

Abstract
Purpose
Survival and progression of mature B-cell malignancies depend on signals from the B-cell antigen receptor, and Bruton tyrosine kinase (BTK) is a critical signaling kinase in this pathway. We evaluated ibrutinib (PCI-32765), a small-molecule irreversible inhibitor of BTK, in patients with B-cell malignancies.
Patients and Methods
Patients with relapsed or refractory B-cell lymphoma and chronic lymphocytic leukemia received escalating oral doses of ibrutinib. Two schedules were evaluated: one, 28 days on, 7 days off; and two, once-daily continuous dosing. Occupancy of BTK by ibrutinib in peripheral blood was monitored using a fluorescent affinity probe. Dose escalation proceeded until either the maximum-tolerated dose (MTD) was achieved or, in the absence of MTD, until three dose levels above full BTK occupancy by ibrutinib. Response was evaluated every two cycles.
Results
Fifty-six patients with a variety of B-cell malignancies were treated over seven cohorts. Most adverse events were grade 1 and 2 in severity and self-limited. Dose-limiting events were not observed, even with prolonged dosing. Full occupancy of the BTK active site occurred at 2.5 mg/kg per day, and dose escalation continued to 12.5 mg/kg per day without reaching MTD. Pharmacokinetic data indicated rapid absorption and elimination, yet BTK occupancy was maintained for at least 24 hours, consistent with the irreversible mechanism. Objective response rate in 50 evaluable patients was 60%, including complete response of 16%. Median progression-free survival in all patients was 13.6 months.
Conclusion
Ibrutinib, a novel BTK-targeting inhibitor, is well tolerated, with substantial activity across B-cell histologies.
INTRODUCTION
Signaling from the B-cell antigen receptor (BCR) regulates multiple cellular processes, including proliferation, differentiation, apoptosis, and cell migration, and is essential for normal B-cell development and survival.1,2 The BCR pathway is implicated in the pathogenesis of several B-cell malignancies, including diffuse large B-cell lymphoma (DLBCL), follicular lymphoma, mantle-cell lymphoma, and B-cell chronic lymphocytic leukemia (CLL).3–10 Bruton tyrosine kinase (BTK), a member of the Tec kinase family, is a signaling molecule positioned early within the BCR signaling cascade, in close proximity to Syk and phosphoinositide 3-kinase delta.11–14 Functional null mutations of BTK in humans lead to the inherited disease X-linked agammaglobulinemia (XLA), which is characterized by a complete lack of mature peripheral B cells and low levels of serum immunoglobulin (Ig).15 Because the phenotype of human XLA is largely restricted to B lymphocytes, BTK is a uniquely attractive target for selective B-cell inhibition.
Ibrutinib (PCI-32765) is a selective and irreversible small-molecule BTK inhibitor (half maximal inhibitory concentration, 0.5 nmol/L) that inhibits BCR signaling in human B cells via specific active-site occupancy.16 In vitro, ibrutinib is selectively cytotoxic to DLBCL cell lines driven by chronic active BCR signaling.17 Orally administered ibrutinib resulted in objective clinical responses in dogs that spontaneously developed non-Hodgkin lymphoma (NHL).16 In this study, an active site–directed affinity probe assay was used to demonstrate that efficacy correlated well with the degree of active-site occupancy of BTK by ibrutinib.
Because of the central role of BCR signaling, the restricted expression of BTK, and promising preclinical activity with this novel agent, we evaluated ibrutinib in a phase I open-label, dose-escalation trial to determine the dose, safety profile, pharmacokinetics (PKs), pharmacodynamics, and tumor response in patients with relapsed or refractory B-cell NHL and B-cell CLL.
PATIENTS AND METHODS
Eligibility
Patients with relapsed or refractory, histologically confirmed NHL, CLL, or Waldenström macroglobulinemia (WM) who had failed at least one previous therapy were eligible. Inclusion criteria included: Eastern Cooperative Oncology Group performance status ≤ 1, age ≥ 18 years, measureable disease (NHL: at least one lymph node 2 cm in any axis; CLL: ≥ 5,000 leukemia cells/μL; WM: IgM level ≥ 1,000 mg/dL with bone marrow infiltration), absolute neutrophil count ≥ 1,500/μL, platelet count ≥ 75,000/μL (unless CLL with cytopenia secondary to marrow involvement), and adequate renal and hepatic function.
Major exclusion criteria included: more than four previous systemic therapies; previous allogeneic stem-cell transplantation; immunotherapy, chemotherapy, radiotherapy, or experimental therapy within 4 weeks of study; major surgery within 4 weeks of study; active infection within 4 weeks of cycle one, day 1; CNS involvement by lymphoma; history of previous cancer < 2 years before study (except skin basal or squamous cell carcinomas, cervical cancer in situ, or other in situ carcinomas); and significant comorbidities.
Study Design
This phase I study was conducted at eight centers in the United States in accordance with Good Clinical Practice guidelines, as provided by the International Conference on Harmonisation and principles of the Declaration of Helsinki. The institutional review board at each participating site approved the study, and all patients provided written informed consent before enrollment.
Patients received ibrutinib capsules orally once daily at 1.25, 2.5, 5, 8.3, or 12.5 mg/kg per day on a 28 days on, 7 days off, schedule (35-day cycle), or received continuous daily dosing of 8.3 mg/kg or 560 mg until disease progression (PD), unacceptable toxicity, or patient or investigator decision to end therapy. To better evaluate BTK occupancy, at least six patients were enrolled in each cohort. Dose escalation continued until MTD was achieved based on protocol-defined dose-limiting toxicities (DLTs), defined as the occurrence in cycle one of any of the following: grade ≥ 3 nonhematologic adverse event (AE), grade ≥ 3 prolongation of the QTc interval, grade ≥ 3 neutropenia, febrile neutropenia, grade 4 thrombocytopenia, or dosing delay because of toxicity for more than 7 days. Dose escalation to next level proceeded after DLT assessment of patients at the end of cycle one. The MTD was defined as the highest dose level at which ≥ 33% of patients in the cohort experienced a DLT or, in the absence of a DLT, until three dose levels above the dose level in which full BTK occupancy could be demonstrated. Patients with complete response (CR), partial response (PR), or stable disease (SD) who had received therapy for 6 months were allowed to continue therapy at a fixed dose of 560 or 420 mg per day on an extension trial until confirmation of PD or at physician's discretion.
Study Assessments
AEs were monitored throughout treatment, and toxicities were assessed by the National Cancer Institute Common Toxicity Criteria for AEs (version 3.0). Physical examination, blood samples for hematology and clinical chemistry, urinalysis, T/B/natural killer cell counts, and serum Ig were assessed on day 1 of each cycle. Disease response assessments included computed tomography scans and physical examination. Review of clinical laboratory results assessments were conducted every two cycles until PD or use of alternative antineoplastic therapy. Computed tomography or positron emission tomography and bone marrow biopsy/aspirate were required to confirm CR. Best clinical response (CR, PR, SD, PD) was determined by investigators based on criteria defined by the International Working Group revised response criteria for malignant lymphoma,18 and the International Workshop on CLL guidelines for diagnosis and treatment of CLL.19 Patients discontinuing therapy for reasons other than progression were observed every 2 to 3 months until PD for a maximum of 6 months after the last study dose.
Blood samples for PKs or BTK occupancy were collected during cycle one on day 1 (predose and up to 6 hours after dosing), day 2 (predose), day 8 (predose and up to 4 hours after dosing), and 24 hours after day-28 dosing. Plasma was analyzed for ibrutinib concentrations by high-performance liquid chromatography with tandem mass spectrometric detection. PK assessments were performed on the plasma concentration-time data obtained on day 1 using noncompartmental analysis (WinNonlin Pro, version 5.3; Pharsight, Cambridge, MA). BTK occupancy in peripheral blood mononuclear cells (PBMCs) was measured using a fluorescent affinity probe (hereafter probe) assay, as described previously.16 Fluorescent probe protein bands were quantified using Molecular Dynamics ImageQuant 5.2 software (GE Healthcare, Waukesha, WI). The relative density of each band was quantified using volume integration and local average background correction. The background for each quantified band was defined by volume integration of a nearby region within the same lane that did not contain a visible band. All background regions on the gel were also used to define a lower limit of quantitation equal to the average volume of background regions plus 3× the standard deviation of the volumes of the background regions. BTK occupancy was compared using GraphPad Prism version 4.0 (GraphPad, San Diego, CA). Statistically significant differences were determined using one-way analysis of variance with Bonferroni's post hoc comparison.
Statistical Methods
This was a dose-escalation trial designed to determine the MTD of ibrutinib and characterize the most frequent AEs and DLTs. The intent-to-treat (ITT) population included all patients receiving any amount of the study drug. The evaluable population included all patients who received at least two cycles of therapy. The best response recorded was used for response analyses. Overall response rate (CR plus PR) was calculated for the entire study population as well as for each treatment cohort and histology. Progression-free survival was measured from the first day of dosing until PD or relapse and determined using the Kaplan-Meier method.20
RESULTS
Patient Characteristics
From March 2009 through September 2010, 56 patients were enrolled and received one or more doses of ibrutinib. Baseline disease characteristics and previous therapies are listed in Table 1. Median age of the patients was 65 years (range, 41 to 82 years). Median number of previous therapies was three (range, one to 10), and all but four patients had previously received rituximab. Five cohorts were treated with the 28 days on, 7 days off, schedule at doses escalating from 1.25 to 12.5 mg/kg (cohorts I to V), and two cohorts were treated on the continuous dosing schedule at 8.3 mg/kg once daily or a fixed dose of 560 mg once daily (Table 2). Median number of cycles received was five.
Table 1.
Baseline Patient Characteristics (N = 56)
| Characteristic | No. | % |
|---|---|---|
| Age, years | ||
| Median | 65 | |
| Range | 41-82 | |
| Sex | ||
| Male | 38 | 68 |
| Female | 18 | 32 |
| Histologic subtype | ||
| Follicular lymphoma | 16 | 29 |
| CLL/SLL | 16 | 29 |
| Mantle-cell lymphoma | 9 | 16 |
| DLBCL | 7 | 13 |
| Marginal zone/mucosal-associated lymphoid tissue lymphoma | 4 | 7 |
| Waldenström macroglobulinemia | 4 | 7 |
| Prior therapy | ||
| Median No. | 3 | |
| Range | 1-10 | |
| Rituximab | 52 | 93 |
| Alkylator based | 47 | 84 |
| Anthracycline based | 25 | 45 |
| Radiotherapy | 15 | 27 |
| Autologous stem-cell transplantation | 3 | 5 |
Abbreviations: CLL, chronic lymphocytic leukemia; DLBCL, diffuse large B-cell lymphoma; SLL, small lymphocytic lymphoma.
Table 2.
Common Adverse Events (regardless of attribution) According to Dosing Cohorts Reported in ≥ 10% of Patients
| Cohort Characteristic and Adverse Event (grade) | Cohort |
% of Total | ||||||
|---|---|---|---|---|---|---|---|---|
| I | II | III | IV | CD-I | V | CD-II | ||
| No. of patients | 7 | 9 | 6 | 8 | 10 | 7 | 9 | |
| Dose | 1.25 mg/kg/d | 2.5 mg/kg/d | 5.0 mg/kg/d | 8.3 mg/kg/d | 8.3 mg/kg/d | 12.5 mg/kg/d | 560 mg/d | |
| Schedule* | 28/7 days | 28/7 days | 28/7 days | 28/7 days | Once daily | 28/7 days | Once daily | |
| Diarrhea | ||||||||
| 1-2 | 0 | 4 | 1 | 4 | 7 | 3 | 5 | 42.9 |
| 3-4 | 0 | 1 | 1 | 0 | 0 | 0 | 0 | 3.6 |
| Nausea/vomiting | ||||||||
| 1-2 | 3 | 3 | 3 | 4 | 4 | 2 | 4 | 41.1 |
| 3-4 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 1.8 |
| Constipation | ||||||||
| 1-2 | 1 | 0 | 2 | 2 | 1 | 1 | 1 | 14.3 |
| 3-4 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Decreased appetite/dyspepsia | ||||||||
| 1-2 | 2 | 3 | 1 | 1 | 5 | 1 | 4 | 30.4 |
| 3-4 | 0 | 0 | 0 | 0 | 2 | 0 | 0 | 3.6 |
| Fatigue | ||||||||
| 1-2 | 4 | 4 | 0 | 3 | 4 | 4 | 2 | 37.5 |
| 3-4 | 0 | 0 | 0 | 0 | 2 | 0 | 0 | 3.6 |
| Insomnia | ||||||||
| 1-2 | 1 | 1 | 1 | 0 | 3 | 0 | 0 | 10.7 |
| 3-4 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Headache | ||||||||
| 1-2 | 1 | 2 | 1 | 4 | 2 | 2 | 2 | 25 |
| 3-4 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Muscle spasms/myalgia | ||||||||
| 1-2 | 2 | 3 | 2 | 3 | 4 | 3 | 4 | 37.5 |
| 3-4 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Other pain | ||||||||
| 1-2 | 8 | 1 | 5 | 4 | 6 | 5 | 6 | 62.5 |
| 3-4 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 1.8 |
| Pyrexia | ||||||||
| 1-2 | 1 | 1 | 2 | 1 | 3 | 2 | 2 | 21.4 |
| 3-4 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 1.8 |
| Rash | ||||||||
| 1-2 | 0 | 1 | 0 | 3 | 3 | 1 | 1 | 16.1 |
| 3-4 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Cough | ||||||||
| 1-2 | 3 | 0 | 3 | 2 | 3 | 3 | 4 | 32.1 |
| 3-4 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Other respiratory | ||||||||
| 1-2 | 1 | 2 | 3 | 5 | 5 | 4 | 8 | 50 |
| 3-4 | 1 | 0 | 1 | 0 | 2 | 0 | 0 | 7.1 |
| Arthralgia | ||||||||
| 1-2 | 1 | 1 | 2 | 0 | 3 | 2 | 0 | 16.1 |
| 3-4 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Edema | ||||||||
| 1-2 | 5 | 2 | 1 | 1 | 4 | 2 | 1 | 28.6 |
| 3-4 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
Abbreviation: CD, continuous dosing.
28/7 days = patients receiving drug for 28 continuous days followed by 7 days off of drug.
Safety Profile and Patient Disposition
The MTD of ibrutinib was not reached. Only two DLTs occurred: one grade 3 allergic hypersensitivity (cohort IV) in a patient with a history of drug hypersensitivity, and one dose interruption for more than 7 days because of transient grade 2 neutropenia (cohort II). Table 2 summarizes the most frequently (≥ 10% of patients) reported AEs by dose cohort. The most common AEs observed in the study were typically grade 1 or 2 in severity; grade 3 or 4 events were infrequent and independent of dose. Grade 3 to 4 hematologic toxicities included neutropenia (12.5%), thrombocytopenia (7.2%), and anemia (7.1%). Additionally, no evidence of cumulative hematologic or nonhematologic toxicity was observed in patients with prolonged dosing. No consistent relationship between dose level and AEs was apparent. At the time of this writing, 20 patients remain on treatment in an extension trial. Thirty-six patients discontinued study treatment: PD (n = 20), patient or physician decision (n = 8), AEs (n = 6), and DLTs (n = 2). The latter AEs included pain (grade 4), cerebrovascular accident (grade 4), small bowel obstruction (grade 3), flare of chronic obstructive pulmonary disease (grade 3), low hemoglobin and thrombocytopenia (grade 2), and hypersensitivity pneumonitis (grade 1).
PKs
Ibrutinib is rapidly absorbed and eliminated after oral administration (Fig 1A). Mean peak plasma concentrations were observed between 1 and 2 hours after dosing, and drug exposure (derived from area under the concentration-time curve [AUC]) increased in a nearly dose-proportional manner (Fig 1B). Plasma concentrations declined biphasically, with an initial mean half-life (determined after time to maximum plasma concentration to 6 hours postdose) of approximately 2 to 3 hours. Mean apparent terminal half-life ranged from 4 to 8 hours. There was no evidence of accumulation of ibrutinib exposure after repeated daily oral dosing.
Fig 1.

Pharmacokinetic and pharmacodynamic studies of ibrutinib. (A) Mean plasma concentration of ibrutinib over time at the 560-mg per day dose and (B) relationship between plasma area under the concentration-time curve (AUC) and dose are shown. Error bars represent SEM; arrow indicates the 560-mg fixed dose, which corresponded to a mean dose of 7.22 mg/kg per day (range, 5.6 to 9.3 mg/kg per day) based on actual patient body weight. (C) Representative probe assay data from peripheral blood mononuclear cell (PBMC) samples collected from patient with chronic lymphocytic leukemia from dose-level cohort IV (8.3 mg/kg per day). The probe (fluorescently tagged derivative of ibrutinib) binds to Bruton tyrosine kinase (BTK; indicated by arrow) with remarkably little specific labeling of other proteins. Total BTK and actin protein levels in each sample are shown to normalize protein in each lane. The BTK-bound fluorescent probe is unable to bind BTK in the samples obtained after drug administration because of occupancy of the binding site by ibrutinib. (D) Normalized intensity of probe labeling is plotted, and the lower limit of detection (LLOD) for this gel is indicated. (E) Comparison of the degree of BTK occupancy by ibrutinib at each dose-level cohort in the study. Dots represent a single BTK occupancy measurement using the probe assay; line indicates median percentage occupancy; asterisks denote P < .001 (analysis of variance). These data suggest that full BTK occupancy was generally achieved in patients at doses ≥ 2.5 mg/kg. (F) Pharmacokinetic and pharmacodynamic relationship of BTK active-site occupancy and ibrutinib exposure. Samples of PBMCs were collected predose and at 4 and 24 hours after administration of ibrutinib for each patient. Data represent the percentage of BTK occupancy before dosing and averaged between 4 and 24 hours postdose for each patient in each group. These values are plotted against the drug exposure (AUC0-24) achieved in the patient after administration of ibrutinib on day 1. Line represents the line of best fit of a simple maximum-effect model to the data. Analysis of pharmacokinetic and pharmacodynamic profiles on day 1 showed that BTK active-site occupancy was saturated or near saturated (> 95%) at AUC values ≥ 160 ng × h/mL.
BTK Occupancy and Pharmacodynamics
Figures 1C and 1D show representative probe assay data from a patient with CLL in dose-level cohort IV (8.3 mg/kg per day). The PBMC sample collected from this patient consisted of 95.7% CD20+ CLL cells, as determined by flow cytometry. At baseline, the probe labeled a robust band corresponding to BTK, and this band decreased significantly within 4 hours of treatment and throughout the entire treatment cycle, suggesting durable and complete inhibition of BTK. Figure 1E shows the dose dependency and durability of BTK occupancy in PBMCs across all patients dosed in the study. BTK occupancy ≥ 95% was achieved 4 hours postdose in all patients in cohort II (2.5 mg/kg per day). Figure 1F plots the average BTK active-site occupancy in PBMCs as a function of ibrutinib exposure (day 1 AUC). Complete or nearly complete BTK occupancy was observed in patients with AUCs exceeding 160 ng × h/mL. No decrease was observed in the level of serum Ig in patients treated over 12 cycles (Appendix Fig A1, online only). A decrease in anti–IgE-stimulated basophil degranulation was observed, consistent with the role of BTK downstream of the high-affinity IgE receptor (Appendix Fig A2, online only).
Tumor Response
Of 50 patients evaluable for tumor response, 60% achieved an objective response (CR or PR), with an overall response rate of 54% in the ITT population. Table 3 indicates the best response according to dose and schedule. Nine of 56 patients had PD at first assessment, of whom three were treated in cohort I, a dose that did not lead to full BTK occupancy. Additionally, responses were observed across all histologies, including seven of nine patients with mantle-cell lymphoma (three CRs), 11 of 16 patients with CLL/small lymphocytic lymphoma (two CRs), six of 16 patients with follicular lymphoma (three CRs), two of seven patients with DLBCL, three of four patients with WM, and one of four patients with marginal zone lymphoma (Appendix Table A1, online only). Of 11 patients with CLL, all experienced rapid reductions in lymphadenopathy during the first cycle of treatment accompanied by an increase in absolute lymphocyte count, suggesting an egress of malignant cells from lymph nodes into peripheral blood. Only one of these patients with CLL did not have an eventual reduction in lymphocytosis sufficient to be categorized as PR. Responses were durable, with median progression-free survival of 13.6 months at the time of data cutoff. Twenty patients remain on study at the time of data cutoff because of continued clinical benefit (Fig 2). Change in aggregate tumor size (data available for 48 patients) and time on study (for all patients) are shown in Figures 2A and 2B, respectively.
Table 3.
Best Clinical Response by Dose-Level Cohort (N = 56)
| Cohort | Dose | No. of Patients | CR | PR |
|---|---|---|---|---|
| I | 1.25 mg/kg/d | 7 | 0 | 2 |
| II | 2.5 mg/kg/d | 9 | 1 | 3 |
| III | 5.0 mg/kg/d | 6 | 2 | 1 |
| IV | 8.3 mg/kg/d | 8 | 3 | 1 |
| CD-I | 8.3 mg/kg/d | 10 | 1 | 5 |
| V | 12.5 mg/kg/d | 7 | 0 | 4 |
| CD-II | 560 mg/d | 9 | 1 | 6 |
| Total | 56* | |||
| No. | 8 | 22 | ||
| % | 14 | 39 |
Abbreviations: CD, continuous dosing; CR, complete response; PR, partial response.
Six of 56 patients were not evaluable. This includes three patients who withdrew consent after 7, 15, and 35 days in the study, respectively; one patient with a serious adverse event of failure to thrive after 14 days in the study; and two patients with dose-limiting toxicities.
Fig 2.
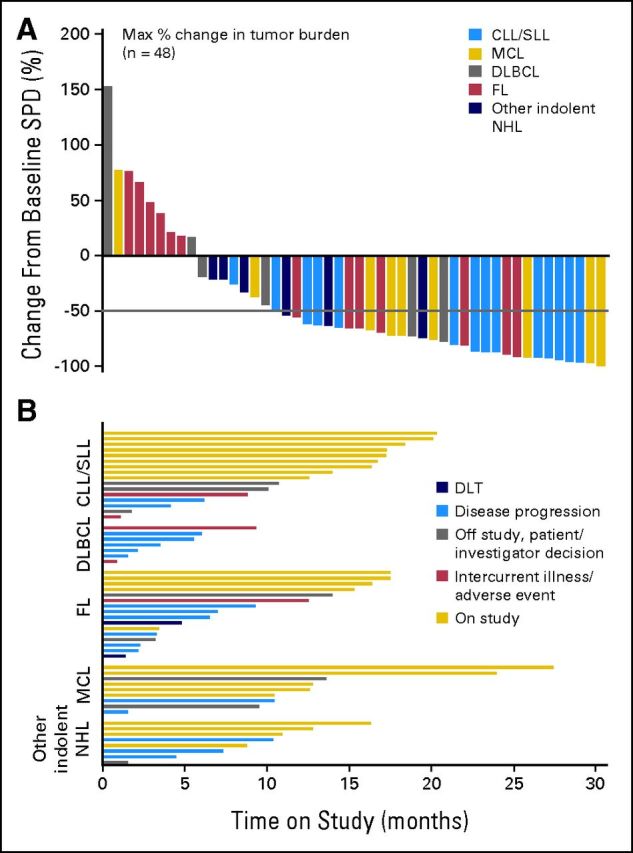
Antitumor response in all evaluable patients. (A) Best responses in the 48 patients evaluated by computed tomography scan for change from baseline in the sum of the largest diameter of each target lesion; negative values indicate tumor shrinkage. (B) Time on study for all 56 patients grouped by histology; bars describe the reason for patient discontinuation. CLL, chronic lymphocytic leukemia; DLBCL, diffuse large B-cell lymphoma; DLT, dose-limiting toxicity; FL, follicular lymphoma; NHL, non-Hodgkin lymphoma; MCL, mantle-cell lymphoma; SLL, small lymphocytic lymphoma; SPD, sum of product of greatest diameters.
DISCUSSION
This study demonstrates that orally administered ibrutinib is well tolerated and induces significant objective responses in patients with B-cell NHL or CLL who have relapsed or were refractory to standard therapy. A majority of AEs were mild (grade 1 or 2) in severity and easily managed or reversible. No significant myelosuppressions or renal or hepatic toxicities were observed. Additionally, no cumulative toxicity was noted with treatment duration over 6 months.
Thirty patients achieved objective responses, with responses seen at each dose level and across all histologies. Responses were durable, with a majority maintaining their responses for at least 10 months; several patients had remained on study for more than 20 months at the time of data cutoff. The highest response rate was observed in patients with mantle-cell lymphoma (78%) and CLL/small lymphocytic lymphoma (ITT, 69%; evaluable, 79%). The response rate in mantle-cell lymphoma is particularly noteworthy, because the outcome of patients with relapsed disease has been modest (32%) with bortezomib, currently the only approved agent.21 In patients with CLL, a characteristic pattern of response included a transient increase in lymphocyte count. This pattern has been reported with other agents affecting BCR signaling22–23 and reflects disruption of BCR-mediated stromal chemotaxis and adhesion, resulting in an egress of tissue-resident malignant lymphocytes.4,24–26
B-cell malignancies are disorders that depend on an intact BCR signaling cascade, of which BTK is a critical mediator.5,13,27,28 BCR signaling, either constitutively or following antigen binding, leads to the activation of several downstream pathways that connect the BCR to proteins involved in cell survival, proliferation, and migration.29 Most B-cell malignancies continue to express a surface BCR, and it has been hypothesized that functional BCR signaling is essential for the survival and proliferation of the malignant cell.5 BTK has been shown to be an essential component of chronic active BCR signaling, which is a key survival pathway in the activated B-cell subtype of DLBCL, because genetic knockdown of BTK results in cell death, presumably resulting from interruption of survival signals from the BCR to nuclear factor-κB.17 In CLL, the BCR is activated, with evidence of ongoing interaction with antigen in vivo.4 Direct evidence of the importance of BCR signaling in CLL comes from gene expression profiling data, where BCR signaling has been shown to be the most prominent activated pathway in CLL cells isolated from lymph nodes.30 The efficacy observed in our study lends further validity to the idea that BCR signaling is critical to the growth of B-cell tumors and suggests that BTK inhibition alone is sufficient to inhibit this growth signal.
The promising safety profile of ibrutinib is likely related to the restricted expression of BTK in hematopoietic cells. Clinical features seen in patients with XLA are primarily restricted to B cells, suggesting that BTK is particularly critical to B-cell development and function. Indeed, ibrutinib has previously been shown to inhibit antigen receptor signaling in B cells but not in T cells.16 Interestingly, in contrast to the complete absence of circulating Igs observed in patients with XLA, ibrutinib treatment did not reduce serum Ig levels, even in patients treated for 12 cycles (Appendix Fig A1, online only). This suggests that daily BTK kinase inhibition is not sufficient to replicate XLA, perhaps because the early developmental role of BTK at the pro–pre–B-cell stage is different from its signaling role in mature B cells. The safety profile of ibrutinib is likely further enhanced by the once-daily administration and rapid absorption and elimination of the compound. Such intermittent exposure limits the duration of off-target effects and is made feasible by the irreversible covalent binding of the compound to BTK. Thus, BTK remained fully occupied by ibrutinib for at least 24 hours in patients in cohorts II to V, despite its rapid clearance from plasma.
A unique aspect of this study is the use of a highly specific fluorescent affinity probe for BTK measuring active-site occupancy. The assay allows for dose escalation based not only on traditional DLT measures but also on an optimal biologic dose, where target binding could be demonstrated. To better define a margin of safety, dose escalation proceeded to three levels above the level where full BTK occupancy was observed. Probe binding has been shown to tightly correlate with the blocking of BCR signaling and in vivo efficacy.16 BTK occupancy > 95% was observed in dose level cohorts II to V (2.5 to 12.5 mg/kg per day), and each of these cohorts had similar response rates, consistent with efficacy deriving from BTK inhibition. Both the intermittent and continuous dosing schedules had similar BTK occupancy and equivalent PKs and toxicity profiles. A fixed continuous dose of 560 mg was well tolerated and also led to full-target occupancy in a range of individual body weights. Interestingly, in patients with CLL, the intermittent dosing schedule was associated with transient reversal of treatment-related lymphocytosis during the 7-days-off drug period, suggesting a reversal of the biologic effect (Appendix Fig A3, online only). On this basis, and given the tolerability of continuous dosing, the latter was selected for phase II studies.
In conclusion, the high level of objective responses observed with ibrutinib in patients with refractory B-cell lymphoma and CLL supports the hypothesis that BTK is a crucial mediator of growth and survival in B-cell malignancies. The data strongly support continued clinical evaluation in various B-cell malignancies.
Appendix
Fig A1.

Serum immunoglobulin (Ig) levels of patients treated with ibrutinib over 12 cycles. Total (A) IgG and (B) IgM levels from cycles one (predose; n = 54), three (n = 18), six (n = 28), nine (n = 10), and 12 (n = 7) were determined for patients and converted to percentage by using the predose level as 100%; shown as mean plus SEM in each bar graph.
Fig A2.

Pharmacodynamic activity of ibrutinib-treated patients in the various cohorts as determined by percentage of CD63+ basophils on α–immunoglobulin E stimulation of whole blood collected at 4 (day 1, day 8) and 24 hours (day 1, day 8, day 15, day 29) after drug administration compared with predose baseline (set at 100%). Patients with < 10% CD63+ expression at baseline after stimulation were excluded.
Fig A3.

Pattern of lymphocytosis seen in patients with chronic lymphocytic leukemia (CLL)/small lymphocytic lymphoma (SLL). Relationship between absolute lymphocyte count (ALC) and the sum of product of greatest diameters (SPD) in patients with CLL or SLL (n = 16). Median percentage change from baseline is shown. Sawtooth pattern suggests a transient reversal of treatment-related lymphocytosis during the 7-days-off-drug period.
Table A1.
Best Response According to Histology
| Histology | No. of Patients | CR | PR | SD | PD | NE | ORR (%) |
|
|---|---|---|---|---|---|---|---|---|
| ITT (N = 56) | Evaluable (n = 50) | |||||||
| CLL/SLL | 16 | 2 | 9 | 3* | 2 | 69 | 79 | |
| MCL | 9 | 3 | 4 | 1 | 1 | 78 | 78 | |
| WM | 4 | 3 | 1 | 75 | 75 | |||
| FL | 16 | 3 | 3 | 4 | 3 | 3 | 38 | 46 |
| MZL/MALT | 4 | 1 | 1 | 1 | 1 | 25 | 33 | |
| DLBCL | 7 | 2 | 1 | 4 | 29 | 29 | ||
| Total | 56 | 8 | 22 | 11 | 9 | 6 | 54 | 60 |
Abbreviations: CLL, chronic lymphocytic leukemia; CR, complete response; DLBCL, diffuse large B-cell lymphoma; FL, follicular lymphoma; ITT, intent to treat; MCL, mantle-cell lymphoma; MZL/MALT, marginal zone/mucosal-associated lymphoid tissue lymphoma; NE, not evaluable; ORR, overall response rate; PD, disease progression; PR, partial response; SD, stable disease; SLL, small lymphocytic lymphoma; WM, Waldenström macroglobulinemia.
One patient with CLL had nodal response with lymphocytosis.
Footnotes
Supported by Pharmacyclics.
Authors' disclosures of potential conflicts of interest and author contributions are found at the end of this article.
Clinical trial information: NCT00849654.
See accompanying article on page 128
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Although all authors completed the disclosure declaration, the following author(s) and/or an author's immediate family member(s) indicated a financial or other interest that is relevant to the subject matter under consideration in this article. Certain relationships marked with a “U” arethose for which no compensation was received; those relationships marked with a “C” were compensated. For a detailed description of the disclosure categories, or for more information about ASCO's conflict of interest policy, please refer to the Author Disclosure Declaration and the Disclosures of Potential Conflicts of Interest section in Information for Contributors.
Employment or Leadership Position: Joseph J. Buggy, Pharmacyclics (C); Betty Y. Chang, Pharmacyclics (C); Juthamas Sukbuntherng, Pharmacyclics (C); Ahmed Hamdy, Pharmacyclics (C); Eric Hedrick, Pharmacyclics (C) Consultant or Advisory Role: Ranjana H.Advani, Pharmacyclics (C); Jeff P. Sharman, Gilead (C), Celgene (C), Seattle Genetics (C); Richard R. Furman, Pharmacyclics (C); Nathan H. Fowler, Pharmacyclics (C) Stock Ownership: Joseph J. Buggy, Pharmacyclics; Sara Rodriguez, Pharamacyclics; Betty Y. Chang, Pharmacyclics; Juthamas Sukbuntherng, Pharmacyclics; Ahmed Hamdy, Pharamcyclics; Eric Hedrick, Pharmacyclics Honoraria: None Research Funding: Ranjana H. Advani, Pharmacyclics, Abbott Laboratories, Seattle Genetics, GlaxoSmithKline, Genentech; Jeff P. Sharman, Genentech, Seattle Genetics, Celgene, Gilead, Pharmacyclics; Barbara Grant, Pharmacyclics, Millennium Pharmaceuticals, Novartis; Nathan H. Fowler, Pharmacyclics, Gilead, Cephalon, Genentech, Millennium Pharmaceuticals, Celgene Expert Testimony: None Other Remuneration: None
AUTHOR CONTRIBUTIONS
Conception and design: Ranjana H. Advani, Joseph J. Buggy, Jeff P. Sharman, Richard R. Furman, Raquel Izumi, Ahmed Hamdy, Nathan H. Fowler
Provision of study materials or patients: Ranjana H. Advani, Sonali M. Smith, Thomas E. Boyd, Barbara Grant, Kathryn S. Kolibaba, Nathan H. Fowler
Collection and assembly of data: Ranjana H. Advani, Joseph J. Buggy, Sonali M. Smith, Thomas E. Boyd, Barbara Grant, Kathryn S. Kolibaba, Richard R. Furman, Sara Rodriguez, Betty Y. Chang, Juthamas Sukbuntherng, Raquel Izumi, Ahmed Hamdy, Nathan H. Fowler
Data analysis and interpretation: Ranjana H. Advani, Joseph J. Buggy, Sonali M. Smith, Richard R. Furman, Sara Rodriguez, Betty Y. Chang, Juthamas Sukbuntherng, Raquel Izumi, Ahmed Hamdy, Eric Hedrick, Nathan H. Fowler
Manuscript writing: All authors
Final approval of manuscript: All authors
REFERENCES
- 1.Niiro H, Clark EA. Regulation of B-cell fate by antigen-receptor signals. Nat Rev Immunol. 2002;2:945–956. doi: 10.1038/nri955. [DOI] [PubMed] [Google Scholar]
- 2.Fuentes-Pananá EM, Bannish G, Monroe JG. Basal B-cell receptor signaling in B lymphocytes: Mechanisms of regulation and role in positive selection, differentiation, and peripheral survival. Immunol Rev. 2004;197:26–40. doi: 10.1111/j.0105-2896.2004.0105.x. [DOI] [PubMed] [Google Scholar]
- 3.Chiorazzi N, Ferrarini M. B cell chronic lymphocytic leukemia: Lessons learned from studies of the B cell antigen receptor. Annu Rev Immunol. 2003;21:841–894. doi: 10.1146/annurev.immunol.21.120601.141018. [DOI] [PubMed] [Google Scholar]
- 4.Stevenson FK, Caligaris-Cappio F. Chronic lymphocytic leukemia: Revelations from the B-cell receptor. Blood. 2004;103:4389–4395. doi: 10.1182/blood-2003-12-4312. [DOI] [PubMed] [Google Scholar]
- 5.Küppers R. Mechanisms of B-cell lymphoma pathogenesis. Nat Rev Cancer. 2005;5:251–262. doi: 10.1038/nrc1589. [DOI] [PubMed] [Google Scholar]
- 6.Gururajan M, Jennings CD, Bondada S. Cutting edge: Constitutive B cell receptor signaling is critical for basal growth of B lymphoma. J Immunol. 2006;176:5715–5719. doi: 10.4049/jimmunol.176.10.5715. [DOI] [PubMed] [Google Scholar]
- 7.Irish JM, Czerwinski DK, Nolan GP, et al. Altered B-cell receptor signaling kinetics distinguish human follicular lymphoma B cells from tumor-infiltrating nonmalignant B cells. Blood. 2006;108:3135–3142. doi: 10.1182/blood-2006-02-003921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pleyer L, Egle A, Hartmann TN, et al. Molecular and cellular mechanisms of CLL: Novel therapeutic approaches. Nat Rev Clin Oncol. 2009;6:405–418. doi: 10.1038/nrclinonc.2009.72. [DOI] [PubMed] [Google Scholar]
- 9.Quiroga MP, Balakrishnan K, Kurtova AV, et al. B-cell antigen receptor signaling enhances chronic lymphocytic leukemia cell migration and survival: Specific targeting with a novel spleen tyrosine kinase inhibitor, R406. Blood. 2009;14:1029–1037. doi: 10.1182/blood-2009-03-212837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lenz G, Staudt LM. Aggressive lymphomas. N Engl J Med. 2010;362:1417–1429. doi: 10.1056/NEJMra0807082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cheng G, Ye ZS, Baltimore D. Binding of Bruton's tyrosine kinase to Fyn, Lyn, or Hck through a Src homology 3 domain-mediated interaction. Proc Natl Acad Sci U S A. 1994;91:8152–8155. doi: 10.1073/pnas.91.17.8152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Afar DE, Park H, Howell BW, et al. Regulation of Btk by Src family tyrosine kinases. Mol Cell Biol. 1996;16:3465–3471. doi: 10.1128/mcb.16.7.3465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Khan WN. Regulation of B lymphocyte development and activation by Bruton's tyrosine kinase. Immunol Res. 2001;23:147–156. doi: 10.1385/IR:23:2-3:147. [DOI] [PubMed] [Google Scholar]
- 14.Humphries LA, Dangelmaier C, Sommer K, et al. Tec kinases mediate sustained calcium influx via site-specific tyrosine phosphorylation of the phospholipase C gamma Src homology 2-Src homology 3 linker. J Biol Chem. 2004;279:37651–37661. doi: 10.1074/jbc.M311985200. [DOI] [PubMed] [Google Scholar]
- 15.Conley ME, Dobbs AK, Farmer DM, et al. Primary B cell immunodeficiencies: Comparisons and contrasts. Annu Rev Immunol. 2009;27:199–227. doi: 10.1146/annurev.immunol.021908.132649. [DOI] [PubMed] [Google Scholar]
- 16.Honigberg LA, Smith AM, Sirisawad M, et al. The Bruton tyrosine kinase inhibitor PCI-32765 blocks B-cell activation and is efficacious in models of autoimmune disease and B-cell malignancy. Proc Natl Acad Sci U S A. 2010;107:13075–13080. doi: 10.1073/pnas.1004594107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Davis RE, Ngo VN, Lenz G, et al. Chronic active B-cell-receptor signalling in diffuse large B-cell lymphoma. Nature. 2010;463:88–92. doi: 10.1038/nature08638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cheson BD, Pfistner B, Juweid ME, et al. Revised response criteria for malignant lymphoma. J Clin Oncol. 2007;25:579–586. doi: 10.1200/JCO.2006.09.2403. [DOI] [PubMed] [Google Scholar]
- 19.Hallek M, Cheson BD, Catovsky D, et al. Guidelines for the diagnosis and treatment of chronic lymphocytic leukemia: A report from the International Workshop on Chronic Lymphocytic Leukemia updating the National Cancer Institute–Working Group 1996 guidelines. Blood. 2008;111:5446–5456. doi: 10.1182/blood-2007-06-093906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kaplan EL, Meier P. Nonparametric estimation from incomplete observations. J Am Stat Assoc. 1958;53:457–481. [Google Scholar]
- 21.Goy A, Bernstein SH, Kahl BS, et al. Bortezomib in patients with relapsed or refractory mantle cell lymphoma: Updated time-to-event analyses of the multicenter phase 2 PINNACLE study. Ann Oncol. 2009;20:520–525. doi: 10.1093/annonc/mdn656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Furman RR, Byrd JC, Flinn IW, et al. Interim results from a phase I study of CAL-101, a selective oral inhibitor of phosphatidylinositol 3-kinase p110d isoform, in patients with relapsed or refractory hematologic malignancies. J Clin Oncol. 2010;28:241s. suppl 15; abstr 3032. [Google Scholar]
- 23.Friedberg JW, Sharman J, Sweetenham J, et al. Inhibition of Syk with fostamatinib disodium has significant clinical activity in non-Hodgkin lymphoma and chronic lymphocytic leukemia. Blood. 2010;115:2578–2585. doi: 10.1182/blood-2009-08-236471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ponader S, Chen S, Buggy JJ, et al. The Bruton tyrosine kinase inhibitor PCI-32765 thwarts chronic lymphocytic leukemia cell survival and tissue homing in vitro and in vivo. Blood. 2012;119:1182–1189. doi: 10.1182/blood-2011-10-386417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.De Rooij MF, Kuil A, Geest CR, et al. The clinically active BTK inhibitor PCI-32765 targets B-cell receptor- and chemokine-controlled adhesion and migration in chronic lymphocytic leukemia. Blood. 2012;119:2590–2594. doi: 10.1182/blood-2011-11-390989. [DOI] [PubMed] [Google Scholar]
- 26.Chang BY, Francesco M, Magadala P, et al. Egress of CD19+CD5+ cells into peripheral blood following treatment with the Bruton tyrosine kinase inhibitor, PCI-32765, in mantle cell lymphoma patients. Blood. 2011:118. doi: 10.1182/blood-2013-02-482125. abstr 954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Anderson JS, Teutsch M, Dong Z, et al. An essential role for Bruton's tyrosine kinase in the regulation of B-cell apoptosis. Proc Natl Acad Sci U S A. 1996;93:10966–10971. doi: 10.1073/pnas.93.20.10966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rui L, Schmitz R, Ceribelli M, et al. Malignant pirates of the immune system. Nat Immunol. 2011;12:933–940. doi: 10.1038/ni.2094. [DOI] [PubMed] [Google Scholar]
- 29.Kurosaki T, Shinohara H, Baba Y. B cell signaling and fate decision. Annu Rev Immunol. 2009;28:21–55. doi: 10.1146/annurev.immunol.021908.132541. [DOI] [PubMed] [Google Scholar]
- 30.Herishanu Y, Pérez-Galán P, Liu D, et al. The lymph node microenvironment promotes B-cell receptor signaling, NF-kappaB activation, and tumor proliferation in chronic lymphocytic leukemia. Blood. 2011;117:563–574. doi: 10.1182/blood-2010-05-284984. [DOI] [PMC free article] [PubMed] [Google Scholar]