

Abstract
Various epigenetic marks at the HIV-1 5′LTR suppress proviral expression and promote latency. Cellular antisense transcripts known as long noncoding RNAs (lncRNAs) recruit the polycomb repressor complex 2 (PRC2) to gene promoters, which catalyzes trimethylation of lysine 27 on histone H3 (H3K27me3), thus promoting nucleosome assembly and suppressing gene expression. We found that an HIV-1 antisense transcript expressed from the 3′LTR and encoding the antisense protein ASP promotes proviral latency. Expression of ASP RNA reduced HIV-1 replication in Jurkat cells. Moreover, ASP RNA expression promoted the establishment and maintenance of HIV-1 latency in Jurkat E4 cells. We show that this transcript interacts with and recruits PRC2 to the HIV-1 5′LTR, increasing accumulation of the suppressive epigenetic mark H3K27me3, while reducing RNA Polymerase II and thus proviral transcription. Altogether, our results suggest that the HIV-1 ASP transcript promotes epigenetic silencing of the HIV-1 5′LTR and proviral latency through the PRC2 pathway.
Keywords: HIV-1, Antisense transcript, ASP, Viral latency, PRC2, H3K27me3, Epigenetic marks
1. Introduction
A unique feature of HIV-1 transcription is the viral transactivator Tat, which allows the virus to regulate its own expression via a positive feedback loop (Karn, 2011). Tat binds the TAR element (Kao et al., 1987) — a stem-loop structure at the 5′ of the nascent viral transcripts — and recruits the elongation factor P-TEFb, comprised of cyclin T1 and CDK9 (Wei et al., 1998). In turn, P-TEFb promotes the removal of the negative elongation factors NELF and DSIF (Fujinaga et al., 2004; Yamaguchi et al., 2002; Jadlowsky et al., 2014; Zhang et al., 2007), and augments processivity of the RNA polymerase II (RNA Pol II) (Czudnochowski et al., 2012). The net outcome is the accumulation of full-length transcripts initiated at the LTR, and the expression of more Tat and other viral proteins. Changes in the cellular environment cause a reduction in the intracellular Tat levels, which progressively reduce the activity of the LTR and viral expression.
A decline in Tat levels is a necessary but not sufficient condition for the establishment and maintenance of viral latency. Indeed, the latent HIV-1 5′LTR is characterized by the presence of specific epigenetic marks, which are not regulated by Tat, but rather are introduced sequentially by cellular enzymes. In vitro studies have shown that CpG islands are heavily methylated within the LTR of latent proviruses, and that inhibitors of DNA methylation potentiate viral reactivation (Blazkova et al., 2009; Kauder et al., 2009). However, it was recently shown that only a fraction of CpG islands are methylated in the LTR of proviruses from infected individuals under suppressive antiretroviral therapy (Blazkova et al., 2012; Palacios et al., 2012; Ho et al., 2013a, 2013b). Two nucleosomes — Nuc-0 and Nuc-1 — are invariably present at the 5′LTR of latent proviruses (Verdin et al., 1993). Histone deacetylases (HDAC) and lysine methyltransferases (HKMT) regulate their assembly and positioning. Members of class I HDACs catalyze deacetylation of lysines in the N-terminal part of core histones (H2A, H2B, H3 and H4) (Keedy et al., 2009). Indeed, HDAC inhibitors such as vorinostat, panobinostat and romidepsin are being tested as therapeutic targets in the context of “kick and kill” cure strategies (Archin et al., 2012; Elliott et al., 2014; Rasmussen, 2014; Sogaard et al., 2015). The histone methyltransferases G9a and Suv39H1 catalyze di- and trimethylation of lysine 9 on histone H3 (H3K9me2 and H3K9me3), respectively, whereas enhancer of zeste homolog 2 (EZH2) is responsible for trimethylation of lysine 27 on histone H3 (H3K27me3) (du Chene et al., 2007; Imai et al., 2010; Marban et al., 2007). EZH2 is the dominant HKMT involved in the assembly of Nuc-0 and Nuc-1, and in turning off transcription from the HIV-1 LTR (Friedman et al., 2011).
EZH2, SUZ12, and EED are core components of the multi-protein complex PRC2, which in higher eukaryotes lacks independent DNA binding activity (Margueron and Reinberg, 2011). In recent years, natural antisense transcripts (NATs) have been shown to suppress the expression of their sense gene through various mechanisms (Khorkova et al., 2014). A group of NATs known as long noncoding RNAs (lncRNA) are present at a very low number of copies per cell, and exert their function by recruiting PRC2 to the promoter of their sense gene, leading to nucleosome assembly, chromatization and silencing (Davidovich and Cech, 2015).
The proviral genomes of human and animal retroviruses direct the expression of antisense transcripts that originate from a promoter within the 3′LTR (Barbeau and Mesnard, 2015). The most studied retroviral antisense transcript is the human T cell leukemia virus 1 (HTLV-1) basic leucine zipper factor (Hbz), which was discovered in 2002 (Gaudray et al., 2002), although minus strand transcription in the HTLV-1 genome had been known for several years (Larocca et al., 1989). The hbz gene expresses two transcription isoforms: a spliced and an unspliced RNA (Gaudray et al., 2002), which differ in their 5′ untranslated regions, and in the 5′ region of the open reading frame that they contain, thus giving rise to two polypeptides that differ slightly in the N-terminus. Both the RNA and the protein expressed by this gene play a role in the virus lifecycle (Ma et al., 2016). In addition, to their protein-coding role, the Hbz RNAs have regulatory roles by promoting cell proliferation and by inhibiting apoptosis, which is supported also by their mostly nuclear localization. The HBZ proteins are also nuclear, and promote persistent HTLV-1 latent infection, T cell proliferation, and inhibit apoptosis and authophagy. Antisense genes have also been identified in the genomes of HTLV-2, 3 and 4 (Barbeau and Mesnard, 2011), and feline immunodeficiency virus (FIV) (Briquet et al., 2001).
The existence of an antisense gene in the HIV-1 genome was first proposed in 1988 as a result of a computational analysis of the proviral sequence (Miller, 1988). Subsequent studies showed that the HIV-1 antisense gene might give rise to several transcripts of different length (Michael et al., 1994; Kobayashi-Ishihara et al., 2012). Several studies have assessed the ratio of sense vs. antisense transcription within the HIV-1 proviral genome (Michael et al., 1994; Kobayashi-Ishihara et al., 2012; Vanhee-Brossollet et al., 1995; Ludwig et al., 2006; Laverdure et al., 2012). In particular, studies by Kobayashi-Ishihara et al. and by Laverdure et al. found that antisense transcripts are 100–2500 times and 1000 times, respectively, less abundant than sense transcripts in lymphoid and myeloid cells infected in vitro (Kobayashi-Ishihara et al., 2012; Laverdure et al., 2012). However, Michael et al. assessed antisense transcription in peripheral blood mononuclear cells of early-stage asymptomatic patients, and showed that it represents as low as 1.3% and as high as 94% (mean 33%) of total HIV-1 proviral transcription (Michael et al., 1994). The HIV-1 antisense transcript contains an open reading frame encoding a putative protein of 189 amino acid residues (Miller, 1988). A few studies have suggested that the product of the HIV-1 antisense transcript is associated with cellular membranes, in line with its predicted hydrophobicity (Laverdure et al., 2012; Briquet and Vaquero, 2002; Clerc et al., 2011; Torresilla et al., 2013). However, the function of this protein remains to be elucidated. Other studies have led to suggest that the HIV-1 transcript encoding the antisense protein (ASP) might play a role in viral replication (Kobayashi-Ishihara et al., 2012; Tagieva and Vaquero, 1997). A possible role of the ASP RNA in its epigenetic silencing was recently inferred by Saayman et al., who showed that this transcript down-regulation led to a decreased recruitment of DMNT3a, EZH2 and HDAC1 to the HIV-1 5′LTR (Saayman et al., 2014). However, the exact mechanism of the ASP RNA acting as a virus-encoded lncRNA remains to be characterized.
Here we show that the HIV-1 ASP transcript inhibits viral replication and promotes the establishment and maintenance of latency by acting as a bona fide lncRNA, through recruitment of PRC2 to the 5′LTR, trimethylation of H3K27, Nuc-1 assembly, and transcriptional silencing. Thus, our data suggest that HIV-1 not only regulates its expression via the Tat positive feedback loop, but also its latency via the ASP transcript.
2. Results
2.1. Design of a highly sensitive strand-specific RT-PCR assay for quantitative measurement of ASP RNA expression
Endogenous RT priming refers to the priming of RNA during reverse transcription by small RNA or DNA fragments that contaminate RNA preparations (Landry et al., 2007). The occurrence of endogenous RT priming is revealed when a PCR signal is obtained with cDNA samples generated without exogenous RT primer. In the case of overlapping transcriptional units encoded on opposite strands of the same genomic region, endogenous RT priming prevents accurate measurement of the expression levels of either transcript, particularly when measuring the expression of a transcript expressed at much lower levels than the other.
Our initial attempts to develop an RT-PCR assay to assess expression of the HIV-1 ASP transcript showed the occurrence of endogenous RT priming, which prevented us from accurately measure the levels of this RNA molecule. To circumvent this issue, we developed an RT-PCR assay that specifically detects the ASP transcript (Fig. 1A). This strand-specific assay makes use of an RT primer that in its 3′ end is complementary to the sequence of the ASP transcript, while its 5′ end contains an unrelated sequence (Kobayashi-Ishihara et al., 2012; Landry et al., 2007). Therefore, the cDNA generated in the RT step are tagged in their 5′ with a non-HIV specific sequence. The tag sequence serves as template for forward primer of the PCR step, while the reverse primer and TaqMan probe are designed on the natural sequence of the ASP RNA molecule. Our strand-specific RT-PCR assay yields a signal only with cDNA samples generated with both tagged RT primer and reverse transcriptase (Fig. 1B), and not with cDNA samples lacking either or both, thus eliminating the possibility of endogenous priming. To generate a standard curve for quantitative analysis of ASP RNA expression, we utilized a linearized plasmid carrying the sequence of the TaqMan amplicon derived from the ASP RNA molecule. Our TaqMan assay allows the detection of 1–10 copies of ASP RNA per reaction (Fig. 1C). Thus, we have developed a highly sensitive, strand-specific RT-PCR assay for quantitative measurement of ASP RNA expression.
Fig. 1.
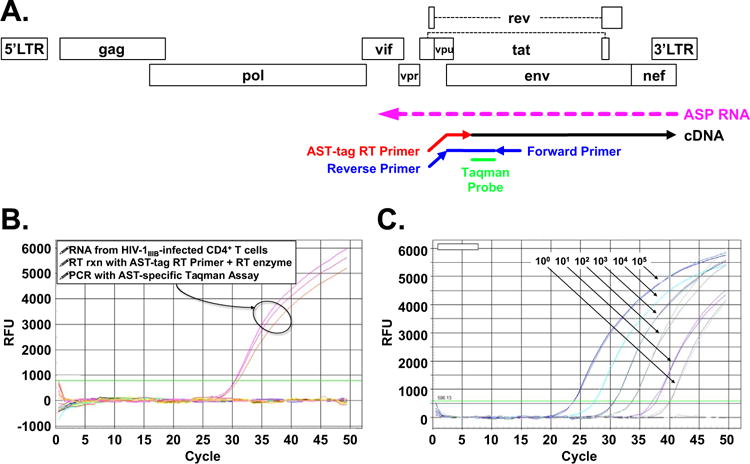
Development of a highly sensitive, strand-specific quantitative RT-PCR assay to detect expression of ASP RNA. Panel A: Scheme of RT-PCR assay for specific detection of the ASP transcript but not HIV-1 sense transcripts. The RT primer (AST-tag) introduces a tag at the 5′ end of the cDNA, which serves as a template for the reverse primer in the subsequence TaqMan PCR step. Panel B: Real time PCR traces show that amplification was obtained only with RNA from infected cells, and only when the RT reaction is carried out in the presence of the AST-tag RT primer and RT enzyme (cDNA was assayed in triplicate PCR reactions). Panel C: PCR traces obtained with 10°–105 copies of a linearized of pUC57-based plasmid carrying the ASP RNA amplicon (all dilutions run in triplicate samples).
2.2. Detection of the HIV-1 ASP RNA in resting CD4+ T cells from HIV-1 infected individuals under suppressive combination antiretroviral therapy
We utilized our RT-PCR assay to carry out a quantitative measurement of ASP RNA expression in various cell systems. We found that expression of the ASP transcript is readily detectable in primary cells infected in vitro (Fig. 2A), but only when cDNA synthesis was carried out in the presence of both AST-tag RT primer and reverse transcriptase. Since ~0.5–1.0% of the cells were infected at the time of RNA collection (3 days post-infection), we estimate that the ASP RNA is expressed at ~1.5–3 copies/infected cell. This is consistent with the observed copy numbers of many functional cellular lncRNA (Palazzo and Lee, 2015; Lee, 2009; Han et al., 2007). The lack of any PCR signal when reverse transcription was carried out in the presence of reverse transcriptase but in the absence of the AST-tag RT primer demonstrates that our assay does not pick up non-specific amplification. Moreover, we detected ASP RNA expression in all chronically infected CD4+ T cell lines tested, but not their uninfected parental cell line (Fig. 2B). The expression levels of ASP RNA varied greatly among these infected cell lines: ~5–25 copies per cell. Different integration sites (and thus local chromatin structure), and baseline levels of key transcription factors may partially account for these differences. These results are in line with previous studies that reported the expression of ASP RNA in chronically infected cell lines and in peripheral blood mononuclear cells acutely infected in vitro (Kobayashi-Ishihara et al., 2012; Vanhee-Brossollet et al., 1995; Tagieva and Vaquero, 1997).
Fig. 2.
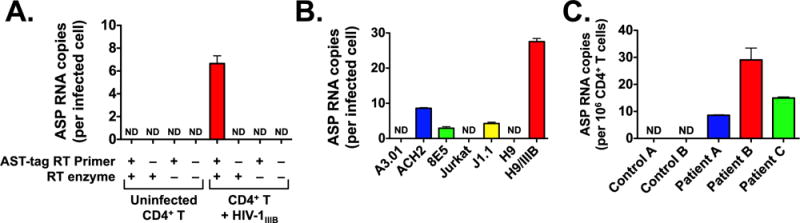
ASP RNA levels in various HIV-1 infected CD4+ T cell systems. Expression levels of the ASP transcript were assessed using the strand-specific, quantitative RT-PCR assay described in Fig. 1. Panel A: Quantification of ASP RNA expression in primary human CD4+ T cells infected in vitro. Total RNA from CD4+ T cells infected with HIV-1IIIB (or uninfected cells) was reverse transcribed with or without AST-tag RT primer, and with or without RT enzyme, and then tested by PCR in triplicate samples. Panel B: Quantification of ASP RNA expression in various chronically infected CD4+ T cell lines (ACH2, 8E5, J1.1, and H9/IIIB) and their uninfected parental cell line (A3.01, Jurkat, and H9). RT reaction was carried with or without AST-tag RT primer, and with or without RT enzyme. Only the +/+ combination is shown. Panel C: Quantification of ASP RNA expression in resting CD4+ T cells from two uninfected donors (Controls A and B) and three infected patients under suppressive antiretroviral therapy for > 24 months (Patients A, B and C). RT reaction was carried with or without AST-tag RT primer, and with or without RT enzyme. Only the +/+ combination is shown. Graphs report average and standard deviation calculated from three replicate samples. ND: not detected.
Next, we sought to determine whether ASP RNA is expressed in vivo. Since the expression of this transcript is independent of — and possibly inhibited by — Tat, we chose to measure expression of ASP RNA in freshly isolated cells without any prior ex vivo stimulation. Thus, we extracted total RNA from freshly isolated, resting CD4+ T cells of three HIV-1 positive individuals under suppressive combination antiretroviral therapy (cART) for at least 24 months. Using our strand-specific RT-qPCR assay, we were able to detect expression of ASP RNA in RNA samples from all three infected individuals, but not in samples from uninfected control individuals (Fig. 2C). Since the frequency of infected cells in these donors is not known, and since a significant proportion of proviral sequences carry large internal deletions (Ho et al., 2013a, 2013b), we cannot estimate the copy number of ASP RNA per cell. These data confirm previous evidence that ASP RNA is expressed in vivo (Michael et al., 1994). However, since the donors included in our study had undetectable viremia for more than 24 months, and since we utilized RNA from resting cells that do not support viral replication, our data demonstrate for the first time that the ASP transcript is expressed during viral latency.
2.3. Expression of ASP RNA suppresses viral replication
A number of studies have demonstrated that the antisense transcript of the human T cell leukemia virus 1 (HTLV-1) antisense gene hbz and its encoded protein play opposite roles in the virus lifecycle (Ma et al., 2016). Previous reports have suggested that the HIV-1 antisense transcript contains an open reading frame directing the expression of a highly hydrophobic 189-aa antisense protein (ASP) (Miller, 1988; Vanhee-Brossollet et al., 1995; Laverdure et al., 2012; Briquet and Vaquero, 2002; Clerc et al., 2011; Champiat et al., 2012). To assess whether expression of ASP RNA plays a role in HIV-1 replication, we transduced Jurkat cells with lentiviruses expressing the ASP transcript. These lentiviruses were generated using the pLVX-Puro vector, which expresses ASP RNA under the CMV promoter/enhancer rather than the lentiviral LTR. Therefore, Tat levels do not affect expression of the ASP transcript. Moreover, to assess effects exclusively due to the HIV-1 ASP RNA, we introduced three single point mutations in the sequence of the ASP transcript so that codons 3, 7 and 10 of the ASP open reading frame were mutated from glutamine, cysteine, and cysteine — respectively — to stop codons. Jurkat cells were stably transduced with purified lentiviral particles carrying the mutated ASP transcript sequence (J-ASTmut) or the empty vector (J-LVX), and then selected with puromycin. First, we performed RT-qPCR to verify expression of the antisense transcript (Fig. 3A). Indeed, we detected expression of ASP RNA in Jurkat cells transduced with the ASTmut lentivirus, but not in Jurkat transduced with the empty lentivirus or in the parental Jurkat cell line. Then, the three cell lines were infected with equal amounts of HIV-1IIIB, and viral replication was monitored over 10 days by measuring p24 antigen in the culture supernatant. We found that Jurkat cells expressing the ASP transcript supported HIV-1 replication at significantly lower levels compared to the other two cell lines (Fig. 3B). Thus, expression of the HIV-1 ASP RNA inhibits viral replication by 70–80%. Moreover, since the sequence of the ASP transcript in these studies carried three early stop codons in the ASP open reading frame, the effects that we observed could be ascribed exclusively to the transcript.
Fig. 3.
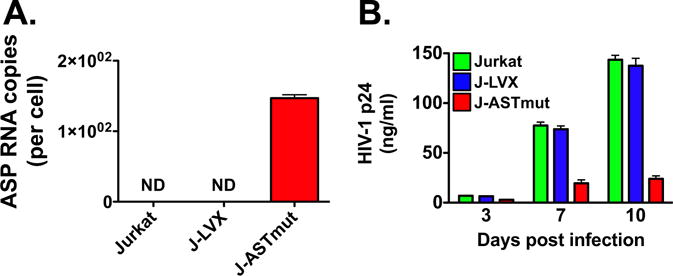
The HIV-1 ASP RNA inhibits viral replication. Jurkat cells were stably transduced with a pLVX-based lentivirus driving CMV promoter-dependent expression of the ASP RNA sequence with stops at codons 3, 7 and 10 of the ORF (J-ASTmut). As a control, we generated a Jurkat cell line stably transduced with the empty lentivirus (J-LVX). Panel A: Quantification of ASP RNA expression in parental Jurkat cells, J-LVX cells, and J-ASTmut cells. Equal amounts of total RNA from the three cell lines was utilized to measure expression of ASP RNA with the strand-specific RT-qPCR assay shown in Fig. 1. ND: not detected. Panel B: Parental Jurkat cells and the two stably transduced cell lines (J-LVX and J-ASTmut) were infected with equal amounts of HIV-1IIIB by spinoculation (m.o.i. 0.01), and cultured in complete medium. Viral replication was assessed by HIV-1 p24 ELISA over 10 days in culture. Graphs report average and standard deviation calculated from three replicate samples.
2.4. Expression of ASP RNA promotes the establishment and maintenance of HIV-1 latency
Next, we assessed whether the ASP RNA suppresses viral replication by promoting viral latency. To that end, we utilized Jurkat E4 (JE4) cells (Friedman et al., 2011; Pearson et al., 2008). This clonal cell line is infected with an HIV-1 provirus that lacks the gag gene, contains the env gene, and carries a destabilized GFP gene in place of nef. Under normal culture condition, the HIV-1 provirus in Jurkat E4 is latent, and ~5% of the cells express GFP. However, viral expression promptly resumes following stimulation with TNFα or latency reversing agents (LRAs), and returns to latency following removal of the stimulus (Friedman et al., 2011). To assess the effect of ASP RNA on HIV-1 latency, we generated Jurkat E4-derived lines stably transduced with the same lentiviruses described above (JE4-ASTmut and JE4-LVX). As shown in Fig. 4A, both Jurkat E4 and JE4-LVX cells expressed antisense transcripts in the HIV-1 provirus. Indeed, our TaqMan PCR assay to detect ASP RNA is designed on a portion of the transcript that overlaps the env gene, and thus it is able to detect antisense transcription in these two cell lines despite the presence of the GFP gene in place of nef. However, we detected significantly higher levels of HIV-1 antisense transcripts in JE4-ASTmut, because the stably transduced lentivirus drives the expression of significant levels of mutated ASP transcript on top of the antisense transcripts driven by the HIV-1 3′LTR (Fig. 4A).
Fig. 4.
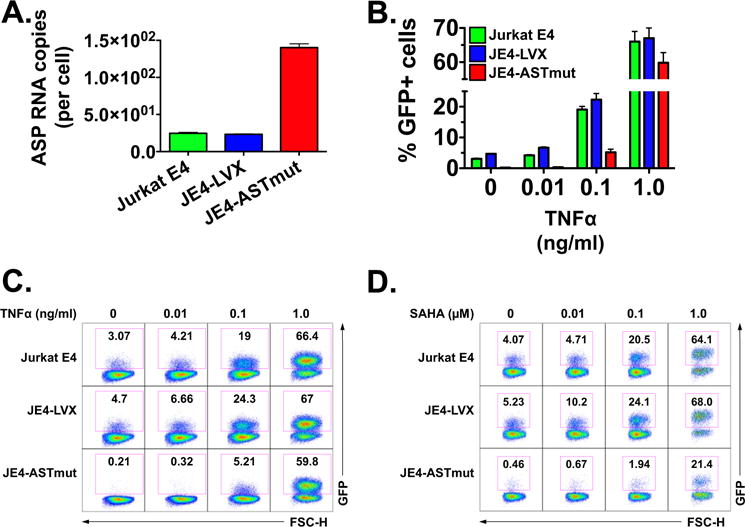
Expression of ASP RNA inhibits HIV-1 reactivation from latency. Latently infected Jurkat E4 cells were stably transduced with a pLVX-based lentivirus driving CMV promoter-dependent expression of the ASP transcript sequence with stops at codons 3, 7 and 10 of the ORF (JE4-ASTmut). As a control, we generated a Jurkat E4 cell line stably transduced with the empty lentivirus (JE4-LVX). Panel A: Quantification of ASP RNA expression in Jurkat E4, JE4-ASTmut and JE4-LVX cells. Equal amounts of total RNA from the three cell lines was utilized to measure ASP RNA expression with the strand-specific RT-qPCR assay shown in Fig. 1. Panel B: HIV-1 reactivation (as measured by GFP expression) in parental Jurkat E4, JE4-LVX and JE4-ASTmut cells exposed to increasing amounts of TNFα (0, 0.01, 0.1 and 1.0 ng/ml) for 24 h. The figure shows the results from three independent experiments, and reports the average and SD. Panel C: Flow cytometry plots from one of the three experiments included in Panel B. Panel D: HIV-1 reactivation (as measured by GFP expression) in parental Jurkat E4, JE4-LVX and JE4-ASTmut cells exposed to increasing amounts of SAHA (0, 0.01, 0.1 and 1.0 μM) for 24 h. We performed three independent experiments, and the figure reports the flow cytometry plots from one of the three experiments.
We utilized these cell lines to test the effect of ASP RNA expression on viral reactivation from latency. We found that in the absence of any stimulation, the percentage of GFP positive cells was markedly lower in the JE4-ASTmut sample than in the two other cell lines (Fig. 4B–C). The three cell lines were then exposed to increasing doses of TNFα (0.01–1.0 ng/ml) or to SAHA (0.01–1.0 μM), and after 24 h we assessed viral reactivation by measuring the percentage of GFP positive cells by flow cytometry. Viral reactivation in response to low doses of TNFα (0.01 and 0.1 ng/ml) was lower in JE4-ASTmut cells compared to the other two cell lines. However, the three cell lines showed similar percentages of GFP positive cells upon treatment with higher TNFα doses (Fig. 4B and C). On the contrary, JE4-ASTmut cells showed markedly reduced viral reactivation in response to all doses of SAHA we tested (Fig. 4D). Next, we performed the reverse experiment: we reactivated viral expression by treating the three cell lines with 10 ng/ml TNFα for 12 h, then we washed away the stimulus and we followed return to latency over two days by measuring the percentage of GFP positive cells every 6 h. Our results show that JE4-ASTmut cells reestablished latency much faster than the other two cell lines (Fig. 5A and B). Altogether, these results suggest that expression of ASP RNA promotes the establishment and maintenance of HIV-1 latency.
Fig. 5.
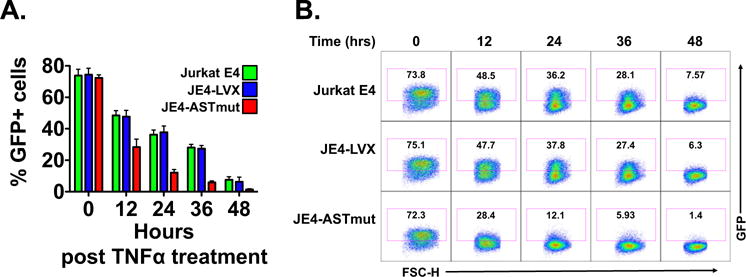
Expression of ASP RNA promotes the establishment of HIV-1 latency. Parental Jurkat E4, JE4-LVX and JE4-ASTmut cells were treated with 10 ng/ml TNFα for 24 h to reactivate HIV-1 from latency. Then the kinetics of return to latency was followed for 48 h by flow cytometry. Panel A: Cells were harvested at time 0 and every 12 h after that, and HIV-1 return to latency was measured by assessing the percentage of GFP+ cells by flow cytometry. The figure shows the results from three independent experiments, and reports the average and SD. Panel B: Flow cytometry plots from one of the three experiments included in Panel A.
2.5. Inhibition of viral expression by ASP RNA does not act at a post-transcriptional step
One of the possible mechanisms of viral suppression by the ASP transcript might entail the formation of double stranded RNA molecules with sense HIV-1 transcripts, and their subsequent degradation by cellular enzymes. To address whether this is the case, we reasoned that the presence of RNA polymerase II (RNA Pol II) ought to be detectable at the HIV-1 promoter and downstream sequences in JE4-ASTmut cells following treatment with LRAs. Thus, JE4, JE4-LVX, and JE4-ASTmut cells were treated with SAHA for 24 h, and then utilized to carry out chromatin immunoprecipitation assays with antibodies to RNA Pol II. Then lysates were probed with primer sets specific for the HIV-1 promoter, Nuc-1 and a sequence immediately before the gag gene. These experiments showed that RNA Pol II is present at the all three locations in unstimulated JE4 and JE4-LVX cells. However, we found that the levels of RNA Pol II are significantly lower in unstimulated JE4-ASTmut cells (Fig. 6A–C). Following treatment with SAHA, we observed that the levels of RNA Pol II augments at all three locations in JE4 and JE4-LVX cells, indicating increased HIV-1 transcription in both cell lines. However, in JE4-ASTmut cells the levels of RNA Pol II remained extremely low at all three locations even after SAHA treatment (Fig. 6A–C). The near absence of RNA Pol II at the HIV-1 promoter, Nuc-1 and a sequence immediately before the gag gene indicates that expression of sense transcripts in JE4-ASTmut cells is also extremely low or absent. Therefore, these results suggest that the ASP transcript suppress HIV-1 expression via a mechanism that acts prior to initiation of HIV-1 transcription rather than a post-transcriptional step such as via degradation of sense transcripts.
Fig. 6.
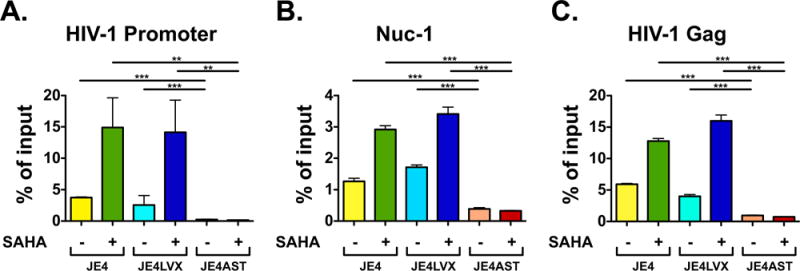
The HIV-1 ASP transcript inhibits HIV-1 expression at the transcription level. ChIP assays measuring the presence of RNA Pol II at the HIV-1 promoter (Panel A), Nuc-1 (Panel B), and at the HIV-1 gag gene (Panel C) in Jurkat E4, JE4-LVX, and JE4-ASTmut before and after treatment with SAHA. Graphs report average and standard deviation calculated from three replicate samples.
2.6. The ASP transcript promotes HIV-1 latency via epigenetic regulation of viral expression
Recent studies have demonstrated that PRC2 and its core member EZH2 are the dominant HKMT involved in the establishment and maintenance of viral latency via nucleosome assembly (Friedman et al., 2011). PRC2 appears to lack DNA binding properties, and its recruitment to the chromatin occurs via NATs (Margueron and Reinberg, 2011; Khorkova et al., 2014). Our data shown above demonstrate that the ASP transcript suppresses HIV-1 expression at the transcriptional level. Thus, we sought to test whether ASP RNA exerts its function by acting as an lncRNA that recruits PRC2 to the HIV-1 LTR, thus promoting H3K27me3 modifications and nucleosome assembly. To test this hypothesis, we first assessed the activity of EZH2 inhibitors on HIV-1 reactivation from latency. We cultured JE4, JE4-LVX, and JE4-ASTmut cells in the presence of increasing doses of EPZ-6438 (2.5–15 μM) for 3 days, and then we assessed viral reactivation by measuring the percentage of GFP positive cells. We found dose-dependent HIV-1 reactivation in both JE4 and JE4-LVX cells (Fig. 7A and B). However, we did not observe any response in JE4-ASTmut cells, where HIV-1 expression remained at baseline levels (Fig. 7A and B). Similar results were obtained with two additional EZH2 inhibitors, CPI-169 and EI1 (Fig. 7C).
Fig. 7.
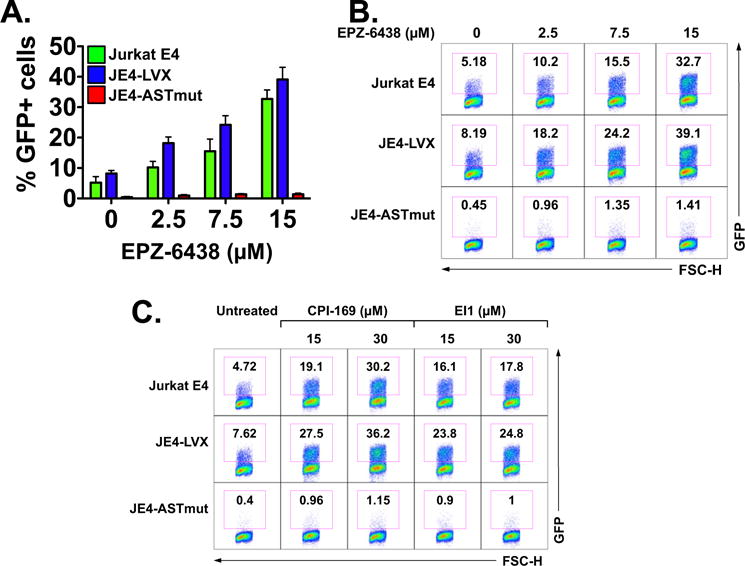
Cells expressing the HIV-1 ASP RNA do not reactivate latent HIV-1 in response to EZH2 inhibitors. Effect of EZH2 inhibitors on reactivation of latent HIV-1 in Jurkat E4, JE4-LVX, and JE4-ASTmut cells. Panel A: Cells were treated with increasing doses of the EZH2 inhibitor, EPZ-6438 (2.5, 7.5 and 15 μM) for 4 days, and then the percentage of GFP positive cells was determined as a measure of viral reactivation. The graph shows the results from three independent experiments and reports average and SD values. Panel B: Flow cytometry plots from one of the three experiments included in Panel A. Panel C: Flow cytometry plots showing response to two additional EZH2 inhibitors, CPI-169 and EI1.
Next, we assessed chromatin changes at Nuc-1 following exposure JE4, JE4-LVX, and JE4-ASTmut to EZH2 inhibitors. To that end, the three cell lines were cultured with 7.5 μM EPZ-6438 for 3 days, and then we performed ChIP with antibodies to EZH2 and to H3K27me3. Samples were probed with Nuc-1 specific probes. We found that prior to exposure to EPZ-6438 the three cell lines had similar levels of EZH2 and H3K27me3 in Nuc-1 (Fig. 8A and B). After treatment with the EZH2 inhibitor, we found a significant ~1.5–2 log decline of EZH2 and H3K27me3 levels in JE4 and JE4-LVX cells. On the other hand, JE4-ASTmut cells maintained unaltered levels of EZH2 at Nuc-1, and showed only a ~4-fold decline of H3K27me3 levels (Fig. 8A and B). In the JE4-ASTmut cell line, the potent CMV promoter/enhancer drives expression of the ASP transcript, leading to RNA levels 5–6 times greater than those JE4 and JE4-LVX cells, in which expression of ASP RNA occurs in the context of the integrated HIV-1 provirus (see Fig. 4A). Moreover, while expression of the native ASP RNA in these two cell lines is driven by the HIV-1 3′LTR and is likely down-regulated following EPZ treatment and induction of Tat expression (Michael et al., 1994; Bentley et al., 2004), this is not the case in JE4-ASTmut that express ASP RNA under the CMV promoter/enhancer. Under these conditions, PRC2 might be recruited to the 5′LTR more efficiently (Fig. 8A), and maintain more sustained levels of H3K27me3 (Fig. 8B) in the JE4-ASTmut cells compared to the other two lines. Altogether, these data show that the ASP transcript suppresses HIV-1 expression by promoting the recruitment and retention of EZH2 to Nuc-1 in the HIV-1 genome, leading to sustained levels of the suppressive epigenetic mark H3K27me3.
Fig. 8.
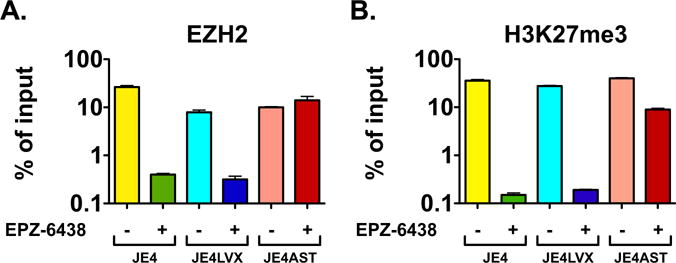
The HIV-1 ASP RNA promotes HIV-1 latency by recruiting PRC2 to the 5′LTR leading to suppressive epigenetic marks at Nuc-1. ChIP assays assessing the presence of the histone lysine methyltransferase EZH2 (Panel A) and histone H3 with trimethylated lysine 27 (H3K27me3; Panel B) at Nuc-1 in Jurkat E4, JE4-LVX, and JE4-ASTmut before and after treatment with the EZH2 specific inhibitor, EPZ-6438. Graphs report average and standard deviation calculated from three replicate samples.
2.7. The HIV-1 ASP transcript interacts with components of the PRC2 complex
Our data suggest that the ASP transcript may promote viral latency by acting as an HIV-encoded lncRNA that recruits PRC2 to the HIV-1 LTR. To conclusively demonstrate that ASP RNA interacts with PRC2, we carried out RNA immunoprecipitation (RIP) assays. Overexpression of the ASP transcript may represent a confounding factor in these studies. Thus, we chose to utilize H9 cells chronically infected with HIV-1IIIB (Fig. 2B), in which the antisense transcript is expressed under its natural promoter within the HIV-1 3′LTR. Cells were fixed with UV, and then cell lysates were immunoprecipitated with antibodies to the catalytic subunit of PRC2, EZH2. Since EZH2 is present in different complexes, we also immunoprecipitated another PRC2 component, SUZ12. As a control, we utilized pre-immune mouse IgG. After extensive washes and elution, the isolated RNA was analyzed by RT-qPCR to assess the presence of ASP RNA. We found significant levels of ASP transcript in immune complexes obtained with antibodies to EZH2 and SUZ12, but not with control mouse IgG (Fig. 9). In addition, we found significant levels of the cellular lncRNA, HOTAΓR that was previously shown to interact with both EZH2 and SUZ12 (Tsai et al., 2010). On the contrary, we found only trace amounts of the snRNA U1, which is routinely used as a negative control in RIP assays (Tsai et al., 2010). Thus, our results demonstrate that the HIV-1 ASP RNA specifically interacts with members of the PRC2 complex, EZH2 and SUZ12.
Fig. 9.
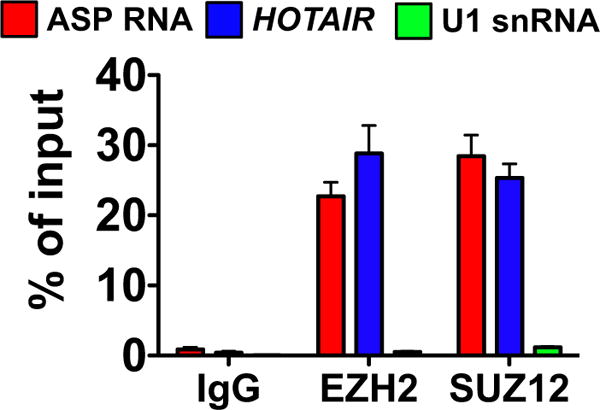
The HIV-1 ASP transcript specifically interacts with two members of the Polycomb Repressor Complex 2 (PRC2), EZH2 and SUZ12. RNA immunoprecipitation assays (RIP) with H9/IIIB cells and antibodies against two PRC2 members (EZH2 and SUZ12), or with control IgG. RNA eluted was analyzed by RT-qPCR to measure the presence of ASP RNA (via the strand-specific assay shown in Fig. 1), the cellular lncRNA HOTAIR and the negative control U1 snRNA. Graph reports average and standard deviation from three replicate samples.
3. Discussion
The entry of HIV-1 into latency requires a series of epigenetic marks at the 5′LTR that lead to the assembly of Nuc-1 around the transcription start site, thus presenting a block to transcription. Several in vitro models have shown a role for DNA methylation at two CpG islands located in the HIV-1 5′LTR (Blazkova et al., 2009; Kauder et al., 2009). However, this has not been confirmed in CD4+ T cells from HIV-1 patients (Blazkova et al., 2012; Palacios et al., 2012; Ho et al., 2013a, 2013b). Our analyses did not include a careful estimate of the frequency of latently infected cells in our three clinical samples. However, if we assume values between 10 and 100 latently infected cells per million CD4+ T cells (Ho et al., 2013a, 2013b), then the number of ASP RNA copies per infected cell with an intact provirus would be < 10. This is in line with the copy number per cell of lncRNAs, often present at < 5 copies/cell (Lee, 2009).
Deacetylation of core histones has been shown to play an important role in HIV-1 latency (He and Margolis, 2002). Indeed, several HDAC inhibitors are being tested clinically for therapeutic reversal of latency (Archin et al., 2012; Elliott et al., 2014; Rasmussen, 2014; Sogaard et al., 2015). Histone H3 methylation at lysine 9 by Suv39H1 and G9a, and lysine 27 by EZH2 are pivotal events for the transcriptional silencing of HIV-1 via epigenetic mechanisms (du Chene et al., 2007; Imai et al., 2010; Marban et al., 2007). Functional studies have shown that EZH2 is the dominant HKMT involved in nucleosome assembly at the HIV-1 LTR (Friedman et al., 2011). In addition to its catalytic activity, EZH2 is a core component of PRC2, which provides a scaffold for other chromatin-modifying enzymes involved in HIV-1 latency, such as HDACs and DNMTs (Tae et al., 2011; Vire et al., 2006; Cheng et al., 2011). Recruitment of HDACs at the 5′LTR is also facilitated by various transcription factors, such as YY1, p50 homodimers, and CBF-1(He and Margolis, 2002; Williams et al., 2006; Tyagi and Karn, 2007).
Several human and animal retroviruses encode an antisense gene on the negative strand of their genome, which is expressed from an antisense promoter within the 3′LTR in opposite direction to the transcripts originating in the 5′LTR (Barbeau and Mesnard, 2015). Here we investigated the expression and function of the antisense RNA encoded into the HIV-1 genome. The development of a highly sensitive, strand-specific, quantitative RT/real time PCR assay allowed us to demonstrate that the ASP transcript is expressed in many different CD4+ T cell systems infected with HIV-1. Previous studies found evidence of ASP RNA expression in chronically infected cell lines, and in primary CD4+ T cells infected in vitro (Kobayashi-Ishihara et al., 2012; Vanhee-Brossollet et al., 1995; Tagieva and Vaquero, 1997). We confirmed these results, but we present new evidence that ASP RNA is also expressed in vivo in resting CD4+ T cells from HIV-1 positive individuals under suppressive cART for at least 24 months.
Our studies demonstrate that expression of ASP RNA inhibits HIV-1 replication during acute infection. Indeed, a Jurkat-derived cell line expressing ASP RNA showed a reduction in the ability to support HIV-1 replication. Constitutive expression of the ASP transcript in this model system was driven under a CMV promoter and enhancer. On one hand this allowed the expression of high levels of ASP RNA, and on the other hand removed expression of ASP RNA from the control of Tat.
We employed the same lentiviral system to stably transduce Jurkat E4 cells, and to address the role of ASP RNA in HIV-1 latency. Our studies demonstrate that the ASP transcript promotes both the establishment and maintenance of latency. Indeed, after reactivation with high doses of TNFα, HIV-1 returned to latency much faster in cells over-expressing ASP RNA. We studied viral reactivation in response to three different classes of compounds. Over-expression of ASP RNA prevents or reduces viral reactivation at low but not high doses of TNFα. Since this cytokine activates the NF-κB pathway, these results suggest that the block imposed by the ASP transcript raises the threshold of Tat needed to achieve full viral expression. Following treatment with the HDAC inhibitor SAHA, we observed a significant reduction in viral reactivation at all doses tested, suggesting that ASP RNA contributes to the recruitment of HDACs to the 5′LTR. Indeed, PRC2 is known to provide a docking platform for HDACs and DNMTs (Tae et al., 2011; Vire et al., 2006; Cheng et al., 2011). However, other cellular factors (such as YY1, p50 homodimers, and CBF-1) (He and Margolis, 2002; Williams et al., 2006; Tyagi and Karn, 2007) participate in this process; thus over-expression of the ASP transcript does not completely suppress viral reactivation in response to HDAC inhibitors. Finally, over-expression of ASP RNA completely suppresses viral reactivation at all doses of EZH2 inhibitors that we tested, suggesting that this transcript promotes viral latency in concert with PRC2. Chromatin immunoprecipitation assays also supported this hypothesis, and showed that in cells over-expressing ASP RNA sustained high levels of EZH2 and H3K27me3 could be found at Nuc-1 following treatment with EZH2 inhibitors. Conclusive evidence came from RIP assays that showed strong, specific interaction between the ASP transcript and two different components of the PRC2 complex, EZH2 and SUZ12. These results are in line with and lend support to a recent report showing that expression of ASP RNA promotes viral latency via DNA methylation at the 5′LTR (Saayman et al., 2014). Indeed, PRC2 contributes to the recruitment of DNMTs, and therefore expression of ASP RNA indirectly promotes their activity at the HIV-1 promoter. Importantly, the report by Saayman et al. utilized the opposite approach (knockdown via siRNA rather than over-expression) to demonstrate that the ASP transcript recruits of DMNT3a, EZH2 and HDAC1 to the HIV-1 5′LTR (Saayman et al., 2014).
Alternative possible mechanisms of action of ASP RNA included transcriptional interference and degradation of sense transcripts. Our experiments employed cell lines stably transduced with lentiviral vectors expressing the ASP transcript under the CMV promoter/enhancer. Since this transcriptional unit is physically separated from the one expressing HIV-1 sense transcript from the 5′LTR, the possibility of transcriptional interference (i.e. collision) between the two transcription machineries can be excluded. Further, the lack of RNA Pol II at the HIV-1 promoter and downstream points in cells over-expressing ASP RNA both before and after stimulation with SAHA indicates that viral suppression by the ASP transcript acts at the transcription level, rather than post-transcriptional level (i.e. via degradation of sense transcripts).
LncRNAs bind their target DNA via formation of RNA:DNA triple helices involving Watson-Crick and Hoogsteen base pairing, which ensures target gene specificity (Mondal et al., 2015; O’Leary et al., 2015; He et al., 2015; Postepska-Igielska et al., 2015; Bierhoff et al., 2010; Schmitz et al., 2010). The first 384 nt at the 5′ end of the ASP RNA are derived from the U3 region of the proviral 3′LTR, and thus are identical to the same region in the 5′LTR. Therefore, it is conceivable that the 5′ end of the ASP transcript mediates its interaction with the 5′LTR of the HIV-1 proviral genome. An additional point to consider is the low copy number of ASP RNA in infected cells, which may contribute to exclude potential off-target effects. For instance, HOTTΓP is an lncRNA that is present at ~1 copy/cell, which allows it to precisely function in cis without affecting other genes (Wang et al., 2011). Thus, while it is unlikely that the HIV-1 antisense transcript may affect the expression of host genes, this possibility cannot be completely ruled out and should be addressed. For instance, it is possible that the ASP transcript may affect the expression of genes encoded by human endogenous retroviruses (HERVs) due to potential homology between the HIV-1 and HERVs long terminal repeats.
Until recently it was thought that HIV-1 does not encode a repressor of viral transcription, and that entry into latency was due to the disappearance of cellular and viral transcription factors along with appearance of suppressive epigenetic marks (Mbonye and Karn, 2014). However, our results suggest that the ASP RNA is an HIV-1 transcript acting as a latency factor. Moreover, a previous report showed that the HIV-1 TAR element is processed by the Dicer enzyme to create a viral miRNA, which is detectable in infected cells and appears to contribute to viral latency (Klase et al., 2007). Thus, the balance between the activities of Tat and ASP RNA could determine the switch between latency and productive infection. In the absence of Tat, the combined effects of NELF, DSIF and ASP RNA restrict HIV-1 expression. However, Tat overcomes such restrictions, achieving transcription elongation and viral expression.
4. Materials and methods
4.1. Cell lines, primary cells, virus infection, and reagents
Jurkat cells were obtained from ATCC (Manassas, VA). All other cell lines were obtained from the NIH AIDS Reagent Program (Germantown, MD). The latently infected cell line Jurkat E4 was a kind gift of Jonathan Karn (Case Western University) (Friedman et al., 2011; Pearson et al., 2008). All cell lines were cultured in RPMI 1640 supplemented with 10% fetal calf serum and antibiotics. Stably transduced cell lines derived from Jurkat and Jurkat E4 were cultured in the same medium containing 1 μg/ml puromycin (Clontech). Lenti-X 293 cells were obtained from Clontech, and cultured in DMEM with 10% tetracycline-free fetal calf serum and antibiotics. Peripheral blood was obtained by venipuncture from HIV-1 positive and negative donors under the University of Maryland, Baltimore Institutional Review Board protocol # HP-00040021. CD4+ T cells were isolated by negative selection using a kit from Miltenyi Biotec. For in vitro infection, cells of healthy donors were activated for 3 days with CD3/CD28 dynabeads (1 bead/cell), harvested, and infected with HIV-1IIIB (0.01 m.o.i.) by spinoculation at 1200×g for 2 h at room temperature. Cells were then washed and cultured in RPMI 1640 supplemented with 10% pooled human serum AB and antibiotics, and 50 units/ml IL-2 (Roche). Infection of Jurkat cells stably transduced with LVX-based lentiviruses was carried out according to the same procedure, except that the cells were not pre-stimulated with CD3/CD28 beads, and they were not cultured in medium containing IL-2 after infection. TNFα was obtained from R & D Systems and solubilized in PBS containing 0.1% BSA. The HDAC inhibitor SAHA was purchased from Sigma-Aldrich, and solubilized in DMSO. The EZH2 inhibitors EPZ-6438, CPI-169 and EI1 were purchased from Selleck Chem and solubilized in DMSO.
4.2. RT-qPCR
Total RNA was isolated from ~5×106 cells using the RNeasy Mini Kit (Qiagen), digested with Turbo DNase (Ambion), and further purified with the RNase Mini Kit. 100–500 ng of RNA were utilized for reverse transcription using the iScript Select cDNA Synthesis Kit (BioRad). The sequence of the AST-tag RT primer was 5′-CTG ATC TAG AGG TAC CGG ATC CAA CAT GTG GCA GGA AGT AGG-3′ (the tag sequence is underlined). For the TaqMan PCR reaction, we utilized the TaqMan Gene Expression Master Mix (Applied Biosystems) according to the manufacturer’s instructions. The sequence of the forward and reverse primers, and the probe were: 5′-TGA TGA ACA TCT AAT TTG TCC ACT GA-3′;5′-CTG ATC TAG AGG TACCGG AT-3′; 5′-CCC AAT GTA TGC CCC TCC CA-3′. For the standard curve, the ASP RNA amplicon carrying the tag sequence was cloned into pUC57. The plasmid was then linearized and utilized at 10°–105 copies to generate a PCR standard curve. The number of copies per 3 μl of cDNA were then normalized to their equivalent number of cells, which were measured manually with a hemacytometer. For RT-qPCR of the U1 snRNA, RT reaction was carried out with the same kit as above, and with the random primers contained in the kit. The sequence of the forward and reverse primers, and the probe were: 5′-GGC GAG GCT TAT CCA TTG CA-3′,5′-GCA GTC GAG TTT CCC ACA TTT G-3′,5′-CCG GAT GTG CTG ACC C-3′. Quantitative TaqMan PCR was carried out as described above.
4.3. Lentiviral construction
We utilized the sequence of the HIV-1 2574-nt antisense transcript reported previously (GenBank accession number JQ866626.1) (Kobayashi-Ishihara et al., 2012). The sequence was mutated at nucleotide 1526 (C > T), 1540 (C > A), and 1549 (C > A) to introduce three stops at codons 3, 7 and 10 in the open reading frame of the HIV-1 antisense protein. The mutated sequence of the ASP transcript (ASTmut) was cloned into the EcoRI and XbaI sites of the pLVX-Puro vector (Clontech). For the generation of lentiviral particles, the pLVX-ASTmut plasmid or the empty pLVX-Puro plasmid were transfected into Lenti-X 293T cells using the Lenti-X Packaging Single Shots (VSV-G) system (Clontech). After 3–4 days, culture supernatants were collected, and viral particles purified and concentrated using the Fast-Trap Lentivirus Purification and Concentration Kit (Millipore). Jurkat and Jurkat E4 cells were transduced with purified lentiviral particles overnight in complete RPMI medium containing 2 μg/ml polybrene. Cells were then selected in fresh medium containing puromycin. Stably transduced cells were maintained in medium containing 1 μg/ml puromycin.
4.4. Flow cytometry
2.5×105 cells were stained with 7-AAD, washed in PBS, and fixed in 2% paraformaldehyde in 1× PBS. Cells were then analyzed with a BD FACSCalibur instrument, and event acquired with Cell Quest software. Data were analyzed using the FlowJo software by gating on the live cell population as determined by forward and side scatter profile, and by excluding 7-AAD positive cells.
4.5. Chromatin and RNA immunoprecipitation assays
ChIP assay was performed using a protocol similar to one described previously (Klase et al., 2007). Briefly, crosslinked samples were sonicated and mono-disomes were used for immunoprecipitation. Antibodies to RNA polymerase II (Santa Cruz Biotechnology sc-899), EZH2 or H3K27me3 (Abcam ab3748 and ab6002, respectively) were added, and the samples were allowed to rotate at 4 °C for 3 h. A 50% (v/v) protein A-Sepharose/protein G-Sepharose mix was then added to the samples, which were rotated overnight at 4 °C. The samples were washed twice with IP Wash Buffer (Sigma) prior to addition of Proteinase K (800 U/ml). After a 15-min incubation at 65 °C, reversing solution (Sigma) was added and the samples incubated at 65 °C for 90 min. DNA was purified and real time qPCR was performed with 2 μl of DNA, 10 μl of SYBR Green (BioRad), 7.84 μl of nuclease free water, and 0.06 μl each of forward and reverse primers for HIV-1 promoter (forward, 5′-AGC TTG CTA CAA GGG ACT TTC C-3′, and reverse, 5′-ACC CAG TACAGG CAA AAA GCA G-3′), the Nuc-1 region (forward, 5′-CTG GGA GCT CTC TGG CTA ACT A-3′, and reverse, 5′-TTA CCA GAG TCA CAC AAC AGA CG-3′); and the gag region (forward 5′-AGG CGT TAC TCG ACA GAG G-3′, and reverse, 5′-AGG CGT TAC TCG ACA GAG GA-3′)(Friedman et al., 2011). For RNA immunoprecipitation (RIP) assays, 2×107 cells were UV-crosslinked twice with 1.25×105 μJ/cm2, washed with ice-cold PBS and lysed in a buffer containing 0.5% NP-40, protease inhibitors, and SUPERase-In in 1× PBS. After incubation on ice for 10 min, 0.5% sodium deoxycholate was added followed by rotation at 4 °C for 20 min, and digestion with Turbo DNase for 15 min at 37 °C. The lysate was then centrifuged at 1350g for 5 min at 4 °C and the supernatant transferred to a fresh tube. One tenth of the volume was saved as input control. For the immunoprecipitation, 100 μlof Protein G dynabeads were pre-adsorbed at 4 °C for 4 h with 10 μg of antibody to EZH2 or SUZ12 (Abcam ab3748 and ab12073, respectively), or control IgG (Abcam). The antibody-dynabeads complexes were then added to the cell lysate and incubated overnight at 4 °C. Beads were then washed Wash Buffer 1 (1% NP-40, 0.5% sodium deoxycholate, 150 mM NaCl, SUPERase-In in PBS), Wash Buffer 1 supplemented with 1 M urea, Wash Buffer 2 (1% NP-40, 0.5% sodium deoxycholate, 150 mM NaCl, 10 mM EDTA, SUPERase-In in PBS), and Wash Buffer 2 supplemented with 1 M urea (three times each). Finally RNA was eluted for 30 min at 55 °C in a buffer containing 100 mM Tris-Cl, 50 mM NaCl, 10 mM EDTA, 0.5% SDS, 100 μg/ml proteinase K and SUPERase-In. After recovery of the supernatant, 300 μl of Trizol were added. The solution was extracted twice with 60 μl of chloroform, and finally the RNA was purified using the RNeasy Mini Kit from Qiagen. After quantification by Nanodrop, 2 μl (~10 ng) of RNA and 3 μl of cDNA were used to quantify ASP RNA, HOTAIR, and U1 snRNA by RT-qPCR as described above for the ASP transcript, and as described previously by Tsai et al. (52) for HOTAIR and U1 snRNA.
Acknowledgments
The authors would like to thank Drs. Rosa Bernardi and Davide Gabellini for insightful discussions and for help with some of the procedures. This work was supported by National Institutes of Health Grants AI106508 and AI120008 (to FR), and National Institutes of Health Grants AI AI078859, AI074410, and AI043894 (to FK).
Footnotes
Conflicts of interest
The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
References
- Archin NM, Liberty AL, Kashuba AD, Choudhary SK, Kuruc JD, Crooks AM, Parker DC, Anderson EM, Kearney MF, Strain MC, Richman DD, Hudgens MG, Bosch RJ, Coffin JM, Eron JJ, Hazuda DJ, Margolis DM. Administration of vorinostat disrupts HIV-1 latency in patients on antiretroviral therapy. Nature. 2012;487:482–485. doi: 10.1038/nature11286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbeau B, Mesnard JM. Making sense out of antisense transcription in human T-cell lymphotropic viruses (HTLVs) Viruses. 2011;3:456–468. doi: 10.3390/v3050456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbeau B, Mesnard JM. Does chronic infection in retroviruses have a sense? Trends Microbiol. 2015;23:367–375. doi: 10.1016/j.tim.2015.01.009. [DOI] [PubMed] [Google Scholar]
- Bentley K, Deacon N, Sonza S, Zeichner S, Churchill M. Mutational analysis of the HIV-1 LTR as a promoter of negative sense transcription. Arch Virol. 2004;149:2277–2294. doi: 10.1007/s00705-004-0386-8. [DOI] [PubMed] [Google Scholar]
- Bierhoff H, Schmitz K, Maass F, Ye J, Grummt I. Noncoding transcripts in sense and antisense orientation regulate the epigenetic state of ribosomal RNA genes. Cold Spring Harb Symp Quant Biol. 2010;75:357–364. doi: 10.1101/sqb.2010.75.060. [DOI] [PubMed] [Google Scholar]
- Blazkova J, Trejbalova K, Gondois-Rey F, Halfon P, Philibert P, Guiguen A, Verdin E, Olive D, Van Lint C, Hejnar J, Hirsch I. CpG methylation controls reactivation of HIV from latency. PLoS Pathog. 2009;5:e1000554. doi: 10.1371/journal.ppat.1000554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blazkova J, Murray D, Justement JS, Funk EK, Nelson AK, Moir S, Chun TW, Fauci AS. Paucity of HIV DNA methylation in latently infected, resting CD4+ T cells from infected individuals receiving antiretroviral therapy. J Virol. 2012;86:5390–5392. doi: 10.1128/JVI.00040-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briquet S, Vaquero C. Immunolocalization studies of an antisense protein in HIV-1-infected cells and viral particles. Virology. 2002;292:177–184. doi: 10.1006/viro.2001.1224. [DOI] [PubMed] [Google Scholar]
- Briquet S, Richardson J, Vanhee-Brossollet C, Vaquero C. Natural antisense transcripts are detected in different cell lines and tissues of cats infected with feline immunodeficiency virus. Gene. 2001;267:157–164. doi: 10.1016/s0378-1119(01)00404-8. [DOI] [PubMed] [Google Scholar]
- Champiat S, Raposo RA, Maness NJ, Lehman JL, Purtell SE, Hasenkrug AM, Miller JC, Dean H, Koff WC, Hong MA, Martin JN, Deeks SG, Spotts GE, Pilcher CD, Hecht FM, Kallas EG, Garrison KE, Nixon DF. Influence of HAART on alternative reading frame immune responses over the course of HIV-1 infection. PLoS One. 2012;7:e39311. doi: 10.1371/journal.pone.0039311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- du Chene I, Basyuk E, Lin YL, Triboulet R, Knezevich A, Chable-Bessia C, Mettling C, Baillat V, Reynes J, Corbeau P, Bertrand E, Marcello A, Emiliani S, Kiernan R, Benkirane M. Suv39H1 and HP1gamma are responsible for chromatin-mediated HIV-1 transcriptional silencing and post-integration latency. EMBO J. 2007;26:424–435. doi: 10.1038/sj.emboj.7601517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng AS, Lau SS, Chen Y, Kondo Y, Li MS, Feng H, Ching AK, Cheung KF, Wong HK, Tong JH, Jin H, Choy KW, Yu J, To KF, Wong N, Huang TH, Sung JJ. EZH2-mediated concordant repression of Wnt antagonists promotes beta-catenin-dependent hepatocarcinogenesis. Cancer Res. 2011;71:4028–4039. doi: 10.1158/0008-5472.CAN-10-3342. [DOI] [PubMed] [Google Scholar]
- Clerc I, Laverdure S, Torresilla C, Landry S, Borel S, Vargas A, Arpin-Andre C, Gay B, Briant L, Gross A, Barbeau B, Mesnard JM. Polarized expression of the membrane ASP protein derived from HIV-1 antisense transcription in T cells. Retrovirology. 2011;8:74. doi: 10.1186/1742-4690-8-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czudnochowski N, Bosken CA, Geyer M. Serine-7 but not serine-5 phosphorylation primes RNA polymerase II CTD for P-TEFb recognition. Nat Commun. 2012;3:842. doi: 10.1038/ncomms1846. [DOI] [PubMed] [Google Scholar]
- Davidovich C, Cech TR. The recruitment of chromatin modifiers by long noncoding RNAs: lessons from PRC2. Rna. 2015;21:2007–2022. doi: 10.1261/rna.053918.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott JH, Wightman F, Solomon A, Ghneim K, Ahlers J, Cameron MJ, Smith MZ, Spelman T, McMahon J, Velayudham P, Brown G, Roney J, Watson J, Prince MH, Hoy JF, Chomont N, Fromentin R, Procopio FA, Zeidan J, Palmer S, Odevall L, Johnstone RW, Martin BP, Sinclair E, Deeks SG, Hazuda DJ, Cameron PU, Sekaly RP, Lewin SR. Activation of HIV transcription with short-course vorinostat in HIV-infected patients on suppressive antiretroviral therapy. PLoS Pathog. 2014;10:e1004473. doi: 10.1371/journal.ppat.1004473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman J, Cho WK, Chu CK, Keedy KS, Archin NM, Margolis DM, Karn J. Epigenetic silencing of HIV-1 by the histone H3 lysine 27 methyltransferase enhancer of Zeste 2. J Virol. 2011;85:9078–9089. doi: 10.1128/JVI.00836-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujinaga K, Irwin D, Huang Y, Taube R, Kurosu T, Peterlin BM. Dynamics of human immunodeficiency virus transcription: P-TEFb phosphorylates RD and dissociates negative effectors from the transactivation response element. Mol Cell Biol. 2004;24:787–795. doi: 10.1128/MCB.24.2.787-795.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaudray G, Gachon F, Basbous J, Biard-Piechaczyk M, Devaux C, Mesnard JM. The complementary strand of the human T-cell leukemia virus type 1 RNA genome encodes a bZIP transcription factor that down-regulates viral transcription. J Virol. 2002;76:12813–12822. doi: 10.1128/JVI.76.24.12813-12822.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han J, Kim D, Morris KV. Promoter-associated RNA is required for RNA-directed transcriptional gene silencing in human cells. Proc Natl Acad Sci USA. 2007;104:12422–12427. doi: 10.1073/pnas.0701635104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He G, Margolis DM. Counterregulation of chromatin deacetylation and histone deacetylase occupancy at the integrated promoter of human immunodeficiency virus type 1 (HIV-1) by the HIV-1 repressor YY1 and HIV-1 activator Tat. Mol Cell Biol. 2002;22:2965–2973. doi: 10.1128/MCB.22.9.2965-2973.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He S, Zhang H, Liu H, Zhu H. LongTarget: a tool to predict lncRNA DNA-binding motifs and binding sites via Hoogsteen base-pairing analysis. Bioinformatics. 2015;31:178–186. doi: 10.1093/bioinformatics/btu643. [DOI] [PubMed] [Google Scholar]
- Ho YC, Shan L, Wang J, Hosmane N, Blankson J, Siliciano R. Characterization of Non-induced HIV-1 Proviruses Dampens the Hope for HIV-1 Eradication; Paper presented at Proceedings of the 20th Conference on Retroviruses and Opportunistic Infections; Atlanta, GA. 2013a. [Google Scholar]
- Ho YC, Shan L, Hosmane NN, Wang J, Laskey SB, Rosenbloom DI, Lai J, Blankson JN, Siliciano JD, Siliciano RF. Replication-competent noninduced proviruses in the latent reservoir increase barrier to HIV-1 cure. Cell. 2013b;155:540–551. doi: 10.1016/j.cell.2013.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai K, Togami H, Okamoto T. Involvement of histone H3 lysine 9 (H3K9) methyltransferase G9a in the maintenance of HIV-1 latency and its reactivation by BIX01294. J Biol Chem. 2010;285:16538–16545. doi: 10.1074/jbc.M110.103531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jadlowsky JK, Wong JY, Graham AC, Dobrowolski C, Devor RL, Adams MD, Fujinaga K, Karn J. Negative elongation factor is required for the maintenance of proviral latency but does not induce promoter-proximal pausing of RNA polymerase II on the HIV long terminal repeat. Mol Cell Biol. 2014;34:1911–1928. doi: 10.1128/MCB.01013-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao SY, Calman AF, Luciw PA, Peterlin BM. Anti-termination of transcription within the long terminal repeat of HIV-1 by tat gene product. Nature. 1987;330:489–493. doi: 10.1038/330489a0. [DOI] [PubMed] [Google Scholar]
- Karn J. The molecular biology of HIV latency: breaking and restoring the Tat-dependent transcriptional circuit. Curr Opin HIV AIDS. 2011;6:4–11. doi: 10.1097/COH.0b013e328340ffbb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauder SE, Bosque A, Lindqvist A, Planelles V, Verdin E. Epigenetic regulation of HIV-1 latency by cytosine methylation. PLoS Pathog. 2009;5:e1000495. doi: 10.1371/journal.ppat.1000495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keedy KS, Archin NM, Gates AT, Espeseth A, Hazuda DJ, Margolis DM. A limited group of class I histone deacetylases act to repress human immunodeficiency virus type-1 expression. J Virol. 2009 doi: 10.1128/JVI.02585-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khorkova O, Myers AJ, Hsiao J, Wahlestedt C. Natural antisense transcripts. Hum Mol Genet. 2014;23:R54–R63. doi: 10.1093/hmg/ddu207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klase Z, Kale P, Winograd R, Gupta MV, Heydarian M, Berro R, McCaffrey T, Kashanchi F. HIV-1 TAR element is processed by Dicer to yield a viral micro-RNA involved in chromatin remodeling of the viral LTR. BMC Mol Biol. 2007;8:63. doi: 10.1186/1471-2199-8-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi-Ishihara M, Yamagishi M, Hara T, Matsuda Y, Takahashi R, Miyake A, Nakano K, Yamochi T, Ishida T, Watanabe T. HIV-1-encoded antisense RNA suppresses viral replication for a prolonged period. Retrovirology. 2012;9:38. doi: 10.1186/1742-4690-9-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landry S, Halin M, Lefort S, Audet B, Vaquero C, Mesnard JM, Barbeau B. Detection, characterization and regulation of antisense transcripts in HIV-1. Retrovirology. 2007;4:71. doi: 10.1186/1742-4690-4-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larocca D, Chao LA, Seto MH, Brunck TK. Human T-cell leukemia virus minus strand transcription in infected T-cells. Biochem Biophys Res Commun. 1989;163:1006–1013. doi: 10.1016/0006-291x(89)92322-x. [DOI] [PubMed] [Google Scholar]
- Laverdure S, Gross A, Arpin-Andre C, Clerc I, Beaumelle B, Barbeau B, Mesnard JM. HIV-1 antisense transcription is preferentially activated in primary monocyte-derived cells. J Virol. 2012;86:13785–13789. doi: 10.1128/JVI.01723-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JT. Lessons from X-chromosome inactivation: long ncRNA as guides and tethers to the epigenome. Genes Dev. 2009;23:1831–1842. doi: 10.1101/gad.1811209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludwig LB, Ambrus JL, Jr, Krawczyk KA, Sharma S, Brooks S, Hsiao CB, Schwartz SA. Human Immunodeficiency Virus-Type 1 LTR DNA contains an intrinsic gene producing antisense RNA and protein products. Retrovirology. 2006;3:80. doi: 10.1186/1742-4690-3-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma G, Yasunaga J, Matsuoka M. Multifaceted functions and roles of HBZ in HTLV-1 pathogenesis. Retrovirology. 2016;13:16. doi: 10.1186/s12977-016-0249-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marban C, Suzanne S, Dequiedt F, de Walque S, Redel L, Van Lint C, Aunis D, Rohr O. Recruitment of chromatin-modifying enzymes by CTIP2 promotes HIV-1 transcriptional silencing. EMBO J. 2007;26:412–423. doi: 10.1038/sj.emboj.7601516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margueron R, Reinberg D. The Polycomb complex PRC2 and its mark in life. Nature. 2011;469:343–349. doi: 10.1038/nature09784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mbonye U, Karn J. Transcriptional control of HIV latency: cellular signaling pathways, epigenetics, happenstance and the hope for a cure. Virology. 2014:454–455. doi: 10.1016/j.virol.2014.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michael NL, Vahey MT, d’Arcy L, Ehrenberg PK, Mosca JD, Rappaport J, Redfield RR. Negative-strand RNA transcripts are produced in human immunodeficiency virus type 1-infected cells and patients by a novel promoter downregulated by Tat. J Virol. 1994;68:979–987. doi: 10.1128/jvi.68.2.979-987.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller RH. Human immunodeficiency virus may encode a novel protein on the genomic DNA plus strand. Science. 1988;239:1420–1422. doi: 10.1126/science.3347840. [DOI] [PubMed] [Google Scholar]
- Mondal T, Subhash S, Vaid R, Enroth S, Uday S, Reinius B, Mitra S, Mohammed A, James AR, Hoberg E, Moustakas A, Gyllensten U, Jones SJ, Gustafsson CM, Sims AH, Westerlund F, Gorab E, Kanduri C. MEG3 long noncoding RNA regulates the TGF-beta pathway genes through formation of RNA-DNA triplex structures. Nat Commun. 2015;6:7743. doi: 10.1038/ncomms8743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Leary VB, Ovsepian SV, Carrascosa LG, Buske FA, Radulovic V, Niyazi M, Moertl S, Trau M, Atkinson MJ, Anastasov N. PARTICLE, a Triplex-forming long ncRNA, regulates locus-specific methylation in response to low-dose irradiation. Cell Rep. 2015;11:474–485. doi: 10.1016/j.celrep.2015.03.043. [DOI] [PubMed] [Google Scholar]
- Palacios JA, Perez-Pinar T, Toro C, Sanz-Minguela B, Moreno V, Valencia E, Gomez-Hernando C, Rodes B. Long-term nonprogressor and elite controller patients who control viremia have a higher percentage of methylation in their HIV-1 proviral promoters than aviremic patients receiving highly active antiretroviral therapy. J Virol. 2012;86:13081–13084. doi: 10.1128/JVI.01741-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palazzo AF, Lee ES. Non-coding RNA: what is functional and what is junk? Front Genet. 2015;6:2. doi: 10.3389/fgene.2015.00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson R, Kim YK, Hokello J, Lassen K, Friedman J, Tyagi M, Karn J. Epigenetic silencing of human immunodeficiency virus (HIV) transcription by formation of restrictive chromatin structures at the viral long terminal repeat drives the progressive entry of HIV into latency. J Virol. 2008;82:12291–12303. doi: 10.1128/JVI.01383-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Postepska-Igielska A, Giwojna A, Gasri-Plotnitsky L, Schmitt N, Dold A, Ginsberg D, Grummt I. LncRNA Khps1 Regulates Expression of the Proto-oncogene SPHK1 via Triplex-Mediated Changes in Chromatin Structure. Mol Cell. 2015;60:626–636. doi: 10.1016/j.molcel.2015.10.001. [DOI] [PubMed] [Google Scholar]
- Rasmussen TA. Panobinostat, a histone deacetylase inhibitor, for latent-virus reactivation in HIV-infected patients on suppressive antiretroviral therapy: a phase 1/2, single group, clinical trial. Lancet HIV. 2014;1:e13–e21. doi: 10.1016/S2352-3018(14)70014-1. [DOI] [PubMed] [Google Scholar]
- Saayman S, Ackley A, Turner AM, Famiglietti M, Bosque A, Clemson M, Planelles V, Morris KV. An HIV-encoded antisense long noncoding RNA epigenetically regulates viral transcription. Mol Ther J Am Soc Gene Ther. 2014;22:1164–1175. doi: 10.1038/mt.2014.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz KM, Mayer C, Postepska A, Grummt I. Interaction of noncoding RNA with the rDNA promoter mediates recruitment of DNMT3b and silencing of rRNA genes. Genes Dev. 2010;24:2264–2269. doi: 10.1101/gad.590910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sogaard OS, Graversen ME, Leth S, Olesen R, Brinkmann CR, Nissen SK, Kjaer AS, Schleimann MH, Denton PW, Hey-Cunningham WJ, Koelsch KK, Pantaleo G, Krogsgaard K, Sommerfelt M, Fromentin R, Chomont N, Rasmussen TA, Ostergaard L, Tolstrup M. The Depsipeptide Romidepsin Reverses HIV-1 Latency In Vivo. PLoS Pathog. 2015;11:e1005142. doi: 10.1371/journal.ppat.1005142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tae S, Karkhanis V, Velasco K, Yaneva M, Erdjument-Bromage H, Tempst P, Sif S. Bromodomain protein 7 interacts with PRMT5 and PRC2, and is involved in transcriptional repression of their target genes. Nucleic Acids Res. 2011;39:5424–5438. doi: 10.1093/nar/gkr170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tagieva NE, Vaquero C. Expression of naturally occurring antisense RNA inhibits human immunodeficiency virus type 1 heterologous strain replication. J Gen Virol. 1997;78(Pt 10):2503–2511. doi: 10.1099/0022-1317-78-10-2503. [DOI] [PubMed] [Google Scholar]
- Torresilla C, Larocque E, Landry S, Halin M, Coulombe Y, Masson JY, Mesnard JM, Barbeau B. Detection ofthe HIV-1 minus-strand-encoded antisense protein and its association with autophagy. J Virol. 2013;87:5089–5105. doi: 10.1128/JVI.00225-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai MC, Manor O, Wan Y, Mosammaparast N, Wang JK, Lan F, Shi Y, Segal E, Chang HY. Long noncoding RNA as modular scaffold of histone modification complexes. Science. 2010;329:689–693. doi: 10.1126/science.1192002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyagi M, Karn J. CBF-1 promotes transcriptional silencing during the establishment of HIV-1 latency. EMBO J. 2007;26:4985–4995. doi: 10.1038/sj.emboj.7601928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanhee-Brossollet C, Thoreau H, Serpente N, D’Auriol L, Levy JP, Vaquero C. A natural antisense RNA derived from the HIV-1 env gene encodes a protein which is recognized by circulating antibodies of HIV+ individuals. Virology. 1995;206:196–202. doi: 10.1016/s0042-6822(95)80034-4. [DOI] [PubMed] [Google Scholar]
- Verdin E, Paras P, Jr, Van Lint C. Chromatin disruption in the promoter of human immunodeficiency virus type 1 during transcriptional activation. EMBO J. 1993;12:3249–3259. doi: 10.1002/j.1460-2075.1993.tb05994.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vire E, Brenner C, Deplus R, Blanchon L, Fraga M, Didelot C, Morey L, Van Eynde A, Bernard D, Vanderwinden JM, Bollen M, Esteller M, Di Croce L, de Launoit Y, Fuks F. The Polycomb group protein EZH2 directly controls DNA methylation. Nature. 2006;439:871–874. doi: 10.1038/nature04431. [DOI] [PubMed] [Google Scholar]
- Wang KC, Yang YW, Liu B, Sanyal A, Corces-Zimmerman R, Chen Y, Lajoie BR, Protacio A, Flynn RA, Gupta RA, Wysocka J, Lei M, Dekker J, Helms JA, Chang HY. A long noncoding RNA maintains active chromatin to coordinate homeotic gene expression. Nature. 2011;472:120–124. doi: 10.1038/nature09819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei P, Garber ME, Fang SM, Fischer WH, Jones KA. A novel CDK9-associated C-type cyclin interacts directly with HIV-1 Tat and mediates its high-affinity, loop-specific binding to TAR RNA. Cell. 1998;92:451–462. doi: 10.1016/s0092-8674(00)80939-3. [DOI] [PubMed] [Google Scholar]
- Williams SA, Chen LF, Kwon H, Ruiz-Jarabo CM, Verdin E, Greene WC. NF-kappaB p50 promotes HIV latency through HDAC recruitment and repression of transcriptional initiation. EMBO J. 2006;25:139–149. doi: 10.1038/sj.emboj.7600900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi Y, Inukai N, Narita T, Wada T, Handa H. Evidence that negative elongation factor represses transcription elongation through binding to a DRB sensitivity-inducing factor/RNA polymerase II complex and RNA. Mol Cell Biol. 2002;22:2918–2927. doi: 10.1128/MCB.22.9.2918-2927.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Klatt A, Gilmour DS, Henderson AJ. Negative elongation factor NELF represses human immunodeficiency virus transcription by pausing the RNA polymerase II complex. J Biol Chem. 2007;282:16981–16988. doi: 10.1074/jbc.M610688200. [DOI] [PubMed] [Google Scholar]