

Abstract
Sleep apnea is a prevalent respiratory disease in which episodic cessation of breathing causes intermittent hypoxia. Patients with sleep apnea and rodents exposed to intermittent hypoxia exhibit hypertension. The carotid body senses changes in blood O2 concentrations and an enhanced carotid body chemosensory reflex contributes to hypertension in sleep apnea patients. Using a rodent model of intermittent hypoxia that simulates blood O2-saturation profiles of patients with sleep apnea, increased generation of reactive oxygen species (ROS) in the carotid body has been shown to enhance the chemosensory reflex and trigger hypertension. Here, we report that ROS inhibited CO generation by heme oxygenase-2 (HO-2), a process that requires Cys265 in the heme regulatory motif of HO-2. CO binds to guanylate cyclase to increase production of cGMP, which stimulates protein kinase G-dependent phosphorylation and inactivation of the H2S-producing enzyme cystathionine-γ-lyase (CSE). We showed that ROS induced by intermittent hypoxia inhibited CO production and increased H2S concentrations, which stimulated carotid body neural activity. Blockade of H2S synthesis by CSE either by pharmacological or genetic approaches inhibited carotid body activation and hypertension induced by intermittent hypoxia. Thus, oxidant-induced inactivation of HO-2 leading to increased CSE-dependent H2S production in the carotid body is a critical trigger of hypertension in rodents exposed to intermittent hypoxia.
ONE-SENTENCE SUMMARY
This study delineates ROS-CO-and H2S signaling cascade underlying carotid body chemosensory reflex mediated hypertension in a rodent model of sleep apnea.
INTRODUCTION
Sleep apnea is a widespread respiratory disease characterized by brief (15–30 sec), repeated episodes of cessation of breathing during sleep, which occur due to defective generation of the respiratory rhythm (central sleep apnea) or due to obstruction of the upper airway (obstructive sleep apnea) (1–3). Repetitive apneas lead to periodic decreases in blood O2 concentrations resulting in intermittent hypoxia. Sleep apnea patients and rodents exposed to intermittent hypoxia which simulates the altered blood O2-saturation profiles caused by sleep apnea, exhibit increased sympathetic nerve activity and hypertension (4, 5). Studies in humans and rodents suggest that the carotid body, the primary sensory organ for detecting hypoxia in arterial blood, mediates a reflex increase in sympathetic nerve activity that results in hypertension (6, 7). Intermittent hypoxia sensitizes the carotid body response to hypoxia (8, 9) and induces sensory long-term facilitation (LTF), which is characterized by a long-lasting increase in sensory nerve activity (10). Sleep apnea patients exhibit marked increases in blood pressureduring apneic episodesdue to intermittent hypoxia-evoked carotid body hypersensitivity to hypoxia, whereas sensory LTF mediates reflex activation of the sympathetic nervous system and persistent daytime hypertension (6). Ablation of the carotid body blocks sympathetic activation and hypertension inintermittent hypoxia subjected rats (11, 12).
Intermittent hypoxia increases the generation of reactive oxygen species (ROS) in the carotid body due to increased expression of genes encoding pro-oxidant enzymes that are regulated by hypoxia-inducible factor 1 (HIF-1) and decreased expression of anti-oxidant enzyme-encoding genes that are regulated by HIF-2 (13–15). Treatment of rats with a ROS scavenger during exposure to intermittent hypoxia normalizes carotid body activity and prevents the development of hypertension (10, 16, 17). Thus, ROS generation in response to intermittent hypoxia triggers carotid body activation and the ensuing cardiovascular pathology. However, the mechanisms by ROS activates the carotid body by intermittent hypoxia have not been fully delineated. Since intermittent hypoxia does not alter carotid body morphology (10), we hypothesized that the augmented carotid body activity reflects effects of ROS on hypoxia signal transduction. Various theories have been proposed to explain hypoxia sensing by the carotid body (18). Emerging evidence suggests that the gaseous messenger hydrogen sulfide (H2S) mediates carotid body activation by hypoxia (19–23). Glomus cells, the primary O2 sensing cells in the carotid body, express at least two major H2S-synthesizing enzymes cystathionine β-synthase (CBS) and cystathionine-γ-lyase (CSE) (19, 24). Intriguingly, CBS is itself induced by chronic hypoxia via transcriptional activation by hypoxia-inducible factors (HIFs) (25). However, CSE null mice display greatly diminished H2S generation and a severely impaired carotid body and physiological responses to acute hypoxia (19), suggesting that CSE-derived H2S is more vital for regulating carotid body activity. We hypothesized that ROS increase the CSE-dependent synthesis of H2S to trigger carotid body activation and hypertension in rodents exposed to IH.
RESULTS
Intermittent hypoxia increases CSE-dependent H2S production through ROS
H2S concentrations were measured in the carotid bodies of adult rats exposed to alternating cycles of 15 sec of hypoxia (5% O2) followed by 5 min of room air (21% O2), nine episodes per hour, and eight hours per day for 10 days. This intermittent hypoxia paradigm decreases arterial blood-O2 saturation from ~97% to ~80% with each episode of hypoxia (12), which is comparable to the O2 desaturation observed in sleep apnea patients (2). H2S concentrations were increased in carotid bodies from intermittent hypoxia-exposed rats as compared with controls (Fig. 1A). To assess the contribution of CSE, an H2S-synthesizing enzyme in the carotid body (19), to the observed increase in H2S, rats were treated with L-propargylglycine (L-PAG), a selective inhibitor of CSE (26). H2S concentrations were not elevated by intermittent hypoxia in carotid bodies from rats treated with L-PAG (Fig. 1A). In addition, intermittent hypoxia increased H2S concentrations in the carotid bodies of wild-type mice, a response that was absent in CSE knockout mice (Fig. 1B). Thus, pharmacologic and genetic approaches in rats and mice, respectively, demonstrate that intermittent hypoxia increases CSE-dependent H2S production.
Fig. 1. Induction of CSE-dependent H2S production by intermittent hypoxia (IH).

(A) H2S concentrations in the carotid bodies of rats exposed to room air (C), room air with L-PAG (C+PAG), IH, or IH with L-PAG (IH+PAG) (n = 3 experiments for each treatment; a total of 12 carotid bodies from 6 rats for each treatment). (B) H2S concentrations in wild-type (WT) and CSE null (Cse−/−) mice exposed to room air (C) or IH (n =3 experiments for each treatment per genotype; a total of 24 carotid bodies from 12 mice for each treatment per genotype). (C to D) Malondialdehyde (MDA; C) and H2S amounts (D) in the carotid bodies of rats exposed to room air (C), room air with MnTMPyP, a ROS scavenger (RS; C+RS), IH or IH with RS (IH+ RS) (n = 3 experiments for each treatment; a total of 12 carotid bodies from 6 rats for each treatment). (E) Reverse transcription and quantitative real-time PCR (RT-qPCR) assay of Cse mRNA normalized to 18S rRNA in carotid bodies of rats exposed to either room air (C) or IH, normalized to C (n = 3 experiments for each treatment; a total of 6 carotid bodies from 3 rats for each treatment). (F) Representative immunoblot of CSE protein abundance in liver tissue of rats exposed to room air (C) or IH (n =3 experiments for each treatment). (G) Representative immunoblot of CSE expression in HEK-293 cells stably transfected with empty vector or vector encoding CSE (n=3 experiments). α-tubulin abundance was assayed in F and G as a loading control. (H) MDA and (I) H2S amounts in CSE-expressing HEK-293 cells exposed to room air (C), room air with RS or IH (n = 4 experiments for each treatment). Data are presented as means ± SEM. **p < 0.01; ***p <0.001; ns, not significant. See fig.S1 for data on concentrations of H2S and MDA and CSE protein abundance in rat liver.
Malondialdehyde (MDA) amounts, which reflect oxidized lipids (27), were measured in the carotid bodies as an index of ROS generation. MDA amounts were increased in carotid bodies from intermittent hypoxia-exposed rats as compared with controls, an effect that was abrogated by treating rats with manganese (III) tetrakis(1-methyl-4-pyridyl)porphyrin pentachloride (MnTMPyP), a membrane-permeable ROS scavenger (28) during the ten days of intermittent hypoxia exposure (Fig. 1C). MnTMPyP treatment also blocked the intermittent hypoxia-induced increase in H2S concentrations in the rat carotid body (Fig. 1D). However, MnTMPyP treatment had no effect either on MDA amounts or on H2S concentrations of carotid bodies from control rats (Fig.1, C and D). These findings indicate that ROS generated during intermittent hypoxia stimulates H2S production in the carotid body.
Reverse transcription and quantitative real-time PCR (RT-qPCR) assays revealed no significant change in Cse mRNA abundance in carotid bodies from intermittent hypoxia-exposed rats (Fig. 1E). As the nature of the carotid body precludes immunoblotting for protein abundance (wet weight of the rat carotid body is ~50 μg), we determined the effects of intermittent hypoxia on CSE protein abundance by assessing rat liver tissue. We believe liver tissue to be a reasonable substitute, as livers of rats exposed to intermittent hypoxia also exhibit increased H2S and MDA amounts, both of which were blocked by MnTMPyP treatment (fig. S1, A and B). In the liver, CSE protein abundance was similar between intermittent hypoxia-exposed and control rats (Fig. 1F; and fig.S1C). Together, these findings suggest that changes in CSE abundance do not account forthe ROS -dependentincrease in H2S in rats exposed to intermittent hypoxia.
We next investigated whether ROS generated by intermittent hypoxia directly affect CSE enzymatic activity in human embryonic kidney (HEK)-293 cells heterologously expressing CSE (Fig. 1G). Cells were exposed to 60 cycles of intermittent hypoxia, with each cycle consisting of 30 sec of hypoxia (1.5% O2) followed by 5 min of room air (20% O2), as described previously (13). This paradigm of in vitro intermittent hypoxia has been shown to induce cellular responses similar to those evoked by in vivo intermittent hypoxia (10, 13). Although intermittent hypoxia increased ROS, as evidenced by increased MDA amounts, H2S concentrations remained unchanged (Fig.1, H and I). H2S concentrations were unaltered by treating control CSE-expressing cells with ROS scavenger MnTMPyP (Fig.1I). These observations rule out a direct effect of intermittent hypoxia-evoked ROS on CSE catalytic activity.
Intermittent hypoxia-induced ROS inhibits CO production to increase H2S
Carotid bodies express heme oxgenase -2 (HO-2), which synthesizes the signaling molecule carbon monoxide (CO) (29). CO inhibits H2S signaling by stimulating protein kinase G (PKG) to phosphorylate and inactivate CSE (21). We hypothesized that intermittent hypoxia- induced ROS may be inhibiting CO synthesis, thereby decreasing the inhibitory actions of PKG on CSE augmenting H2S production from CSE. To test this possibility, CO concentrations were measured in carotid bodies and liver tissue of intermittent hypoxia-exposed rats, and PKG activity and CSE phosphorylation were analyzed only in liver tissue due to the limited availability of the carotid body tissue. CO concentrations were decreased in the carotid bodies and liver tissue of intermittent hypoxia-exposed rats (Fig.2, A and B) and these effects were associated with reduced PKG activity and CSE phosphorylation (Fig.2, C and D). All of these responses to intermittent hypoxia were abolished by administration of the ROS scavenger MnTMPyP (Fig.2 A to D). However, ROS scavenger had no effect on basal CO concentration, PKG activity and CSE phosphorylation of carotid bodies and liver tissue from control rats (Fig. 2, Ato D). Neither HO-2-mRNA norHO -2 protein abundanceswere altered in the carotid bodies and liver tissue, respectively, of rats exposed to intermittent hypoxia (fig.S2, A and B). These observations indicate that ROS generated during intermittent hypoxia inhibits HO-2- CO production without affecting HO-2 expression, ultimately leading to an increase in H2S production by CSE.
Fig. 2. Intermittent hypoxia (IH) increases H2S concentrations through ROS-dependent inhibition of CO production.

(A) CO concentrations in the carotid bodies of rats exposed to room air (C), room air with MnTMPyP, a ROS scavenger (RS; C+RS), IH, or IH with RS (n = 3 experiments for each treatment; a total of 12 carotid bodies from 6 rats for each treatment). (B to D) CO concentrations (B), protein kinase G (PKG) activity (C) and serine phosphorylation of CSE (D) in liver samples from rats exposed to room air (C), room air with RS (C+RS), IH, or IH with RS. (n = 4 experiments for each treatment). (E to I) Concentrations of MDA (E), CO (F), H2S (G), activity of PKG (H), and serine phosphorylation of CSE (I) in HO-2- and CSE- expressing HEK-293 cells exposed to room air (C), room air with RS (C+RS), IH, or IH with RS (IH+RS) (n = 5 experiments for each treatment). In D and I, representative immunoblots (upper panel) and densitometry analysis (lower panel) are shown. I.P., immunoprecipitation. Data are presented as means ± SEM. *p < 0.05; **p < 0.01; ***p< 0.001; ns, not significant. See fig.S2 for data on HO-2 mRNA in the rat carotid body and HO-2 protein abundance in the rat liver.
Intermittent hypoxia reduces CO production through direct inhibition of HO-2 enzyme activity by ROS
We next investigated whether intermittent hypoxia reduces CO production by direct inactivation of HO-2 through ROS. As the limited mass of carotid body precludes rigorous biochemical analysis, we tested whether we could observe inhibitory effects of intermittent hypoxia on CO production similar to that of the carotid body in HEK-293 cells heterologously expressing both HO-2 and CSE (21) (fig.S3A). Intermittent hypoxia increased ROS as monitored by MDA amounts (Fig. 2E), decreased CO concentrations (Fig.2F), enhanced H2S concentrations (Fig.2G), and diminished both PKG activity (Fig.2H) and CSE phosphorylation (Fig.2I). The ROS scavenger MnTMPyP had no effects in control cells, but blocked the increase in MDA amounts and abolished intermittent hypoxia-induced changes in the amounts of CO, H2S, PKG activity and CSE phosphorylation (Fig.2 E to I) without affecting the protein abundance of either HO-2 or CSE (fig.S3A). These findings suggest that effects of intermittent hypoxia on carotid bodies can be at least partially recapitulated in HEK-293 cells.
To assess the mechanism by which ROS generated in response to intermittent hypoxia inhibit CO production, HO-2 enzyme activity was analyzed as a function of one of its substrates, hemin. In HEK-293 cells co-expressing HO-2 and CSE, the apparent Km of HO-2 for hemin was not altered by exposure to intermittent hypoxia (fig.S3B). However, intermittent hypoxia reduced maximal CO synthesis (Vmax), an effect prevented by the ROS scavenger MnTMPyP (Fig.3A and B). The C-terminal region of HO-2 contains three heme regulatory motifs at cysteine residues (Cys127, Cys265, and Cys282) (30) (Fig.3C, upper panel). Oxidation and reduction of cysteine thiols are thought to be a major mechanism for redox regulation of proteins/enzymes (30). We reasoned that one (or more) of these cysteines is required for ROS-dependent inhibition of CO synthesis by HO-2. Substituting alanine for Cys265 (C265A), but not Cys127 (C127A) or Cys282 (C282A) abolished the decrease in Vmax and CO generation in response to intermittent hypoxia (Fig.3, C and D and fig.S3C). Cells co-expressing CSE and HO-2 (C265A) still exhibited an increase in ROS during intermittent hypoxia, but no longer responded with changes in PKG activity, CSE phosphorylation or H2S concentrations (Fig.3, E to H). Unlike HO-2, HO-1, an inducible CO synthesizing enzyme, does not have cysteine containing heme regulatory motifs (30). Cells expressing HO-1 exhibited elevated ROS levels during intermittent hypoxia without inhibition of CO concentration (fig.S4, Aand B).
Fig. 3. Intermittent hypoxia (IH) alters the kinetic properties of HO-2.

(A to B) CO production as a function of hemin concentration (A) and maximal reaction rate of CO production (Vmax; B) in HO-2- and CSE-expressing HEK-293 cells exposed to room air (C), room air with ROS scavenger (RS; C+RS), IH, or IH with RS (n = 4 experiments for each treatment). (C to D) Effects of mutation of cysteine residues in HO-2 on Vmax (C) and CO generation (D) in HEK-293 cells exposed to room air (C) or IH. Upper panel in C shows schematic location of cysteine residues in heme regulatory motifs (HRMs) of HO-2. MSR, membrane spanning region (n= 5 experiments for each treatment). (E to H) MDA amounts (E), PKG activity (F), serine phosphorylation of CSE (G), and H2S concentrations (H) in CSE and HO-2 (C265A)-expressing HEK-293 cells exposed to room air (C), room air with RS (C+RS), IH, or IH with RS (n = 4 experiments for each treatment). In G representative immunoblots (upper panel) and densitometry analysis (lower panel) are shown. I.P., immunoprecipitation. (I) CO production in wild-type HO-2-expressing HEK-293 cells exposed to room air (C), room air with PEG-catalase (C+CAT), IH, and IH with CAT (IH+CAT) (n = 4 experiments for each treatment. (J) Effect of hydrogen peroxide (H2O2) on CO production in HEK-293 cells expressing either wild-type HO-2 (WT) or C265A mutant HO-2 (n = 5 experiments in each group). Data are presented as means ± SEM. *p < 0.05; **p < 0.01; ***p< 0.001; ns, not significant. See fig.S3 for data on CSE and HO-2 protein abundance and hemin binding affinity (Km) for HO-2. See fig.S4 for data on concentrations of MDA and CO in HEK-293 cells expressing HO-1.
We next sought to identify particular species of ROS mediating the effects of intermittent hypoxia on HO-2. Hydrogen peroxide (H2O2), derived from superoxide anion (O2.-), has been implicated in mediating, in part, the effects of intermittent hypoxia on the carotid body (31). Polyethylene glycol (PEG)-catalase, a scavenger of H2O2 , had no effect on CO concentration in control cells expressing HO-2 and CSE, but rescued inhibition of CO concentration by intermittent hypoxia (Fig.3I). Moreover, exogenous H2O2, like intermittent hypoxia, inhibited CO concentration in cells expressing wild-type HO-2 but not in cells expressing intermittent hypoxia-insensitive C265A mutant HO-2 (Fig.3J).
H2S mediates carotid body activation by intermittent hypoxia
We next investigated whether increased H2S production contributes to carotid body activation during intermittent hypoxia. Carotid bodies were harvested and sensory nerve activity was recorded ex vivo to exclude the confounding influence of cardiovascular changes caused by intermittent hypoxia in intact animals. Action potentials of the same height, duration, and shape (i.e. single unit) were selected for analysis (fig.S5, A to H). To simulate the apneic episodes, carotid bodies were challenged ex vivo with 10 episodes of alternating cycles of 30 sec of hypoxia (5% O2) and five minutes of room air (20% O2). Carotid bodies isolated from intermittent hypoxia-exposed rats exhibited higher baseline sensory nerve activity, augmented responses to each episode of hypoxia, and a progressive increase in baseline activity after the termination of repetitive hypoxia (that is, sensory LTF). All of these effects were lost in rats treated with L-PAG during the last two days of a 10 day exposure to intermittent hypoxia (Fig. 4, A to D). Carotid bodies from wild-type mice, but not those from CSE-null mice, showed similar responses to intermittent hypoxia (Fig.4, E to H). Similar to wild-type mice exposed to intermittent hypoxia, carotid bodies of HO-2 null mice exhibited elevated baseline, augmented responses to each episode of hypoxia, and sensory long-term facilitation, and 10 day exposure to intermittent hypoxia had no further effect on these responses (fig. S6, A to D).
Fig. 4. Increased H2S production mediates activation of the carotid body by intermittent hypoxia (IH).

(A) Examples of ex vivo carotid body sensory nerve responses during ten 30-sec episodes of acute IH (AIH; at arrows) and post-AIH for 60 min (sensory LTF; sLTF) from rats exposed to room air (C), room air with L-PAG (C+PAG), IH, or IH with L-PAG (IH+PAG). (B to D) Average data of pre-AIH baseline activity (B), hypoxia (Hx) response (mean of 10 episodes of AIH; C), and sensory long-term facilitation (sLTF; averaged over 60 min of post-AIH; D) (n = 8 experiments for each treatment; 16 single units from 8 carotid bodies for each treatment). (E) Examples of ex vivo carotid body sensory nerve responses during five 30-sec episodes of AIH (at arrows) and post-AIH for 60 min (sensory LTF; sLTF) from wild-type mice exposed to room air (WT-C) or IH (WT-IH) and Cse−/− mice exposed to room air (Cse−/−-C) or IH (Cse−/−-IH). (Fto H) Average data of pre-AIH baseline activity (F), hypoxia (Hx) response (mean of 5 episodes of AIH; G), and sLTF (averaged over 60 min of post-AIH; H) (n = 8 experiments for each treatment per genotype; 16 single units from 8 carotid bodies for each treatment per genotype. Data are presented as means ± SEM. *p < 0.05; ***p < 0.001; ns, not significant. See fig.S5 for analysis of carotid body single unit sensory nerve activity.
Inhibiting H2S synthesis prevents intermittent hypoxia-induced sympathetic activation and hypertension
The chemosensory reflex arising from the carotid body plays a critical role in mediating sympathetic activation and hypertension in intermittent hypoxia-exposed rodents (6). We tested whether blocking H2S synthesis prevents intermittent hypoxia-induced sympathetic activation and hypertension. Blood pressure and splanchnic sympathetic nerve activity (SNA) were recorded in anesthetized rats. Intermittent hypoxia-exposed rats showed increased basal blood pressure and SNA (Fig.5, A to C). To simulate apneas, rats were challenged with brief episodes of hypoxia (12% inspired O2 for 30 sec). In response to brief hypoxia, blood pressure and SNA were markedly increased in intermittent hypoxia-exposed rats, whereas these responses were nearly absent in control rats (Fig.5A and D to E). L-PAG treatment during the last two days of a 10-day exposure to intermittent hypoxia normalized basal blood pressure and SNA, and also prevented the increases in blood pressure and SNA during brief hypoxic challenges in intermittent hypoxia-exposed rats, without affecting baseline blood pressure and SNA in control rats (Fig.5, A to E).
Fig. 5. H2S mediates chemosensory reflex-dependent sympathetic activation and hypertension in intermittent hypoxia-exposed rodents.

(A) Examples of arterial blood pressure, (BP; upper panels) and splanchnic sympathetic nerve activity (SNA; middle and lower panels) responses to acute hypoxia (12% O2 for 30 sec, indicated by black bars) of rats exposed to room air (C), room air with L-PAG (C+PAG), IH, or IH with L-PAG (IH+PAG). Arrows denote baseline BP (upper panel) and SNA (lower panel). Lower panels show SNA on an expanded time scale of 200 milliseconds as compared to 2 minutes in middle pannels to demonstrate action potentials of the splanchnic nerve under normoxia (Nx) and hypoxia (Hx) conditions. (B to E) Average data of baseline BP (B), baseline SNA (C), Hx-induced change in BP (D), and SNA (E) of rats exposed to room air (C), room air with L-PAG (C+PAG), IH, or IH with L-PAG (IH+PAG)( n = 8 rats for each treatment). (F to G) Effect of intermittent hypoxia on baseline BP (F) and plasma catecholamine concentrations (G) of wild-type (WT) and CSE-null (Cse−/−) mice exposed either to room air (C) or IH (n = 7 to 9 mice for each treatment per genotype). Data are presented as means ± SEM. *p < 0.05; **p < 0.01; ***p < 0.001; ns, not significant.
To further investigate the role of CSE-derived H2S in intermittent hypoxia-induced hypertension, we compared the effects of intermittent hypoxia between four-week old wild-type and CSE null mice. Blood pressure was monitored by the tail-cuff method before and after intermittent hypoxia, and plasma catecholamine (epinephrine and norepinephrine) concentrations were measured as an index of sympathetic activation. Intermittent hypoxia-exposed wild-type mice exhibited elevated blood pressure and plasma catecholamines, responses that were absent in intermittent hypoxia-exposedCSE -nullmice (Fig. 5, F and G).
DISCUSSION
Sleep apnea, a clinically prevalent disorder, elicits intermittent hypoxia which often leads to hypertension. Considerable evidence suggests that a hyperactive carotid body and the ensuing chemosensory reflex can drive this hypertension through increased sympathetic nerve activity, but the underlying molecular mechanisms have been elusive. Our results establish a fundamental mechanism contributing to carotid body activation and the resulting cardiovascular pathology in a rodent model of sleep apnea. Specifically, we find that ROS generated during intermittent hypoxia in the carotid body depresses CO signaling, ultimately elevating stimulatory H2S signaling (Fig. 6).
Fig. 6. Intermittent hypoxia activates H2S signaling in the carotid body.
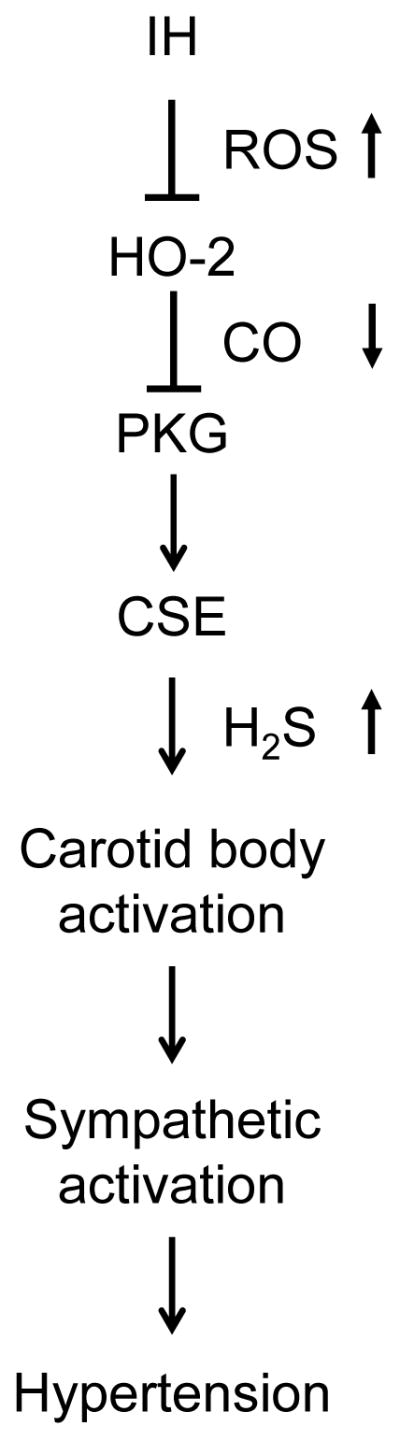
Intermittent hypoxia (IH) induces ROS-dependent inhibition of CO production by HO-2, which leads to decreased PKG activity and increased H2S production by CSE that triggers the carotid body chemosensory reflex, leading to sympathetic activation and hypertension.
Challenging rodents with intermittent hypoxia to simulate the blood-O2 saturation profiles encountered during sleep apnea increased CSE-dependent H2S production in the carotid body. Scavenging ROS was sufficient to abrogate the increase in H2S concentrations in the carotid body in response to intermittent hypoxia, demonstrating the epistatic importance of ROS. We observed that H2S signaling was elevated during intermittent hypoxia due to a decrease in inhibitory CO signaling and not due to changes in CSE expression. Intermittent hypoxia reduced the rate of CO production through the direct inhibition of HO-2 activity via ROS. Attenuated CO production in turn decreased the level of inhibitory phosphorylation of CSE by PKG, resulting in increased H2S synthesis.
Analysis of the molecular mechanism underlying HO-2 inactivation by intermittent hypoxia revealed that ROS reduced the rate of CO production (Vmax) from the catalysis of hemin without altering the binding of hemin to HO-2. Furthermore, we identified Cys265 in the heme regulatory motif as a critical redox-sensing residue. A role for Cys265 was further supported by the finding that ROS produced during intermittent hypoxia had no inhibitory effect on CO production in cells expressing HO-1, which lacks cysteine containing heme regulatory motif (30). Our results further suggest that H2O2 is one of the species of ROS that mediate HO-2 inactivation during intermittent hypoxia. Due to technical limitations these analyses were performed in HEK-293 cells with heterologous expression of HO-2 and CSE. However, similar signaling is likely to occur in the carotid bodies because the effects of intermittent hypoxia on ROS, CO, and H2S concentrations were similar between HEK-293 cells and the carotid body.
Acute hypoxia also activates H2S generation through decreased CO production via HO-2 and reduced serine phosphorylation of CSE by PKG (21). However, the reduced CO production under conditions of acute hypoxia is primarily due to the low affinity of HO-2 for O2, rather than an effect on Vmax, which enables the enzyme to transduce changes in O2 availability into changes in CO production (21). Moreover, the effects of acute hypoxia are transient and reversed after a few minutes of re-oxygenation (21). In contrast, the effects of intermittent hypoxia on CO production, serine phosphorylation of CSE and H2S concentrations persist for several hours after terminating the intermittent hypoxia exposure. Our results suggest that the long-lasting effects of intermittent hypoxia on CO-regulated H2S production are due to increased ROS generation during intermittent hypoxia, which is further accentuated by imbalanced changes in the expression of anti-oxidant and pro-oxidant enzymes (14, 31).
A major finding of the present study is that blockade of H2S synthesis is sufficient to prevent carotid body activation and hypertension in intermittent hypoxia-exposed rodents. Although exogenous and endothelial H2S has been found to yield local blood vessel relaxation (32–34), we find that H2S exerts excitatory effects on carotid body activity during intermittent hypoxia. Consistent with previous reports (8-10), carotid bodies from intermittent hypoxia exposed rats displayed exaggerated hypoxic sensitivity and a persistent increase in baseline activity after repetitive hypoxia (sensory LTF). The enhanced carotid body activity in rats exposed to intermittent hypoxia was reflected in the heightened chemosensory reflex, as evidenced by increased baseline sympathetic activity and BP, as well as marked hypertension and sympathetic activation during simulated apneas. Treating rats with a CSE inhibitor during the last two days of intermittent hypoxia exposure restored normal carotid body function, sympathetic nerve activity and blood pressure, and blocked hypertensive responses to simulated apneas. It is important to note that CSE is also expressed in the brainstem neuronal populations important for regulating blood pressure (35). While our results illustrate that CSE null carotid bodies exhibit a tissue-autonomous reduction in activity, the full blood pressure responses of CSE null mice may be due to altered signaling from the brainstem regions as well as the carotid body.
Anti-oxidant treatment also normalized carotid body function (10) and prevented hypertension (16), demonstrating that ROS is epistatic to H2S in driving hypertension. However, anti-oxidants must be administered during the entire ten days of intermittent hypoxia exposure, and they are ineffective in normalizing carotid body function when given during the last day of intermittent hypoxia (10). Previous studies have implicated endothelin-1 (ET-1) in causing the augmented hypoxic sensitivity (36-38) and 5-hydroxytryptamine (5-HT) in inducing sensory LTF (31) in carotid bodies of intermittent hypoxia-exposed rodents. Since ET-1 and 5-HT also increase ROS (39, 40), the effects of these transmitters on the carotid body are presumably mediated through ROS-dependent activation of H2S production. Such a mechanism might explain why blocking H2S production by administration of a CSE inhibitor was sufficient to prevent augmented hypoxic sensitivity and sensory LTF as well as the ensuing chemosensory reflex-mediated hypertension. We recently discovered that HO-2 null mice upregulate another gaseous messenger, nitric oxide (NO), and its synthesizing enzyme, neural nitric oxide synthase (nNOS), to maintain oxygen sensing (21). However, carotid bodies of HO-2 null mice exhibited phenotype including elevated baseline sensory nerve activity, augmented response to acute hypoxia, and sensory LTF in response to acute intermittent hypoxia, similar to wild-type mice exposed to ten days of intermittent hypoxia. Given that ten days of intermittent hypoxia had no further effect on the carotid body sensory activity in HO-2 null mice, NO signaling from nNOS is unlikely play a major role in mediating sensory LTF and hypertension, a possibility that requires further investigation.
Population based studies have shown a strong correlation between the severity of sleep apnea and hypertension (1, 41). At least one third of patients with obstructive sleep apnea exhibit a heightened chemosensory reflex that adversely contributes to the severity of their hypertension (42). Although continuous positive airway pressure (CPAP) is presently the principal treatment for obstructive sleep apnea, a substantial number of patients do not respond to CPAP (43). In some patients, CPAP induces central apnea and periodic breathing, which places patients at increased risk of hypertension (44). Idiopathic central sleep apnea is also characterized by an exaggerated chemosensory reflex and greatly increased risk of hypertension (45), and treatment options for central apnea are even more limited. In both central and obstructive sleep apnea, the acute elevations in blood pressure associated with apneic episodes may predispose patients to hemorrhagic stroke, while chronic hypertension increases the risk of heart failure. Thus, controlling hypertension in sleep apnea patients is a major clinical problem. Our results, which models both obstructive and central sleep apnea, suggest that inhibiting CSE to reduce H2S signaling in the carotid body may be a novel approach to treat hypertension in patients with sleep apnea.
MATERIALS AND METHODS
Preparation of Animals
Experiments were approved by the Institutional Animal Care and Use Committee of the University of Chicago and were performed on age-matched male rats or age-matched male C57/BL6 wild-type and Cse−/ − mice (32) unless otherwise noted. Acute experiments were performed on rats and mice anesthetized with urethane (1.2 g/kg i.p.). At the end of the experiment, rats and mice were euthanized by intra-cardiac injection (0.1 mL) of euthanasia solution (Beuthanasia-D Special, Schering-Plough). Where indicated, rats were treated by i.p. injection of MnTMPyP (Enzo Life Sciences; 5 mg/kg/day) or L-PAG (Chem-Impex International Inc;30 mg/kg/d ay).
Exposure to Intermittent Hypoxia
The protocols for intermittent hypoxia exposure were described previously (10, 12). Briefly, conscious rats or mice were exposed to alternating cycles of hypoxia (5% O2 for 15 sec) and room air (21% O2 for 5 min), between 09:00 and 17:00 h for 10 days. Control experiments were performed on rats and mice exposed to alternating cycles of room air instead of hypoxia. Sixteen hours after terminating the 10th day of intermittent hypoxia exposure, carotid bodies and livers were collected underterminal ur ethane anesthesia.
Measurements of BPand SNA
BP was monitored by the tail-cuff method in conscious mice using a non-invasive BP system (IITC Life Science Inc.) as described (12). Acute experiments were performed on rats anesthetized with urethane. After tracheal intubation, the femoral artery and vein were cannulated. BP was monitored via the femoral artery catheter. The rectal temperature was maintained at 37.5°C ± 1°C by means of a heating pad. The rats were mechanically ventilated (Harvard Apparatus). The left splanchnic nerve was isolated retroperitoneally, transected and desheathed. The central cut ends of the nerve were placed on bipolar platinum-iridium electrodes for recording electrical activity as described previously (12). For quantification of SNA, hexamethonium (25 mg/kg i.v.) was administered at the end of the experiment to block the electrical activity for calibration.
Ex VivoCarotid Body Recording
Sensory nerve activity was recorded from ex vivo carotid bodies harvested from anesthetized rats and mice as previously described (10, 27). Briefly, the carotid sinus nerve was treated with collagenase, and several nerve bundles were isolated. Action potentials from one of the nerve bundles were recorded using a suction electrode (~20-μm-diameter tip). In general, 2-3 action potentials of varying size and amplitude were seen in a given nerve bundle. For analysis of sensory nerve activity, action potentials of the same height, duration, and shape (i.e. single unit) were selected using Spike histogram software (Labchart 7 Pro; fig.S5). To obtain stable baseline sensory nerve activity, carotid bodies were first superfused for 1 h with room air-equilibrated medium. Baseline sensory nerve activity under normoxia was recorded for 5 min, followed by acute intermittent hypoxia (30 sec of hypoxia followed by 5 min of room air for each cycle and a total of 10 cycles in rats and 5 cycles in mice) as described previously (31). Sensory nerve activity was recorded for 60 min after the last cycle of acute intermittent hypoxia. Baseline sensory nerve activity and hypoxic response (cumulative response during the entire cycles of AIH) were expressed as impulses/second. Sensory nerve activity during the entire 60 min following the termination of acute intermittent hypoxia was expressed as percentage of baseline activity.
Plasmids, Cell Culture and Transfection
cDNA encoding rat HO-2 was ligated into pcDNA3.1(Thermo Fisher; #V790-20) directly 3’ to a nucleotide sequence encoding an N-terminal Myc tag. cDNA encoding mouse CSE was ligated into pCMV- Myc-N (Clontech; #635689). Missense mutations of HO-2 were generated using the QuikChange II XL mutagenesis kit (Agilent Technologies; #200521) according to the manufacturer’s instructions. Mutations were verified by nucleotide sequencing at the University of Chicago Core sequencing facility. HEK-293 cells were cultured in Dulbecco’s modified Eagle’s medium (Life Technologies; #11995-065), 10% fetal bovine serum, 100 units/mL penicillin, and 100 units/mL streptomycin in 95% air and 5% CO2 (20% O2) at 37°C. Cells were seeded on 60-mm plates and transfected with 0.5 μg of plasmid DNA encoding either wild-type or mutant HO-2 and/or 2μg of plasmid DNA encoding CSE using Lipofectamine (Invitrogen; #18324-012) according to the manufacturer’s protocol. HEK-293 cell cultures were exposed to intermittent hypoxia as described previously (13). Where indicated, HEK-293 cells were pre-treated with MnTMPyP (50 μM) for 30 min prior to intermittent hypoxia exposure.
Immunoblot Assaysfor HO -2 and CSE
Immunoblot assays for HO-2 and CSE were performed as described (21). The following primary antibodies (and dilution) were used: HO-2 (Abcam; #ab90492, 1:1000), CSE (Novus Biologicals; #NBP1-52849, 1:1000), and α-tubulin (Sigma-Aldrich; #T6199, 1:1000).
Immunoprecipitation Assay forCSE phosphorylation
To assess the relative abundance of phosphorylated CSE, either HEK-293 cell or liver extracts were treated with anti-CSE antibody (2 μg; Novus Biologicals; #NBP1-52849) at 4oC for 18 h. The resulting CSE-antibody complex was pull down with 50 μl suspension of Protein G magnetic beads (Millipore; #LSKMAGG10) by incubating at room temperature for 20 minutes. The immunoprecipitates were washed with phosphate-buffered-saline containing 0.1% Tween-20 and extracted with Laemmli buffer for Western blot analysis. The immunoblots were probed with either anti-phosphoserine antibody (Millipore, #AB1603,1:1000) or anti-CSE antibody (Novus Biologicals; #NBP1-52849, 1:1000).
Measurement of H2S
H2S generation in the carotid bodies, liver tissues and HEK-293 cells were determined by in vitro methylene blue assay, as described previously (19) with following modifications. The absorbance of H2S trapping solution was monitored at 670 nm using a Shimadzu UV-VIS Spectrophotometer (UV-2401PC). H2S concentrations were calculated using a molar extinction coefficient of 71,089 M−1cm−1 at 670 nm and expressed as nanomoles per hour per milligram of protein.
Measurement of CO
CO production in the carotid bodies, liver tissues and HEK-293 cells was determined using in vitro crystal violet assay, as described previously (20, 46) except that CO concentrations were calculated using a molar extinction coefficient of 87,000 M−1.cm−1 at 590 nm and expressed as nanomoles per hour per milligram of protein.
Measurement of MDAlevels
Carotid bodies, liver tissue, or HEK-293 cells were homogenized in 10 volumes of 20 mM phosphate buffer (pH 7.4) at 4°C and the resulting homogenate was centrifuged at 500 g for 10 min at 4°C. MDA levels were analyzed in the supernatant as previously described (27) and were reported in nanomoles of MDA formed per milligram of protein.
Measurement of PKG activity
cGMP concentrations were determined by a chemiluminescent ELISA assay (Cell Biolabs; #STA-506) according to the manufacturer’s instructions. Prior to the experiment, cells were treated with 1 mM 3-isobutyl-1-methylxanthine to inhibit degradation of cGMP by phosphodiesterases. cGMP concentrations were normalized to protein concentration as determined by bicinchoninic acid assay and were reported in milliunits per milligram of protein.
Measurement of plasma catecholamine concentrations
Blood samples ( 300 μl) were collected from anesthetized mice by cardiac puncture, and placed in heparinized (30 U per ml of blood) ice-cold microcentrifuge tubes. Plasma was separated by centrifugation and stored at 80°C. Plasma norepinephrine and epinephrine were determined by high pressure liquid chromatography combined with electrochemical detection using 3,4-dihydroxy-benzyl-amine hydrobromide (DHBA; Sigma-Aldrich) as an internal standard (27) and expressed as nanogramsof catecholamines per m illiliterof plasma.
Quantitation of HO-2 and CsemRNA
HO-2 and Cse mRNA levels in the carotid bodies were analyzed by RT-qPCR with SYBR GreenER (#11764-100, Invitrogen) as previously described (20). Briefly, RNA was extracted from rat carotid bodies (two carotid bodies from a single rat) using TRIzol (Thermo Fisher) and was reverse transcribed using SuperScript III reverse transcriptase (Thermo Fisher). Relative mRNA quantification, expressed as fold change (F) was calculated using the formula = 2−Δ ΔCt where ΔCT is the difference between the threshold cycles of the given target cDNA and 18S rRNA, and Δ (ΔCT) is the difference between the ΔCT values under normoxia and intermittent hypoxia. PCR specificity was confirmed by omitting the template and by performing a standard melting curve analysis. The nucleotide sequences of primers used for qPCR are as follows: rat CSE: 5’-AGC GAT CAC ACC ACA GAC CAA-3’ and 5’-ATC AGC ACC CAG AGC CAA AGG-3’; rat HO-2: 5’-ACT GGG AGG AGC AGG TGA AG-3’ and 5’-GGT AGA ACT GGG TCC CTT CCC-3’; 18S rRNA: 5’-GTA ACC CGT TGA ACC CCA TT-3’ and 5’-CCA TCC AATCGG TAG TAG CG-3’.
Determination of Kinetic Properties of HO-2
HEK-293 cells co-expressing HO-2 and CSE were homogenized at 4°C with 3 volumes of 0.05 M Tris HCl buffer (pH 7.4) with 10 mM EDTA and 20% (Vol/Vol) glycerol and centrifuged at 100,000 g for 70 min at 4°C. CO production was determined as a function of hemin concentration (from 0.01 to 20 μM). The apparent hemin binding affinity (Km) and maximum velocity (Vmax) were analyzed as previously described (20).
Drugs
The following drugs were used: MnTMPyP (Enzo Life Sciences, #ALX-430-070-M050), L-PAG (Chem-Impex International Inc., #02726), H2O2 (Sigma-Aldrich, #323381) and Catalase-polyethylene glycol (PEG-Catalase, Sigma-Aldrich, #C4963). All drugs were dissolved in physiological saline, and prepared fresh during the experiment. The following concentrations of drugs were used: MnTMPyP, 50 μM; PEG-Catalase, 10units/ml and H2O2, 100 μM. In experiments involving HEK-293 cells, cells were treated with desired concentration of drugs 30 min before the experiment. Rats and mice were treated with MnTMPyP (5mg/kg intraperitoneal route) every day prior to exposure to intermittent hypoxia. In the experiments with L-PAG, rats and mice were treated with L-PAG (30 mg/kg intraperitoneal route) daily during the last two days of exposure to intermittent hypoxia.
Data Analysis and Statistics
All data are reported as mean ± SEM derived from three independent biological experiments, unless otherwise stated in the figure legends. Statistical analysis was performed with either one-way or two-way analysis of variance (ANOVA) with repeated measures followed by post-hoc Tukey’s test. For the analysis of normalized data, Mann-Whitney’s test was employed. All p values < 0.05 were considered significant.
Supplementary Material
Fig. S1. Effect of IH on H2S and MDA concentrations and CSE protein abundance in rat liver. (A to B) H2S (A) and MDA (B) amounts in liver tissue of rats exposed to room air (C), room air with ROS scavenger (RS; C+RS), IH, or IH treatment with RS (IH+RS) (n = 3 rats for each treatment). (C) Densitometry analysis of CSE protein abundance in liver tissue of rats exposed to room air (C) or IH (n = 3 rats for each treatment) presented as ratio of CSE/α-tubulin (loading control). Data are presented as means± SEM. ***p < 0.001; ns, not significant.
Fig. S2. Effect of IH on HO-2 mRNA levels and protein abundance. (A) The ratio of HO-2 mRNA to 18S rRNA in carotid bodies of rats exposed to room air (C) or IH was determined by RT-qPCR and normalized to C (n = 5 for each treatment). Data are presented as means ±SEM. ns, not significant. (B) Representative immunoblot of HO-2 protein abundance in liver tissue from room air (C) and IH-exposed rats (n = 3 rats for each treatment). α-tubulin abundance was assayed as a loading control.
Fig. S3. Analysis of CSE and HO-2 protein abundance in HEK-293 cells. (A) Representative immunoblots of CSE and HO-2 protein abundance in CSE and HO-2-expressing HEK-293 cells exposed to room air (20% O2), (C), room air with ROS scavenger (C+RS), IH, or IH with RS (IH+RS) (n = 5 independent experiments). (B) Hemin binding affinity (Km) of HO-2 in HEK-293 cells expressing wild-type HO-2 treated with room air (20% O2), (C) or IH (n = 4 experiments for each treatment). Data are presented as means ±SEM. ns, not significant. (C) HO-2 protein abundance in HEK-293 cells expressing either wild-type (WT) or mutant (C265A, C127A, or C282A) HO-2 exposed to room air (20% O2), (C) or IH (n = 4 independent experiments for each treatment). α-tubulin abundance was assayed as a loading control.
Fig. S4 Effect of IH on CO concentration in HO-1-expressing HEK-293 cells. (A) MDA and (B) CO concentrations in HO-1 expressing HEK-293 cells exposed to room air (20% O2), (C) or IH (n = 4 independent experiments for each treatment. Data are presented as means ±SEM. *p < 0.05.
Fig. S5 Analysis of carotid body sensory nerve activity. (A to B) Upper panels show examples of raw action potentials recorded from the carotid sinus nerve of carotid bodies harvested from rats exposed to room air (CON, A) and intermittent hypoxia (IH, B) and their response to acute hypoxia (Hx). (C to D) Lower panels show identification of two “single units” with differing amplitudes in the control (C) and IH (D) treated carotid bodies. (E to F) Analysis of spike frequencies of Unit 1 (E and F) and Unit 2 (Gand H).
Fig. S6 Carotid body response to intermittent hypoxia in heme oxygenase-2 (HO-2) null mice (A) Examples of ex vivo carotid body sensory nerve responses during five 30-sec episodes of AIH (at arrows) and post-AIH for 60 min (sensory LTF; sLTF) from wild-type and HO-2 null mice exposed to room air (WT-C, HO-2 −/− -C) or HO-2 null mice exposed to intermittent hypoxia (HO-2 −/− -IH). (B to D) Average data of pre-AIH baseline activity (B), hypoxia (Hx) response (mean of 5 episodes of AIH; C), and sLTF (averaged over 60 min of post-AIH; D) (n = 6 experiments for each treatment per genotype; 12 single units from 6 carotid bodies for each treatment per genotype. Data are presented as means ± SEM. *p < 0.05; ***p < 0.001; ns, not significant.
SIGNIFICANCE STATEMENT.
Sleep apnea, a clinically prevalent disorder, often leads to hypertension. We demonstrate a signaling cascade that underlies this disorder. Sleep apnea causes intermittent hypoxia that activates carotid body signaling, which increases blood pressure. Here, we report that intermittent hypoxia generates reactive oxygen species (ROS) in the carotid body, which inactivates heme oxygenase-2, an enzyme that generates carbon monoxide (CO), ultimately increasing hydrogen sulfide (H2S) generation from cystathionine-γ-lyase to stimulate the carotid body neural activity triggering chemosensory reflex-mediated hypertension. These studies delineate epistatic molecular interactions between ROS, CO, and H2S production that control carotid body activation and the pathogenesis of hypertension in a rodent model of sleep apnea. These findings account for the pathophysiology of sleep apnea-associated hypertension and suggest that cystathionine-γ-lyase inhibitors may be useful in the treatment of hypertension in sleep apnea patients.
Acknowledgments
We are grateful to Professor Dan Nicolae for help with the statistical analysis and Professor Rui Wang for providing CSE knockout mice.
FUNDING: This work was supported by National Institutes of Health grants P01-HL-90554 and UH2-HL-123610.
Footnotes
AUTHOR CONTRIBUTIONS: NRP conceived the study, NRP and GKK designed research; GY, Y-JP, SAK, JN, AS, GKK performed experiments; GY, Y-JP, SAK, AS, JN, and GK analyzed the data; CV and SHS contributed to reagents; NRP, GK, GLS and SHS wrote the paper.
COMPETING INTERESTS: The authors declare that they have no competing interests.
REFERENCES AND NOTES
- 1.Nieto FJ, Young TB, Lind BK, Shahar E, Samet JM, Redline S, D'Agostino RB, Newman AB, Lebowitz MD, Pickering TG. Association of sleep-disordered breathing, sleep apnea, and hypertension in a large community-based study. Sleep Heart Health Study. JAMA. 2000;283:1829–1836. doi: 10.1001/jama.283.14.1829. [DOI] [PubMed] [Google Scholar]
- 2.Young T, Palta M, Dempsey J, Skatrud J, Weber S, Badr S. The occurrence of sleep-disordered breathing among middle-aged adults. N Engl J Med. 1993;328:1230–1235. doi: 10.1056/NEJM199304293281704. [DOI] [PubMed] [Google Scholar]
- 3.Ramirez JM, Garcia AJ, 3rd, Anderson TM, Koschnitzky JE, Peng YJ, Kumar G, Prabhakar N. Central and peripheral factors contributing to obstructive sleep apneas. Respir Physiol Neurobiol. 2013;189:344–353. doi: 10.1016/j.resp.2013.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Narkiewicz K, Somers VK. The sympathetic nervous system and obstructive sleep apnea: implications for hypertension. J Hypertens. 1997;15:1613–1619. doi: 10.1097/00004872-199715120-00062. [DOI] [PubMed] [Google Scholar]
- 5.Carlson JT, Hedner J, Elam M, Ejnell H, Sellgren J, Wallin BG. Augmented resting sympathetic activity in awake patients with obstructive sleep apnea. Chest. 1993;103:1763–1768. doi: 10.1378/chest.103.6.1763. [DOI] [PubMed] [Google Scholar]
- 6.Prabhakar NR, Peng YJ, Kumar GK, Nanduri J. Peripheral chemoreception and arterial pressure responses to intermittent hypoxia. Compr Physiol. 2015;5:561–577. doi: 10.1002/cphy.c140039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Narkiewicz K, van de Borne PJ, Pesek CA, Dyken ME, Montano N, Somers VK. Selective potentiation of peripheral chemoreflex sensitivity in obstructive sleep apnea. Circulation. 1999;99:1183–1189. doi: 10.1161/01.cir.99.9.1183. [DOI] [PubMed] [Google Scholar]
- 8.Peng YJ, Prabhakar NR. Effect of two paradigms of chronic intermittent hypoxia on carotid body sensory activity. J Appl Physiol. 2004;96:1236–1242. doi: 10.1152/japplphysiol.00820.2003. discussion 1196. [DOI] [PubMed] [Google Scholar]
- 9.Rey S, Del Rio R, Alcayaga J, Iturriaga R. Chronic intermittent hypoxia enhances cat chemosensory and ventilatory responses to hypoxia. J Physiol. 2004;560:577–586. doi: 10.1113/jphysiol.2004.072033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Peng YJ, Overholt JL, Kline D, Kumar GK, Prabhakar NR. Induction of sensory long-term facilitation in the carotid body by intermittent hypoxia: implications for recurrent apneas. Proc Natl Acad Sci U S A. 2003;100:10073–10078. doi: 10.1073/pnas.1734109100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fletcher EC, Lesske J, Behm R, Miller CC, 3rd, Stauss H, Unger T. Carotid chemoreceptors, systemic blood pressure, and chronic episodic hypoxia mimicking sleep apnea. J Appl Physiol. 1992;72:1978–1984. doi: 10.1152/jappl.1992.72.5.1978. [DOI] [PubMed] [Google Scholar]
- 12.Peng YJ, Yuan G, Khan S, Nanduri J, Makarenko VV, Reddy VD, Vasavda C, Kumar GK, Semenza GL, Prabhakar NR. Regulation of hypoxia-inducible factor-α isoforms and redox state by carotid body neural activity in rats. J Physiol. 2014;592:3841–3858. doi: 10.1113/jphysiol.2014.273789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yuan G, Khan SA, Luo W, Nanduri J, Semenza GL, Prabhakar NR. Hypoxia-inducible factor 1 mediates increased expression of NADPH oxidase-2 in response to intermittent hypoxia. J Cell Physiol. 2011;226:2925–2933. doi: 10.1002/jcp.22640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nanduri J, Wang N, Yuan G, Khan SA, Souvannakitti D, Peng YJ, Kumar GK, Garcia JA, Prabhakar NR. Intermittent hypoxia degrades HIF-2alpha via calpains resulting in oxidative stress: implications for recurrent apnea-induced morbidities. Proc Natl Acad Sci U S A. 2009;106:1199–1204. doi: 10.1073/pnas.0811018106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nanduri J, Peng YJ, Yuan G, Kumar GK, Prabhakar NR. Hypoxia-inducible factors and hypertension: lessons from sleep apnea syndrome. J Mol Med (Berl) 2015;93:473–480. doi: 10.1007/s00109-015-1274-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kumar GK, Rai V, Sharma SD, Ramakrishnan DP, Peng YJ, Souvannakitti D, Prabhakar NR. Chronic intermittent hypoxia induces hypoxia-evoked catecholamine efflux in adult rat adrenal medulla via oxidative stress. J Physiol. 2006;575:229–239. doi: 10.1113/jphysiol.2006.112524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Del Rio R, Moya EA, Iturriaga R. Carotid body and cardiorespiratory alterations in intermittent hypoxia: the oxidative link. Eur Respir J. 2010;36:143–150. doi: 10.1183/09031936.00158109. [DOI] [PubMed] [Google Scholar]
- 18.Kumar P, Prabhakar NR. Peripheral chemoreceptors: function and plasticity of the carotid body. Comprehensive Physiology. 2012;2:141–219. doi: 10.1002/cphy.c100069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Peng YJ, Nanduri J, Raghuraman G, Souvannakitti D, Gadalla MM, Kumar GK, Snyder SH, Prabhakar NR. H2S mediates O2 sensing in the carotid body. Proc Natl Acad Sci U S A. 2010;107:10719–10724. doi: 10.1073/pnas.1005866107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Peng YJ, Makarenko VV, Nanduri J, Vasavda C, Raghuraman G, Yuan G, Gadalla MM, Kumar GK, Snyder SH, Prabhakar NR. Inherent variations in CO-H2S- mediated carotid body O2 sensing mediate hypertension and pulmonary edema. Proc Natl Acad Sci U S A. 2014;111:1174–1179. doi: 10.1073/pnas.1322172111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yuan G, Vasavda C, Peng YJ, Makarenko VV, Raghuraman G, Nanduri J, Gadalla MM, Semenza GL, Kumar GK, Snyder SH, Prabhakar NR. Protein kinase G- regulated production of H2S governs oxygen sensing. Sci Signal. 2015;8:ra37. doi: 10.1126/scisignal.2005846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li Q, Sun B, Wang X, Jin Z, Zhou Y, Dong L, Jiang LH, Rong W. A crucial role for hydrogen sulfide in oxygen sensing via modulating large conductance calcium-activated potassium channels. Antioxid Redox Signal. 2010;12:1179–1189. doi: 10.1089/ars.2009.2926. [DOI] [PubMed] [Google Scholar]
- 23.Telezhkin V, Brazier SP, Cayzac SH, Wilkinson WJ, Riccardi D, Kemp PJ. Mechanism of inhibition by hydrogen sulfide of native and recombinant BKCa channels. Respir Physiol Neurobiol. 2010;172:169–178. doi: 10.1016/j.resp.2010.05.016. [DOI] [PubMed] [Google Scholar]
- 24.Kemp PJ, Telezhkin V. Oxygen sensing by the carotid body: is it all just rotten eggs? Antioxid Redox Signal. 2014;20:794–804. doi: 10.1089/ars.2013.5377. [DOI] [PubMed] [Google Scholar]
- 25.Takano N, Peng YJ, Kumar GK, Luo W, Hu H, Shimoda LA, Suematsu M, Prabhakar NR, Semenza GL. Hypoxia–inducible factors regulate human and rat cystathionine β-synthase gene expression. Biochem J. 2014;458:203–211. doi: 10.1042/BJ20131350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Asimakopoulou A, Panopoulos P, Chasapis CT, Coletta C, Zhou Z, Cirino G, Giannis A, Szabo C, Spyroulias GA, Papapetropoulos A. Selectivity of commonly used pharmacological inhibitors for cystathionine beta synthase (CBS) and cystathionine gamma lyase (CSE) Br J Pharmacol. 2013;169:922–932. doi: 10.1111/bph.12171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Peng YJ, Yuan G, Ramakrishnan D, Sharma SD, Bosch-Marce M, Kumar GK, Semenza GL, Prabhakar NR. Heterozygous HIF-1alpha deficiency impairs carotid body-mediated systemic responses and reactive oxygen species generation in mice exposed to intermittent hypoxia. J Physiol. 2006;577:705–716. doi: 10.1113/jphysiol.2006.114033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gardner PR, Nguyen DD, White CW. Superoxide scavenging by Mn(II/III) tetrakis (1-methyl-4-pyridyl) porphyrin in mammalian cells. Arch Biochem Biophys. 1996;325:20–28. doi: 10.1006/abbi.1996.0003. [DOI] [PubMed] [Google Scholar]
- 29.Prabhakar NR, Dinerman JL, Agani FH, Snyder SH. Carbon monoxide: a role in carotid body chemoreception. Proc Natl Acad Sci U S A. 1995;92:1994–1997. doi: 10.1073/pnas.92.6.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McCoubrey WK, Huang TJ, Maines MD. Heme oxygenase-2 is a hemoprotein and binds heme through heme regulatory motifs that are not involved in heme catalysis. J Biol Chem. 1997;272:12568–12574. doi: 10.1074/jbc.272.19.12568. [DOI] [PubMed] [Google Scholar]
- 31.Peng YJ, Nanduri J, Yuan G, Wang N, Deneris E, Pendyala S, Natarajan V, Kumar GK, Prabhakar NR. NADPH oxidase is required for the sensory plasticity of the carotid body by chronic intermittent hypoxia. J Neurosci. 2009;29:4903–4910. doi: 10.1523/JNEUROSCI.4768-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yang G, Wu L, Jiang B, Yang W, Qi J, Cao K, Meng Q, Mustafa AK, Mu W, Zhang S, Snyder SH, Wang R. H2S as a physiologic vasorelaxant: hypertension in mice with deletion of cystathionine gamma-lyase. Science. 2008;322:587–590. doi: 10.1126/science.1162667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhong G, Chen F, Cheng Y, Tang C, Du J. The role of hydrogen sulfide generation in the pathogenesis of hypertension in rats induced by inhibition of nitric oxide synthase. J Hypertens. 2003;21:1879–1885. doi: 10.1097/00004872-200310000-00015. [DOI] [PubMed] [Google Scholar]
- 34.Lu M, Liu YH, Goh HS, Wang JJ, Yong QC, Wang R, Bian JS. Hydrogen sulfide inhibits plasma renin activity. J Am Soc Nephrol. 2010;21:993–1002. doi: 10.1681/ASN.2009090949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Paul BD, Sbodio JI, Xu R, Vandiver MS, Cha JY, Snowman AM, Snyder SH. Cystathionine γ-lyase deficiency mediates neurodegeneration in Huntington's disease. Nature. 2014;509:96–100. doi: 10.1038/nature13136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rey S, Del Rio R, Iturriaga R. Contribution of endothelin-1 to the enhanced carotid body chemosensory responses induced by chronic intermittent hypoxia. Brain Res. 2006;1086:152–159. doi: 10.1016/j.brainres.2006.02.082. [DOI] [PubMed] [Google Scholar]
- 37.Peng YJ, Nanduri J, Raghuraman G, Wang N, Kumar GK, Prabhakar NR. Role of oxidative stress-induced endothelin-converting enzyme activity in the alteration of carotid body function by chronic intermittent hypoxia. Exp Physiol. 2013;98:1620–1630. doi: 10.1113/expphysiol.2013.073700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Iturriaga R. Intermittent hypoxia: endothelin-1 and hypoxic carotid body chemosensory potentiation. Exp Physiol. 2013;98:1550–1551. doi: 10.1113/expphysiol.2013.075820. [DOI] [PubMed] [Google Scholar]
- 39.Gorąca A, Kleniewska P, Skibska B. ET-1 mediates the release of reactive oxygen species and TNF-α in lung tissue by protein kinase C α and β1. Pharmacol Rep. 2016;68:121–126. doi: 10.1016/j.pharep.2015.07.007. [DOI] [PubMed] [Google Scholar]
- 40.Peng YJ, Yuan G, Jacono FJ, Kumar GK, Prabhakar NR. 5-HT evokes sensory long-term facilitation of rodent carotid body via activation of NADPH oxidase. J Physiol. 2006;576:289–295. doi: 10.1113/jphysiol.2006.116020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Peppard PE, Young T, Palta M, Skatrud J. Prospective study of the association between sleep-disordered breathing and hypertension. N Engl J Med. 2000;342:1378–1384. doi: 10.1056/NEJM200005113421901. [DOI] [PubMed] [Google Scholar]
- 42.Eckert DJ, White DP, Jordan AS, Malhotra A, Wellman A. Defining phenotypic causes of obstructive sleep apnea. Identification of novel therapeutic targets. Am J Respir Crit Care Med. 2013;188:996–1004. doi: 10.1164/rccm.201303-0448OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Thomas RJ, Terzano MG, Parrino L, Weiss JW. Obstructive sleep-disordered breathing with a dominant cyclic alternating pattern--a recognizable polysomnographic variant with practical clinical implications. Sleep. 2004;27:229–234. doi: 10.1093/sleep/27.2.229. [DOI] [PubMed] [Google Scholar]
- 44.Gilmartin G, McGeehan B, Vigneault K, Daly RW, Manento M, Weiss JW, Thomas RJ. Treatment of positive airway pressure treatment-associated respiratory instability with enhanced expiratory rebreathing space (EERS) J Clin Sleep Med. 2010;6:529–538. [PMC free article] [PubMed] [Google Scholar]
- 45.Solin P, Roebuck T, Johns DP, Walters EH, Naughton MT. Peripheral and central ventilatory responses in central sleep apnea with and without congestive heart failure. Am J Respir Crit Care Med. 2000;162:2194–2200. doi: 10.1164/ajrccm.162.6.2002024. [DOI] [PubMed] [Google Scholar]
- 46.Lambert JL, Wiens RE. Induced colorimetric method for carbon monoxide. Anal Chem. 1974;46:929–930. doi: 10.1021/ac60343a021. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Effect of IH on H2S and MDA concentrations and CSE protein abundance in rat liver. (A to B) H2S (A) and MDA (B) amounts in liver tissue of rats exposed to room air (C), room air with ROS scavenger (RS; C+RS), IH, or IH treatment with RS (IH+RS) (n = 3 rats for each treatment). (C) Densitometry analysis of CSE protein abundance in liver tissue of rats exposed to room air (C) or IH (n = 3 rats for each treatment) presented as ratio of CSE/α-tubulin (loading control). Data are presented as means± SEM. ***p < 0.001; ns, not significant.
Fig. S2. Effect of IH on HO-2 mRNA levels and protein abundance. (A) The ratio of HO-2 mRNA to 18S rRNA in carotid bodies of rats exposed to room air (C) or IH was determined by RT-qPCR and normalized to C (n = 5 for each treatment). Data are presented as means ±SEM. ns, not significant. (B) Representative immunoblot of HO-2 protein abundance in liver tissue from room air (C) and IH-exposed rats (n = 3 rats for each treatment). α-tubulin abundance was assayed as a loading control.
Fig. S3. Analysis of CSE and HO-2 protein abundance in HEK-293 cells. (A) Representative immunoblots of CSE and HO-2 protein abundance in CSE and HO-2-expressing HEK-293 cells exposed to room air (20% O2), (C), room air with ROS scavenger (C+RS), IH, or IH with RS (IH+RS) (n = 5 independent experiments). (B) Hemin binding affinity (Km) of HO-2 in HEK-293 cells expressing wild-type HO-2 treated with room air (20% O2), (C) or IH (n = 4 experiments for each treatment). Data are presented as means ±SEM. ns, not significant. (C) HO-2 protein abundance in HEK-293 cells expressing either wild-type (WT) or mutant (C265A, C127A, or C282A) HO-2 exposed to room air (20% O2), (C) or IH (n = 4 independent experiments for each treatment). α-tubulin abundance was assayed as a loading control.
Fig. S4 Effect of IH on CO concentration in HO-1-expressing HEK-293 cells. (A) MDA and (B) CO concentrations in HO-1 expressing HEK-293 cells exposed to room air (20% O2), (C) or IH (n = 4 independent experiments for each treatment. Data are presented as means ±SEM. *p < 0.05.
Fig. S5 Analysis of carotid body sensory nerve activity. (A to B) Upper panels show examples of raw action potentials recorded from the carotid sinus nerve of carotid bodies harvested from rats exposed to room air (CON, A) and intermittent hypoxia (IH, B) and their response to acute hypoxia (Hx). (C to D) Lower panels show identification of two “single units” with differing amplitudes in the control (C) and IH (D) treated carotid bodies. (E to F) Analysis of spike frequencies of Unit 1 (E and F) and Unit 2 (Gand H).
Fig. S6 Carotid body response to intermittent hypoxia in heme oxygenase-2 (HO-2) null mice (A) Examples of ex vivo carotid body sensory nerve responses during five 30-sec episodes of AIH (at arrows) and post-AIH for 60 min (sensory LTF; sLTF) from wild-type and HO-2 null mice exposed to room air (WT-C, HO-2 −/− -C) or HO-2 null mice exposed to intermittent hypoxia (HO-2 −/− -IH). (B to D) Average data of pre-AIH baseline activity (B), hypoxia (Hx) response (mean of 5 episodes of AIH; C), and sLTF (averaged over 60 min of post-AIH; D) (n = 6 experiments for each treatment per genotype; 12 single units from 6 carotid bodies for each treatment per genotype. Data are presented as means ± SEM. *p < 0.05; ***p < 0.001; ns, not significant.