

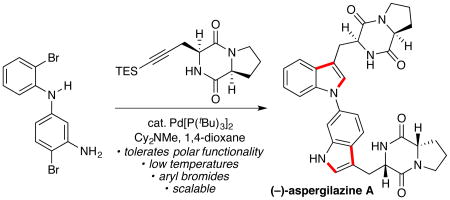
Abstract
A mild and general protocol for the Pd(0)-catalyzed heteroannulation of o-bromoanilines and alkynes is described. Application of a Pd(0)/P(tBu)3 catalyst system enables the efficient coupling of o-bromoanilines at 60 °C, mitigating deleterious side reactions and enabling access to a broad range of useful unnatural tryptophans. The utility of this new protocol is demonstrated in the highly convergent total synthesis of the bisindole natural product (−)-aspergilazine A.
Graphical abstract
The development of new methods to prepare unnatural tryptophan derivatives is an important endeavor with interdisciplinary applications. In the context of natural product synthesis, a large number of alkaloids contain tryptophan subunits embedded explicitly or implicitly within their structures;1 in chemical biology, functionalized tryptophan derivatives have emerged as important molecules for studying protein function and dynamics.2 Although subtle perturbations to this core amino acid may seem trivial, the preparation of the requisite tryptophan building block often constitutes a substantial synthetic undertaking. The Pd-catalyzed heteroannulation reaction between o-bromoanilines and serine-derived alkynes represents a convergent approach to unnatural tryptophan derivatives; yet, its versatility has thus far been limited due to challenges of reactivity and functional group tolerance. Herein, we describe the development and application of a Pd-catalyzed synthesis of structurally complex and synthetically useful tryptophan derivatives from widely-available o-bromoanilines. Using this method, a concise synthesis of the bisindole alkaloid (−)-aspergilazine A is reported.
The Pd(0)-catalyzed heteroannulation of disubstituted alkynes with o-haloanilines,3 commonly known as the Larock indole synthesis, is a powerful method for the preparation of 2,3-disubstituted indoles.4,5,6 Larock's original conditions, which employ a “ligand-less” Pd-catalyst in conjunction with an inorganic base and chloride salt additive, were developed for o-iodoanilines and remain the most widely utilized (Figure 1A).7 In 2004, Senanayake and co-workers expanded the scope of this transformation to include o-chloro- and o-bromoanilines by employing a bidentate phosphine ligand, 1,1′-bis-di-tert-butylphosphinoferrocene (dtbpf), at elevated temperatures (110–130 °C), providing simple indoles in good yield and high regioselectivity (not shown).8
Figure 1. The Larock Indole Synthesis.

Despite these advances, efforts to employ the Larock indole synthesis in complex settings, particularly using alkynyl substrates containing peptide functionality and o-bromoanilines, reveal significant limitations in the state of the art. For example, Baran's elegant synthesis of (+)-kapakahine B necessitated prolonged reaction times (24 h) and increased catalyst loadings (20 mol %) under Larock's standard conditions (Scheme 1A).5c Similarly, Boger's landmark synthesis of the chloropeptins, which employed an intramolecular Larock macrocyclization of an aryl bromide, required excess [Pd] and dtbpf (1.1 and 1.3 equiv, respectively) to effect high yields (Scheme 1B).6a
Scheme 1. Challenging Larock Indolizations in Natural Product Synthesis.

We also encountered challenges during our efforts to implement a late-stage indole annulation for the total syntheses of the C3-arylpyrroloindolines (+)-naseseazines A and B (Scheme 1C). Specifically, poor catalyst turnover and low yields were obtained using substoichiometric [Pd], while the use of higher catalyst loadings or higher temperatures resulted in hydrodehalogenation, epimerization of the diketopiperazine, poor regioselectivity, and low mass recovery.9 Following extensive experimentation, we determined that use of 25 mol % Pd[P(o-tol)3]2 in conjunction with Na2CO3 provided the product in 56% isolated yield. Although only two turnovers of [Pd] were achieved, this report represents the first Larock indole synthesis using an o-bromoaniline with substoichiometric Pd in the context of a complex polypeptide substrate.10
Given our general interest in the synthesis of indole alkaloids,11 we sought to develop a mild and reliable Pd-catalyzed synthesis of tryptophan derivatives that addresses the challenges outlined above. Specifically, we hoped to identify conditions that would 1) enable the general use of widely available o-bromoanilines, 2) proceed with synthetically useful catalyst loadings, and 3) deliver tryptophan products at lower temperatures in order to mitigate deleterious side reactivity.
In assessing the existing limitations of the Larock indole synthesis, we hypothesized that the poor reactivity of o-bromoanilines under the originally disclosed ligandless conditions was likely due to slow rates of oxidative addition. Although this elementary step could be enabled by the use of an electron-donating phosphine ligand,12 it was recognized that such ligands might slow the rate of subsequent alkyne insertion. We reasoned that sterically demanding phosphines, which have been demonstrated to favor Pd-monophosphine complexes,13 could serve to balance these opposing effects by providing a vacant coordination site to facilitate alkyne insertion.
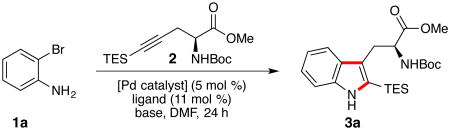
Our studies commenced with the coupling between o-bromoaniline (1a) and alkyne 214 to afford tryptophan 3a. Treatment of a mixture of 1a and 2 with 5 mol % Pd(OAc)2 and Na2CO3 at 100 °C, Larock's original conditions,3 provided 3a in 27% yield (Table 1, entry 1). A survey of conditions previously reported in the literature to promote Larock indolizations, including the addition of 11 mol % PPh3, PCy3, DavePhos, or dtbpf (the optimal ligand in Senanayake's report)8 suppressed the reaction (entries 2–5). The preformed complex Pd[P(o-tol)3]2, our most successful catalyst in the synthesis of the naseseazines,9 delivered 3a in 70% yield.
Table 1. Optimization Studies.
| |||||
|---|---|---|---|---|---|
|
| |||||
| entry | [Pd cat.] | ligand | base | t (°C) | yield (%)b |
| 1 | Pd(OAc)2 | – | Na2CO3 | 100 | 27 |
| 2 | Pd(OAc)2 | PPh3 | Na2CO3 | 100 | 17 |
| 3 | Pd(OAc)2 | DavePhos | Na2CO3 | 100 | 8 |
| 4 | Pd(OAc)2 | PCy3 | Na2CO3 | 100 | <5 |
| 5 | Pd(OAc)2 | dtbpf | Na2CO3 | 100 | <5 |
| 6 | Pd[P(o-tol)3]2 | – | Na2CO3 | 100 | 70 |
| 7 | Pd[P(tBu)3]2 | – | Na2CO3 | 100 | 78 |
| 8 | Pd[P(tBu)3]2 | – | Na2CO3 | 60 | 85 |
| 9 | Pd[P(tBu)3]2 | – | Cy2NMe | 60 | 85 |
| 10 e | Pd[P(tBu)3]2 | – | Cy2NMe | 60 | 84 (87) d |
| 11 c,e | Pd2(dba)3 | P(tBu)3 | Cy2NMe | 60 | 83 |
Reactions conducted on 0.1 mmol scale with 2.0 equiv alkyne 2 and 2.5 equiv base in DMF (0.5 mL)
Yield determined by 1H NMR analysis of the crude reaction mixture relative to an internal standard.
1:1 [Pd]/ligand used.
Isolated yield on 0.3 mmol scale.
Reaction performed in 1,4-dioxane.
Increasing the steric demand of the ligand through the use of Pd[P(tBu)3]213 further increased the yield (Table 1, entry 7). Pleasingly, lowering the temperature to 60 °C enabled a clean reaction and provided the product in 85% yield (entry 8). Use of the soluble organic base Cy2NMe in 1,4-dioxane furnished 3a with comparable efficiency (entries 9 and 10). Generation of the catalyst from a 1:1 [Pd]:ligand ratio was also feasible and improved the initial rate of the reaction (entry 11). To the best of our knowledge, this reaction represents the lowest temperature Larock indolization of any o-haloaniline reported to date.
As shown in Figure 2, the reaction exhibits excellent scope: both electron-rich (3b–3d) and electron-deficient (3e–3l) o-bromoanilines react efficiently to provide an array of unnatural tryptophan derivatives. Substitution is readily tolerated at all positions of the aniline substrate, including at nitrogen, although the preparation of 4-substituted indoles requires slightly elevated temperatures to achieve acceptable reaction rates (3c and 3i).
Figure 2.

The substrate scope of the o-bromoaniline. Reactions were conducted with 1 (1.0 equiv), 2 (2.0 equiv), Cy2NMe (2.5 equiv) in 1,4-dioxane (0.2 M) at 60 °C; the isolated yields of 3 are reported. For compound 3c and 3i, the reaction was performed at 80 °C. For compounds 3h, 3s, and 3u, desilylation with 1 M TBAF or 1 N HCl in MeOH was performed prior to chromatography. The synthesis of 3o was conducted on 5.0 mmol scale with 2.5 mol % Pd[P(tBu)3]2 and 1.5 equiv 2, then desilylated with 1.0 M TBAF.
Halogenated substrates react with excellent chemoselectivity for the aryl bromide over the aryl chloride; a variety of useful chlorinated (3f, 3g, 3i) and fluorinated (3e, 3j) tryptophans are readily prepared. Lewis-basic heterocycles also perform well under these conditions (3n, 3o). It is noteworthy that tryptophan 3o, prepared here in two steps from commercially available materials, has recently been reported as a new fluorescent probe with useful photophysical properties.15 Finally, these conditions can be extended to o-bromophenol to provide direct access to a substituted benzofuran derivative (3u). Importantly, chiral SFC analysis of the products verifies that this reaction proceeds without racemization, providing all products in enantiopure form.16 The 2-triethylsilyl group is easily removed using aqueous acid or fluoride sources, or alternatively can serve as a useful functional handle for a variety of transformations.17
To investigate the scope of the alkyne, several dipeptide-and diketopiperazine-based substrates were prepared and subjected to the reaction conditions (Figure 3). In all cases, the products are obtained in good yields and with no observed epimerization of the α-stereocenters.
Figure 3.

The substrate scope of the alkyne. Reactions were conducted with 1 (1.0 equiv), 4 (2.0 equiv), Cy2NMe (2.5 equiv) in 1,4-dioxane (0.2 M) at 60 °C; isolated yields of 5 are reported. For compound 5e, the reaction was performed at 80 °C.
The synthetic studies described above utilize 5 mol % catalyst for ease-of-operation; however, individual couplings can be conducted on preparatively useful scales with lower catalyst loadings. For example, the coupling between 2-amino-3-bromopyridine (1n) and alkyne 2 was carried out on 5 mmol scale using 2.5 mol % Pd[P(tBu)3]2 and 1.5 equiv alkyne, which upon desilylation with 1M TBAF in THF, provided 1.28 g (80% yield) of 7-aza-tryptophan (Figure 2, 3o).
To further highlight the utility of this protocol, we sought to complete a total synthesis of the dimeric diketopiperazine natural product (−)-aspergilazine A (10),19,20 which is characterized by its novel C6-to-N1 bisindole linkage (Scheme 2). We envisioned that 10 would be ideally suited for a sequential indolization strategy that would allow assembly of the natural product in a direct and convergent manner. To this end, the requisite dibromide (8) was prepared via Buchwald-Hartwig coupling of 1-bromo-2-iodobenzene (6) with diamine 7.21 Subjection of a mixture of dibromide 8 and alkyne 9 to 10 mol % Pd[P(tBu)3]2 and 2.5 equiv of Cy2NMe in 1,4-dioxane at 80 °C furnished bis-triethylsilyl-(−)-aspergilazine A in 62% isolated yield, representing an average reaction efficiency of 79% per indolization. Subsequent HCl-mediated desilylation cleanly provided the natural product. Importantly, the success of this strategy hinges largely on the ability of this new protocol to enable the coupling of 2-bromoanilines; the preparation of the diiodinated analog of diarylamine 8 via C–N bond formation is a considerably more challenging synthetic undertaking. This highly convergent synthesis proceeds in an overall yield of 21% from commercially available starting materials and underscores the utility of this methodology in the direct preparation of complex molecular scaffolds.
Scheme 2. Total Synthesis of (−)-Aspergilazine A.

In summary, we have developed a mild and general protocol for the Pd-catalyzed synthesis of functionalized tryptophan derivatives. The reaction proceeds with low catalyst loadings, displays excellent substrate scope, and is readily scalable to provide gram quantities of synthetically useful unnatural tryptophans. Furthermore, the synthetic utility of this transformation has been demonstrated in the concise synthesis of the natural product (−)-aspergilazine A.
Supplementary Material
Acknowledgments
We thank Dr. Scott Virgil of Caltech for helpful discussions, the Caltech Center for Catalysis and Chemical Synthesis for access to screening facilities and analytical equipment, and Sigma-Aldrich for a kind donation of chemicals. We gratefully acknowledge Jordan Beck and Lauren Chapman of Caltech for their assistance in the preparation of this manuscript. Fellowship support was provided by the National Science Foundation (Graduate Research Fellowship, M. E. K. and K. V. C. Grant No. DGE-1144469). S.E.R. is an American Cancer Society Research Scholar and a Heritage Medical Research Foundation investigator. Financial support from the NIH (NIGMS RGM097582A) and the donors of the ACS Petroleum Research Foundation is gratefully acknowledged.
Footnotes
Supporting Information: The Supporting Information is available free of charge on the ACS Publications website.
Experimental data, characterization information, and spectra (PDF)
References
- 1.For representative reviews: Veale CGL, Davies-Coleman MT. The Alkaloids: Chemistry and Biology. 2014;73:1–64. doi: 10.1016/B978-0-12-411565-1.00001-9.Li SM. Nat Prod Rep. 2010;27:57–58. doi: 10.1039/b909987p.
- 2.For the use of tryptophan derivatives as biological probes: Royer CA. Chem Rev. 2006;106:1769. doi: 10.1021/cr0404390.Lepthien S, Hoesl MG, Merkel L, Budisa N. Proc Natl Acad Sci, U S A. 2008;105:16095. doi: 10.1073/pnas.0802804105.Zhong W, Gallivan JP, Zhang Y, Li L, Lester HA, Dougherty DA. Proc Natl Acad Sci, U S A. 1998;95:12088. doi: 10.1073/pnas.95.21.12088.
- 3.(a) Larock RC, Yum EK. J Am Chem Soc. 1991;113:6689. [Google Scholar]; (b) Larock RC, Yum EK, Refvik MD. J Org Chem. 1998;63:7652. [Google Scholar]; (c) Chen Y, Markina NA, Yao T, Larock RC. Org Synth. 2012;88:377. [Google Scholar]
- 4.(a) Ma C, Yu S, He X, Liu X, Cook JM. Tetrahedron Lett. 2000;41:2781. [Google Scholar]; (b) Ma C, Liu X, Li X, Flippen-Anderson J, Yu S, Cook JM. J Org Chem. 2001;66:4525. doi: 10.1021/jo001679s. [DOI] [PubMed] [Google Scholar]; (c) Liu X, Deschamp JR, Cook JM. Org Lett. 2002;4:3339. doi: 10.1021/ol020101x. [DOI] [PubMed] [Google Scholar]
- 5.(a) Newhouse T, Baran PS. J Am Chem Soc. 2008;130:10886. doi: 10.1021/ja8042307. [DOI] [PubMed] [Google Scholar]; (b) Newhouse T, Lewis CA, Baran PS. J Am Chem Soc. 2009;131:6360. doi: 10.1021/ja901573x. [DOI] [PubMed] [Google Scholar]; (c) Newhouse T, Lewis CA, Baran PS. J Am Chem Soc. 2010;132:7119. doi: 10.1021/ja1009458. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Foo K, Newhouse T, Mori I, Takayama H, Baran PS. Angew Chem Int Ed. 2011;50:2716. doi: 10.1002/anie.201008048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.(a) Garfunkle J, Kimball FS, Trzupek JD, Takizawa S, Shimamura H, Tomishima M, Boger DL. J Am Chem Soc. 2009;131:16036. doi: 10.1021/ja907193b. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Shimamura H, Breazzano SP, Garfunkle J, Kimball FS, Trzupek JD, Boger DL. J Am Chem Soc. 2010;132:7776. doi: 10.1021/ja102304p. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Breazzano SP, Boger DL. J Am Chem Soc. 2011;133:18495. doi: 10.1021/ja208570q. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Breazzano SP, Poudel YB, Boger DL. J Am Chem Soc. 2013;135:1600. doi: 10.1021/ja3121394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.For recent reports utilizing Larock's original conditions, see: Shan D, Gao Y, Jia Y. Angew Chem Int Ed. 2013;52:4902. doi: 10.1002/anie.201300571.Danner P, Morkunas M, Maier ME. Org Lett. 2013;15:2474. doi: 10.1021/ol4009409.Tao P, Liang J, Jia Y. Eur J Org Chem. 2014;2014:5735.Miyamoto H, Hirano T, Okawa Y, Nakazaki A, Kobayashi S. Tetrahedron. 2013;69:9481.
- 8.(a) Shen M, Li G, Lu BZ, Hossain A, Roschangar F, Farina V, Senanayake CH. Org Lett. 2004;6:4129. doi: 10.1021/ol048114t. [DOI] [PubMed] [Google Scholar]; (b) Liu J, Shen M, Zhang Y, Li G, Khodabocus A, Rodriguez S, Qu B, Farina V, Senanayake C, Lu BZ. Org Lett. 2008;8:3573. doi: 10.1021/ol061440j. [DOI] [PubMed] [Google Scholar]
- 9.Kieffer ME, Chuang KV, Reisman SE. J Am Chem Soc. 2013;135:5557. doi: 10.1021/ja4023557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.For elegant syntheses in complex systems using o-iodoanilines, see ref. 5a–5d. For examples using stoichiometric [Pd] o-bromoanilines see ref. 6a–6c.
- 11.Wang H, Reisman SE. Angew Chem Int Ed. 2014;53:6206. doi: 10.1002/anie.201402571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hartwig JF. Organotransition Metal Chemistry: From Bonding to Catalysis. University Science Books; Sausalito, CA: 2010. [Google Scholar]
- 13.(a) Littke A, Fu GC. Angew Chem, Int Ed. 1998;37:3387. doi: 10.1002/(SICI)1521-3773(19981231)37:24<3387::AID-ANIE3387>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]; (b) Littke A, Fu GC. J Org Chem. 1999;64:10. doi: 10.1021/jo9820059. [DOI] [PubMed] [Google Scholar]; (c) Littke A, Fu GC. Angew Chem, Int Ed. 1999;38:2411. doi: 10.1002/(sici)1521-3773(19990816)38:16<2411::aid-anie2411>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]; (d) Littke AF, Dai C, Fu GC. J Am Chem Soc. 2000;122:4020. [Google Scholar]; (e) Littke A, Fu GC. J Am Chem Soc. 2001;123:6989. doi: 10.1021/ja010988c. [DOI] [PubMed] [Google Scholar]; (f) Littke AF, Schwarz L, Fu GC. J Am Chem Soc. 2002;124:6343. doi: 10.1021/ja020012f. [DOI] [PubMed] [Google Scholar]; (g) Littke AF, Fu GC. Angew Chem, Int Ed. 2002;41:4176. doi: 10.1002/1521-3773(20021115)41:22<4176::AID-ANIE4176>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 14.Readily prepared on decagram scale following the protocol of Baran and co-workers, see ref. 5b.
- 15.For an eight step synthesis of the analogous o-nitrobenzyl carbamate protected compound, see: Talukder P, Chen S, Arce PM, Hecht SM. Org Lett. 2014;16:556. doi: 10.1021/ol403429e.
- 16.See Supporting Information for additional details.
- 17.Toutov AA, Liu WB, Betz KN, Fedorov A, Stoltz BM, Grubbs RH. Nature. 2015;518:80. doi: 10.1038/nature14126. [DOI] [PubMed] [Google Scholar]
- 18.The previously reported yield was 63%; see ref. 8a.
- 19.Cai S, Kong X, Wang W, Zhou H, Zhu T, Li D, Gu Q. Tetrahedron Lett. 2012;53:2615. [Google Scholar]
- 20.For a recent total synthesis of (–)-aspergilazine A, see: Boyd EM, Sperry J. Org Lett. 2014;16:5056. doi: 10.1021/ol5024097.
- 21.Garcia-Fortanet J, Kessler F, Buchwald SL. J Am Chem Soc. 2009;131:6676. doi: 10.1021/ja9025193. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.