

Abstract
Both Type 1 and Type 2 diabetes exhibit features of inflammation associated with alterations in pancreatic islet function and mass. These immunological disruptions, if unresolved, contribute to the overall pathogenesis of disease onset. This review presents the emerging role of pancreatic islet chemokine production as a critical factor regulating immune cell entry into pancreatic tissue as well as an important facilitator of changes in tissue resident leukocyte activity. Signaling through two specific chemokine receptors (i.e., CXCR2 and CXCR3) is presented to illustrate key points regarding ligand-mediated regulation of innate and adaptive immune cell responses. The prospective roles of chemokine ligands and their corresponding chemokine receptors to influence the onset and progression of autoimmune- and obesity-associated forms of diabetes are discussed.
Keywords: Cytokine, Chemokine, Diabetes, Islet, Inflammation
Introduction
Development of diabetes mellitus is currently classified based on the route by which hyperglycemia develops. Currently, Type 1 diabetes (T1D) is described as an autoimmune disease that results when the function and/or mass of the insulin-producing β-cells is reduced to a degree that produces clinical symptoms [e.g., hyperglycemia, polyuria, etc.; see (Atkinson, et al. 2014)]. Type 2 diabetes (T2D) is described as a more slowly progressing disease characterized first by insulin resistance and glucose intolerance, two conditions that indicate ‘pre-diabetes’(Cefalu 2016; Cefalu, et al. 2014; Johnson and Olefsky 2013). The pre-diabetes stage progresses to overt clinical onset of diabetes when the function and mass of the islet β-cells decrease to the point where the ability to maintain glucose homeostasis is ultimately lost (Doria, et al. 2008; Kahn 1998; Muoio and Newgard 2008). At present, the hallmark of each disease appears to be dysfunction of the islet β-cell population, a reduction in total numbers of insulin-producing cells, or both outcomes.
While the specific subclasses of diabetes are delineated by their individual etiological associations accompanying islet β-cell death and dysfunction, there are changes in the genetic programming of β-cells during progression to each form of diabetes that contribute to and influence disease progression (Burke and Collier 2015; Cnop, et al. 2014; Donath 2014; Lopes, et al. 2014; Sanchez, et al. 2015). This transcriptional reprogramming is occasionally overlooked although it is an important contributor to disease progression and outcome. Precise signaling inputs control these genetic effects with the major alterations in gene transcription connected to the production of molecules that regulate cellular viability, influence immune cell recruitment, impact glucose-stimulated calcium dynamics, and eventually reduce maximal glucose-mediated insulin secretion. The end result is an overall diminution in either insulin-positive cell mass, circulating insulin levels, or both. Despite distinct etiologies for T1D and T2D, understanding the pathophysiological mechanisms of each disease will likely benefit the development of therapies to treat both diseases. Thus, we begin by briefly outlining some of the noteworthy similarities between T1D and T2D to illustrate that significant parallels exist between these common endocrine diseases.
Parallels between T1D and T2D
Autoimmunity is a key driver of T1D (Castano and Eisenbarth 1990), while obesity is one of the most important risk factors for the development of T2D (Bray 1992). However, there are a number of commonalities between T1D and T2D that are worth considering. First, insulin resistance is a risk factor for both T1D (Donga, et al. 2015; Fourlanos, et al. 2004) and T2D (Kahn 1998; Samuel and Shulman 2016). Second, pro-inflammatory cytokines (e.g., IL-1β, IFN-γ, TNF-α, etc.) contribute to the disease phenotype in both T1D and T2D (Burke, et al. 2015b; Dogan, et al. 2006; Donath 2014). Third, advanced glycation end products are present in both T1D (Coughlan, et al. 2011; Forbes, et al. 2011) and T2D (Nowotny, et al. 2015). Fourth, insulin therapy is indicated for therapeutic intervention during specific stages of each disease (Atkinson et al. 2014; Home, et al. 2014; Kreider and Lien 2015). Fifth, there are altered circulating levels of various chemokines in both T1D and T2D (Burke and Collier 2015; Corrado, et al. 2014; Hanifi-Moghaddam, et al. 2006; Nunemaker, et al. 2014; Sajadi, et al. 2013; Shigihara, et al. 2006; Takahashi, et al. 2011). Sixth, there are defects in insulin secretion and calcium usage in rodent and human islets associated with T1D and T2D (Boucher, et al. 2004; Burke et al. 2015b; Do, et al. 2014; Dula, et al. 2010; Kenty and Melton 2015; Qureshi, et al. 2015; Ramadan, et al. 2011). Seventh, immune cells infiltrate the islets in both T1D and T2D (Boni-Schnetzler, et al. 2008; Burke, et al. 2016; Ehses, et al. 2007; Gepts 1965; Richardson, et al. 2009; Willcox, et al. 2009). With this last point in mind, we note that excellent reviews exist discussing the immunology of diabetes (Boldison and Wong 2016; Castano and Eisenbarth 1990; Wallberg and Cooke 2013). Therefore, we will focus this review around pancreatic islet β-cell chemokine production with a discussion of two important chemokine receptor signaling paradigms that fundamentally impact specific subsets of immune cells known to participate in the pathogenesis of T1D and T2D.
Chemokines: soluble secreted proteins that regulate immune cell movement and activity
Chemotactic cytokines (aka chemokines) are a family of small (8–10 kDa), secreted, signaling proteins that have biological impact through activation of their specific cell surface receptors. These receptor-mediated actions include the directed chemotaxis of a responsive target cell, changes in intracellular second messengers, and the ability to influence gene expression, protein localization, protein production, and secretion of molecules relevant to immune cell function (Charo and Ransohoff 2006). Chemokines are currently classified by their structural characteristics and are divided into different families based on the spacing of their N-terminal cysteines (i.e., CC, CXC, CX3C, or C). Chemokines as a group are usually sub-classified into homeostatic or pro-inflammatory categories, with many present in both groups. These proteins direct leukocyte migration and influence immune cell activity. Their contribution to development, onset, maintenance, and resolution of various disease processes constitute active research areas.
Chemokines are protein ligands for their cognate chemokine receptors, which are members of the much larger G protein coupled receptor (GPCR) superfamily. Upon ligand binding to the chemokine receptor, a conformational change leads to the exchange of GDP with GTP and the activation of the heterotrimeric G proteins (Pierce, et al. 2002). These heterotrimeric G proteins consist of 21, 6, and 12 different Gα, β, γ subunits, respectively, which contribute to the complexity of signaling outputs. The downstream effects of GPCR activation is dictated by signal strength and subsequent effects on the receptors: internalization (Marchese, et al. 2008), intracellular location (Jiao, et al. 2005), hetero/homo-dimerization(Martinez Munoz, et al. 2009; Trettel, et al. 2003; Wilson, et al. 2005), and G protein subunit usage (Khan, et al. 2013; Syrovatkina, et al. 2016). In addition, the desensitization and inactivation of the intracellular signal also affects signaling outcomes. G protein receptor kinases (GRKs) and β–arrestins turn off the signaling of individual chemokine receptors.
Interestingly, β-arrestins can also induce their own downstream signaling outcomes (Defea 2008; Luttrell and Lefkowitz 2002; Shenoy and Lefkowitz 2005), with the levels of the desensitizing proteins and their isoforms affecting the overall signaling mechanisms. For example, CXCR1 and CXCR2 bind to GRK2 and GRK6, respectively, which produce differences in neutrophil activation (Raghuwanshi, et al. 2012). Discrete activation mechanisms allow for fine-tuning of the chemokine signaling pathways, an important observation bearing in mind there are ~50 associated chemokine ligands that impact ~20 different chemokine receptors. Most of the chemokine receptors display promiscuous interaction with multiple chemokines, which was initially thought to be redundancy in the biological setting. However, in light of recent data on chemokine receptor activation linked to specific biological responses, a more complex picture has formed showing multiple different stimuli at the same receptor trigger differential downstream effects(Zweemer, et al. 2014). In other words, one receptor can produce distinct outcomes depending on which ligand occupies the binding site. This updated paradigm is often referred to as biased signaling, biased agonism, or functional selectivity (Karin, et al. 2016; Wisler, et al. 2014).
In the case of both T1D and T2D, we postulate that the most logical biochemical and immunological explanation for the initial and sustained entry of one or more immune cell populations into pancreatic tissue is directed chemotaxis by specific signaling molecules (e.g., chemokines). Because increased immune cell presence within pancreatic tissue occurs as part of the phenotype underlying T1D and T2D (Boni-Schnetzler et al. 2008; Donath, et al. 2008; Foulis, et al. 1991; Foulis and Stewart 1984; Hanafusa and Imagawa 2008), we and others have focused a significant portion of our research efforts into understanding the inflammation-associated processes related to islet chemokine production and secretion. Below, we provide a brief discussion of signaling through two representative chemokine receptors relevant to innate and adaptive immunity.
Chemokine Ligand Activation of Chemokine Receptors in Innate and Adaptive Immunity
Pancreatic β-cells synthesize and secrete a variety of chemokines capable of recruiting leukocytes into pancreatic tissue (Sarkar, et al. 2012). The expression of many of these chemokines is markedly enhanced by β-cell exposure to inflammatory signals (Burke and Collier 2015; Cnop et al. 2014; Lopes et al. 2014; Sarkar et al. 2012). Below, we focus on ligand-mediated signaling events associated with CXCR2 and CXCR3, which are two specific chemokine receptors reported to influence onset and progression of diabetes. These two receptors are also important because they exemplify enriched expression in neutrophils (CXCR2) and T-cells (CXCR3). Because β-cell exposure to inflammatory mediators induces various chemokine ligands that bind to CXCR2 and CXCR3 (Burke et al. 2016; Burke, et al. 2015a; Burke, et al. 2014; Burke et al. 2015b), we discuss what is known about the activation pathways of these two receptors and the downstream effector mechanisms that contribute to immune activation and subsequent β cell destruction.
CXCR2
CXCR2 is highly expressed on neutrophils (Liu, et al. 2010; Murphy and Tiffany 1991) and oligodendrocytes (Veenstra and Ransohoff 2012). It is expressed less consistently on T cells, basophils, mast cells, and epithelial cells during wound healing. CXCR2 binds ELR-CXC chemokines (CXCL1, CXCL2, CXCL3, CXCL5, CXCL6, CXCL7, and CXCL8) with high affinity). CXCR1 binds CXCL8 with high affinity, while CXCL6 and CXCL7 bind less well to CXCR1 than to CXCR2 (Waugh and Wilson 2008). In the case of CXCR2, this biased agonism (or functional selectivity) was initially thought of as redundancy for receptor activation. However, distinct and complex signaling crosstalk produces different phenotypic responses and is now thought to be important for physiological and pathophysiological outcomes (Zweemer et al. 2014).
For CXCR2 activation, specific chemokines bind and activate the receptor via the amino terminus and first extracellular loop. Site directed mutagenesis in these regions generates different activation and functional consequences (Katancik, et al. 2000; Katancik, et al. 1997). The receptor-associated signal is transduced to the second intracellular loop where the G proteins are bound to the aspartic acid, arginine, and tyrosine (DRY) box and leads to the GDP to GTP exchange and G protein activation. After activation the carboxyl terminus is phosphorylated on the LLKIL motif, which leads to activation of the β-arrestins and internalization/desensitization (Raman, et al. 2014). The receptor is then targeted to different endosomal compartments based on its phosphorylation state for degradation or recycling. These varied signals from CXCR2 stimulation initiate intracellular Ca++ flux, inhibition of apoptosis, migration, priming, and adhesion depending on the downstream signaling pathway that is activated (Mocsai 2013). Neutrophil recruitment via CXCR2 is critical for the autoimmune responses targeting islet β-cells; inhibiting CXCR2 partially blocks this outcome (Citro, et al. 2012; Diana and Lehuen 2014).
Moreover, neutrophils and CXCR2 signaling during diabetes development was shown in the NOD mouse model, which develops T1D spontaneously with many features of the human disease. When CXCR2 was inhibited or neutrophils were depleted, disease progression was limited in these mice (Citro, et al. 2015; Diana, et al. 2013). Additional evidence showing that allosteric inhibitors of CXCR2/CXCR1 lead to prolonged survival of islet transplants in both mice and humans confirms the importance of neutrophil chemokine/chemokine receptor activation during disease onset (Citro et al. 2012). Using neutrophil elastase inhibitors, Talukdar et al. demonstrated that neutrophils also impact obesity-induced insulin resistance (Talukdar, et al. 2012). Taken together, these data illustrate the promise of CXCR2- and/or neutrophil-based immunomodulatory strategies as possible treatments for pathological responses that contribute to the onset of T1D and T2D.
CXCR3
The interferon (IFN)-inducible, non-ELR CXC chemokine subgroup includes CXCL9 (Mig), CXCL10 (IP10) and CXCL11 (I-TAC), which are proteins that bind to CXCR3 (Groom and Luster 2011). This receptor is mainly found on NK cells and T cells and has a role in a variety of diseases, especially those with a Th component (Van Raemdonck, et al. 2015). When CXCL10 is expressed in the islets of transgenic mice, an accelerated autoreactive immunological response was observed (Rhode, et al. 2005). In addition, islet isografts were protected when CXCL10 expression was diminished (Bender, et al. 2017). Serum CXCL10 levels are increased in both human subjects and in the NOD mouse model, linking elevated chemokine production with diabetes (Antonelli, et al. 2014; Christen, et al. 2003; Corrado et al. 2014). When CXCR3 is knocked out, there is a delay in the onset of virally induced diabetes (Frigerio, et al. 2002) and in diabetes induced by multiple low doses of streptozotocin (Burke et al. 2016). By contrast, when CXCR3 null mice are crossed onto the NOD background, TID development is accelerated (Yamada, et al. 2012). What appear to be contrary findings could be due to genetic differences in the different mouse strains, the distinct models of T1D, or functional selectivity of CXCR3 activation that is related to strain and/or the models of diabetes.
It is important to note that mice on a C57BL/6 background do not produce CXCL11 due to an insertion of 2 bases after nucleotide 39, which shifts the open reading frame of the CXCL11-encoding gene to a premature stop codon (Sierro, et al. 2007). By contrast, the NOD mice retain the ability to produce CXCL11, which could explain differences in whole body phenotypes observed between knockouts of CXCR3 when compared with C57BL/6 mice. In the viral model of T1D, knocking out ligands binding to CXCR3 showed redundancy instead of biased agonism, i.e., knocking out one of the CXCR3 ligands had no effect (Coppieters, et al. 2013), leaving us to speculate how the individual ligands contribute to onset and progression diabetes. Along these lines, the different chemokine ligands that bind CXCR3 show biased agonism during in vitro studies. Watts et al. used impedance measurements, BRET, and CRE luciferase assays to show that CXCL9 induces β-arrestin binding and produces higher impedance measurements when compared with CXCL10 and CXCL11 (Watts, et al. 2012). Conversely, Rajagopal et al. showed bias only for CXCR3 for internalization, with CXCL11 the more potent agonist when compared with CXCL9 and CXCL10 (Rajagopal 2013; Rajagopal, et al. 2013). Although the signaling pathways activated after differential ligand binding have not been fully elucidated, there are some clues about CXCR3 signal transduction cascades that are informative. For example, the differential downstream signaling is due, at least in part, to differential Gα subunit usage (Kouroumalis, et al. 2005; Thompson, et al. 2007). CXCL10 stimulation of CXCR3 also leads to activation of the ERK/Ras pathway and an increase in migration and signaling (Bonacchi, et al. 2001).
Decoding the chemokine receptor signaling pathways could allow for drugs that have functional selectivity for some of the beneficial phenotypes while ideally eliminating the detrimental outcomes. One clue to the importance of specific pathways on diabetes developments came from nucleotide-binding leucine-rich repeat and pyrin domain containing protein 3 (NLRP3) knockout mice; NLRP3 functions as part of the innate immunity system via pattern recognition responses (Martinon 2008). In NLRP3−/− mice, expression of CXCR3, and the corresponding ligands, CXCL9 and CXCL10, were decreased (Hu, et al. 2015). The expression of other chemokines and their receptors was also reduced. Importantly, diabetes took longer to develop in NLRP3−/− mice, implying that these chemokines and associated receptors contribute to T1D development.
On a related note, the small molecule CXCR3 antagonist, SCH 546738, was shown to prevent autoimmune diseases (Jenh, et al. 2012), while another CXCR3 inhibitor, NBI-74330, had distinct potencies for the different CXCR3 ligands (Heise, et al. 2005). Whether this information regarding functional selectivity will be useful for the design of new therapeutics aimed at modulating human CXCR3 pathways during autoimmune diseases remains to be revealed. It will be of particular interest to determine whether any of the existing molecules targeting chemokine receptors directly have therapeutic benefit in the different models of diabetes.
Islet-derived chemokines in T1D
Genes encoding chemokines are located within the known diabetes susceptibility locus Idd4 in the NOD mouse, which is associated with T1D development (Grattan, et al. 2002). While the initial trigger(s) prompting the onset of β-cell directed autoimmunity is unknown, enhanced production and release of chemokines from pancreatic islets coupled with the associated chemokine receptor activation on individual leukocytes is a very plausible component of the disease course. The following components are proposed to be contributors to T1D development: 1) Secretion of cytokines and chemokines by resident immune cells (e.g., macrophages) in response to a specific pathogen or pathogenic signal (e.g., death of surrounding cells, viral infection, LPS, etc.). 2) The production of islet-derived chemokines increase in response to such signals, promoting the recruitment of additional immune cell populations (e.g, neutrophils, T-cells, etc.) into the pancreatic islets. 3) T-cell priming for one or more antigens (typically β-cell specific). 4) Chemokine-mediated entry of primed lymphocytes into the pancreatic tissue. 5) Continued production and release of cytokines (e.g., IL-1β, IFN-γ, etc.) that regulate inflammatory responses, including immune cell activity, within pancreatic islets. 6) Sustained production of chemokines, including from pancreatic β-cells, creating a vicious feed forward cycle intended to either clear infection or promote tissue repair (physiological outcome) that transitions during chronic disease states (e.g., autoimmunity) to become pathophysiological. Portions of this model have been validated using transgenic mouse models where production of CCL2 or CXCL10 directly from β-cells stimulates immune cell entry into the pancreatic tissue (Grewal, et al. 1997; Martin, et al. 2008b; Rhode et al. 2005) and by reducing chemokine action via decoy receptor expression in NOD mice (Martin, et al. 2008a).
T1D arises through multiple immune cell interactions, including macrophages, neutrophils, and T-cells (Calderon, et al. 2006; Diana et al. 2013). This means various innate and adaptive immune cells, and molecules secreted from such cells, all participate and contribute in some capacity to the disease pathology. Thus, signals contributing to immune cell recruitment, such as chemokines, are critical players in initiating, accelerating, and/or maintaining disease trajectory. Consistent with this idea, IL-1α and TNF-α are elevated in ‘at risk’ and new onset T1D subjects when compared with healthy controls (Chatzigeorgiou, et al. 2010; Rosa, et al. 2008; Zak, et al. 2010). IL-1α and TNF-α signal through the IL-1 receptor and TNF receptor, respectively, both of which activate the NF-κB pathway. NF-κB activation drives production of multiple chemokines in islet β-cells (Burke and Collier 2014, 2015) and reduces insulin secretion in rodent and human islets (Burke et al. 2015b; Giannoukakis, et al. 2000; Rehman, et al. 2003; Rink, et al. 2012). Conversely, restricting NF-κB activity in the multiple low dose streptozotocin (MLDS) models reduces chemokine production and protects against hyperglycemia (Eldor, et al. 2006). In an alternative approach, blocking chemokine action using decoy receptors also revealed key roles for chemokine proteins in the pathogenesis of diabetes development (Martin, et al. 2007; Martin et al. 2008a).
When examining rodent models of T1D (see Tables 1 and 2), the production and release of CXCL9 and CXCL10 protein in islets from 4 and 10 week old NOD mice correlated with the degree of insulitis (Welzen-Coppens, et al. 2013). Blocking CXCL10 activity with neutralizing antibodies prevented re-infiltration of pancreatic islets after a course of anti-CD3 therapy (Lasch, et al. 2015). Because T-cells primed with autoantigens typically produce and secrete IFN-γ, targeted upregulation of CXCL10 by IFN-γ signaling in β-cells is likely contributing to T1D disease pathology (Burke, et al. 2013a; Burke et al. 2016; Lundberg, et al. 2016). The enhanced transcription of the CXCL10 gene is also consistent with heightened STAT1 activity in islet β-cells (Burke et al. 2013a; Lundberg et al. 2016). Moreover, pro-inflammatory cytokines (e.g., IL-1β, IFN-γ, etc.) increase the expression of CXCL9 and CXCL11 in mouse, rat, and human β-cells using activated STAT1 as a key component of the signaling response (Burke et al. 2016). STAT1 is therefore a key driver of chemokine production (Burke et al. 2013a; Burke et al. 2016) and inducible nitric oxide synthase abundance and activity (Burke et al. 2015b; Burke, et al. 2013b; Corbett, et al. 1993; Corbett, et al. 1992; Heitmeier, et al. 1997). Increased STAT1 expression is also strongly correlated with HLA class I (i.e. HLA-ABC) and HLA-F isoforms in insulin-containing islets (Richardson, et al. 2016). Thus, NF-κB and STAT1 likely cooperate to control inflammatory responses within pancreatic islets, which ultimately determine the autoimmune and auto-inflammatory outcomes (Figure 1). The model shown in Figure 1 offers an attractive conceptual framework from which to explain why individual immunomodulatory approaches may have underachieved from a therapeutic perspective.
Table 1.
Islet Chemokines in Monogenic Rodent Models of Obesity and Diabetes.
| Animal Model (species) | Relevant Human Condition | Islet Chemokine Production | References |
|---|---|---|---|
| Akita (mouse) | ER stress/Insulin insufficiency | No* | Burke, Karlstad, & Collier, unpublished |
| db/db (mouse) | Obesity/T2D | Yes | (Burke et al. 2015a; Eguchi et al. 2012) |
| KKAy (mouse) | Obesity/T2D | Yes | (Eguchi et al. 2012) |
| ZDF (rat) | Obesity/T2D | Yes | (Jourdan, et al. 2013) |
male mice at 8 weeks of age.
Table 2.
Islet Chemokines in Polygenic Rodent Models of Obesity and Diabetes.
| Animal Model (species) | Relevant Human Condition | Islet Chemokine Production | References |
|---|---|---|---|
| BB (rat) | Autoimmunity/T1D | Yes | (Kuttler, et al. 2007) |
| GK (rat) | Insulin Resistance/T2D | Yes | (Ehses et al. 2009) |
| MLDS (mouse/rat)* | Insulin insufficiency/T1D | Yes | (Burke et al. 2016; Martin et al. 2007; Martin et al. 2008a) |
| NOD (mouse) | Autoimmunity/T1D | Yes | (Burke et al. 2016; Martin et al. 2008a; Welzen-Coppens et al. 2013) |
, MLDS, multiple low doses of streptozotocin (STZ). STZ impacts multiple mouse and rat strains; thus, we have included it under the polygenic models.
Figure 1. Signals that activate NF-κB and STAT1 enhance chemokine production within pancreatic islets that contribute to the inflammatory response influencing both insulin secretion and total β-cell mass.
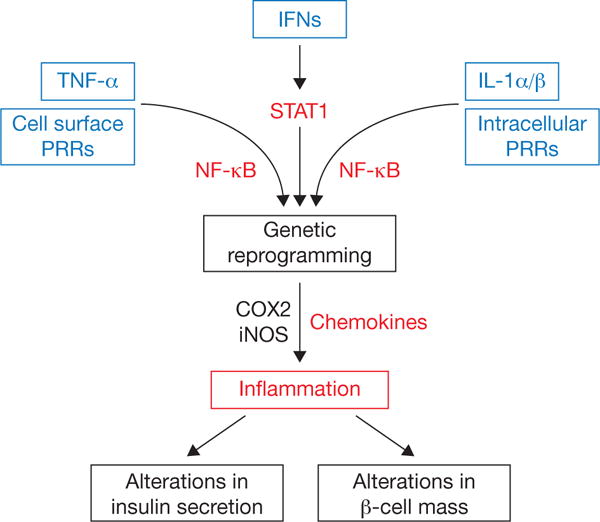
Many signals converge on the NF-κB and STAT1 pathways to coordinately reprogram beta cells at a transcriptional level, leading to inflammation-based changes in insulin secretion, β-cell mass, or both. PRRs can be either cell surface based, such as Toll-like receptor-2 and -4 or positioned intracellularly (e.g., Toll-like receptor-3, NOD1, NOD2, etc.). TNF-α and IL-1 signal through specific cell surface receptors (receptor not shown) and converge on the NF-κB pathway. The interferon family of cytokines signals through cell surface receptors that activate JAK-STAT pathways. Activation of COX2 produces prostaglandins, which may influence leukocyte activity. Cytokine-mediated increases in iNOS elevate intracellular production of nitric oxide, which acts as a rheostat for insulin secretion. COX2: cyclooxygenase-2; IFNs: interferons alpha, beta, and gamma; iNOS: inducible nitric oxide synthase; PRR: pattern recognition receptors.
One such example of a promising single immunomodulatory strategy that has thus far failed to produce remission, or prevent T1D, in mice is anti-IL-1β neutralization (Gill, et al. 2015). Although it was promising that human subjects with recent onset T1D receiving a targeted anti-IL-1 therapeutic (Anakinra) displayed reduced insulin requirements one and four months after diagnosis compared to controls (Sumpter, et al. 2011), in a larger study, IL-1 neutralization strategies were not effective at meeting the clinical endpoints (Moran, et al. 2013). It is worth noting however, that one month after diagnosis the insulin-dose-adjusted A1c in patients given Anakinra was lower than controls (Sumpter et al. 2011).
Multiple explanations might exist to explain these initially disappointing findings. The first is that the neutralization or trap strategies do not completely block signaling through the IL-1 receptor, which has two known agonist ligands (i.e., IL-1α and IL-1β). The second explanation is that other signals, such as TNF-α or ligands that activate pattern recognition receptors (PRR), can still activate NF-κB despite interventions that restrict signaling through the IL-1 receptor (Figure 1). The third explanation is that signaling through specific receptors, such as IL-1R1, may have both physiological and pathological outcomes and that receptor antagonism eliminates both positive and negative outcomes.
We note that NF-κB activation by a variety of inflammatory pathways, in conjunction with elevated levels of STAT1 and/or enhanced STAT1 transcriptional responses, would likely support sustained production of soluble factors (e.g., chemokines) that influence immune cell recruitment. In addition, the powerful combination of NF-κB and STAT1 activation regulates other key genes relevant to islet β-cell function and dysfunction (Figure 1). Consequently, in our view, combination therapies may end up providing the most benefit to prevention of autoimmune diseases, such as T1D, by restricting signaling of multiple pathways that collectively contribute to chemokine production, and other important mediators of inflammation, which ultimately regulate islet β-cell function and mass.
Islet-derived chemokines in T2D
Multiple models of rodent obesity and diabetes display elevations in islet chemokine production (Tables 1 and 2). In addition, staining of human pancreatic tissue revealed high levels of CXCL10 in islets from subjects with T2D (Schulthess, et al. 2009) while RNA sequencing approaches showed the expression of multiple chemokines in human islets exposed to the fatty acid palmitate (Cnop et al. 2014). These data are consistent with enhanced leukocyte presence in human pancreatic tissue during obesity (Boni-Schnetzler et al. 2008; Donath et al. 2008). Schultess and colleagues suggest that CXCL10 signals through TLR4 on β-cells as a mechanism for deleterious effects during obesity (Schulthess et al. 2009). While an intriguing possibility, these observations still await independent verification.
On the other hand, genetic deletion of the CX3CR1 receptor reduces insulin secretion in response to the physiological signals glucose and GLP-1(Lee, et al. 2013). The chemokine CX3CL1 (aka fractalkine), which signals through CX3CR1, is decreased in islets during aging and obesity (Lee et al. 2013). It is therefore possible that particular chemokines promote β-cell health, either by recruiting specific immune cells, through direct effects on β-cells, or both. In addition to mechanisms that support leukocyte recruitment and production of pro-inflammatory cytokines, loss of these “protective” or homeostatic chemokines might be a mechanism which also contributes to β-cell dysfunction during progression to T2D.
Obesity and Islet Inflammation
How does obesity trigger islet inflammation? Since no one mechanism has emerged to explain all observations, we put forth two conceptual possibilities. The first is that obesity negatively influences gut barrier function, allowing translocation of bacterial products initially into the mesenteric lymphatic system and then into the systemic circulation (Figure 2A). This elevation in lipopolysaccharide (LPS), or other molecules that signal through pattern recognition receptors, may contribute to the subclinical, low grade inflammation that drives insulin resistance and associated tissue dysfunction (Carneiro, et al. 2008; Cox, et al. 2015; Purohit, et al. 2013). With this in mind, it is conceivable that islet resident macrophages become activated during obesity due to elevated levels of circulating LPS derived from increased intestinal permeability (i.e., “leaky gut”). The presence of LPS increases macrophage cytokine production and also enhances macrophage sensitivity to IFN-γ (Held, et al. 1999).
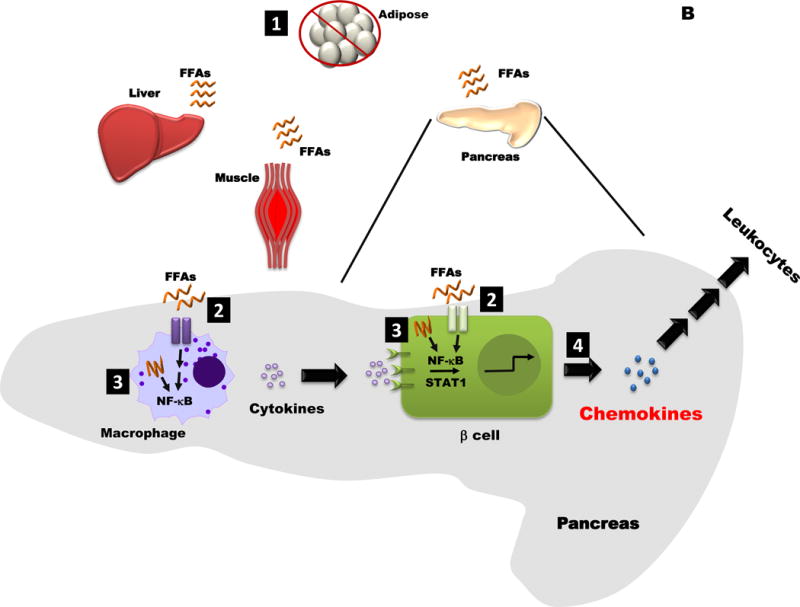
Figure 2. Conceptual Models for how Obesity may influence Islet Inflammation and Chemokine Production.
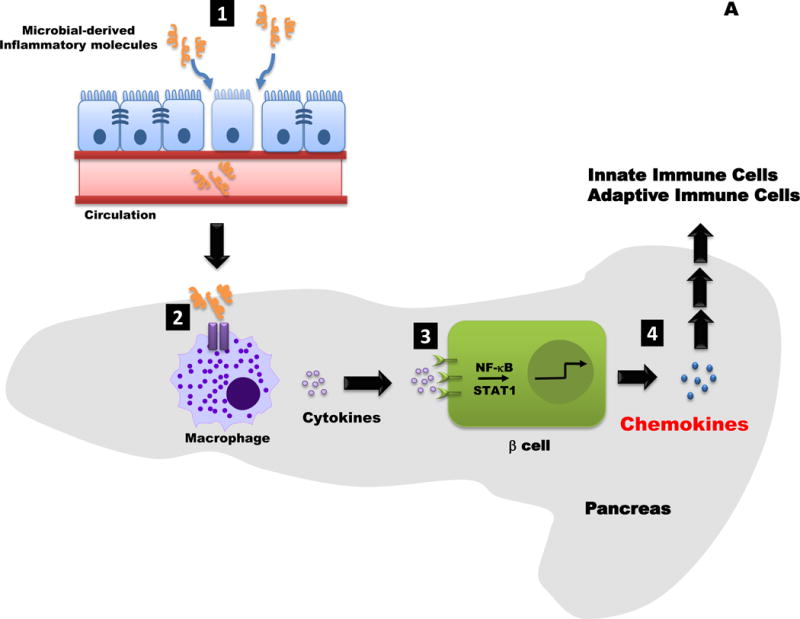
A. 1. Increased intestinal permeability allows microbial products (e.g., LPS) to enter the circulation. 2. These molecules are “sensed” by pattern recognition receptor enriched in immune cells, such as neutrophils (not shown) and macrophages (shown). An increase in macrophage activation leads to production and release of cytokines (e.g., IL-1β). 3. Distinct pro-inflammatory cytokines (e.g., IL-1β, IFN-γ, TNF-α, etc.) promote transcriptionally active NF-κB and STAT1 proteins. 4. The NF-κB and STAT1 pathways regulate chemokine production in beta-cells; release of β-cell derived chemokines influences innate and adaptive immune responses.
B. 1. When consumption of free fatty acids (FFAs) exceeds storage capacity in adipose tissue, these lipids begin to be stored in lean tissues (e.g. liver, muscle, and pancreas). 2. FFAs cell surface receptors, altering intracellular signaling pathways. 3. Incomplete fatty acid oxidation may also activate signaling pathways, such as NF-kB, that are linked with transcriptional changes. 4. Chemokine production is elevated in the obese, insulin resistant state, which would impact both tissue resident and infiltrating leukocytes.
IFN-γ is a cytokine that activates macrophages and also promotes the differentiation of immature DCs into effector DCs that heavily influence Th1 responses (Boehm, et al. 1997). These biological responses are intended to induce robust anti-bacterial activity, but may be dysregulated during obesity. Indeed, CXCL8, CXCL10, and IFN-γ increase in circulation with excess body weight (Sharabiani, et al. 2011; Straczkowski, et al. 2002). Consequently, it is plausible that bacterial translocation from a leaky gut results in a low level of LPS that, in combination with elevations in other circulating factors linked with inflammation, promotes detrimental changes in pancreatic islets. Macrophage cytokine production is a key component that impacts pancreatic islet function and mass (Figure 2A). While speculative, this mechanism fits with existing observations put forth in the literature (Balzan, et al. 2007; Cani, et al. 2007; Cani and Delzenne 2007).
The second conceptual possibility explaining how obesity leads to islet inflammation is lipid overload and associated metabolic trauma (Figure 2B). In this model, lipids accumulate in lean tissues once storage in adipose tissue has been exceeded (Unger 2003; Unger and Orci 2000). Lean tissue responses to surplus fatty acids may vary. For example, fatty acids promote macrophage activation as well as chemokine production in pancreatic islets (Eguchi, et al. 2012), potentially via cell surface (e.g., TLR4) or intracellular (e.g., NOD1, NOD2, etc.) pattern recognition receptor signaling (Figure 1). Moreover, incomplete fatty acid oxidation products drive enhanced NF-κB activity in macrophages, promoting pro-inflammatory actions (Rutkowsky, et al. 2014). If such incomplete fatty acid oxidation mechanisms also occur in β-cells, it could help to explain lipid-induced changes in β-cell insulin secretion as well as chemokine production that occur in obesity (Burke et al. 2015b; Eguchi et al. 2012; Ehses, et al. 2009). This is important because the increase in β-cell chemokine production by inflammatory signals coincides with diminutions in insulin secretion (Burke et al. 2015b).
Moreover, accumulation of excess lipid within β-cells impairs insulin secretion, at least in part, by interfering with metabolic coupling steps, such as pyruvate cycling (Boucher et al. 2004). Reducing the lipid burden or restoring pyruvate cycling improves islet β-cell secretory function (Boucher et al. 2004; Shimabukuro, et al. 1998). Finally, intracellular lipid accumulation in the islet β-cell may also result in ER stress. Induction of mild ER stress induces β-cell proliferation (Sharma, et al. 2015), while lipid signaling through pattern recognition receptors likely reduces β-cell function and promotes production of molecules, such as chemokines and cytokines, that modulate immune cell recruitment and inflammation (Boni-Schnetzler, et al. 2009).
Thus, multiple signaling mechanisms may exist to limit insulin secretion while augmenting chemokine production within pancreatic islets (and other tissues), driving increased immune cell infiltration and therefore influencing the status of pancreatic inflammation. Because increased numbers of immune cells have been observed within pancreatic islets from both rodents and humans during obesity validate these findings as conserved, important phenotypes (Boni-Schnetzler et al. 2008; Donath et al. 2008; Ehses et al. 2007). However, it is important to note that any immune cell entry into the pancreatic tissue is often heterogeneous. In addition, changes in the tissue resident immune cell activation state (e.g., M2 → M1), without enhancements in immune cell number, could also drive inflammation-mediated changes in tissue function (Burke and Collier 2014).
Consistent with changes in leukocyte number or activation state, increased presence of chemokine ligands has been documented in pancreatic islets of mice, rats, and humans during obesity (Burke et al. 2015a; Donath et al. 2008; Eguchi et al. 2012; Sajadi et al. 2013; Schulthess et al. 2009). While there is no doubt that obesity is a major risk factor for the development of T2D, the molecular determinants explaining the increased risk are still being investigated. Our view is that pancreatic islet-derived chemokines play critical roles by regulating immune cell flux into the pancreatic islets, influencing the activation state of resident immune cells, or both. These outcomes ultimately impact β-cell substrate metabolism linked with insulin secretion as well as influencing proliferation of insulin-producing cells.
Summary and Conclusion
A role for elevated chemokine production within β-cells as a critical mechanistic determinant controlling islet inflammation during T1D and T2D development is emerging (Table 1 and Table 2). Chemokines, and other proteins that regulate inflammation, can be induced by a variety of signals that activate either STAT1, NF-κB, or both signaling pathways (see Figure 1). The increased synthesis and secretion of discrete chemokines induce receptor-mediated signals that regulate immune cell entry into, and activity within, pancreatic tissue (Figure 2). Importantly, chemokine production in pancreatic islets appears to be a common phenomenon between T1D and T2D. While rodent work has been incredibly informative towards understanding the genetics and phenotypes of the human disease, much remains to be uncovered. It is our hope that understanding the inflammatory signaling-induced upregulation of chemokine production and chemokine/chemokine receptor interactions will assist in the design of therapeutics relevant to improving β-cell mass and secretory function as a way to combat the growing incidence of T1D and T2D.
Acknowledgments
Funding
Work in the authors’ laboratories was supported by NIH grants P20 - GM103528 (J.J.C.), P30 - GM118430 (J.J.C.), R44 - GM099207 (J.J.C. and M.D.K.) and R01 - AI071042 (T.E.S.).
Footnotes
Disclosures: None.
References
- Antonelli A, Ferrari SM, Corrado A, Ferrannini E, Fallahi P. CXCR3, CXCL10 and type 1 diabetes. Cytokine Growth Factor Rev. 2014;25:57–65. doi: 10.1016/j.cytogfr.2014.01.006. [DOI] [PubMed] [Google Scholar]
- Atkinson MA, Eisenbarth GS, Michels AW. Type 1 diabetes. Lancet. 2014;383:69–82. doi: 10.1016/S0140-6736(13)60591-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balzan S, de Almeida Quadros C, de Cleva R, Zilberstein B, Cecconello I. Bacterial translocation: overview of mechanisms and clinical impact. J Gastroenterol Hepatol. 2007;22:464–471. doi: 10.1111/j.1440-1746.2007.04933.x. [DOI] [PubMed] [Google Scholar]
- Bender C, Christen S, Scholich K, Bayer M, Pfeilschifter JM, Hintermann E, Christen U. Islet-Expressed CXCL10 Promotes Autoimmune Destruction of Islet Isografts in Mice With Type 1 Diabetes. Diabetes. 2017;66:113–126. doi: 10.2337/db16-0547. [DOI] [PubMed] [Google Scholar]
- Boehm U, Klamp T, Groot M, Howard JC. Cellular responses to interferon-gamma. Annu Rev Immunol. 1997;15:749–795. doi: 10.1146/annurev.immunol.15.1.749. [DOI] [PubMed] [Google Scholar]
- Boldison J, Wong FS. Immune and Pancreatic beta Cell Interactions in Type 1 Diabetes. Trends Endocrinol Metab. 2016;27:856–867. doi: 10.1016/j.tem.2016.08.007. [DOI] [PubMed] [Google Scholar]
- Bonacchi A, Romagnani P, Romanelli RG, Efsen E, Annunziato F, Lasagni L, Francalanci M, Serio M, Laffi G, Pinzani M, et al. Signal transduction by the chemokine receptor CXCR3: activation of Ras/ERK, Src, and phosphatidylinositol 3-kinase/Akt controls cell migration and proliferation in human vascular pericytes. J Biol Chem. 2001;276:9945–9954. doi: 10.1074/jbc.M010303200. [DOI] [PubMed] [Google Scholar]
- Boni-Schnetzler M, Boller S, Debray S, Bouzakri K, Meier DT, Prazak R, Kerr-Conte J, Pattou F, Ehses JA, Schuit FC, et al. Free fatty acids induce a proinflammatory response in islets via the abundantly expressed interleukin-1 receptor I. Endocrinology. 2009;150:5218–5229. doi: 10.1210/en.2009-0543. [DOI] [PubMed] [Google Scholar]
- Boni-Schnetzler M, Ehses JA, Faulenbach M, Donath MY. Insulitis in type 2 diabetes. Diabetes Obes Metab. 2008;10(Suppl 4):201–204. doi: 10.1111/j.1463-1326.2008.00950.x. [DOI] [PubMed] [Google Scholar]
- Boucher A, Lu D, Burgess SC, Telemaque-Potts S, Jensen MV, Mulder H, Wang MY, Unger RH, Sherry AD, Newgard CB. Biochemical mechanism of lipid-induced impairment of glucose-stimulated insulin secretion and reversal with a malate analogue. J Biol Chem. 2004;279:27263–27271. doi: 10.1074/jbc.M401167200. [DOI] [PubMed] [Google Scholar]
- Bray GA. Obesity increases risk for diabetes. Int J Obes Relat Metab Disord. 1992;16(Suppl 4):S13–17. [PubMed] [Google Scholar]
- Burke SJ, Collier JJ. Insulitis and Diabetes: A Perspective on Islet Inflammation. Immunome Research. 2014;10 Special Issue: Cytokine Biology. [Google Scholar]
- Burke SJ, Collier JJ. Transcriptional Regulation of Chemokine Genes: A Link to Pancreatic Islet Inflammation? Biomolecules. 2015;5:1020–1034. doi: 10.3390/biom5021020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke SJ, Goff MR, Lu D, Proud D, Karlstad MD, Collier JJ. Synergistic Expression of the CXCL10 Gene in Response to IL-1beta and IFN-gamma Involves NF-kappaB, Phosphorylation of STAT1 at Tyr701, and Acetylation of Histones H3 and H4. J Immunol. 2013a;191:323–336. doi: 10.4049/jimmunol.1300344. [DOI] [PubMed] [Google Scholar]
- Burke SJ, Karlstad MD, Eder AE, Regal KM, Lu D, Burk DH, Collier JJ. Pancreatic beta-Cell production of CXCR3 ligands precedes diabetes onset. Biofactors. 2016;42:703–715. doi: 10.1002/biof.1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke SJ, Karlstad MD, Regal KM, Sparer TE, Lu D, Elks CM, Grant RW, Stephens JM, Burk DH, Collier JJ. CCL20 is elevated during obesity and differentially regulated by NF-kappaB subunits in pancreatic beta-cells. Biochim Biophys Acta. 2015a;1849:637–652. doi: 10.1016/j.bbagrm.2015.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke SJ, Lu D, Sparer TE, Masi T, Goff MR, Karlstad MD, Collier JJ. NF-kappaB and STAT1 control CXCL1 and CXCL2 gene transcription. Am J Physiol Endocrinol Metab. 2014;306:E131–149. doi: 10.1152/ajpendo.00347.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke SJ, Stadler K, Lu D, Gleason E, Han A, Donohoe DR, Rogers RC, Hermann GE, Karlstad MD, Collier JJ. IL-1beta reciprocally regulates chemokine and insulin secretion in pancreatic beta-cells via NF-kappaB. Am J Physiol Endocrinol Metab. 2015b;309:E715–726. doi: 10.1152/ajpendo.00153.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke SJ, Updegraff BL, Bellich RM, Goff MR, Lu D, Minkin SC, Jr, Karlstad MD, Collier JJ. Regulation of iNOS Gene Transcription by IL-1beta and IFN-gamma Requires a Coactivator Exchange Mechanism. Mol Endocrinol. 2013b;27:1724–1742. doi: 10.1210/me.2013-1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calderon B, Suri A, Unanue ER. In CD4+ T-cell-induced diabetes, macrophages are the final effector cells that mediate islet beta-cell killing: studies from an acute model. Am J Pathol. 2006;169:2137–2147. doi: 10.2353/ajpath.2006.060539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cani PD, Amar J, Iglesias MA, Poggi M, Knauf C, Bastelica D, Neyrinck AM, Fava F, Tuohy KM, Chabo C, et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes. 2007;56:1761–1772. doi: 10.2337/db06-1491. [DOI] [PubMed] [Google Scholar]
- Cani PD, Delzenne NM. Gut microflora as a target for energy and metabolic homeostasis. Curr Opin Clin Nutr Metab Care. 2007;10:729–734. doi: 10.1097/MCO.0b013e3282efdebb. [DOI] [PubMed] [Google Scholar]
- Carneiro LA, Magalhaes JG, Tattoli I, Philpott DJ, Travassos LH. Nod-like proteins in inflammation and disease. J Pathol. 2008;214:136–148. doi: 10.1002/path.2271. [DOI] [PubMed] [Google Scholar]
- Castano L, Eisenbarth GS. Type-I diabetes: a chronic autoimmune disease of human, mouse, and rat. Annu Rev Immunol. 1990;8:647–679. doi: 10.1146/annurev.iy.08.040190.003243. [DOI] [PubMed] [Google Scholar]
- Cefalu WT. “Prediabetes”: Are There Problems With This Label? No, We Need Heightened Awareness of This Condition! Diabetes Care. 2016;39:1472–1477. doi: 10.2337/dc16-1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cefalu WT, Petersen MP, Ratner RE. The alarming and rising costs of diabetes and prediabetes: a call for action! Diabetes Care. 2014;37:3137–3138. doi: 10.2337/dc14-2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charo IF, Ransohoff RM. The many roles of chemokines and chemokine receptors in inflammation. N Engl J Med. 2006;354:610–621. doi: 10.1056/NEJMra052723. [DOI] [PubMed] [Google Scholar]
- Chatzigeorgiou A, Harokopos V, Mylona-Karagianni C, Tsouvalas E, Aidinis V, Kamper EF. The pattern of inflammatory/anti-inflammatory cytokines and chemokines in type 1 diabetic patients over time. Ann Med. 2010;42:426–438. doi: 10.3109/07853890.2010.495951. [DOI] [PubMed] [Google Scholar]
- Christen U, McGavern DB, Luster AD, von Herrath MG, Oldstone MB. Among CXCR3 chemokines, IFN-gamma-inducible protein of 10 kDa (CXC chemokine ligand (CXCL) 10) but not monokine induced by IFN-gamma (CXCL9) imprints a pattern for the subsequent development of autoimmune disease. J Immunol. 2003;171:6838–6845. doi: 10.4049/jimmunol.171.12.6838. [DOI] [PubMed] [Google Scholar]
- Citro A, Cantarelli E, Maffi P, Nano R, Melzi R, Mercalli A, Dugnani E, Sordi V, Magistretti P, Daffonchio L, et al. CXCR1/2 inhibition enhances pancreatic islet survival after transplantation. J Clin Invest. 2012;122:3647–3651. doi: 10.1172/JCI63089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Citro A, Valle A, Cantarelli E, Mercalli A, Pellegrini S, Liberati D, Daffonchio L, Kastsiuchenka O, Ruffini PA, Battaglia M, et al. CXCR1/2 inhibition blocks and reverses type 1 diabetes in mice. Diabetes. 2015;64:1329–1340. doi: 10.2337/db14-0443. [DOI] [PubMed] [Google Scholar]
- Cnop M, Abdulkarim B, Bottu G, Cunha DA, Igoillo-Esteve M, Masini M, Turatsinze JV, Griebel T, Villate O, Santin I, et al. RNA sequencing identifies dysregulation of the human pancreatic islet transcriptome by the saturated fatty acid palmitate. Diabetes. 2014;63:1978–1993. doi: 10.2337/db13-1383. [DOI] [PubMed] [Google Scholar]
- Coppieters KT, Amirian N, Pagni PP, Baca Jones C, Wiberg A, Lasch S, Hintermann E, Christen U, von Herrath MG. Functional redundancy of CXCR3/CXCL10 signaling in the recruitment of diabetogenic cytotoxic T lymphocytes to pancreatic islets in a virally induced autoimmune diabetes model. Diabetes. 2013;62:2492–2499. doi: 10.2337/db12-1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbett JA, Sweetland MA, Wang JL, Lancaster JR, Jr, McDaniel ML. Nitric oxide mediates cytokine-induced inhibition of insulin secretion by human islets of Langerhans. Proc Natl Acad Sci U S A. 1993;90:1731–1735. doi: 10.1073/pnas.90.5.1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbett JA, Wang JL, Sweetland MA, Lancaster JR, Jr, McDaniel ML. Interleukin 1 beta induces the formation of nitric oxide by beta-cells purified from rodent islets of Langerhans. Evidence for the beta-cell as a source and site of action of nitric oxide. J Clin Invest. 1992;90:2384–2391. doi: 10.1172/JCI116129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corrado A, Ferrari SM, Ferri C, Ferrannini E, Antonelli A, Fallahi P. Type 1 diabetes and (C-X-C motif) ligand (CXCL) 10 chemokine. Clin Ter. 2014;165:e181–185. doi: 10.7471/CT.2014.1706. [DOI] [PubMed] [Google Scholar]
- Coughlan MT, Yap FY, Tong DC, Andrikopoulos S, Gasser A, Thallas-Bonke V, Webster DE, Miyazaki J, Kay TW, Slattery RM, et al. Advanced glycation end products are direct modulators of beta-cell function. Diabetes. 2011;60:2523–2532. doi: 10.2337/db10-1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox AJ, West NP, Cripps AW. Obesity, inflammation, and the gut microbiota. Lancet Diabetes Endocrinol. 2015;3:207–215. doi: 10.1016/S2213-8587(14)70134-2. [DOI] [PubMed] [Google Scholar]
- Defea K. Beta-arrestins and heterotrimeric G-proteins: collaborators and competitors in signal transduction. Br J Pharmacol. 2008;153(Suppl 1):S298–309. doi: 10.1038/sj.bjp.0707508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diana J, Lehuen A. Macrophages and beta-cells are responsible for CXCR2-mediated neutrophil infiltration of the pancreas during autoimmune diabetes. EMBO Mol Med. 2014;6:1090–1104. doi: 10.15252/emmm.201404144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diana J, Simoni Y, Furio L, Beaudoin L, Agerberth B, Barrat F, Lehuen A. Crosstalk between neutrophils, B-1a cells and plasmacytoid dendritic cells initiates autoimmune diabetes. Nat Med. 2013;19:65–73. doi: 10.1038/nm.3042. [DOI] [PubMed] [Google Scholar]
- Do OH, Low JT, Gaisano HY, Thorn P. The secretory deficit in islets from db/db mice is mainly due to a loss of responding beta cells. Diabetologia. 2014;57:1400–1409. doi: 10.1007/s00125-014-3226-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dogan Y, Akarsu S, Ustundag B, Yilmaz E, Gurgoze MK. Serum IL-1beta, IL-2, and IL-6 in insulin-dependent diabetic children. Mediators Inflamm. 2006;2006:59206. doi: 10.1155/MI/2006/59206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donath MY. Targeting inflammation in the treatment of type 2 diabetes: time to start. Nat Rev Drug Discov. 2014;13:465–476. doi: 10.1038/nrd4275. [DOI] [PubMed] [Google Scholar]
- Donath MY, Schumann DM, Faulenbach M, Ellingsgaard H, Perren A, Ehses JA. Islet inflammation in type 2 diabetes: from metabolic stress to therapy. Diabetes Care. 2008;31(Suppl 2):S161–164. doi: 10.2337/dc08-s243. [DOI] [PubMed] [Google Scholar]
- Donga E, Dekkers OM, Corssmit EP, Romijn JA. Insulin resistance in patients with type 1 diabetes assessed by glucose clamp studies: systematic review and meta-analysis. Eur J Endocrinol. 2015;173:101–109. doi: 10.1530/EJE-14-0911. [DOI] [PubMed] [Google Scholar]
- Doria A, Patti ME, Kahn CR. The emerging genetic architecture of type 2 diabetes. Cell Metab. 2008;8:186–200. doi: 10.1016/j.cmet.2008.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dula SB, Jecmenica M, Wu R, Jahanshahi P, Verrilli GM, Carter JD, Brayman KL, Nunemaker CS. Evidence that low-grade systemic inflammation can induce islet dysfunction as measured by impaired calcium handling. Cell Calcium. 2010;48:133–142. doi: 10.1016/j.ceca.2010.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eguchi K, Manabe I, Oishi-Tanaka Y, Ohsugi M, Kono N, Ogata F, Yagi N, Ohto U, Kimoto M, Miyake K, et al. Saturated fatty acid and TLR signaling link beta cell dysfunction and islet inflammation. Cell Metab. 2012;15:518–533. doi: 10.1016/j.cmet.2012.01.023. [DOI] [PubMed] [Google Scholar]
- Ehses JA, Lacraz G, Giroix MH, Schmidlin F, Coulaud J, Kassis N, Irminger JC, Kergoat M, Portha B, Homo-Delarche F, et al. IL-1 antagonism reduces hyperglycemia and tissue inflammation in the type 2 diabetic GK rat. Proc Natl Acad Sci U S A. 2009;106:13998–14003. doi: 10.1073/pnas.0810087106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehses JA, Perren A, Eppler E, Ribaux P, Pospisilik JA, Maor-Cahn R, Gueripel X, Ellingsgaard H, Schneider MK, Biollaz G, et al. Increased number of islet-associated macrophages in type 2 diabetes. Diabetes. 2007;56:2356–2370. doi: 10.2337/db06-1650. [DOI] [PubMed] [Google Scholar]
- Eldor R, Yeffet A, Baum K, Doviner V, Amar D, Ben-Neriah Y, Christofori G, Peled A, Carel JC, Boitard C, et al. Conditional and specific NF-kappaB blockade protects pancreatic beta cells from diabetogenic agents. Proc Natl Acad Sci U S A. 2006;103:5072–5077. doi: 10.1073/pnas.0508166103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forbes JM, Soderlund J, Yap FY, Knip M, Andrikopoulos S, Ilonen J, Simell O, Veijola R, Sourris KC, Coughlan MT, et al. Receptor for advanced glycation end-products (RAGE) provides a link between genetic susceptibility and environmental factors in type 1 diabetes. Diabetologia. 2011;54:1032–1042. doi: 10.1007/s00125-011-2058-z. [DOI] [PubMed] [Google Scholar]
- Foulis AK, McGill M, Farquharson MA. Insulitis in type 1 (insulin-dependent) diabetes mellitus in man–macrophages, lymphocytes, and interferon-gamma containing cells. J Pathol. 1991;165:97–103. doi: 10.1002/path.1711650203. [DOI] [PubMed] [Google Scholar]
- Foulis AK, Stewart JA. The pancreas in recent-onset type 1 (insulin-dependent) diabetes mellitus: insulin content of islets, insulitis and associated changes in the exocrine acinar tissue. Diabetologia. 1984;26:456–461. doi: 10.1007/BF00262221. [DOI] [PubMed] [Google Scholar]
- Fourlanos S, Narendran P, Byrnes GB, Colman PG, Harrison LC. Insulin resistance is a risk factor for progression to type 1 diabetes. Diabetologia. 2004;47:1661–1667. doi: 10.1007/s00125-004-1507-3. [DOI] [PubMed] [Google Scholar]
- Frigerio S, Junt T, Lu B, Gerard C, Zumsteg U, Hollander GA, Piali L. Beta cells are responsible for CXCR3-mediated T-cell infiltration in insulitis. Nat Med. 2002;8:1414–1420. doi: 10.1038/nm1202-792. [DOI] [PubMed] [Google Scholar]
- Gepts W. Pathologic anatomy of the pancreas in juvenile diabetes mellitus. Diabetes. 1965;14:619–633. doi: 10.2337/diab.14.10.619. [DOI] [PubMed] [Google Scholar]
- Giannoukakis N, Rudert WA, Trucco M, Robbins PD. Protection of human islets from the effects of interleukin-1beta by adenoviral gene transfer of an Ikappa B repressor. J Biol Chem. 2000;275:36509–36513. doi: 10.1074/jbc.M005943200. [DOI] [PubMed] [Google Scholar]
- Gill RG, Pagni PP, Kufper T, Wasserfall CH, Deng S, Posgai A, Manenkova Y, Hani AB, Straub L, Bernstein P, et al. A Preclinical Consortium Approach for Assessing the Efficacy of Combined Anti-CD3 Plus IL-1 Blockade in Reversing New-Onset Autoimmune Diabetes in NOD Mice. Diabetes. 2015 doi: 10.2337/db15-0492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grattan M, Mi QS, Meagher C, Delovitch TL. Congenic mapping of the diabetogenic locus Idd4 to a 5.2-cM region of chromosome 11 in NOD mice: identification of two potential candidate subloci. Diabetes. 2002;51:215–223. doi: 10.2337/diabetes.51.1.215. [DOI] [PubMed] [Google Scholar]
- Grewal IS, Rutledge BJ, Fiorillo JA, Gu L, Gladue RP, Flavell RA, Rollins BJ. Transgenic monocyte chemoattractant protein-1 (MCP-1) in pancreatic islets produces monocyte-rich insulitis without diabetes: abrogation by a second transgene expressing systemic MCP-1. J Immunol. 1997;159:401–408. [PubMed] [Google Scholar]
- Groom JR, Luster AD. CXCR3 ligands: redundant, collaborative and antagonistic functions. Immunol Cell Biol. 2011;89:207–215. doi: 10.1038/icb.2010.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanafusa T, Imagawa A. Insulitis in human type 1 diabetes. Ann N Y Acad Sci. 2008;1150:297–299. doi: 10.1196/annals.1447.052. [DOI] [PubMed] [Google Scholar]
- Hanifi-Moghaddam P, Kappler S, Seissler J, Muller-Scholze S, Martin S, Roep BO, Strassburger K, Kolb H, Schloot NC. Altered chemokine levels in individuals at risk of Type 1 diabetes mellitus. Diabet Med. 2006;23:156–163. doi: 10.1111/j.1464-5491.2005.01743.x. [DOI] [PubMed] [Google Scholar]
- Heise CE, Pahuja A, Hudson SC, Mistry MS, Putnam AL, Gross MM, Gottlieb PA, Wade WS, Kiankarimi M, Schwarz D, et al. Pharmacological characterization of CXC chemokine receptor 3 ligands and a small molecule antagonist. J Pharmacol Exp Ther. 2005;313:1263–1271. doi: 10.1124/jpet.105.083683. [DOI] [PubMed] [Google Scholar]
- Heitmeier MR, Scarim AL, Corbett JA. Interferon-gamma increases the sensitivity of islets of Langerhans for inducible nitric-oxide synthase expression induced by interleukin 1. J Biol Chem. 1997;272:13697–13704. doi: 10.1074/jbc.272.21.13697. [DOI] [PubMed] [Google Scholar]
- Held TK, Weihua X, Yuan L, Kalvakolanu DV, Cross AS. Gamma interferon augments macrophage activation by lipopolysaccharide by two distinct mechanisms, at the signal transduction level and via an autocrine mechanism involving tumor necrosis factor alpha and interleukin-1. Infect Immun. 1999;67:206–212. doi: 10.1128/iai.67.1.206-212.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Home P, Riddle M, Cefalu WT, Bailey CJ, Bretzel RG, Del Prato S, Leroith D, Schernthaner G, van Gaal L, Raz I. Insulin therapy in people with type 2 diabetes: opportunities and challenges? Diabetes Care. 2014;37:1499–1508. doi: 10.2337/dc13-2743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu C, Ding H, Li Y, Pearson JA, Zhang X, Flavell RA, Wong FS, Wen L. NLRP3 deficiency protects from type 1 diabetes through the regulation of chemotaxis into the pancreatic islets. Proc Natl Acad Sci U S A. 2015;112:11318–11323. doi: 10.1073/pnas.1513509112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenh CH, Cox MA, Cui L, Reich EP, Sullivan L, Chen SC, Kinsley D, Qian S, Kim SH, Rosenblum S, et al. A selective and potent CXCR3 antagonist SCH 546738 attenuates the development of autoimmune diseases and delays graft rejection. BMC immunology. 2012;13:2. doi: 10.1186/10.1186/1471-2172-13-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao X, Zhang N, Xu X, Oppenheim JJ, Jin T. Ligand-induced partitioning of human CXCR1 chemokine receptors with lipid raft microenvironments facilitates G-protein-dependent signaling. Mol Cell Biol. 2005;25:5752–5762. doi: 10.1128/MCB.25.13.5752-5762.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson AM, Olefsky JM. The origins and drivers of insulin resistance. Cell. 2013;152:673–684. doi: 10.1016/j.cell.2013.01.041. [DOI] [PubMed] [Google Scholar]
- Jourdan T, Godlewski G, Cinar R, Bertola A, Szanda G, Liu J, Tam J, Han T, Mukhopadhyay B, Skarulis MC, et al. Activation of the Nlrp3 inflammasome in infiltrating macrophages by endocannabinoids mediates beta cell loss in type 2 diabetes. Nat Med. 2013;19:1132–1140. doi: 10.1038/nm.3265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahn BB. Type 2 diabetes: when insulin secretion fails to compensate for insulin resistance. Cell. 1998;92:593–596. doi: 10.1016/s0092-8674(00)81125-3. [DOI] [PubMed] [Google Scholar]
- Karin N, Wildbaum G, Thelen M. Biased signaling pathways via CXCR3 control the development and function of CD4+ T cell subsets. J Leukoc Biol. 2016;99:857–862. doi: 10.1189/jlb.2MR0915-441R. [DOI] [PubMed] [Google Scholar]
- Katancik JA, Sharma A, de Nardin E. Interleukin 8, neutrophil-activating peptide-2 and GRO-alpha bind to and elicit cell activation via specific and different amino acid residues of CXCR2. Cytokine. 2000;12:1480–1488. doi: 10.1006/cyto.2000.0742. [DOI] [PubMed] [Google Scholar]
- Katancik JA, Sharma A, Radel SJ, De Nardin E. Mapping of the extracellular binding regions of the human interleukin-8 type B receptor. Biochem Biophys Res Commun. 1997;232:663–668. doi: 10.1006/bbrc.1997.6352. [DOI] [PubMed] [Google Scholar]
- Kenty JH, Melton DA. Testing pancreatic islet function at the single cell level by calcium influx with associated marker expression. PLoS One. 2015;10:e0122044. doi: 10.1371/journal.pone.0122044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan SM, Sleno R, Gora S, Zylbergold P, Laverdure JP, Labbe JC, Miller GJ, Hebert TE. The expanding roles of Gbetagamma subunits in G protein-coupled receptor signaling and drug action. Pharmacol Rev. 2013;65:545–577. doi: 10.1124/pr.111.005603. [DOI] [PubMed] [Google Scholar]
- Kouroumalis A, Nibbs RJ, Aptel H, Wright KL, Kolios G, Ward SG. The chemokines CXCL9, CXCL10, and CXCL11 differentially stimulate G alpha i-independent signaling and actin responses in human intestinal myofibroblasts. J Immunol. 2005;175:5403–5411. doi: 10.4049/jimmunol.175.8.5403. [DOI] [PubMed] [Google Scholar]
- Kreider KE, Lien LF. Transitioning safely from intravenous to subcutaneous insulin. Curr Diab Rep. 2015;15:23. doi: 10.1007/s11892-015-0595-4. [DOI] [PubMed] [Google Scholar]
- Kuttler B, Wanka H, Kloting N, Gerstmayer B, Volk HD, Sawitzki B, Ritter T. Ex vivo gene transfer of viral interleukin-10 to BB rat islets: no protection after transplantation to diabetic BB rats. J Cell Mol Med. 2007;11:868–880. doi: 10.1111/j.1582-4934.2007.00059.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasch S, Muller P, Bayer M, Pfeilschifter JM, Luster AD, Hintermann E, Christen U. Anti-CD3/anti-CXCL10 antibody combination therapy induces a persistent remission of type 1 diabetes in two mouse models. Diabetes. 2015 doi: 10.2337/db15-0479. [DOI] [PubMed] [Google Scholar]
- Lee YS, Morinaga H, Kim JJ, Lagakos W, Taylor S, Keshwani M, Perkins G, Dong H, Kayali AG, Sweet IR, et al. The fractalkine/CX3CR1 system regulates beta cell function and insulin secretion. Cell. 2013;153:413–425. doi: 10.1016/j.cell.2013.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Belkadi A, Darnall L, Hu T, Drescher C, Cotleur AC, Padovani-Claudio D, He T, Choi K, Lane TE, et al. CXCR2-positive neutrophils are essential for cuprizone-induced demyelination: relevance to multiple sclerosis. Nat Neurosci. 2010;13:319–326. doi: 10.1038/nn.2491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopes M, Kutlu B, Miani M, Bang-Berthelsen CH, Storling J, Pociot F, Goodman N, Hood L, Welsh N, Bontempi G, et al. Temporal profiling of cytokine-induced genes in pancreatic beta-cells by meta-analysis and network inference. Genomics. 2014;103:264–275. doi: 10.1016/j.ygeno.2013.12.007. [DOI] [PubMed] [Google Scholar]
- Lundberg M, Krogvold L, Kuric E, Dahl-Jorgensen K, Skog O. Expression of Interferon-Stimulated Genes in Insulitic Pancreatic Islets of Patients Recently Diagnosed With Type 1 Diabetes. Diabetes. 2016;65:3104–3110. doi: 10.2337/db16-0616. [DOI] [PubMed] [Google Scholar]
- Luttrell LM, Lefkowitz RJ. The role of beta-arrestins in the termination and transduction of G-protein-coupled receptor signals. J Cell Sci. 2002;115:455–465. doi: 10.1242/jcs.115.3.455. [DOI] [PubMed] [Google Scholar]
- Marchese A, Paing MM, Temple BR, Trejo J. G protein-coupled receptor sorting to endosomes and lysosomes. Annu Rev Pharmacol Toxicol. 2008;48:601–629. doi: 10.1146/annurev.pharmtox.48.113006.094646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin AP, Alexander-Brett JM, Canasto-Chibuque C, Garin A, Bromberg JS, Fremont DH, Lira SA. The chemokine binding protein M3 prevents diabetes induced by multiple low doses of streptozotocin. J Immunol. 2007;178:4623–4631. doi: 10.4049/jimmunol.178.7.4623. [DOI] [PubMed] [Google Scholar]
- Martin AP, Grisotto MG, Canasto-Chibuque C, Kunkel SL, Bromberg JS, Furtado GC, Lira SA. Islet expression of M3 uncovers a key role for chemokines in the development and recruitment of diabetogenic cells in NOD mice. Diabetes. 2008a;57:387–394. doi: 10.2337/db07-1309. [DOI] [PubMed] [Google Scholar]
- Martin AP, Rankin S, Pitchford S, Charo IF, Furtado GC, Lira SA. Increased expression of CCL2 in insulin-producing cells of transgenic mice promotes mobilization of myeloid cells from the bone marrow, marked insulitis, and diabetes. Diabetes. 2008b;57:3025–3033. doi: 10.2337/db08-0625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez Munoz L, Lucas P, Navarro G, Checa AI, Franco R, Martinez AC, Rodriguez-Frade JM, Mellado M. Dynamic regulation of CXCR1 and CXCR2 homo- and heterodimers. J Immunol. 2009;183:7337–7346. doi: 10.4049/jimmunol.0901802. [DOI] [PubMed] [Google Scholar]
- Martinon F. Detection of immune danger signals by NALP3. J Leukoc Biol. 2008;83:507–511. doi: 10.1189/jlb.0607362. [DOI] [PubMed] [Google Scholar]
- Mocsai A. Diverse novel functions of neutrophils in immunity, inflammation, and beyond. J Exp Med. 2013;210:1283–1299. doi: 10.1084/jem.20122220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran A, Bundy B, Becker DJ, DiMeglio LA, Gitelman SE, Goland R, Greenbaum CJ, Herold KC, Marks JB, Raskin P, et al. Interleukin-1 antagonism in type 1 diabetes of recent onset: two multicentre, randomised, double-blind, placebo-controlled trials. Lancet. 2013;381:1905–1915. doi: 10.1016/S0140-6736(13)60023-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muoio DM, Newgard CB. Mechanisms of disease: molecular and metabolic mechanisms of insulin resistance and beta-cell failure in type 2 diabetes. Nat Rev Mol Cell Biol. 2008;9:193–205. doi: 10.1038/nrm2327. [DOI] [PubMed] [Google Scholar]
- Murphy PM, Tiffany HL. Cloning of complementary DNA encoding a functional human interleukin-8 receptor. Science. 1991;253:1280–1283. doi: 10.1126/science.1891716. [DOI] [PubMed] [Google Scholar]
- Nowotny K, Jung T, Hohn A, Weber D, Grune T. Advanced glycation end products and oxidative stress in type 2 diabetes mellitus. Biomolecules. 2015;5:194–222. doi: 10.3390/biom5010194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunemaker CS, Chung HG, Verrilli GM, Corbin KL, Upadhye A, Sharma PR. Increased serum CXCL1 and CXCL5 are linked to obesity, hyperglycemia, and impaired islet function. J Endocrinol. 2014;222:267–276. doi: 10.1530/JOE-14-0126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce KL, Premont RT, Lefkowitz RJ. Seven-transmembrane receptors. Nat Rev Mol Cell Biol. 2002;3:639–650. doi: 10.1038/nrm908. [DOI] [PubMed] [Google Scholar]
- Purohit J, Hu P, Burke SJ, Collier JJ, Chen J, Zhao L. The effects of NOD activation on adipocyte differentiation. Obesity (Silver Spring) 2013;21:737–747. doi: 10.1002/oby.20275. [DOI] [PubMed] [Google Scholar]
- Qureshi FM, Dejene EA, Corbin KL, Nunemaker CS. Stress-induced dissociations between intracellular calcium signaling and insulin secretion in pancreatic islets. Cell Calcium. 2015;57:366–375. doi: 10.1016/j.ceca.2015.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raghuwanshi SK, Su Y, Singh V, Haynes K, Richmond A, Richardson RM. The chemokine receptors CXCR1 and CXCR2 couple to distinct G protein-coupled receptor kinases to mediate and regulate leukocyte functions. J Immunol. 2012;189:2824–2832. doi: 10.4049/jimmunol.1201114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajagopal S. Quantifying biased agonism: understanding the links between affinity and efficacy. Nat Rev Drug Discov. 2013;12:483. doi: 10.1038/nrd3954-c1. [DOI] [PubMed] [Google Scholar]
- Rajagopal S, Bassoni DL, Campbell JJ, Gerard NP, Gerard C, Wehrman TS. Biased agonism as a mechanism for differential signaling by chemokine receptors. J Biol Chem. 2013;288:35039–35048. doi: 10.1074/jbc.M113.479113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramadan JW, Steiner SR, O’Neill CM, Nunemaker CS. The central role of calcium in the effects of cytokines on beta-cell function: implications for type 1 and type 2 diabetes. Cell Calcium. 2011;50:481–490. doi: 10.1016/j.ceca.2011.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raman D, Sai J, Hawkins O, Richmond A. Adaptor protein2 (AP2) orchestrates CXCR2-mediated cell migration. Traffic. 2014;15:451–469. doi: 10.1111/tra.12154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehman KK, Bertera S, Bottino R, Balamurugan AN, Mai JC, Mi Z, Trucco M, Robbins PD. Protection of islets by in situ peptide-mediated transduction of the Ikappa B kinase inhibitor Nemo-binding domain peptide. J Biol Chem. 2003;278:9862–9868. doi: 10.1074/jbc.M207700200. [DOI] [PubMed] [Google Scholar]
- Rhode A, Pauza ME, Barral AM, Rodrigo E, Oldstone MB, von Herrath MG, Christen U. Islet-specific expression of CXCL10 causes spontaneous islet infiltration and accelerates diabetes development. J Immunol. 2005;175:3516–3524. doi: 10.4049/jimmunol.175.6.3516. [DOI] [PubMed] [Google Scholar]
- Richardson SJ, Rodriguez-Calvo T, Gerling IC, Mathews CE, Kaddis JS, Russell MA, Zeissler M, Leete P, Krogvold L, Dahl-Jorgensen K, et al. Islet cell hyperexpression of HLA class I antigens: a defining feature in type 1 diabetes. Diabetologia. 2016;59:2448–2458. doi: 10.1007/s00125-016-4067-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson SJ, Willcox A, Bone AJ, Foulis AK, Morgan NG. Islet-associated macrophages in type 2 diabetes. Diabetologia. 2009;52:1686–1688. doi: 10.1007/s00125-009-1410-z. [DOI] [PubMed] [Google Scholar]
- Rink JS, Chen X, Zhang X, Kaufman DB. Conditional and specific inhibition of NF-kappaB in mouse pancreatic beta cells prevents cytokine-induced deleterious effects and improves islet survival posttransplant. Surgery. 2012;151:330–339. doi: 10.1016/j.surg.2011.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosa JS, Flores RL, Oliver SR, Pontello AM, Zaldivar FP, Galassetti PR. Sustained IL-1alpha, IL-4, and IL-6 elevations following correction of hyperglycemia in children with type 1 diabetes mellitus. Pediatr Diabetes. 2008;9:9–16. doi: 10.1111/j.1399-5448.2007.00243.x. [DOI] [PubMed] [Google Scholar]
- Rutkowsky JM, Knotts TA, Ono-Moore KD, McCoin CS, Huang S, Schneider D, Singh S, Adams SH, Hwang DH. Acylcarnitines activate proinflammatory signaling pathways. Am J Physiol Endocrinol Metab. 2014;306:E1378–1387. doi: 10.1152/ajpendo.00656.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sajadi SM, Khoramdelazad H, Hassanshahi G, Rafatpanah H, Hosseini J, Mahmoodi M, Arababadi MK, Derakhshan R, Hasheminasabzavareh R, Hosseini-Zijoud SM, et al. Plasma levels of CXCL1 (GRO-alpha) and CXCL10 (IP-10) are elevated in type 2 diabetic patients: evidence for the involvement of inflammation and angiogenesis/angiostasis in this disease state. Clin Lab. 2013;59:133–137. doi: 10.7754/clin.lab.2012.120225. [DOI] [PubMed] [Google Scholar]
- Samuel VT, Shulman GI. The pathogenesis of insulin resistance: integrating signaling pathways and substrate flux. J Clin Invest. 2016;126:12–22. doi: 10.1172/JCI77812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez I, Reynoso-Camacho R, Salgado LM. The diet-induced metabolic syndrome is accompanied by whole-genome epigenetic changes. Genes Nutr. 2015;10:471. doi: 10.1007/s12263-015-0471-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar SA, Lee CE, Victorino F, Nguyen TT, Walters JA, Burrack A, Eberlein J, Hildemann SK, Homann D. Expression and regulation of chemokines in murine and human type 1 diabetes. Diabetes. 2012;61:436–446. doi: 10.2337/db11-0853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulthess FT, Paroni F, Sauter NS, Shu L, Ribaux P, Haataja L, Strieter RM, Oberholzer J, King CC, Maedler K. CXCL10 impairs beta cell function and viability in diabetes through TLR4 signaling. Cell Metab. 2009;9:125–139. doi: 10.1016/j.cmet.2009.01.003. [DOI] [PubMed] [Google Scholar]
- Sharabiani MT, Vermeulen R, Scoccianti C, Hosnijeh FS, Minelli L, Sacerdote C, Palli D, Krogh V, Tumino R, Chiodini P, et al. Immunologic profile of excessive body weight. Biomarkers. 2011;16:243–251. doi: 10.3109/1354750X.2010.547948. [DOI] [PubMed] [Google Scholar]
- Sharma RB, O’Donnell AC, Stamateris RE, Ha B, McCloskey KM, Reynolds PR, Arvan P, Alonso LC. Insulin demand regulates beta cell number via the unfolded protein response. J Clin Invest. 2015;125:3831–3846. doi: 10.1172/JCI79264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shenoy SK, Lefkowitz RJ. Receptor regulation: beta-arrestin moves up a notch. Nat Cell Biol. 2005;7:1159–1161. doi: 10.1038/ncb1205-1059. [DOI] [PubMed] [Google Scholar]
- Shigihara T, Oikawa Y, Kanazawa Y, Okubo Y, Narumi S, Saruta T, Shimada A. Significance of serum CXCL10/IP–10 level in type 1 diabetes. J Autoimmun. 2006;26:66–71. doi: 10.1016/j.jaut.2005.09.027. [DOI] [PubMed] [Google Scholar]
- Shimabukuro M, Zhou YT, Lee Y, Unger RH. Troglitazone lowers islet fat and restores beta cell function of Zucker diabetic fatty rats. J Biol Chem. 1998;273:3547–3550. doi: 10.1074/jbc.273.6.3547. [DOI] [PubMed] [Google Scholar]
- Sierro F, Biben C, Martinez-Munoz L, Mellado M, Ransohoff RM, Li M, Woehl B, Leung H, Groom J, Batten M, et al. Disrupted cardiac development but normal hematopoiesis in mice deficient in the second CXCL12/SDF-1 receptor, CXCR7. Proc Natl Acad Sci U S A. 2007;104:14759–14764. doi: 10.1073/pnas.0702229104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straczkowski M, Dzienis-Straczkowska S, Stepien A, Kowalska I, Szelachowska M, Kinalska I. Plasma interleukin-8 concentrations are increased in obese subjects and related to fat mass and tumor necrosis factor-alpha system. J Clin Endocrinol Metab. 2002;87:4602–4606. doi: 10.1210/jc.2002-020135. [DOI] [PubMed] [Google Scholar]
- Sumpter KM, Adhikari S, Grishman EK, White PC. Preliminary studies related to anti-interleukin-1beta therapy in children with newly diagnosed type 1 diabetes. Pediatr Diabetes. 2011;12:656–667. doi: 10.1111/j.1399-5448.2011.00761.x. [DOI] [PubMed] [Google Scholar]
- Syrovatkina V, Alegre KO, Dey R, Huang XY. Regulation, Signaling, and Physiological Functions of G-Proteins. J Mol Biol. 2016;428:3850–3868. doi: 10.1016/j.jmb.2016.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K, Ohara M, Sasai T, Homma H, Nagasawa K, Takahashi T, Yamashina M, Ishii M, Fujiwara F, Kajiwara T, et al. Serum CXCL1 concentrations are elevated in type 1 diabetes mellitus, possibly reflecting activity of anti-islet autoimmune activity. Diabetes Metab Res Rev. 2011;27:830–833. doi: 10.1002/dmrr.1257. [DOI] [PubMed] [Google Scholar]
- Talukdar S, Oh da Y, Bandyopadhyay G, Li D, Xu J, McNelis J, Lu M, Li P, Yan Q, Zhu Y, et al. Neutrophils mediate insulin resistance in mice fed a high-fat diet through secreted elastase. Nat Med. 2012;18:1407–1412. doi: 10.1038/nm.2885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson BD, Jin Y, Wu KH, Colvin RA, Luster AD, Birnbaumer L, Wu MX. Inhibition of G alpha i2 activation by G alpha i3 in CXCR3-mediated signaling. J Biol Chem. 2007;282:9547–9555. doi: 10.1074/jbc.M610931200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trettel F, Di Bartolomeo S, Lauro C, Catalano M, Ciotti MT, Limatola C. Ligand-independent CXCR2 dimerization. J Biol Chem. 2003;278:40980–40988. doi: 10.1074/jbc.M306815200. [DOI] [PubMed] [Google Scholar]
- Unger RH. Lipid overload and overflow: metabolic trauma and the metabolic syndrome. Trends Endocrinol Metab. 2003;14:398–403. doi: 10.1016/j.tem.2003.09.008. [DOI] [PubMed] [Google Scholar]
- Unger RH, Orci L. Lipotoxic diseases of nonadipose tissues in obesity. Int J Obes Relat Metab Disord. 2000;24(Suppl 4):S28–32. doi: 10.1038/sj.ijo.0801498. [DOI] [PubMed] [Google Scholar]
- Van Raemdonck K, Van den Steen PE, Liekens S, Van Damme J, Struyf S. CXCR3 ligands in disease and therapy. Cytokine Growth Factor Rev. 2015;26:311–327. doi: 10.1016/j.cytogfr.2014.11.009. [DOI] [PubMed] [Google Scholar]
- Veenstra M, Ransohoff RM. Chemokine receptor CXCR2: physiology regulator and neuroinflammation controller? J Neuroimmunol. 2012;246:1–9. doi: 10.1016/j.jneuroim.2012.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallberg M, Cooke A. Immune mechanisms in type 1 diabetes. Trends Immunol. 2013;34:583–591. doi: 10.1016/j.it.2013.08.005. [DOI] [PubMed] [Google Scholar]
- Watts AO, Scholten DJ, Heitman LH, Vischer HF, Leurs R. Label-free impedance responses of endogenous and synthetic chemokine receptor CXCR3 agonists correlate with Gi-protein pathway activation. Biochem Biophys Res Commun. 2012;419:412–418. doi: 10.1016/j.bbrc.2012.02.036. [DOI] [PubMed] [Google Scholar]
- Waugh DJ, Wilson C. The interleukin-8 pathway in cancer. Clin Cancer Res. 2008;14:6735–6741. doi: 10.1158/1078-0432.CCR-07-4843. [DOI] [PubMed] [Google Scholar]
- Welzen-Coppens JM, van Helden-Meeuwsen CG, Leenen PJ, Drexhage HA, Versnel MA. The kinetics of plasmacytoid dendritic cell accumulation in the pancreas of the NOD mouse during the early phases of insulitis. PLoS One. 2013;8:e55071. doi: 10.1371/journal.pone.0055071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willcox A, Richardson SJ, Bone AJ, Foulis AK, Morgan NG. Analysis of islet inflammation in human type 1 diabetes. Clin Exp Immunol. 2009;155:173–181. doi: 10.1111/j.1365-2249.2008.03860.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson S, Wilkinson G, Milligan G. The CXCR1 and CXCR2 receptors form constitutive homo- and heterodimers selectively and with equal apparent affinities. J Biol Chem. 2005;280:28663–28674. doi: 10.1074/jbc.M413475200. [DOI] [PubMed] [Google Scholar]
- Wisler JW, Xiao K, Thomsen AR, Lefkowitz RJ. Recent developments in biased agonism. Curr Opin Cell Biol. 2014;27:18–24. doi: 10.1016/j.ceb.2013.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada Y, Okubo Y, Shimada A, Oikawa Y, Yamada S, Narumi S, Matsushima K, Itoh H. Acceleration of diabetes development in CXC chemokine receptor 3 (CXCR3)-deficient NOD mice. Diabetologia. 2012;55:2238–2245. doi: 10.1007/s00125-012-2547-8. [DOI] [PubMed] [Google Scholar]
- Zak KP, Popova VV, Mel’nichenko SV, Tron’ko EN, Man’kovskii BN. The level of circulating cytokines and chemokines in the preclinical and early clinical stages of type IA diabetes mellitus development. Ter Arkh. 2010;82:10–15. [PubMed] [Google Scholar]
- Zweemer AJ, Toraskar J, Heitman LH, AP IJ. Bias in chemokine receptor signalling. Trends Immunol. 2014;35:243–252. doi: 10.1016/j.it.2014.02.004. [DOI] [PubMed] [Google Scholar]