

Abstract
(+)-Ryanodine and (+)-ryanodol are complex diterpenoids that modulate intracellular Ca2+ release at ryanodine receptors, ion channels critical for skeletal and cardiac muscle excitation–contraction coupling and synaptic transmission. Chemical derivatization of these diterpenoids has demonstrated that certain peripheral structural modifications can alter binding affinity and selectivity among ryanodine receptor isoforms. Here we report a short chemical synthesis of (+)-ryanodol that proceeds in only 15 steps from the commercially available terpene (S)-pulegone. The efficiency of the synthesis derives from the use of a Pauson-Khand reaction to rapidly build the carbon framework, and a remarkable SeO2-mediated oxidation to install three oxygen atoms in single step. This work highlights how strategic C–O bond constructions can streamline the synthesis of poly-hydroxylated terpenes by minimizing protecting group and redox adjustments.
Terpenes are a large and structurally diverse family of natural products that range from simple hydrocarbons associated with flavors and fragrances, to complex, highly oxidized polycyclic molecules such as the anti-malarial drug artemisinin, and the anticancer compounds ingenol and taxol (1). Although terpenes are isolated from natural sources, it can be challenging to translate their biological activity into a practical application (2). In some cases the hurdle is low natural abundance; other times, it is the difficulty encountered by chemists seeking to precisely edit a terpene's molecular structure in order to improve its drug-like properties or interrogate its role in modulating disease pathways. The development of concise chemical syntheses of terpenes can transform our ability to use these molecules and their synthetic derivatives as biological probes or as lead compounds for the development of new medicines (3– 5). Furthermore, these scientific efforts often innovate chemical reactivity or synthetic design concepts (6).
The natural product ryanodine (1) (7,8) and its hydrolysis product ryanodol (2) (8,9) are among the most highly oxidized and synthetically challenging diterpenoids reported to date (Fig 1-A). Isolated from the tropical shrub Ryania speciosa Vahl in connection with its insecticidal properties (7), ryanodine is the namesake ligand of the ryanodine receptors (RyRs) (10), an important family of ion channels that regulate intracellular Ca2+ release and play a critical role in signal transduction (11). In mammalian cells, these receptors exist in multiple isoforms (RyR1, RyR2, and RyR3) that serve to mediate both movement and cognitive function. Mutations of RyRs are associated with genetic diseases such as malignant hyperthermia and central core disease (12), while altered expression of RyR2 and RyR3 has been associated with the pathogenesis of neurodegenerative disorders such as Alzheimer's disease (13). Ryanodine binds with high affinity to the conducting state of RyRs, exerting concentration dependent modulation of Ca2+ release: at nanomolar concentrations, ryanodine locks RyRs in an open, subconductance state, whereas at higher concentrations, ryanodine causes closure of the channels (14). The deacylated compound ryanodol binds with lower affinity than 1 to mammalian RyRs; however, it still induces a subconductance state and has been reported as a reversible probe of RyR-mediated Ca2+ release in cells (15).
Fig. 1. Ryanodine and selected ryanoids.

(A) Chemical structures of (+)-ryanodine (1), (+)-ryanodol (2), (+)-anhydroryanodol (3), (+)-3-epi-ryanodol (4), and (+)-3-epi-ryanodine (5). (B) Carbon numbering and ring system letter assignment. (C) Retrosynthetic analysis of anhydroryanodol.
Since the initial reports describing the isolation of ryanodine from Ryania, a number of congeners (known as ryanoids) that vary in oxidation pattern have been isolated (16–20). Whereas ryanodol – the compound obtained by hydrolysis of ryanodine – has not yet been isolated directly from a natural source, the closely related compound C3-epi-ryanodol (4) was isolated by Gonzaléz-Coloma from Persea indica (18). Indeed, due to their structural similarities, C3-epi-ryanodol (4) was initially erroneously reported to be ryanodol (2); however, the structure of the Coloma-Gonzaléz isolate (21) was recently reassigned through the synthetic efforts of Inoue (vide infra) (22). These subtle differences in structure exert a pronounced effect on RyR-binding: C3-epi-ryanodine (5), prepared from 4, binds 100 fold more weakly to RyRs than 1 (23).
Given the biological importance of the RyRs, the ryanoids have been the focus of both total synthesis and derivatization efforts. In 1979, Deslongchamps and coworkers disclosed a total synthesis of (+)-2, which hinged on a key Diels–Alder cycloaddition and a series of elegantly designed intramolecular aldol reactions to assemble the tetracyclic ABCD framework (Fig 1-B) (24-28). In a classic example of relay synthesis, the degradation product (+)-anhydroryanodol (3) was converted to (+)-ryanodol (2) in a final two-step sequence (28), providing the target molecule in 41 total steps (37 steps in its longest linear sequence). A more recent series of studies from Inoue and coworkers highlighted the utility of several radical-based C–O and C–C bond-forming reactions in a total synthesis that furnishes (±)-2 (29), and (±)-4 (22), each in 35 total steps. These studies allowed Inoue to correctly reassign the structure of the natural product isolated from Persea indica (18) as C3-epi-ryanodol (4). Inoue and coworkers subsequently reported that their synthesis could be rendered enantioselective through a catalytic asymmetric desymmetrization process, and that an appropriately protected derivative of (+)-2 could be elaborated to (+)-1 in five synthetic steps (30,31). In addition to these total syntheses, several medicinal chemistry studies have focused on the derivatization of ryanodine and ryanodol (32–34). Here, we disclose a direct and concise strategy to access the central ryanoid ring system, which has enabled the chemical synthesis of (+)-ryanodol (2) in only 15 steps from commercially available starting materials. These studies provide a synthetic platform from which it will be possible to prepare previously inaccessible ryanoid derivatives.
Perhaps the most substantial synthetic challenge embedded within the structure of 2 is the highly oxidized five-membered A-ring, which bears an all-carbon quaternary center and four additional stereogenic carbons bonded to oxygen (Fig. 1-A). A concise synthesis of 2 requires a carefully choreographed introduction of these moieties in order to minimize redox, protecting group, and functional group transformations. Guided by the landmark studies of Deslongchamps and coworkers (28), we identified the C1–C15 bond of ryanodol (2) as the most simplifying and strategic initial retrosynthetic disconnection (Fig 1-B). This analysis revealed the tetracyclic lactone anhydroryanodol (3) as our initial synthetic target (Fig 1-C), with the isopropyl group introduced at a late stage through a transition metal-catalyzed cross-coupling reaction of enol triflate 6; this approach would also allow for versatile incorporation of alternative carbon-based fragments to facilitate long-term goals of preparing ryanoid derivatives. We anticipated that the requisite A-ring oxidation pattern could be accessed via chemo- and stereoselective functionalization of cyclopentenone 7, whereby the enone would provide a functional group handle to install the critical C3, C4, and C12 alcohols of 2. We envisioned preparing cyclopentenone 7 by an intramolecular Pauson–Khand reaction (35), a transformation that would be facilitated by the conformational rigidity of bicyclic lactone 8. Lactone 8 could arise through a series of transformations from (S)-pulegone, a commercially available terpene. By this analysis, the oxygen atoms at C6, C10, and C11 would be incorporated early in the synthesis, while the oxygen atoms at C3, C4, and C12 would be introduced at a late stage. With the implementation of appropriate protecting groups, we anticipated that this strategy would minimize synthetic manipulations ancillary to the direct construction of the natural product.
We first investigated the stereoselective oxidation of (S)-pulegone (10) to install the C6 and C10 alcohols of the C-ring (Fig. 2), envisioning functionalization at both carbons via enolates accessed by γ- or α′;-deprotonation of the ketone, respectively. Preliminary experiments confirmed that the C6-alcohol could be installed with 2.5:1 diastereomeric ratio (dr) through generation of the thermodynamic dienolate with potassium hexamethyldisilazide (KHMDS) followed by quenching with oxaziridine 11 (36). Noting that KHMDS did not appear to directly react with oxaziridine 11 at −78 °C, we hypothesized that a one-step protocol involving excess KHMDS and 11 might simultaneously install both alcohols. Indeed, treatment of 10 with 2.5 equiv of KHMDS at −78 °C followed by dropwise addition of 2.4 equiv of 11 provided α,α′-diol 12, which could be isolated as a single diastereomer in 42% yield (120 mmol scale). Although the yield is modest, this single transformation converts a simple terpene to a valuable building block with the requisite ryanodol C-ring oxidation pattern. We improved the efficiency of this transformation to 50% yield by modification of the standard procedure on a smaller scale (10 mmol, see Supplementary Material). However, given the operational simplicity of the former procedure on larger scales, this protocol was preferentially employed for increased material throughput.
Fig. 2. Complete synthetic sequence for the chemical synthesis of (+)-anhydroryanodol and (+)-ryanodol.
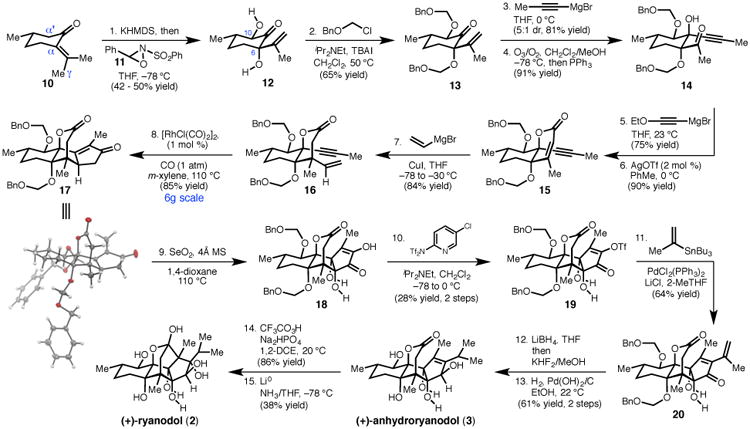
Reagents and conditions as follows: 1. potassium hexamethyldisilazide (2.5 equiv), THF −78 °C; then 11 (2.4 equiv), 42-50% yield. 2. benzyl chloromethyl ether (5.0 equiv), iPr2NEt (8.0 equiv), nBu4NI (2.0 equiv), CH2Cl2, 50 °C, 65% yield. 3. Propynylmagnesium bromide, THF, 0 °C, 81% yield, 5:1 dr. 4. O3,/O2, CH2Cl2/MeOH (4:1), −78 °C; then PPh3, 91% yield. 5. Ethoxyethynylmagnesium bromide (5.0 equiv), THF, 0 °C, 75% yield. 6. AgOTf (2 mol %), PhMe, 0 °C, 90% yield. 7. CuI (3.0 equiv), vinylmagnesium bromide (6.0 equiv), THF, −78 to −30 °C, 84% yield. 8. [RhCl(CO)2]2 (1 mol %), CO (1 atm), m-xylene, 110 °C, 85% yield. 9. SeO2 (10 equiv), 4Å MS, 1,4-dioxane, 110 °C. 10. Comins' reagent, iPr2NEt, CH2Cl2, −78 to 0 °C, 28% yield, 2-steps. 11. PdCl2(PPh3)2, tributyl(prop-1-en-2-yl)stannane, LiCl, 2-MeTHF, 85 °C, 64% yield. 12. LiBH4, THF, −15 to −10 °C; then KHF2/MeOH. 13. H2, Pd(OH)2/C, EtOH, 61% yield, 2-steps. 14. Trifluoroacetic anhydride, urea hydrogen peroxide, Na2HPO4, 86% yield. 15. Li0 wire, NH3/THF, −78 °C, 38% yield.
Treatment of diol 12 with excess benzyl chloromethyl ether effected protection of both alcohols as benzyloxymethyl ethers to give 13. At this stage, the D-ring was constructed by an efficient four-step sequence. First, addition of propynylmagnesium bromide to 13 at 0 °C proceeded in 5:1 dr, providing the equatorially-disposed alkyne in 81% isolated yield. Ozonolytic cleavage of the 1,1-disubstituted olefin proceeded in high yield to afford methyl ketone 14. Although a N, N′-dicyclohexylcarbodiimide-mediated phosphonoacylation/intramolecular Horner–Wadsworth–Emmons lactone synthesis had proven moderately effective in model studies lacking C10-oxidation, the tertiary alkynol of 14 proved resistant to acylation under a number of conditions. Presumably, the increased steric encumbrance and inductively withdrawing C10-ether dramatically decreases the nucleophilicity of the tertiary alkynol. Instead, ketone 14 was efficiently converted to α,β-unsaturated lactone 15 via 1,2-addition of ethoxyethynylmagnesium bromide followed by an Ag-catalyzed cyclization/elimination cascade (37).
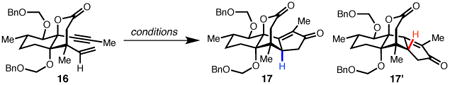
With lactone 15 in hand, 1,4-addition of magnesium divinyl cuprate provided the corresponding enyne 16 as a single diastereomer in 84% yield, forging the critical all-carbon quaternary center at C5 while simultaneously setting the stage for a key Pauson–Khand cyclization. A number of Pauson–Khand procedures were screened for their ability to deliver 17 in both high yield and diastereoselectivity (Table 1). Although standard conditions (38,39) using stoichiometric amounts of Co2(CO)8 produced the desired product, separation of diastereomeric enone 17′ proved challenging and indicated that a more selective protocol was necessary. Monometallic mediators were found to improve the dr, with Mo(CO)3(DMF)3 (40) providing 17 as a single diastereomer in 67% yield. Despite the serviceable yield and scalability of this transformation, we sought a more efficient protocol that could obviate the need for stoichiometric metals and their attendant byproducts. We were pleased to find that treatment of 16 with 1 mol% [RhCl(CO)2]2 (41) under an atmosphere of carbon monoxide provided the desired product in 85% yield, again as a single diastereomer. This reaction has been conducted on multigram scale, providing 5.7 g of enone 17 in a single pass. Single crystal X-ray diffraction analysis of 17 confirmed both the absolute configuration and structural assignment of this key tetracyclic intermediate.
Table 1.
Evaluation of Pauson–Khand reaction conditions.
| |||
|---|---|---|---|
|
| |||
| entry | conditions* | dr† | yield (%)‡ |
| 1 | Co2(CO)8 (1.2 equiv), THF, 12 h; then DMSO, 65 °C | 2.2 : 1 | 46 |
| 2 | Co2(CO)8 (1.2 equiv), CH2Cl2, 9 h; then NMO, 23 °C | 4.5 : 1 | 78 |
| 3 | Mo(CO)6 (1.2 equiv), DMSO, PhMe, 110 °C | -- | trace |
| 4 | Mo(CO)3(DMF)3 (1.1 equiv), CH2Cl2, 23 °C | > 20 : 1 | 67 |
| 5 | [RhCl(CO)2]2 (1 mol %), CO (1 atm), m-xylene, 110 °C | >20 : 1 | 85 |
Reactions conducted on 0.2 mmol scale.
Determined by 1H NMR spectroscopy.
Isolated yield after purification by silica gel chromatography.
The successful realization of the Pauson–Khand cyclization led us to the next phase of our synthetic studies: chemo- and stereoselective functionalization of the A-ring through the introduction of the C3, C4, and C12-alcohols, as well as the C2-isopropyl unit. To this end, initial investigations were aimed at oxidation of the C4 allylic methine of 17, but efforts in this vein were thwarted by undesired reactivity. For example, established methods for allylic oxidation via reactive radical species (e.g. Pd(OH)2/C/t-BuOOH (42) or Rh2(cap)4/t-BuOOH (43)) were unfruitful due to competitive functionalization of the C1–C12 olefin (i.e. epoxidation, 1,2-difunctionalization). Although we entertained the possibility of advancing these unanticipated products further in the synthesis, it was unclear how to achieve efficient transposition to the requisite oxidation pattern.
Instead, we turned to C3-functionalization of 17 using SeO2 (44), anticipating that the enone might be readily oxidized to the corresponding α-diketone (Fig. 3). To our delight, we found that treatment of 17 with excess SeO2 in wet 1,4-dioxane at 110 °C not only effected C3-oxidation, but also the formal hydration of the enone, thereby installing the necessary C12-alcohol and providing diosphenol 21. Hypothesizing that rigorous exclusion of water might promote C4-allylic hydroxylation prior to diketone formation, enone 17 was subjected to SeO2 under anhydrous conditions in the presence of 4Å molecular sieves (4Å MS). Prolonged heating at 110 °C in 1,4-dioxane provided a distinct product, which had incorporated an additional oxygen based on mass spectrometry analysis. Rigorous structural assignment of this compound revealed it to be the fully oxidized diosphenol 18, a compound with the C4, C12 syn-vicinal diol of 2. This astonishing single-step transformation installs the necessary oxygen atoms at C3, C4, and C12 while simultaneously providing a functional group handle for incorporation of the C2-isopropyl group.
Fig. 3. Selenium dioxide-mediated A-ring oxidation.

In the presence of water, SeO2-mediated oxidation of enone 17 provides 21. In the absence of water, further oxidation occurs to deliver 18.
Because C4-deoxy analogue of ryanodine is itself a natural product, deoxyspiganthine (20), and could be of interest for future biological studies, we optimized procedures to prepare both 18 and 21 (Fig. 3). Given concerns that contamination by 1H NMR silent, red selenium byproducts resulted in artificially inflated isolated yields, our optimization efforts were guided by 1H NMR yields determined by integration versus an added standard. Isolated yields were determined after conversion to corresponding enol triflates 19 and 22, which could be obtained as analytically pure compounds. Thus, subjection of tetracycle 17 to 10 equiv of SeO2 in wet 1,4-dioxane at 110 °C for 1 h provided 21 in 67-69% 1H NMR yield, and the corresponding vinyl triflate 22 in 56% isolated yield over two steps. Alternatively, treatment of 17 with 10 equiv of SeO2 in anhydrous 1,4-dioxane in the presence of freshly activated 4Å MS at 110 °C for 9 h provided 18 in 33-35% 1H NMR yield, and vinyl triflate 19 in 28% isolated yield over two steps. Despite the modest yield, this sequence accomplishes the stereospecific incorporation of three oxygen atoms, proceeds with an average efficiency of approximately 70% yield per transformation, and fares well in comparison to conceivable multiple step protocols to achieve the same reactivity. Although the precise mechanism of the SeO2-mediated oxidation remains unclear at this time, investigations are ongoing and should aid the development of a more efficient protocol.
In the final stages of the synthesis, advancement of 19 to (+)-anhydroryanodol was achieved by a three-step sequence. Palladium-catalyzed cross-coupling between 19 and tributyl(2-propenyl)stannane installed the final three carbons, delivering 20 in 64% yield. LiBH4-mediated 1,2-reduction stereoselectively furnished the C3 alcohol, which was subjected to H2 and Pd(OH)2/C to simultaneously reduce the disubstituted olefin and remove the benzyloxymethyl groups, providing (+)-3 in 61% yield over two steps. Using this route, we have prepared >400 mg of (+)-anhydroryanodol to date. Conversion of this material to (+)-ryanodol was achieved via a slight modification of Deslongchamps' two-step protocol (28). Treatment of 3 with trifluoroperacetic acid, freshly prepared from trifluoroacetic anhydride and urea hydrogen peroxide (in place of the originally reported concentrated hydrogen peroxide), cleanly afforded epianhydroryanodol epoxide in 86% yield. Subjection of this material to Li0 in NH3 (distilled from Na0) at −78 °C resulted in reductive cyclization to produce (+)-2 in 38% yield (lit. 60% yield). In our hands, the reaction profile was highly dependent on the purity of the ammonia. Specifically, independent control reactions conducted with ammonia condensed directly from the gas cylinder, or using redistilled ammonia with either added H2O (10 equiv), or exogenous Fe-salts (45), revealed that these parameters all significantly affect the ratio of 2 to carbonyl-reduction products, as well as the formation of minor unidentified degradation products.
The concise synthesis of (+)-ryanodol described here proceeds in only 15 synthetic steps (0.42% overall yield) from (S)-pulegone (10), fewer than half the steps of the previously disclosed syntheses by Deslongchamps (37 linear steps, 0.23% yield) and Inoue (35 linear steps, 0.008% yield). The efficiency of our approach derives from the development of a direct and scalable route to key cyclopentenone 17, which can be prepared on multigram scale in only eight steps and rapidly elaborated to (+)-anhydroryanodol. The strategic use of C–O bond forming reactions minimizes redox adjustments and the use of protecting groups. Indeed, the five alcohols found in (+)-3 are incorporated using just two transformations: the dihydroxylation of 10, and the SeO2-mediated oxidation of enone 17. Moreover, all but the C3-alcohol are introduced with the correct carbon oxidation level. We anticipate that the brevity of the synthesis will render feasible the design and preparation of ryanoid derivatives for studying RyR function.
Supplementary Material
Acknowledgments
The Caltech Center for Catalysis and Chemical Synthesis is gratefully acknowledged for access to analytical equipment. We thank Dr. Scott Virgil and Ms. Julie Hofstra for assistance in obtaining X-ray quality crystals and solving the structure of 15, respectively. Dr. Michael Takase and Mr. Larry Henling are acknowledged for acquiring the X-ray diffraction data for 15 (CCDC deposition #1478621; the data are available free of charge from The Cambridge Crystallographic Data Centre). Dr. Madeleine Kieffer is gratefully acknowledged for critical feedback and helpful suggestions. Fellowship support was provided by the National Science Foundation (graduate research fellowship to K. V. C., Grant No. DGE-1144469), and the Shenzhen UV-ChemTech Inc. (postdoctoral fellowship to C. X.). S. E. R. is an American Cancer Society Research Scholar and Heritage Medical Research Institute investigator. Financial support from the NIH (NIGMS RGM097582-01), Eli Lilly, and Novartis is gratefully acknowledged. The California Institute of Technology has filed a provisional patent on this work (application # 62/269,760).
References and Notes
- 1.Breitmaier E. Terpenes: Flavors, Fragrances, Pharmaca, Pheromones. Wiley; Weinheim, Germany: 2006. [Google Scholar]
- 2.Koehn FE, Carter GT. Nat Rev Drug Discov. 2005;4:206–220. doi: 10.1038/nrd1657. [DOI] [PubMed] [Google Scholar]
- 3.Jørgensen L, McKerrall SJ, Kuttruff CA, Ungehuer F, Felding J, Baran PS. Science. 2013;341:878–882. doi: 10.1126/science.1241606. [DOI] [PubMed] [Google Scholar]
- 4.Kawamura S, Chu H, Felding J, Baran PS. Nature. 2016;532:90–93. doi: 10.1038/nature17153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lu HH, Martinez MD, Shenvi RA. Nature Chem. 2015;7:604–607. doi: 10.1038/nchem.2283. [DOI] [PubMed] [Google Scholar]
- 6.Maimone TJ, Baran PS. Nature Chem Biol. 2007;3:396–407. doi: 10.1038/nchembio.2007.1. [DOI] [PubMed] [Google Scholar]
- 7.Rogers EF, Koniuszy FR, Shavel J, Folkers K. J Am Chem Soc. 1948;70:3086–3088. doi: 10.1021/ja01189a074. [DOI] [PubMed] [Google Scholar]
- 8.Wiesner K, Valenta Z, Findlay JA. Tetrahedron Lett. 1968;8:221–223. [Google Scholar]
- 9.Srivastava SN, Przybylska M. Can J Chem. 1968;46:795–797. [Google Scholar]
- 10.Pessah IN, Waterhouse AL, Casida JE. Biochem Biophys Res Commun. 1985;128:449–456. doi: 10.1016/0006-291x(85)91699-7. [DOI] [PubMed] [Google Scholar]
- 11.Lanner JT, Georgiou DK, Joshi AD, Hamilton SL. Cold Spring Harb Perspect Biol. 2010;2:1–21. doi: 10.1101/cshperspect.a003996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McCarthy TV, Quane KA, Lynch PJ. Hum Mutat. 2000;15:410–417. doi: 10.1002/(SICI)1098-1004(200005)15:5<410::AID-HUMU2>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 13.Kelliher M, Fastbom J, Cowburn RF, Bonkale W. Neuroscience. 1999;92:499–513. doi: 10.1016/s0306-4522(99)00042-1. [DOI] [PubMed] [Google Scholar]
- 14.Meissner G. J Biol Chem. 1986;261:6300–6306. [PubMed] [Google Scholar]
- 15.Ramos-Franco J, Gomez AM, Nani A, Liu Y, Copello JA, Fill M. Eur J Physiol. 2010;460:767–776. doi: 10.1007/s00424-010-0839-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Isogai A, Murakoshi S, Suzuki A, Tamura S. Agric Biol Chem. 1977;41:1779–1784. [Google Scholar]
- 17.Nohara T, Tokubuchi N, Kuroiwa M, Nishioka I. Chem Pharm Bull. 1980;28:2682–2686. [Google Scholar]
- 18.González-Coloma A, Hernandez MG, Perales A, Fraga BM. J Chem Ecol. 1990;16:2723–2733. doi: 10.1007/BF00988081. [DOI] [PubMed] [Google Scholar]
- 19.González-Coloma A, Terrero D, Perales A, Escoubas P, Fraga BM. J Agric Food Chem. 1996;44:296–300. [Google Scholar]
- 20.Achenbach H, Hubner H, Vierling W, Brandt W, Reiter M. J Nat Prod. 1995;58:1092–1096. doi: 10.1021/np50121a019. [DOI] [PubMed] [Google Scholar]
- 21.This compound has been referred to by Inoue et. al. as “natural ryanodol.” See ref. 22.
- 22.Koshimizu M, Nagatomo M, Inoue M. Angew Chem Int Ed. 2016;55:2493–2497. doi: 10.1002/anie.201511116. [DOI] [PubMed] [Google Scholar]
- 23.Welch W, Williams AJ, Tinker A, Mitchell KE, Deslongchamps P, Lamothe J, Gerzon K, Bidasee KR, Besch HR, Airey JA, Sutko JL, Ruest L. Biochemistry. 1997;36:2939–2950. doi: 10.1021/bi9623901. [DOI] [PubMed] [Google Scholar]
- 24.Belanger A, Berney DJF, Borschberg HJ, Brousseau R, Doutheau A, Durand R, Katayama H, Lapalme R, Leturc DM, Liao CC, MacLachlan FN, Maffrand JP, Marazza F, Martino R, Moreau C, Saint-Laurent L, Saintonge R, Soucy P, Ruest L, Deslongchamps P. Can J Chem. 1979;57:3348–3354. [Google Scholar]
- 25.Deslongchamps P, Belanger A, Berney DJF, Borschberg HJ, Brousseau R, Doutheau A, Durand R, Katayama H, Lapalme R, Leturc DM, Liao CC, MacLachlan FN, Maffrand JP, Marazza F, Martino R, Moreau C, Saint-Laurent L, Saintonge R, Soucy P. Can J Chem. 1990;68:115–126. [Google Scholar]
- 26.Deslongchamps P, Belanger A, Berney DJF, Borschberg HJ, Brousseau R, Doutheau A, Durand R, Katayama H, Lapalme R, Leturc DM, Liao CC, MacLachlan FN, Maffrand JP, Marazza F, Martino R, Moreau C, Saint-Laurent L, Saintonge R, Soucy P. Can J Chem. 1990;68:127–152. [Google Scholar]
- 27.Deslongchamps P, Belanger A, Berney DJF, Borschberg HJ, Brousseau R, Doutheau A, Durand R, Katayama H, Lapalme R, Leturc DM, Liao CC, MacLachlan FN, Maffrand JP, Marazza F, Martino R, Moreau C, Saint-Laurent L, Saintonge R, Soucy P. Can J Chem. 1990;68:153–185. [Google Scholar]
- 28.Deslongchamps P, Belanger A, Berney DJF, Borschberg HJ, Brousseau R, Doutheau A, Durand R, Katayama H, Lapalme R, Leturc DM, Liao CC, MacLachlan FN, Maffrand JP, Marazza F, Martino R, Moreau C, Saint-Laurent L, Saintonge R, Soucy P. Can J Chem. 1990;68:186–192. [Google Scholar]
- 29.Nagatomo M, Koshimizu M, Masuda K, Tabuchi T, Urabe D, Inoue M. J Am Chem Soc. 2014;136:5916–5919. doi: 10.1021/ja502770n. [DOI] [PubMed] [Google Scholar]
- 30.Nagatomo M, Hagiwara K, Masuda K, Koshimizu M, Inoue M. Chem Eur J. 2016;22:222–229. doi: 10.1002/chem.201503640. [DOI] [PubMed] [Google Scholar]
- 31.Masuda K, Koshimizu M, Nagatomo M, Inoue M. Chem Eur J. 2016;22:230–236. doi: 10.1002/chem.201503641. [DOI] [PubMed] [Google Scholar]
- 32.Waterhouse AL, Pessah IN, Francini AO, Casida JE. J Med Chem. 1987;30:710–716. doi: 10.1021/jm00387a022. [DOI] [PubMed] [Google Scholar]
- 33.Welch W, Ahmed S, Airey JA, Gerzon K, Humerickhouse RA, Besch HR, Ruest L, Deslongchamps P, Sutko JL. Biochemistry. 1994;33:6074–6085. doi: 10.1021/bi00186a006. [DOI] [PubMed] [Google Scholar]
- 34.Sutko JL, Airey JA, Welch W, Ruest L. Pharmacol Rev. 1997;49:53–98. [PubMed] [Google Scholar]
- 35.Khand U, Knox GR, Pauson PL, Watts WE, Forman MI. J Chem Soc, Perkin Trans I. 1973;1973:977–981. [Google Scholar]
- 36.Vishwakarma LC, Stringer OD, Davis FA. Org Synth. 1988;66:203–207. [Google Scholar]
- 37.Egi M, Ota Y, Nishimura Y, Shimizu K, Azechi K, Akai S. Org Lett. 2013;15:4150–4153. doi: 10.1021/ol401824v. [DOI] [PubMed] [Google Scholar]
- 38.Chung YK, Lee BY, Jeong N, Hudecek M, Pauson PL. Organometallics. 1993;12:220–223. [Google Scholar]
- 39.Shambayani S, Crowe WE, Schreiber SL. Tetrahedron Lett. 1990;31:5289–5292. [Google Scholar]
- 40.Adrio MR, Rivero JC, Carretero Org Lett. 2005;7:431–434. doi: 10.1021/ol047678u. [DOI] [PubMed] [Google Scholar]
- 41.Koga Y, Kobayashi T, Narasaka K. Chem Lett. 1998;27:249–250. [Google Scholar]
- 42.Yu JQ, Corey EJ. Org Lett. 2002;4:2727–2730. doi: 10.1021/ol0262340. [DOI] [PubMed] [Google Scholar]
- 43.Cantino J, Forslund RE, Doyle MP. J Am Chem Soc. 2004;126:13622–13623. doi: 10.1021/ja045330o. [DOI] [PubMed] [Google Scholar]
- 44.Riley R, Morley JF, Friend NAC. J Chem Soc. 1932;1932:1875–1883. [Google Scholar]
- 45.Harvey RG, Urberg K. J Org Chem. 1968;33:2570–2571. [Google Scholar]
- 46.Hill AJ, Keach DT. J Am Chem Soc. 1926;48:257–262. [Google Scholar]
- 47.Jeong N, Lee SJ, Lee BY, Chung YK. Tetrahedron Lett. 1990;34:4027–4030. [Google Scholar]
- 48.Emmons WD, Pagano AS. J Am Chem Soc. 1955;77:4557–4559. [Google Scholar]
- 49.APEX2, Version 2 User Manual, M86-E01078, Bruker Analytical X-ray Systems. Madison, WI: Jun, 2006. [Google Scholar]
- 50.Sheldrick GM. SADABS (version 2008/1): Program for Absorption Correction for Data from Area Detector Frames. University of Göttingen; Göttingen, Germany: 2008. [Google Scholar]
- 51.Sheldrick GM. Acta Cryst. 2008;A-64:112–122. doi: 10.1107/S0108767307043930. [DOI] [PubMed] [Google Scholar]
- 52.Müller P. Crystallogr Rev. 2009;15:57–83. [Google Scholar]
- 53.Parsons S, Flack HD, Wagner T. Acta Cryst. 2013;B69:249–259. doi: 10.1107/S2052519213010014. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.