosentan had a lower microclot burden compared to controls (5 versus 23 microclots/mm3) (Fig. 5). The average microclot burden was higher among animals receiving clazosentan alone compared to controls (33 versus 23 microclots/mm3). Differences in microclot burden were statistically significant (p<0.05) among all groups, except between scFv/uPA-T and scFv/uPA-T+clazosentan groups.
Fig. 5.

Microclot burden (#/mm3) for each experimental group. Compared to controls (23 microclots/mm3), microclot burden was lower in the scFv/uPA-T and scFv/uPA-T+clazosentan groups (each approximately 5 microclots/mm3 and higher in the clazosentan group (33 microclots/mm3). Significant differences were observed between all groups except scFv/uPA-T and scFv/uPA-T+clazosentan. On the x-axis, uPA refers to scFv/uPA-T.
Discussion
Our findings show that microclots form diffusely within the cerebral vasculature in a delayed fashion after filament perforation-induced SAH in mice and that the microclot burden changes over time, with a peak at 48 hours among surviving mice. We are the first to demonstrate a safe reduction of microclot burden using a red blood cell-targeted scFv/uPA-T administered after hemorrhage. Furthermore, clazosentan, an endothelin antagonist that decreases vasospasm without improving outcome, was associated with an increased microclot burden. Taken together, these findings support thrombosis and thromboembolism as a novel pathway in the development of DCI and as a potential target for therapeutic intervention.
The spatial distribution of microclots observed in our study is consistent with the pattern of delayed ischemia and infarction that occurs after SAH. Following aneurysm rupture, there is a rapid increase in intracranial pressure and a subsequent decrease in cerebral perfusion pressure (Sehba and Bederson, 2006). We observed microclots bilaterally, which is consistent with a global insult. Similarly, in SAH patients, TCD and MRI studies have detected microembolic signals and delayed ischemic lesions in both hemispheres, regardless of the site of aneurysm rupture or vasospasm (Rabinstein et al., 2005; Romano et al., 2008; Kivisaari et al., 2001). We noted microclots both ipsilateral and contra-lateral to the site of filament puncture, indicating that either endothelial damage from filament insertion or emboli originating from the clot at the puncture site are unlikely to fully account for the pattern of observed microclots. Autopsy studies have revealed widespread and multifocal infarct patterns more consistent with diffuse microangiopathy associated with thrombosis or thromboembolism, rather than large vessel spasm (Neil-Dwyer et al., 1994). We likewise noted a scattered distribution of microclots within each hemisphere and in various brain regions. Moreover, microclots found in the cortex and hypothalamus, regions with large microclot burden in our study, correlate with brain regions of ischemia noted in autopsy studies (Neil-Dwyer et al., 1994; Crompton, 1964). Although multiple regions of small vessel spasm may be an additional explanation (Hijdra et al., 1986), our findings support the role of a diffuse process, such as microclots.
An understanding of the temporal pattern of microclot formation following SAH helps to identify the optimal time for administration of novel microclot-targeted therapy. The activation of coagulation occurs early after SAH, and an elevated concentration of fibrinopeptide A, a marker of thrombin generation, is present in the CSF within two days of hemorrhage (Kasuya et al., 1998). Microclot formation has been documented in traumatic brain injury and reaches a maximum at 1–3 days (Lu et al., 2004a). In experimental SAH, we identified the peak microclot burden to occur within this time frame. Peak microclot burden occurred at 48 hours in our mouse model, which is consistent with the delayed onset of DCI in humans, which commonly occurs between 4 and 10 days after hemorrhage in SAH patients (Hijdra et al., 1986; Roos et al., 2000). Other investigators have shown that platelets aggregate acutely after SAH, reaching a peak at 24 hours, and are undetectable at 48 hours (Sehba et al., 2005). One reason for the discrepancy between the timing of peak microclot burden and platelet aggregation is that platelets may be so enmeshed in fibrin by 48 hours that a platelet-specific antibody is unable to reach the GPIIb/IIIa receptor (Sehba et al., 2005). Although the mechanism is unknown by which acute injury and sub-arachnoid blood leads to intravascular coagulation, clots forming in a delayed fashion within the microvasculature may mechanically obstruct the lumen and decrease blood flow. Other animal studies of SAH have implicated mechanical obstruction by platelet aggregates as a contributing factor to the development of DCI (Sehba et al., 2005). Thus, drugs administered early after hemorrhage, in order to prevent ensuing clot formation, may act to decrease mechanical obstruction and subsequent decreases in cerebral perfusion.
Despite reports of intra-cisternal delivery of a fibrinolytics after SAH (Amin-Hanjani et al., 2004), few studies have investigated IV administration of a fibrinolytic agent for fear of hemorrhage in the case of an unsecured aneurysm or after a recent craniotomy for aneurysm clipping. Enoxaparin, a low molecular weight heparin that has been used safely after aneurysm occlusion surgery, was administered within 72 hours of SAH in two studies, yielding positive (Wurm et al., 2004) and negative (Siironen et al., 2003) results. Authors of the latter study suggested that the drug may have improved outcomes had it been administered at a higher dose, rather than the low dose used for prophylaxis. To target microclot burden in SAH patients, a therapeutic must possess enough fibrinolytic strength to lyse newly forming pathological intravascular clots that may occlude vessels, without affecting the preexisting hemostatic plug formed at the site of aneurysm rupture, and remain in the circulation for extended periods of time. Current thrombolytics are difficult to use prophylactically because of their short duration of action due to unfavorable pharmacokinetics and their inability to discriminate between pathological and hemostatic clots. We have developed an agent to address these concerns by creating a mutant form of a low molecular weight single chain uPA activated by thrombin and fused to an antibody fragment that binds to the surface of RBCs upon a single IV injection, scFv/uPA-T. Carriage by RBCs prolongs circulation time by orders of magnitude and prevents scFv/uPA-T from acting in hemostatic plugs and extravascular tissue including the CNS parenchyma, whereas activation by thrombin limits scFv/uPA-T activity to the sites of ongoing intravascular thrombosis and growing thromboemboli. We tested the hypothesis that the fusion protein would safely reduce microclot burden after experimental SAH.
RBC-targeted scFv/uPA-T given shortly after the time of hemorrhage was associated with a significantly decreased microclot burden compared to control animals receiving only saline, without an associated increase in mortality. Microclot burden was measured at 48 hours, the time of peak burden as determined by the time course study. The coupling of scFv/uPA-T to RBC prevents the fibrinolytic agent from penetrating into surrounding tissues and into existing hemostatic clots, while delivering plasminogen activator to the interior of nascent thrombi (Murciano et al., 2003; Murciano and Muzykantov, 2003; Zaitsev et al., 2010b). Unlike tissue type plasminogen activator (tPA), uPA does not possess constitutive activity (Zaitsev et al., 2010a) and was therefore chosen as the fibrinolytic agent to be used in this study, as mutation of its activation site renders thrombin specificity. In addition, the lifespan of scFv/uPA-T is increased due to the long circulation time of RBC. In our model, the clot securing the site of filament puncture was not compromised in a fatal manner when given at 3 hours after hemorrhage.
In addition to known fibrinolytic agents, we investigated the effect of the ET-antagonist clazosentan on microclot burden. Several drugs that decrease the incidence of DCI, without affecting vasospasm, have anticoagulant effects. For example, nimodipine is associated with increased tPA activity and decreased plasminogen activator inhibitor levels (Roos et al., 2001; Vergouwen et al., 2007), and HMG-CoA reductase inhibitors, such as simvastatin, have anticoagulant effects in models of SAH (Wang et al., 2010; Guven et al., 2006) and TBI (Lu et al., 2004b). There is crosstalk between inflammation and coagulation, and the inflammatory cascade set off after aneurysm rupture may promote the formation of microthrombi (Levi et al., 2004; Vergouwen et al., 2008). Despite the involvement of ET in pro-inflammatory pathways, we found that clazosentan was associated with increased microclot formation compared to controls and shams. Pro-thrombotic properties of clazosentan have not been previously studied, yet such pleiotropic effects may explain the lack of improved overall outcome in clinical trials (Macdonald et al., 2008), as microclot formation may contribute to DCI. Anti-fibrinolytic agents, such as tranexamic acid, were associated with no improvement in overall outcome after SAH as a result of an increased incidence of DCI and impeded recovery from DCI (Roos, 2000; Roos et al., 2003; Roos et al., 2003; Tsementzis et al., 1990). The result of co-administration of scFv/uPA-T and clazosentan in the current study indicates that scFv/uPA-T-mediated fibrinolysis overpowered the pro-thrombotic nature of clazosentan with respect to microclot burden. These preclinical findings suggest that clazosentan may be administered without impeding the reduction of microclot burden by scFv/uPA-T.
In the time course study, the finding that premature deaths most commonly occurred among animals intended to be sacrificed at 72 or 96 hours suggests that these animals may have died from severe micro-thrombosis. The inherent requirement to sacrifice animals in order to histologically investigate microclot burden limited our conclusions regarding the time of peak microclot burden to surviving animals only. In the second study, our interest in the intravascular compartment precluded the use of previously established methods for vasospasm measurement, such as measuring vessel diameter after India Ink/gelatin casting of cerebral blood vessels (Parra et al., 2002). As in prior studies involving filament puncture of the anterior circulation (Parra et al., 2002), we found no significant difference between groups with respect to the average diameter of the basilar artery, a vessel not contiguous with the hemorrhage (data not shown). Vasospasm measurements are necessary to determine the relative contribution of spasm and microclots to DCI, and a correlation between microclots and outcome, such as neuronal injury or behavioral assessment, was therefore not performed in this exploratory study. In addition, the severity of the initial hemorrhage was assessed qualitatively rather than quantitatively. Also, although potential vasodilator effects of clazosentan, such as hypotension, were not measured in this study, such effects were not of sufficient magnitude to result in differences in mortality among animals with and without exposure to clazosentan. Finally, we did not determine whether animals had episodes of rebleeding after filament puncture. Despite a small sample size, the mortality rate was highest among controls, indicating that any additional bleeds were unlikely to be associated with the administration of scFv/uPA-T or clazosentan.
Conclusion
The presence of microclots after experimental SAH suggests a contribution of thrombosis and thromboembolism in the development of DCI. Peak microclot burden occurs in a delayed manner after SAH and can be reduced safely in an animal model by a RBC-bound fibrinolytic agent. Clazosentan has pro-thrombotic properties, but its effect on microclot formation is outweighed by RBC-targeted scFv/uPA-T. Although micro-thrombosis may contribute to DCI in parallel with vasospasm or occur in only a subset of SAH patients, microclots represent a potential target for therapeutic intervention. Further study is required of the association between microclot reduction and a surrogate marker of DCI in animals, such as neuronal injury.
Acknowledgments
We thank Kevin Browne, B.S., Kaitlin Kariko, Ph.D., Robin Armstrong B.S., and Emma Meagher, M.D. for their support, expertise, and guidance.
Abbreviations
- CSF
cerebrospinal fluid
- DCI
delayed cerebral ischemia
- ET
endothelin
- H&E
hematoxylin and eosin
- RBCs
red blood cells
- SAH
subarachnoid hemorrhage
- TCD
transcranial Doppler
- tPA
tissue plasminogen activator
- uPA
urokinase-type plasminogen activator
References
- Allen GS, Ahn HS, Preziosi TJ, Battye R, Boone SC, Boone SC, Chou SN, Kelly DL, Weir BK, Crabbe RA, Lavik PJ, Rosenbloom SB, Dorsey FC, Ingram CR, Mellits DE, Bertsch LA, Boisvert DP, Hundley MB, Johnson RK, Strom JA, Transou CR. Cerebral arterial spasm—a controlled trial of nimodipine in patients with sub-arachnoid hemorrhage. N Engl J Med. 1983;308:619–624. doi: 10.1056/NEJM198303173081103. [DOI] [PubMed] [Google Scholar]
- Amin-Hanjani S, Ogilvy CS, Barker FG., II Does intracisternal thrombolysis prevent vasospasm after aneurysmal subarachnoid hemorrhage? A meta-analysis Neurosurgery. 2004;54:326–334. doi: 10.1227/01.neu.0000103488.94855.4f. discussion 334–5. [DOI] [PubMed] [Google Scholar]
- Antovic J, Bakic M, Zivkovic M, Ilic A, Blomback M. Blood coagulation and fibrinolysis in acute ischaemic and haemorrhagic (intracerebral and subarachnoid haemorrhage) stroke: does decreased plasmin inhibitor indicate increased fibrinolysis in subarachnoid haemorrhage compared to other types of stroke? Scand J Clin Lab Invest. 2002;62:195–199. doi: 10.1080/003655102317475452. [DOI] [PubMed] [Google Scholar]
- Collen D, Lu HR, Lijnen HR, Nelles L, Stassen JM. Thrombolytic and pharmacokinetic properties of chimeric tissue-type and urokinase-type plasminogen activators. Circulation. 1991;84:1216–1234. doi: 10.1161/01.cir.84.3.1216. [DOI] [PubMed] [Google Scholar]
- Crompton MR. The pathogenesis of cerebral infarction following the rupture of cerebral berry aneurysms. Brain. 1964;87:491–510. doi: 10.1093/brain/87.3.491. [DOI] [PubMed] [Google Scholar]
- Denton IC, Robertson JT, Dugdale M. An assessment of early platelet activity in experimental subarachnoid hemorrhage and middle cerebral artery thrombosis in the cat. Stroke. 1971;2:268–272. doi: 10.1161/01.str.2.3.268. [DOI] [PubMed] [Google Scholar]
- Ecker A, Riemenschneider PA. Arteriographic demonstration of spasm of the intracranial arteries, with special reference to saccular arterial aneurysms. J Neurosurg. 1951;8:660–667. doi: 10.3171/jns.1951.8.6.0660. [DOI] [PubMed] [Google Scholar]
- Feigin VL, Rinkel GJ, Algra A, Vermeulen M, van Gijn J. Calcium antagonists in patients with aneurysmal subarachnoid hemorrhage: a systematic review. Neurology. 1998;50:876–883. doi: 10.1212/wnl.50.4.876. [DOI] [PubMed] [Google Scholar]
- Giller CA, Giller AM, Landreneau F. Detection of emboli after surgery for intracerebral aneurysms. Neurosurgery. 1998;42:490–493. doi: 10.1097/00006123-199803000-00010. discussion 493–4. [DOI] [PubMed] [Google Scholar]
- Guven GS, Atalar E, Yavuz B, Beyazit Y, Kekilli M, Kilicarslan A, Sahiner L, Oz G, Ozer N, Aksoyek S, Haznedaroglu IC, Sozen T. Simvastatin treatment improves endothelial function and increases fibrinolysis in patients with hypercholestrolemia. J Natl Med Assoc. 2006;98:627–630. [PMC free article] [PubMed] [Google Scholar]
- Hijdra A, Van Gijn J, Stefanko S, Van Dongen KJ, Vermeulen M, Van Crevel H. Delayed cerebral ischemia after aneurysmal subarachnoid hemorrhage: clinicoanatomic correlations. Neurology. 1986;36:329–333. doi: 10.1212/wnl.36.3.329. [DOI] [PubMed] [Google Scholar]
- Hirashima Y, Nakamura S, Endo S, Kuwayama N, Naruse Y, Takaku A. Elevation of platelet activating factor, inflammatory cytokines, and coagulation factors in the internal jugular vein of patients with subarachnoid hemorrhage. Neurochem Res. 1997;22:1249–1255. doi: 10.1023/a:1021985030331. [DOI] [PubMed] [Google Scholar]
- Ishikawa M, Kusaka G, Yamaguchi N, Sekizuka E, Nakadate H, Minamitani H, Shinoda S, Watanabe E. Platelet and leukocyte adhesion in the microvasculature at the cerebral surface immediately after subarachnoid hemorrhage. Neurosurgery. 2009;64:546–553. doi: 10.1227/01.NEU.0000337579.05110.F4. discussion 553–4. [DOI] [PubMed] [Google Scholar]
- Juvela S, Hillbom M, Kaste M. Platelet thromboxane release and delayed cerebral ischemia in patients with subarachnoid hemorrhage. J Neurosurg. 1991;74:386–392. doi: 10.3171/jns.1991.74.3.0386. [DOI] [PubMed] [Google Scholar]
- Juvela S, Siironen J, Varis J, Poussa K, Porras M. Risk factors for ischemic lesions following aneurysmal subarachnoid hemorrhage. J Neurosurg. 2005;102:194–201. doi: 10.3171/jns.2005.102.2.0194. [DOI] [PubMed] [Google Scholar]
- Kasuya H, Shimizu T, Takakura K. Thrombin activity in CSF after SAH is correlated with the degree of SAH the persistence of subarachnoid clot and the development of vasospasm. Acta Neurochir (Wien) 1998;140:579–584. doi: 10.1007/s007010050143. [DOI] [PubMed] [Google Scholar]
- Kivisaari RP, Salonen O, Servo A, Autti T, Hernesniemi J, Ohman J. MR imaging after aneurysmal subarachnoid hemorrhage and surgery: a long-term follow-up study. AJNR Am J Neuroradiol. 2001;22:1143–1148. [PMC free article] [PubMed] [Google Scholar]
- Levi M, van der Poll T, Buller HR. Bidirectional relation between inflammation and coagulation. Circulation. 2004;109:2698–2704. doi: 10.1161/01.CIR.0000131660.51520.9A. [DOI] [PubMed] [Google Scholar]
- Lu D, Mahmood A, Goussev A, Qu C, Zhang ZG, Chopp M. Delayed thrombosis after traumatic brain injury in rats. J Neurotrauma. 2004a;21:1756–1766. doi: 10.1089/neu.2004.21.1756. [DOI] [PubMed] [Google Scholar]
- Lu D, Mahmood A, Goussev A, Schallert T, Qu C, Zhang ZG, Li Y, Lu M, Chopp M. Atorvastatin reduction of intravascular thrombosis, increase in cerebral micro-vascular patency and integrity, and enhancement of spatial learning in rats subjected to traumatic brain injury. J Neurosurg. 2004b;101:813–821. doi: 10.3171/jns.2004.101.5.0813. [DOI] [PubMed] [Google Scholar]
- Macdonald RL, Pluta RM, Zhang JH. Cerebral vasospasm after subarachnoid hemorrhage: the emerging revolution. Nat Clin Pract Neurol. 2007;3:256–263. doi: 10.1038/ncpneuro0490. [DOI] [PubMed] [Google Scholar]
- Macdonald RL, Kassell NF, Mayer S, Ruefenacht D, Schmiedek P, Weidauer S, Frey A, Roux S, Pasqualin A CONSCIOUS-1 Investigators. Clazosentan to overcome neurological ischemia and infarction occurring after subarachnoid hemorrhage (CONSCIOUS-1): randomized, double-blind, placebo-controlled phase 2 dose-finding trial. Stroke. 2008;39:3015–3021. doi: 10.1161/STROKEAHA.108.519942. [DOI] [PubMed] [Google Scholar]
- McGirt MJ, Parra A, Sheng H, Higuchi Y, Oury TD, Laskowitz DT, Pearlstein RD, Warner DS. Attenuation of cerebral vasospasm after subarachnoid hemorrhage in mice overexpressing extracellular superoxide dismutase. Stroke. 2002;33:2317–2323. doi: 10.1161/01.str.0000027207.67639.1e. [DOI] [PubMed] [Google Scholar]
- Mesis RG, Wang H, Lombard FW, Yates R, Vitek MP, Borel CO, Warner DS, Laskowitz DT. Dissociation between vasospasm and functional improvement in a murine model of subarachnoid hemorrhage. Neurosurg Focus. 2006;21:E4. doi: 10.3171/foc.2006.21.3.4. [DOI] [PubMed] [Google Scholar]
- Minhas PS, Menon DK, Smielewski P, Czosnyka M, Kirkpatrick PJ, Clark JC, Pickard JD. Positron emission tomographic cerebral perfusion disturbances and transcranial Doppler findings among patients with neurological deterioration after subarachnoid hemorrhage. Neurosurgery. 2003;52:1017–1022. discussion 1022–4. [PubMed] [Google Scholar]
- Murciano JC, Muzykantov VR. Coupling of anti-thrombotic agents to red blood cells offers safer and more effective management of thrombosis. Discov Med. 2003;3:28–29. [PubMed] [Google Scholar]
- Murciano JC, Medinilla S, Eslin D, Atochina E, Cines DB, Muzykantov VR. Prophylactic fibrinolysis through selective dissolution of nascent clots by tPA-carrying erythrocytes. Nat Biotechnol. 2003;21:891–896. doi: 10.1038/nbt846. [DOI] [PubMed] [Google Scholar]
- Neil-Dwyer G, Lang DA, Doshi B, Gerber CJ, Smith PW. Delayed cerebral ischaemia: the pathological substrate. Acta Neurochir (Wien) 1994;131:137–145. doi: 10.1007/BF01401464. [DOI] [PubMed] [Google Scholar]
- Nolan CP, Macdonald RL. Can angiographic vasospasm be used as a surrogate marker in evaluating therapeutic interventions for cerebral vasospasm? Neurosurg Focus. 2006;21:E1. doi: 10.3171/foc.2006.21.3.1. [DOI] [PubMed] [Google Scholar]
- Ohkuma H, Manabe H, Tanaka M, Suzuki S. Impact of cerebral microcirculatory changes on cerebral blood flow during cerebral vasospasm after aneurysmal subarachnoid hemorrhage. Stroke. 2000;31:1621–1627. doi: 10.1161/01.str.31.7.1621. [DOI] [PubMed] [Google Scholar]
- Parra A, McGirt MJ, Sheng H, Laskowitz DT, Pearlstein RD, Warner DS. Mouse model of subarachnoid hemorrhage associated cerebral vasospasm: methodological analysis. Neurol Res. 2002;24:510–516. doi: 10.1179/016164102101200276. [DOI] [PubMed] [Google Scholar]
- Peltonen S, Juvela S, Kaste M, Lassila R. Hemostasis and fibrinolysis activation after subarachnoid hemorrhage. J Neurosurg. 1997;87:207–214. doi: 10.3171/jns.1997.87.2.0207. [DOI] [PubMed] [Google Scholar]
- Petruk KC, West M, Mohr G, Weir BK, Benoit BG, Gentili F, Disney LB, Khan MI, Grace M, Holness RO. Nimodipine treatment in poor-grade aneurysm patients, results of a multicenter double-blind placebo-controlled trial. J Neurosurg. 1988;68:505–517. doi: 10.3171/jns.1988.68.4.0505. [DOI] [PubMed] [Google Scholar]
- Rabinstein AA, Weigand S, Atkinson JL, Wijdicks EF. Patterns of cerebral infarction in aneurysmal subarachnoid hemorrhage. Stroke. 2005;36:992–997. doi: 10.1161/01.STR.0000163090.59350.5a. [DOI] [PubMed] [Google Scholar]
- Rabinstein AA, Friedman JA, Weigand SD, McClelland RL, Fulgham JR, Manno EM, Atkinson JL, Wijdicks EF. Predictors of cerebral infarction in aneurysmal sub-arachnoid hemorrhage. Stroke. 2004;35:1862–1866. doi: 10.1161/01.STR.0000133132.76983.8e. [DOI] [PubMed] [Google Scholar]
- Rinkel GJ, Feigin VL, Algra A, van den Bergh WM, Vermeulen M, van Gijn J. Calcium antagonists for aneurysmal subarachnoid haemorrhage. Cochrane Database Syst Rev. 2005;1:CD000277. doi: 10.1002/14651858.CD000277.pub2. [DOI] [PubMed] [Google Scholar]
- Romano JG, Rabinstein AA, Arheart KL, Nathan S, Campo-Bustillo I, Koch S, Forteza AM. Microemboli in aneurysmal subarachnoid hemorrhage. J Neuroimaging. 2008;18:396–401. doi: 10.1111/j.1552-6569.2007.00215.x. [DOI] [PubMed] [Google Scholar]
- Romano JG, Forteza AM, Concha M, Koch S, Heros RC, Morcos JJ, Babikian VL. Detection of microemboli by transcranial Doppler ultrasonography in aneurysmal subarachnoid hemorrhage. Neurosurgery. 2002;50:1026–1030. doi: 10.1097/00006123-200205000-00016. discussion 1030–1. [DOI] [PubMed] [Google Scholar]
- Roos Y. Antifibrinolytic treatment in subarachnoid hemorrhage: a randomized placebo-controlled trial. STAR Study Group. Neurology. 2000;54:77–82. doi: 10.1212/wnl.54.1.77. [DOI] [PubMed] [Google Scholar]
- Roos YB, Rinkel GJ, Vermeulen M, Algra A, van Gijn J. Antifibrinolytic therapy for aneurysmal subarachnoid haemorrhage. Cochrane Database Syst Rev. 2003;2:CD001245. doi: 10.1002/14651858.CD001245. [DOI] [PubMed] [Google Scholar]
- Roos YB, Levi M, Carroll TA, Beenen LF, Vermeulen M. Nimodipine increases fibrinolytic activity in patients with aneurysmal subarachnoid hemorrhage. Stroke. 2001;32:1860–1862. doi: 10.1161/01.str.32.8.1860. [DOI] [PubMed] [Google Scholar]
- Roos YB, de Haan RJ, Beenen LF, Groen RJ, Albrecht KW, Vermeulen M. Complications and outcome in patients with aneurysmal subarachnoid haemorrhage: a prospective hospital based cohort study in the Netherlands. J Neurol Neurosurg Psychiatry. 2000;68:337–341. doi: 10.1136/jnnp.68.3.337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux S, Breu V, Giller T, Neidhart W, Ramuz H, Coassolo P, Clozel JP, Clozel M. Ro 61–1790, a new hydrosoluble endothelin antagonist: general pharmacology and effects on experimental cerebral vasospasm. J Pharmacol Exp Ther. 1997;283:1110–1118. [PubMed] [Google Scholar]
- Rowe JG, Soper N, Ouwerkerk R, Kerr RS, Radda GK, Rajagopalan B. Delayed ischaemia after subarachnoid haemorrhage: a role for small vessel changes. J Neurol Neurosurg Psychiatry. 1995;59:451–452. doi: 10.1136/jnnp.59.4.451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sehba FA, Bederson JB. Mechanisms of acute brain injury after subarachnoid hemorrhage. Neurol Res. 2006;28:381–398. doi: 10.1179/016164106X114991. [DOI] [PubMed] [Google Scholar]
- Sehba FA, Mostafa G, Friedrich V, Jr, Bederson JB. Acute microvascular platelet aggregation after subarachnoid hemorrhage. J Neurosurg. 2005;102:1094–1100. doi: 10.3171/jns.2005.102.6.1094. [DOI] [PubMed] [Google Scholar]
- Siironen J, Juvela S, Varis J, Porras M, Poussa K, Ilveskero S, Hernesniemi J, Lassila R. No effect of enoxaparin on outcome of aneurysmal subarachnoid hemorrhage: a randomized, double-blind, placebo-controlled clinical trial. J Neurosurg. 2003;99:953–959. doi: 10.3171/jns.2003.99.6.0953. [DOI] [PubMed] [Google Scholar]
- Stein SC, Levine JM, Nagpal S, LeRoux PD. Vasospasm as the sole cause of cerebral ischemia: how strong is the evidence? Neurosurg Focus. 2006a;21:E2. doi: 10.3171/foc.2006.21.3.2. [DOI] [PubMed] [Google Scholar]
- Stein SC, Browne KD, Chen XH, Smith DH, Graham DI. Thromboembolism and delayed cerebral ischemia after subarachnoid hemorrhage: an autopsy study. Neurosurgery. 2006b;59:781–787. doi: 10.1227/01.NEU.0000227519.27569.45. discussion 787–8. [DOI] [PubMed] [Google Scholar]
- Suzuki M, Kudo A, Otawara Y, Hirashima Y, Takaku A, Ogawa A. Extrinsic pathway of blood coagulation and thrombin in the cerebrospinal fluid after sub-arachnoid hemorrhage. Neurosurgery. 1999;44:487–493. doi: 10.1097/00006123-199903000-00029. discussion 493–4. [DOI] [PubMed] [Google Scholar]
- Suzuki S, Kimura M, Souma M, Ohkima H, Shimizu T, Iwabuchi T. Cerebral microthrombosis in symptomatic cerebral vasospasm—a quantitative histological study in autopsy cases. Neurol Med Chir (Tokyo) 1990;30:309–316. doi: 10.2176/nmc.30.309. [DOI] [PubMed] [Google Scholar]
- Treggiari MM, Walder B, Suter PM, Romand JA. Systematic review of the prevention of delayed ischemic neurological deficits with hypertension, hypervolemia, and hemodilution therapy following subarachnoid hemorrhage. J Neurosurg. 2003;98:978–984. doi: 10.3171/jns.2003.98.5.0978. [DOI] [PubMed] [Google Scholar]
- Tsementzis SA, Hitchcock ER, Meyer CH. Benefits and risks of antifibrinolytic therapy in the management of ruptured intracranial aneurysms, A double-blind placebo-controlled study. Acta Neurochir (Wien) 1990;102:1–10. doi: 10.1007/BF01402177. [DOI] [PubMed] [Google Scholar]
- Vajkoczy P, Meyer B, Weidauer S, Raabe A, Thome C, Ringel F, Breu V, Schmiedek P. Clazosentan (AXV-034343), a selective endothelin A receptor antagonist, in the prevention of cerebral vasospasm following severe aneurysmal subarachnoid hemorrhage: results of a randomized, double-blind, placebo-controlled, multicenter phase IIa study. J Neurosurg. 2005;103:9–17. doi: 10.3171/jns.2005.103.1.0009. [DOI] [PubMed] [Google Scholar]
- Vergouwen MD, Vermeulen M, Coert BA, Stroes ES, Roos YB. Microthrombosis after aneurysmal subarachnoid hemorrhage: an additional explanation for delayed cerebral ischemia. J Cereb Blood Flow Metab. 2008;28:1761–1770. doi: 10.1038/jcbfm.2008.74. [DOI] [PubMed] [Google Scholar]
- Vergouwen MD, Vermeulen M, de Haan RJ, Levi M, Roos YB. Dihydropyridine calcium antagonists increase fibrinolytic activity: a systematic review. J Cereb Blood Flow Metab. 2007;27:1293–1308. doi: 10.1038/sj.jcbfm.9600431. [DOI] [PubMed] [Google Scholar]
- Wang Z, Chen G, Zhu WW, Bian JY, Shen XO, Zhou D. Influence of simvastatin on microthrombosis in the brain after subarachnoid hemorrhage in rats: a preliminary study. Ann Clin Lab Sci. 2010;40:32–42. [PubMed] [Google Scholar]
- Weidauer S, Lanfermann H, Raabe A, Zanella F, Seifert V, Beck J. Impairment of cerebral perfusion and infarct patterns attributable to vasospasm after aneurysmal subarachnoid hemorrhage: a prospective MRI and DSA study. Stroke. 2007;38:1831–1836. doi: 10.1161/STROKEAHA.106.477976. [DOI] [PubMed] [Google Scholar]
- Woertgen C, Ullrich OW, Rothoerl RD, Brawanski A. Comparison of the Claassen and Fisher CT classification scale to predict ischemia after aneurysmatic SAH? Zentralbl Neurochir. 2003;64:104–108. doi: 10.1055/s-2003-41880. [DOI] [PubMed] [Google Scholar]
- Wurm G, Tomancok B, Nussbaumer K, Adelwohrer C, Holl K. Reduction of ischemic sequelae following spontaneous subarachnoid hemorrhage: a double-blind, randomized comparison of enoxaparin versus placebo. Clin Neurol Neurosurg. 2004;106:97–103. doi: 10.1016/j.clineuro.2004.01.006. [DOI] [PubMed] [Google Scholar]
- Zaitsev S, Spitzer D, Murciano JC, Ding BS, Tliba S, Kowalska MA, Marcos-Contreras OA, Kuo A, Stepanova V, Atkinson JP, Poncz M, Cines DB, Muzykantov VR. Sustained thromboprophylaxis mediated by an RBC-targeted pro-urokinase zymogen activated at the site of clot formation. Blood. 2010a;115:5241–5248. doi: 10.1182/blood-2010-01-261610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaitsev S, Spitzer D, Murciano JC, Ding BS, Tliba S, Kowalska MA, Bdeir K, Kuo A, Stepanova V, Atkinson JP, Poncz M, Cines DB, Muzykantov VR. Targeting of a mutant plasminogen activator to circulating red blood cells for prophylactic fibrinolysis. J Pharmacol Exp Ther. 2010b;332:1022–1031. doi: 10.1124/jpet.109.159194. [DOI] [PMC free article] [PubMed] [Google Scholar]