

Abstract
Identifying which neuropsychological measures detect early cognitive changes associated with Alzheimer disease (AD) brain pathology would be helpful clinically for the diagnosis of early AD and for the design of clinical trials. We evaluated which neuropsychological measures in our cognitive battery are most strongly associated with cerebrospinal fluid (CSF) biomarkers of AD brain pathology. We studied a large cohort (n=233) of middle- to older-aged community-dwelling individuals (mean age 61 years) who had no clinical symptoms of dementia and underwent baseline CSF collection at baseline. Participants completed a battery of nine neuropsychological measures at baseline and then every one to three years. CSF tau/Aβ42 was associated with baseline performance on 5/9 neuropsychological measures, especially measures of episodic memory, and longitudinal performance on 7/9 neuropsychological measures, especially measures of global cognition. The free recall portion of the Free and Cued Selective Reminding Task (FCSRT-free) detected declining cognition in the high CSF tau/Aβ42 group the earliest, followed by another measure of episodic memory and a sequencing task.
Keywords: Cerebrospinal fluid, preclinical Alzheimer disease, neuropsychological measures, subtle cognitive decline
1. INTRODUCTION
The brain pathology of Alzheimer disease (AD) begins 10–20 years before the onset of slowly progressive cognitive decline that eventually culminates in dementia (Price, et al., 2009, Price and Morris, 1999). Individuals with biomarker evidence of AD brain pathology but no symptoms of dementia are considered to have “preclinical AD” (Fagan, et al., 2007, Morris, et al., 2009,Skoog, et al., 2003,Stomrud, et al., 2007,Vos, et al., 2013). As the disease progresses, individuals with preclinical AD develop subtle cognitive decline that is detectable by sensitive neuropsychological measures (Albert, et al., 2001,Amieva, et al., 2008,Grober, et al., 2008,Twamley, et al., 2006). When cognitive impairment becomes noticeable to individuals and/or their close family or friends but does not yet significantly affect function, mild cognitive impairment (MCI) is diagnosed (Albert, et al., 2011). Progressive cognitive decline eventually causes impairment of daily function and individuals then meet criteria for AD dementia. The boundaries between preclinical AD, subtle cognitive decline, MCI and dementia are not always clear, especially since symptoms range along a continuum (see Fig. 1).
Fig. 1.
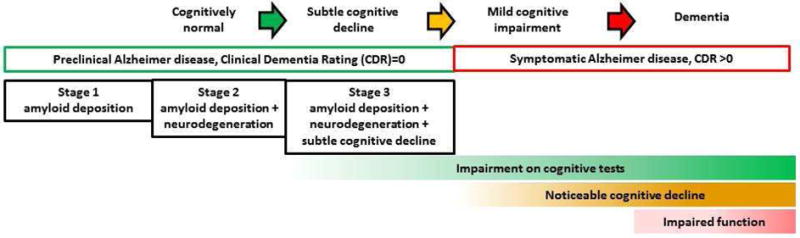
The cognitive spectrum of individuals with Alzheimer disease brain pathology.
The pathological hallmarks of AD are amyloid plaques, comprised primarily of amyloid β-peptide (Aβ), and neurofibrillary tangles, comprised of tau, including phosphorylated tau (ptau). Fluid and imaging biomarkers can identify individuals with significant AD brain pathology. Brain deposition of amyloid is correlated with low levels of cerebrospinal fluid (CSF) Aβ42 and can be imaged with amyloid positron emission tomography (PET). Tau-related neuronal pathology is correlated with high levels of CSF tau and ptau and can also be imaged by PET. CSF Aβ42, tau and ptau, and especially the ratios of CSF tau/Aβ42 and ptau/Aβ42, differentiate between individuals with dementia and cognitively normal controls (Duits, et al., 2014) and predict the future cognitive decline of non-demented adults (Fagan, et al., 2007) and individuals with mild cognitive impairment (MCI) (Brys, et al., 2009,Duits, et al., 2014).
There is great interest in identifying neuropsychological measures that are sensitive to the very earliest cognitive changes caused by AD and that mark the transition from preclinical AD to symptomatic AD (MCI due to AD or early AD dementia). Many studies of neuropsychological measures in AD research have compared the performance of individuals based on diagnosis, e.g. MCI or AD dementia versus cognitively normal. This approach is problematic, because some individuals clinically diagnosed with MCI and AD dementia later are discovered to have non-AD etiologies and cognitively normal controls may have preclinical AD (Hassenstab, et al., 2016). Even at Alzheimer Disease Centers, the sensitivity for clinical AD diagnosis is 71–87% and the specificity is 44–71% when compared against neuropathological diagnosis (Beach, et al., 2012). Additionally, a potential confound may occur when comparing neuropsychological measures by diagnosis because many studies include the same neuropsychological measures in formulating their diagnosis.
The goals of this study were 1) to examine the associations between baseline CSF AD biomarkers and baseline and longitudinal performance on neuropsychological measures in middle aged and older adults without symptoms of dementia at baseline and 2) to identify those neuropsychological measures that are sensitive to the earliest cognitive changes in biomarker-defined preclinical AD. Instead of comparing neuropsychological measures between diagnostic groups, we directly examined the relationship between CSF biomarkers of AD brain pathology and neuropsychological measures. We studied 233 individuals who underwent CSF collection and had a baseline Clinical Dementia Rating (CDR) rating of 0, which indicates no noticeable cognitive decline or impairment of function. Clinicians assigned CDR based on a detailed interview of the participant and their study partner and were unaware of the participant’s performance on the neuropsychological battery or their CSF biomarkers. This approach was utilized to eliminate confounds between diagnosis, performance on the cognitive battery, and CSF biomarkers. Notably, since normal performance on the neuropsychological battery was not required for inclusion, our cohort included individuals with normal cognition and subtle cognitive decline at baseline. We examined the participants’ performance at baseline and over time on nine different neuropsychological measures.
2. METHODS
2.1 Participants, standard protocol approvals and consents
Data were obtained at the Knight Alzheimer Disease Research Center (ADRC) at Washington University in St. Louis from participants in the Adult Children Study (ACS). The ACS is designed to collect longitudinal cognitive and biomarker data on community-dwelling, middle- to older-aged research volunteers. A substantial proportion of ACS participants are the adult children of at least one parent with AD dementia. The Washington University Human Research Protection Office approved the study, and written informed consent was obtained from all participants.
At enrollment, all ACS participants underwent a comprehensive clinical assessment that included semi-structured interviews with the participant and their study partner (typically a spouse, adult child, or close friend) that allowed assignment of a Clinical Dementia Rating (CDR) (Morris, 1993). Approximately two weeks following the clinical assessment, participants completed a baseline neuropsychological battery. Participants then returned every year (for individuals age 65 and older) or every three years (for individuals under age 65) for repeat clinical and cognitive assessments, which included the assignment of a CDR and follow-up testing with the neuropsychological battery. Importantly, clinicians assigning the CDR did not have access to the results of prior clinical assessments, the results of the neuropsychological battery or any biomarker findings.
For the current analyses, we included data from all individals who: 1) had no clinical symptoms of dementia (i.e., were rated CDR 0) at baseline; 2) underwent CSF collection within one year of the baseline neuropsychological battery; and 3) completed at least one follow-up clinical assessment and neuropsychological battery.
2.2 Cognitive measures
The two hour neuropsychological battery was conducted by experienced psychometrists who were unaware of the participant’s CDR or diagnosis. The neuropsychological battery for ACS was designed to capture the early subtle changes in cognition that characterize preclinical AD and the transition from cognitive normality to symptomatic AD. Thus, the battery does not contain measures of confrontation naming, motor skills, or visuospatial ability that typically show little change in the earlier stages of disease. Instead, there is a particular focus on measures of episodic and working memory and executive functioning that are sensitive to early disease. Our episodic memory measures included the picture version of the Free and Cued Selective Reminding Test with immediate recall, free recall portion (FCSRT-free) (Grober, et al., 1988); the Wechsler Memory Scale-III (WMS-III) Logical Memory Immediate and Delayed Recall (logical memory); and Verbal Paired Associates (associate learning) (Wechsler and Stone, 1973). Measures of working memory and executive function included the following: the Letter-Number Sequencing task from the Wechsler Adult Intelligence Scale-III (Wechsler, 1997) (sequencing); the Trailmaking Test, Parts A & B (Armitage, 1946) (trails A and B); and the Animal Naming task (Goodglass and Kaplan, 1972) (category). The Mini-Mental State Examination (MMSE) (Folstein, et al., 1975), a test of global cognitive function, was completed as a part of the clinical assessment. A global cognitive composite average z-score (global cognition) was formed from all cognitive measures using the baseline mean and standard deviation (SD) of all participants. Logical memory and associate learning were only included in the battery for individuals age 65 and older. Sequencing was introduced to the battery later than other measures (in 2005).
2.3 CSF collection, processing and analysis
Participants underwent lumbar puncture (LP) at 8 am following overnight fasting for collection of 20–30 ml of CSF. The CSF was gently inverted to disrupt potential gradient effects, centrifuged at low speed to pellet any cellular debris, aliquoted into polypropylene tubes and stored at −80° C as previously described (Fagan, et al., 2006). Aβ42, total tau, and phopho-tau181 (ptau) were measured by enzyme-linked immunosorbent assay ELISA (INNOTEST, Fujirebio [formerly Innogenetics] Ghent, Belgium).
2.4 Defining a CSF tau/Aβ42 cut-off for AD brain pathology
High amyloid ligand binding by PET is a well-established biomarker for AD brain pathology (Sperling, et al., 2014), but only some of the participants in this study had undergone amyloid PET. Because CSF data was available from all individuals in this study, and the CSF tau/Aβ42 ratio strongly correlates with amyloid PET binding (Fagan, et al., 2009), we determined the CSF tau/Aβ42 ratio that best correlates with a positive amyloid PET scan. Another group recently used a similar approach (Zwan, et al., 2015). We analyzed data from the subset of participants (n=76) who underwent amyloid PET imaging with Pittsburgh Compound B (PiB) (Mintun, et al., 2006) within one year of CSF collection. The area under the curve for the Receiver Operating Characteristic analysis of CSF tau/Aβ42 versus amyloid PET positivity was 0.94, p<0.0001. A CSF tau/Aβ42 ratio of >0.46 had the highest Youden Index and displayed 90% sensitivity and specificity for PET positivity. Similar CSF tau/Aβ42 ratios have been shown to distinguish between cognitively normal individuals and individuals with AD dementia (>0.52) (Duits, et al., 2014) and cognitively normal individuals who declined on cognitive testing (≥0.31) (Steenland, et al., 2014).
2.5 Statistical analyses
Baseline characteristics (shown in Table 1) were compared using t-tests for continuous variables and Chi-square tests for categorical variables. For each individual, the first cognitive assessment that occurred within a ±365-day window of CSF collection was considered the baseline. To allow comparison of different CSF biomarkers and cognitive measures, the biomarkers and cognitive measures were standardized into z-scores based on the sample mean and standard deviation at baseline.
Table 1.
Demographic and biomarker information for study participants.
| All participants | Low CSF Tau/Aβ42 | High CSF Tau/Aβ42 | Cognitively Stable | Progressors | |
|---|---|---|---|---|---|
| n= | 233 | 167 | 66 | 218 | 15 |
| Age (years)a | 60.7 ± 8.4 | 59.1 ± 8.5 | 64.9 ± 6.7**** | 60.2 ± 8.4 | 68.3 ± 5.4*** |
| Sex (% female) | 64% | 62% | 67% | 65% | 40% |
| APOE ε4 (% positive) | 36% | 27% | 61%**** | 36% | 47% |
| Family history (% positive)b | 55% | 56% | 55% | 55% | 60% |
| Education (years)a | 16.1 ± 2.5 | 16.1 ± 2.4 | 16.1 ± 2.8 | 16.1 ± 2.5 | 15.9 ± 2.9 |
| Follow-up (years)a | 5.9 ± 2.5 | 5.9 ± 2.5 | 6.0 ± 2.7 | 5.9 ± 2.5 | 6.6 ± 3.0 |
| Test interval (months)a,c | 2.9 ± 1.9 | 2.8 ± 1.9 | 3.2 ± 2.0 | 2.9 ± 1.9 | 3.4 ± 2.4 |
| CSF Aβ42 (pg/ml)a | 642 ± 226 | 706 ± 200 | 483 ± 212**** | 657 ± 222 | 437 ± 199*** |
| CSF Tau (pg/ml)a | 254 ± 122 | 206 ± 70 | 378 ± 139**** | 247 ± 116 | 369 ± 156**** |
| CSF pTau181 (pg/ml)a | 49 ± 19 | 43 ± 13 | 64 ± 24**** | 48 ± 18 | 66 ± 30*** |
| Logical memoryd | 29 ± 7 (84) | 29 ± 7 (66) | 29 ± 7 (18) | 29 ± 7 (80) | 30 ± 8 (4) |
| FCSRT - freed | 32 ± 6 (227) | 33 ± 5 (163) | 30 ± 6 (64)* | 32 ± 5 (214) | 26 ± 6 (13)*** |
| Global cognitiond | 0.0 ± 0.5 (233) | 0.1 ± 0.5 (167) | −0.1 ± 0.6 (66)* | 0.0 ± 0.5 (218) | −0.4 ± 0.7 (15)* |
| Sequencingd | 11 ± 3 (95) | 11 ± 3 (73) | 10 ± 3 (22) | 11 ± 3 (91) | 12 ± 6 (4) |
| MMSEd | 29 ± 1 (233) | 29 ± 1 (167) | 29 ± 1 (66) | 29 ± 1 (218) | 28 ± 2 (15) |
| Associate learningd | 19 ± 8 (84) | 18 ± 8 (66) | 20 ± 7 (18) | 19 ± 8 (80) | 20 ± 5 (4) |
| Trails Bd | 69 ± 27 (232) | 66 ± 25 (167) | 75 ± 31 (65)* | 67 ± 26 (218) | 99 ± 34 (14)**** |
| Trails Ad | 29 ± 10 (233) | 29 ± 10 (167) | 30 ± 10 (66) | 29 ± 10 (218) | 35 ± 8 (15)* |
| Categoryd | 23 ± 6 (232) | 23 ± 6 (167) | 22 ± 6 (65) | 23 ± 6 (218) | 22 ± 5 (14) |
Statistical comparisons are between low CSF tau/Aβ42 versus high CSF tau/Aβ42 groups or cognitively stable versus progressor groups,
p<0.05,
p<0.01,
p<0.001,
p<0.0001,
a mean ± standard deviation, b Positive family history is having 1 or more parent who developed AD dementia before age 80; negative family history is having both parents live to age 70 or greater without dementia, c Test interval is the time from baseline cognitive battery until CSF collection, d raw measure score mean ± standard deviation (n=).
General linear mixed models, with random intercepts and slopes (Laird and Ware, 1982) at the subject level, were employed to statistically evaluate the relationship between baseline biomarker concentrations and subsequent changes in cognitive test performance over time. On visual inspection of spaghetti plots, there was no apparent non-linearity of temporal trends. Unstructured covariance was used for all models for the random intercept. For modeling any potential covariation of the deviations from each individual’s estimated longitudinal trajectory, a first-order autoregressive process (AR1) and Toeplitz covariance structures were considered for each model. Akaike’s information criterion (Akaike, 1974) was subsequently used to select, among the two candidate structures, the best fitting covariance structure for each model. The best covariance structure varied from model to model. The fixed effects of these models included age at baseline, gender, years of education, presence of at least one APOE ε4 allele, family history of AD, time in years from the baseline cognitive assessment, as well as its two way interactions with all the other factors. The estimates of the intercepts, which captured the association between baseline biomarkers and cognitive measurements at baseline, and the slopes, which captured the association between baseline biomarkers and longitudinal cognitive change, were tested relative to zero with approximate t tests.
Comparisons between the high (> 0.46) and low (≤ 0.46) CSF tau/Aβ42 groups were made through approximate t-tests on appropriate contrasts derived from the fixed effects in the models. Also, predicted longitudinal trajectories based on these models were plotted for the dichotomized groups, with the standard error represented by dotted lines. For illustrative purposes, predicted trajectories represent two hypothetical high and low risk individuals who were women, APOE ε4 negative, lacked a family history and had baseline age and education fixed at the sample averages, as these were the most representative values of the covariates included in the models used to derive the trajectories.
All general linear mixed models were estimated using restricted maximum likelihood estimation, with the approximate F test denominator degrees-of-freedom based on the Satterthwaite method (Satterthwaite, 1941). All statistical analyses were performed using SAS version 9.4 (SAS Institute, Inc., Cary, NC, USA), and statistical significance was defined as p < 0.05. Due to exploratory nature of the study, we did not control for multiple comparisons.
3. RESULTS
3.1 Participants
Participant characteristics are shown in Table 1. The mean clinical follow-up period for all participants was 5.9 ± 2.5 years. Sixty-six (28%) of the 233 individuals in the study had high CSF tau/Aβ42 (> 0.46). They were older (64.9 ± 6.7 versus 59.1 ± 8.5 years, p<0.0001), more likely to carry an APOE ε4 allele (61% versus 27%, p<0.0001), and had worse raw scores (not adjusted for covariates) on three neuropsychological measures at baseline: FCSRT-free, global cognition, and trails B (Table 1).
Most participants (n=218, 94%) did not develop clinical symptoms of dementia (i.e., remained CDR 0) at their most recent clinical assessment and are referred to as cognitively stable. Fifteen participants (6%) developed clinically significant cognitive impairment (CDR>0) and were rated either CDR 0.5 (very mild dementia or MCI) or CDR 1 (mild dementia) at their most recent assessment and are referred to as “progressors.” The average time to progression (CDR>0) was 3.9 ± 2.8 years following baseline assessment. The progressors were older than participants who remained CDR 0 (68.3 ± 5.4 versus 60.2 ± 8.4 years, p<0.001). Twelve of the fifteen progressors (80%) had high CSF tau/Aβ42 at baseline. On their baseline neuropsychological battery, the progressors had worse raw scores (not adjusted for co-variates) on FCSRT-free, trails A, trails B, and the global cognition composite (Table 1).
3.2 High CSF tau/Aβ42 predicts cognitive impairment and cognitive decline on multiple neuropsychological measures
We first examined whether CSF biomarkers, evaluated as continuous variables, were associated with cognitive performance on the baseline neuropsychological battery administered within one year of CSF collection. Compared to other CSF biomarkers (Aβ42, tau, ptau, and ptau/Aβ42), tau/Aβ42 was significantly associated with the greatest number of neuropsychological measures. High CSF tau/Aβ42 was associated with impairment (significantly worse performance) on 5/9 measures at a p<0.05 level of statistical significance, 3/9 measures at p<0.01, and 1/9 measures (logical memory) at p<0.001 (Table 2). At this baseline timepoint, when all participants were rated CDR 0 and AD brain pathology would be exerting its very earliest effects on cognition, episodic memory performance (e.g., on the FCSRT-free and logical memory measures) were the most highly associated with CSF tau/Aβ42.
Table 2. CSF biomarkers predict cognitive performance at baseline.
Numbers listed are the linear mixed model coefficient ± standard error. Each unit indicates a one standard deviation change from the mean. A negative value indicates that a higher CSF biomarker measure is associated with worse performance.
| Aβ42 | Tau | pTau | Tau/Aβ42 | pTau/Aβ42 | |
|---|---|---|---|---|---|
| Logical memory | 0.154 ± 0.07* | −0.084 ± 0.06 | −0.093 ± 0.06 | −0.232 ± 0.07*** | −0.229 ± 0.07*** |
| FCSRT-free | 0.028 ± 0.06 | −0.169 ± 0.06* | −0.116 ± 0.06 | −0.185 ± 0.06** | −0.130 ± 0.06* |
| Global cognition | 0.062 ± 0.03* | −0.019 ± 0.03 | −0.020 ± 0.03 | −0.086 ± 0.03** | −0.077 ± 0.03* |
| Sequencing | 0.232 ± 0.07*** | −0.082 ± 0.07 | 0.117 ± 0.07 | −0.162 ± 0.07* | −0.029 ± 0.07 |
| MMSE | 0.089 ± 0.07 | −0.040 ± 0.06 | 0.015 ± 0.06 | −0.132 ± 0.06* | −0.062 ± 0.07 |
| Associate learning | 0.009 ± 0.06 | 0.034 ± 0.06 | 0.047 ± 0.06 | −0.036 ± 0.06 | −0.003 ± 0.06 |
| Trails B | 0.190 ± 0.06*** | 0.117 ± 0.05* | 0.098 ± 0.05 | −0.035 ± 0.06 | −0.046 ± 0.06 |
| Trails A | −0.008 ± 0.06 | 0.035 ± 0.06 | −0.011 ± 0.06 | 0.033 ± 0.06 | 0.010 ± 0.06 |
| Category | −0.028 ± 0.07 | 0.000 ± 0.07 | −0.082 ± 0.06 | −0.013 ± 0.07 | −0.057 ± 0.07 |
p<0.05 (yellow),
p<0.01 (orange),
p<0.001 (red)
Next, we examined whether baseline CSF biomarkers, evaluated as continuous variables, predicted longitudinal cognitive performance. Again, CSF tau/Aβ42 was significantly associated with more neuropsychological measures than other CSF biomarkers. High CSF tau/Aβ42 was associated with decline (a negative slope that was significantly different from zero) on 7/9 measures at p<0.05, 3/9 measures at p<0.01, and 1/9 measures (MMSE) at p<0.001 (Table 3). During the follow-up period during which fifteen participants progressed to clinically significant cognitive impairment (CDR>0), measures of global cognition (e.g. the MMSE and global cognition composite) were most highly associated with CSF tau/Aβ42.
Table 3. CSF biomarkers predict longitudinal cognitive performance.
Numbers listed are the linear mixed model coefficient ± standard error. Each unit indicates a one standard deviation change from the mean. A negative value indicates that a higher CSF biomarker measure is associated with declining performance over time.
| Aβ42 | Tau | pTau | Tau/Aβ42 | pTau/Aβ42 | |
|---|---|---|---|---|---|
| MMSE | 0.047 ± 0.02* | −0.032 ± 0.02 | −0.019 ± 0.02 | −0.073 ± 0.02*** | −0.064 ± 0.02*** |
| Associate learning | 0.014 ± 0.01 | −0.023 ± 0.01** | −0.033 ± 0.01*** | −0.028 ± 0.01** | −0.032 ± 0.01*** |
| Global cognition | 0.009 ± 0.01 | −0.008 ± 0.004* | −0.008 ± 0.004* | −0.013 ± 0.004** | −0.013 ± 0.004** |
| FCSRT-free | 0.024 ± 0.01* | −0.007 ± 0.01 | −0.011 ± 0.01 | −0.030 ± 0.01* | −0.035 ± 0.01** |
| Category | 0.014 ± 0.01 | −0.013 ± 0.01 | −0.005 ± 0.01 | −0.026 ± 0.01* | −0.019 ± 0.01 |
| Trails B | 0.023 ± 0.01* | −0.010 ± 0.01 | 0.000 ± 0.01 | −0.024 ± 0.01* | −0.017 ± 0.1 |
| Trails A | 0.021 ± 0.01 | −0.006 ± 0.01 | −0.003 ± 0.01 | −0.022 ± 0.01* | −0.022 ± 0.01* |
| Sequencing | −0.015 ± 0.02 | −0.019 ± 0.02 | −0.025 ± 0.02 | −0.021 ± 0.02 | −0.024 ± 0.02 |
| Logical memory | −0.003 ± 0.01 | −0.013 ± 0.01 | −0.018 ± 0.01 | −0.003 ± 0.01 | −0.004 ± 0.01 |
Obs is the total number of observations per measure,
p<0.05 (yellow),
p<0.01 (orange),
p<0.001 (red)
3.3 The temporal sequence in which neuropsychological measures become sensitive to AD brain pathology
We next sought to determine the temporal sequence in which neuropsychological measures become sensitive to AD brain pathology. Individuals were dichotomized into groups with high CSF tau/Aβ42 (>0.46) that is consistent with AD brain pathology or low CSF tau/Aβ42 (≤ 0.46) that is inconsistent with AD brain pathology. The longitudinal cognitive performance for the high and low CSF tau/Aβ42 groups was estimated using general linear mixed models and the standardized difference on each neuropsychological measure was estimated for each time point (years 0 through 6), which corresponds to the average follow-up period of 6 years (Table 4). Using this approach, we found that FCSRT-free was impaired (significantly worse) at baseline in the high CSF tau/Aβ42 group. By one year following baseline assessment, the high CSF tau/Aβ42 group developed significant decline on sequencing and logical memory tasks. By two years following baseline assessment, the high CSF tau/Aβ42 group developed significant decline on tests of global cognition, including the MMSE and the global cognition composite.
Table 4. Temporal sequence of cognitive change.
Longitudinal cognitive performance for the high and low CSF tau/Aβ42 groups was estimated using general linear mixed models and the standardized difference on each neuropsychological measure was calculated for each time point. Each unit indicates a one standard deviation from the mean.
| Baseline | Year 1 | Year 2 | Year 3 | Year 4 | Year 5 | Year 6 | |
|---|---|---|---|---|---|---|---|
| FCSRT-free | −0.336 ± 0.14* | −0.381 ± 0.14** | −0.427 ± 0.14** | −0.472 ± 0.15** | −0.518 ± 0.16** | −0.563 ± 0.17** | −0.608 ± 0.19** |
| Sequencing | −0.301 ± 0.16 | −0.320 ± 0.15* | −0.339 ± 0.14* | −0.358 ± 0.14* | −0.377 ± 0.15* | −0.396 ± 0.17* | −0.415 ± 0.20* |
| Logical memory | −0.288 ± 0.15 | −0.313 ± 0.14* | −0.339 ± 0.14* | −0.364 ± 0.14* | −0.390 ± 0.15** | −0.415 ± 0.16** | −0.441 ± 0.17** |
| MMSE | −0.092 ± 0.14 | −0.226 ± 0.13 | −0.360 ± 0.13** | −0.494 ± 0.14*** | −0.628 ± 0.17*** | −0.761 ± 0.20*** | −0.896 ± 0.23*** |
| Global cognition | −0.087 ± 0.07 | −0.114 ± 0.07 | −0.142 ± 0.07* | −0.169 ± 0.07* | −0.197 ± 0.07** | −0.224 ± 0.08** | −0.252 ± 0.08** |
| Trails B | −0.020 ± 0.13 | −0.058 ± 0.13 | −0.096 ± 0.13 | −0.134 ± 0.13 | −0.172 ± 0.14 | −0.210 ± 0.16 | −0.248 ± 0.17 |
| Associate learning | 0.165 ± 0.14 | 0.101 ± 0.13 | 0.036 ± 0.13 | −0.028 ± 0.13 | −0.093 ± 0.13 | −0.157 ± 0.14 | −0.222 ± 0.15 |
| Category naming | 0.154 ± 0.16 | 0.098 ± 0.15 | 0.042 ± 0.15 | −0.014 ± 0.15 | −0.071 ± 0.15 | −0.127 ± 0.16 | −0.183 ± 0.17 |
| Trails A | 0.093 ± 0.14 | 0.081 ± 0.13 | 0.069 ± 0.13 | 0.057 ± 0.13 | 0.045 ± 0.13 | 0.033 ± 0.14 | 0.021 ± 0.15 |
p<0.05 (yellow),
p<0.01,(orange),
p<0.001(red)
Estimated slopes of cognition for the measures that significantly varied between the high and low CSF tau/Aβ42 groups were plotted (Fig. 2). Participants underwent neuropsychological testing multiple times, which can produce practice effects whereby cognition improves with the number of testing sessions. Cognitively normal individuals with biomarker evidence of AD pathology have been shown to have blunted practice effects on episodic memory (Hassenstab, et al., 2015). The high CSF tau/Aβ42 group (red) exhibited stable performance on FCSRT-free and logical memory over six years, but declining performance on sequencing, global cognition and the MMSE. In contrast, the low CSF tau/Aβ42 group (blue) showed stable performance on the sequencing and global cognition measures and improvement on FCSRT-free, logical memory and the MMSE.
Fig. 2. Estimated longitudinal cognitive performance of individuals with high and low CSF tau/Aβ42.
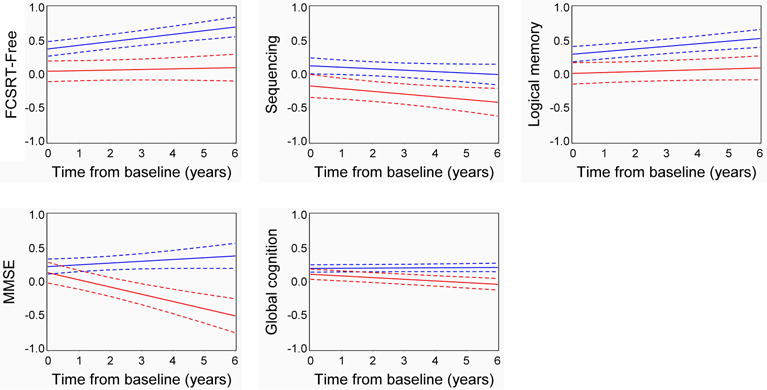
The trajectories represent two hypothetical individuals, one with low CSF tau/Aβ42 (≤0.46, blue) and one with high CSF tau/Aβ42 (>0.46, red), who were women, APOE ε4 negative, lacked a family history of AD and had a baseline age and education fixed at the sample averages. Dashed lines represent the standard error. Each unit indicates one standard deviation from the mean.
3.4 Examining the performance of cognitively stable individuals
We suspected that participants who developed clinically significant cognitive impairment (CDR>0) during follow-up may have driven the associations between baseline CSF biomarkers and longitudinal cognitive performance. To test this hypothesis, we excluded the fifteen progressors and modeled the longitudinal cognitive performance of cognitively stable individuals with high and low CSF tau/Aβ42 (Table 5). There were significant associations with CSF tau/Aβ42 on only three neuropsychological measures in the cognitively stable individuals. The high tau/Aβ42 group performed worse on sequencing, which reached significance at years 1–3 but was only a trend in later years. More interestingly, cognitively stable individuals with high CSF tau/Aβ42 declined on FCSRT-free and the MMSE over time, which became significant at 4 years following CSF collection and persisted through 6 years following CSF collection.
Table 5. Temporal sequence of cognitive change for cognitively stable individuals.
Longitudinal cognitive performance for the high and low CSF tau/Aβ42 groups was estimated using general linear mixed models and the standardized difference on each neuropsychological measure was calculated for each time point. Each unit indicates a one standard deviation from the mean.
| Baseline | Year 1 | Year 2 | Year 3 | Year 4 | Year 5 | Year 6 | |
|---|---|---|---|---|---|---|---|
| FCSRT-free | −0.203 ± 0.15 | −0.223 ± 0.14 | −0.243 ± 0.13 | −0.263 ± 0.13 | −0.283 ± 0.14* | −0.303 ± 0.15* | −0.322 ± 0.16* |
| Sequencing | −0.316 ± 0.16 | −0.307 ± 0.15* | −0.299 ± 0.14* | −0.29 ± 0.15* | −0.281 ± 0.16 | −0.272 ± 0.18 | −0.263 ± 0.20 |
| Logical memory | −0.118 ± 0.15 | −0.132 ± 0.14 | −0.146 ± 0.14 | −0.160 ± 0.14 | −0.173 ± 0.14 | −0.187 ± 0.15 | −0.201 ± 0.16 |
| MMSE | −0.091 ± 0.13 | −0.123 ± 0.12 | −0.155 ± 0.10 | −0.187 ± 0.10 | −0.219 ± 0.09* | −0.252 ± 0.09** | −0.284 ± 0.10** |
| Global cognition | −0.020 ± 0.07 | −0.033 ± 0.07 | −0.047 ± 0.07 | −0.061 ± 0.07 | −0.074 ± 0.07 | −0.088 ± 0.07 | −0.102 ± 0.08 |
| Trails B | 0.050 ± 0.13 | 0.035 ± 0.13 | 0.019 ± 0.13 | 0.004 ± 0.14 | −0.012 ± 0.14 | −0.027 ± 0.15 | −0.043 ± 0.16 |
| Associate learning | 0.197 ± 0.15 | 0.158 ± 0.14 | 0.119 ± 0.13 | 0.080 ± 0.13 | 0.041 ± 0.13 | 0.002 ± 0.13 | −0.037 ± 0.14 |
| Category naming | 0.151 ± 0.17 | 0.120 ± 0.16 | 0.090 ± 0.16 | 0.059 ± 0.16 | 0.029 ± 0.16 | −0.002 ± 0.17 | −0.032 ± 0.18 |
| Trails A | 0.078 ± 0.14 | 0.084 ± 0.14 | 0.090 ± 0.13 | 0.095 ± 0.13 | 0.101 ± 0.13 | 0.107 ± 0.14 | 0.113 ± 0.14 |
p<0.05 (yellow),
p<0.01 (orange)
3.5 The effects of covariates
We examined the fixed effects of covariates in our models. Covariates included age at baseline, gender, years of education, presence of at least one APOE ε4 allele, family history of AD, time from the baseline cognitive assessment, as well as the interaction of time from the baseline cognitive assessment with the other covariates. The model summaries are shown in Supplemental Tables 1–5. For most models of baseline performance on neuropsychological measures as a function of each CSF biomarker (Aβ42, tau, ptau, tau/Aβ42 and ptau/Aβ42), older age was significantly associated with worse performance. This was not true for logical memory and the MMSE—for models of these measures, age did not add additional predictive value as compared to the CSF biomarker alone. In contrast, female gender and more years of education were significantly associated with better performance on nearly all models of baseline neuropsychological measures as a function of CSF biomarker. The covariates had relatively few significant effects on models of neuropsychological measures over time as a function of CSF biomarkers.
Since covariates—especially age, gender, and education—were significantly associated with performance on neuropsychological measures, valid models require inclusion of these covariates. However, to evaluate the robustness of our models, we re-ran models of baseline and longitudinal performance on neuropsychological measures as a function of CSF biomarkers with the covariates excluded (Supplemental Tables 6–7). We found that, in general, the significance of the association between CSF biomarkers and neuropsychological measures increased when covariates were excluded. This is likely because individuals with worse cognitive performance were more likely to have high tau/Aβ42 and were more likely to be older (see Table 1). Ultimately, the most significant associations between CSF tau/Aβ42 and neuropsychological measures were the same in both the covariate adjusted and unadjusted models.
4. DISCUSSION
As expected from prior studies, tau/Aβ42 was the CSF biomarker measure that predicted performance on the largest number of neuropsychological measures, both at baseline and longitudinally (Duits, et al., 2014, Fagan, et al., 2009, Fagan, et al., 2007). This ratio was most strongly associated with episodic memory measures (logical memory and FCSRT-free) at baseline, when AD brain pathology exerts its earliest effects on cognition. An analysis of the temporal sequence in which neuropsychological measures detected early cognitive changes associated with AD brain pathology demonstrated that a measure of episodic memory (FCSRT-free) declined the earliest, followed by another measure of episodic memory (logical memory) and working memory (sequencing). This finding is congruent with reports that multiple memory measures are impaired early in participants who later develop AD dementia (Albert, et al., 2001,Amieva, et al., 2008,Bondi, et al., 2008,Grober, et al., 2008,Grober, et al., 2000,Storandt, et al., 2006) or who are cognitively normal but have biomarkers consistent with AD brain pathology (Racine, et al., 2016).
Over the follow-up period during which fifteen participants developed clinically significant cognitive impairment (CDR>0), CSF tau/Aβ42 was more strongly associated with measures of global cognition (the MMSE and global cognition composite) than episodic memory. This suggests that while the earliest cognitive changes associated with AD brain pathology are in episodic memory, global cognitive measures correlate with clinically significant cognitive impairment. This agrees with previous reports that global deficits follow memory deficits and parallel worsening clinical symptoms of dementia (Amieva, et al., 2008). One recent study that formulated composites incorporating many neuropsychological and functional measures found that the MMSE was a dominant predictor in distinguishing between subjects with and without evidence of AD brain pathology (Insel, et al., 2016). Further studies are needed to examine which questions in the MMSE are driving the association with cognitive impairment.
Our study has some important limitations. The neuropsychological battery did not include some neuropsychological measures and/or domains that are typically impaired in symptomatic AD (Rentz, et al., 2013). Subjects completed all neuropsychological measures included in the cognitive battery at each testing session, so there is no missing data per se. However, not all measures were included in every cognitive battery at every testing session: logical memory and associate learning were only included in the battery for individuals age 65 and older. Additionally, sequencing was introduced to the battery later than other measures (in 2005). Consequently, we have a lower number of individuals represented and less power to detect significant changes in the sequencing, logical memory and associate learning measures. Finally, the CSF collection and baseline neuropsychological battery were not perfectly synchronized—the average interval between CSF collection and the baseline neuropsychological battery was 2.9 months—but this interval was not different between groups (see Table 1).
A major strength of this study is that we directly evaluated the effect of AD brain pathology as indicated by CSF biomarkers on neuropsychological measures. This is in contrast to many other studies where neuropsychological measures and CSF biomarkers were compared between diagnostic groups (controls, MCI, AD dementia) that may not be completely accurate or truly reflective of underlying AD brain pathology. Many other studies have correlated CSF biomarkers to diagnosis (Brys, et al., 2009, Fagan, et al., 2009,Stomrud, et al., 2007), but not to baseline and longitudinal performance on a battery of neuropsychological measures. We included only individuals without clinical symptoms of dementia (CDR 0) at baseline, allowing us to examine the early cognitive changes associated with AD brain pathology. Finally, the large cohort (n=233) and relatively long average period of follow-up (6 years) are strengths of this study.
Stages for preclinical AD have been proposed (Sperling, et al., 2011) and each successive stage denotes greater risk for progression to AD dementia (Knopman, et al., 2012, Vos, et al., 2013). Individuals with biomarker evidence of AD but without symptoms of AD dementia are categorized as follows: abnormal amyloid markers only (stage 1), abnormal amyloid and neuronal injury markers (stage 2) and abnormal amyloid and neuronal injury markers along with subtle cognitive decline (stage 3). What constitutes subtle cognitive decline has not been clearly defined, but it could be considered to be impaired performance on neuropsychological measures in the absence of clinically significant cognitive impairment (CDR 0 based on the participant and study partner interview). Studies that include neuropsychological measures in their assessment of MCI or dementia may be classifying some individuals as MCI, although these individuals and their study partners deny any significant decline in their memory or other cognitive domains. Based on our results, impaired performance on FCSRT-free, logical memory and sequencing may help to define subtle cognitive decline. Future studies could compare FCSRT-free, logical memory, and working memory to other measures and/or define the specific test cut-offs that correspond with higher risk for progression to AD dementia. Cognitive composites have been proposed to determine the presence and severity of subtle cognitive decline (Amariglio, et al., 2015,Donohue, et al., 2014,Insel, et al., 2016). We suggest that FCSRT-free, logical memory, and a sequencing task would be helpful to include in a cognitive composite for subtle cognitive decline.
Supplementary Material
HIGHLIGHTS.
The associations between CSF biomarkers and neuropsychological measures were examined
High CSF tau/Aβ42 was associated with baseline impairment on episodic memory measures
High CSF tau/Aβ42 was associated with decline on global cognitive measures
The Free and Cued Selective Reminding Task detected cognitive decline the earliest
Acknowledgments
FUNDING
This study was supported by National Institute on Aging grant P01AG026276 (JC Morris, PI).
DISCLOSURES
Dr. Schindler has been supported by 5K12 HD001459-15 and is currently supported by UL1 TR00448, Sub-Award KL2 TR000450 and R03AG050921. Dr. Schindler has a family member with stock in Eli Lilly. Mr. Jasielec, Dr. Weng and Dr. McCue report no disclosures. Dr. Hassenstab is supported by a NIH career development award (K23 DK094982) and consults for Biogen. Dr. Grober receives a small royalty for commercial use of the FCSRT with immediate recall. Dr. Morris has served as a consultant for Lilly USA and Takeda Pharmaceuticals. Dr. Morris receives research support from Eli Lilly/Avid Radiopharmaceuticals and is funded by NIH grants P50AG005681, P01AG003991, P01AG026276 and UF01AG032438. Dr. Holtzman is supported by NIH grants P01AG03991, P01AG026276, and a research grant from Eli Lilly. Dr. Holtzman co-founded C2N Diagnostics and is on the scientific advisory boards of Genentech, Denali, AstraZeneca, and Neurophage. Dr. Holtzman consults for AbbVie, Biogen, and Eli Lilly. Dr. Xiong is supported by NIH grants including P50AG005681, P01AG003991, P01AG026276, UF01AG032438, and AG034119 and AG038656. Dr. Fagan is supported by NIH grants including P50AG005681, P01AG003991, P01AG026276 and UF01AG032438. Dr. Fagan is on the Scientific Advisory Boards for Roche, IBL International, and Genentech, and consults for DiamiR and AbbVie.
ABBREVIATIONS
- AD
Alzheimer disease
- CSF
cerebrospinal fluid
- FCSRT-free
free recall portion of the Free and Cued Selective Reminding Task
- Aβ
amyloid β-peptide
- ptau
phosphorylated tau
- PET
positron emission tomography
- MCI
mild cognitive impairment
- CDR
clinical dementia rating
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
AUTHOR CONTRIBUTIONS
Drs. Fagan, Schindler, and Xiong were responsible for study design. Dr. Schindler drafted the manuscript with comments and editing from all co-authors. Mr. Jasielec, Dr. Weng and Dr. McCue provided statistical analyses. Drs. Grober and Hassenstab provided critical review of the manuscript for important intellectual content. Dr. Morris was responsible for collection of clinical data. Dr. Holtzman provided critical review of the manuscript and study design for important intellectual content. Dr. Xiong oversaw the statistical analyses. Dr. Fagan was responsible for acquisition and quality control of the CSF biomarker data. Dr. Xiong had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
References
- Akaike H. A new look at the statistical model identification. IEEE Transactions on Automatic Control. 1974;19:716–23. [Google Scholar]
- Albert MS, DeKosky ST, Dickson D, Dubois B, Feldman HH, Fox NC, Gamst A, Holtzman DM, Jagust WJ, Petersen RC, Snyder PJ, Carrillo MC, Thies B, Phelps CH. The diagnosis of mild cognitive impairment due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s & dementia: the journal of the Alzheimer’s Association. 2011;7(3):270–9. doi: 10.1016/j.jalz.2011.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albert MS, Moss MB, Tanzi R, Jones K. Preclinical prediction of AD using neuropsychological tests. Journal of the International Neuropsychological Society: JINS. 2001;7(5):631–9. doi: 10.1017/s1355617701755105. [DOI] [PubMed] [Google Scholar]
- Amariglio RE, Donohue MC, Marshall GA, Rentz DM, Salmon DP, Ferris SH, Karantzoulis S, Aisen PS, Sperling RA, Alzheimer’s Disease Cooperative, S. Tracking early decline in cognitive function in older individuals at risk for Alzheimer disease dementia: the Alzheimer’s Disease Cooperative Study Cognitive Function Instrument. JAMA neurology. 2015;72(4):446–54. doi: 10.1001/jamaneurol.2014.3375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amieva H, Le Goff M, Millet X, Orgogozo JM, Peres K, Barberger-Gateau P, Jacqmin-Gadda H, Dartigues JF. Prodromal Alzheimer’s disease: successive emergence of the clinical symptoms. Annals of neurology. 2008;64(5):492–8. doi: 10.1002/ana.21509. [DOI] [PubMed] [Google Scholar]
- Armitage SG. An analysis of certain psychological tests used for the evaluation of brain injury. Psychological monographs. 1946;60:i-68. [Google Scholar]
- Beach TG, Monsell SE, Phillips LE, Kukull W. Accuracy of the clinical diagnosis of Alzheimer disease at National Institute on Aging Alzheimer Disease Centers, 2005–2010. Journal of neuropathology and experimental neurology. 2012;71(4):266–73. doi: 10.1097/NEN.0b013e31824b211b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondi MW, Jak AJ, Delano-Wood L, Jacobson MW, Delis DC, Salmon DP. Neuropsychological contributions to the early identification of Alzheimer’s disease. Neuropsychology review. 2008;18(1):73–90. doi: 10.1007/s11065-008-9054-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brys M, Pirraglia E, Rich K, Rolstad S, Mosconi L, Switalski R, Glodzik-Sobanska L, De Santi S, Zinkowski R, Mehta P, Pratico D, Saint Louis LA, Wallin A, Blennow K, de Leon MJ. Prediction and longitudinal study of CSF biomarkers in mild cognitive impairment. Neurobiology of aging. 2009;30(5):682–90. doi: 10.1016/j.neurobiolaging.2007.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donohue MC, Sperling RA, Salmon DP, Rentz DM, Raman R, Thomas RG, Weiner M, Aisen PS, Australian Imaging B, Lifestyle Flagship Study of, A., Alzheimer’s Disease Neuroimaging, I., Alzheimer’s Disease Cooperative, S. The preclinical Alzheimer cognitive composite: measuring amyloid-related decline. JAMA neurology. 2014;71(8):961–70. doi: 10.1001/jamaneurol.2014.803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duits FH, Teunissen CE, Bouwman FH, Visser PJ, Mattsson N, Zetterberg H, Blennow K, Hansson O, Minthon L, Andreasen N, Marcusson J, Wallin A, Rikkert MO, Tsolaki M, Parnetti L, Herukka SK, Hampel H, De Leon MJ, Schroder J, Aarsland D, Blankenstein MA, Scheltens P, van der Flier WM. The cerebrospinal fluid “Alzheimer profile”: easily said, but what does it mean? Alzheimer’s & dementia: the journal of the Alzheimer’s Association. 2014;10(6):713–23 e2. doi: 10.1016/j.jalz.2013.12.023. [DOI] [PubMed] [Google Scholar]
- Fagan AM, Mintun MA, Mach RH, Lee SY, Dence CS, Shah AR, LaRossa GN, Spinner ML, Klunk WE, Mathis CA, DeKosky ST, Morris JC, Holtzman DM. Inverse relation between in vivo amyloid imaging load and cerebrospinal fluid Abeta42 in humans. Annals of neurology. 2006;59(3):512–9. doi: 10.1002/ana.20730. [DOI] [PubMed] [Google Scholar]
- Fagan AM, Mintun MA, Shah AR, Aldea P, Roe CM, Mach RH, Marcus D, Morris JC, Holtzman DM. Cerebrospinal fluid tau and ptau(181) increase with cortical amyloid deposition in cognitively normal individuals: implications for future clinical trials of Alzheimer’s disease. EMBO molecular medicine. 2009;1(8–9):371–80. doi: 10.1002/emmm.200900048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fagan AM, Roe CM, Xiong C, Mintun MA, Morris JC, Holtzman DM. Cerebrospinal fluid tau/beta-amyloid(42) ratio as a prediction of cognitive decline in nondemented older adults. Archives of neurology. 2007;64(3):343–9. doi: 10.1001/archneur.64.3.noc60123. [DOI] [PubMed] [Google Scholar]
- Folstein MF, Folstein SE, McHugh PR. “Mini-mental state”. A practical method for grading the cognitive state of patients for the clinician. Journal of psychiatric research. 1975;12(3):189–98. doi: 10.1016/0022-3956(75)90026-6. [DOI] [PubMed] [Google Scholar]
- Goodglass H, Kaplan E. The assessment of aphasia and related disorders. Lea & Febiger; Philadelphia: 1972. [Google Scholar]
- Grober E, Buschke H, Crystal H, Bang S, Dresner R. Screening for dementia by memory testing. Neurology. 1988;38(6):900–3. doi: 10.1212/wnl.38.6.900. [DOI] [PubMed] [Google Scholar]
- Grober E, Hall CB, Lipton RB, Zonderman AB, Resnick SM, Kawas C. Memory impairment, executive dysfunction, and intellectual decline in preclinical Alzheimer’s disease. Journal of the International Neuropsychological Society: JINS. 2008;14(2):266–78. doi: 10.1017/S1355617708080302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grober E, Lipton RB, Hall C, Crystal H. Memory impairment on free and cued selective reminding predicts dementia. Neurology. 2000;54(4):827–32. doi: 10.1212/wnl.54.4.827. [DOI] [PubMed] [Google Scholar]
- Hassenstab J, Chasse R, Grabow P, Benzinger TL, Fagan AM, Xiong C, Jasielec M, Grant E, Morris JC. Certified normal: Alzheimer’s disease biomarkers and normative estimates of cognitive functioning. Neurobiology of aging. 2016;43:23–33. doi: 10.1016/j.neurobiolaging.2016.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassenstab J, Ruvolo D, Jasielec M, Xiong C, Grant E, Morris JC. Absence of Practice Effects in Preclinical Alzheimer’s Disease. Neuropsychology. 2015 doi: 10.1037/neu0000208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Insel PS, Donohue MC, Mackin RS, Aisen PS, Hansson O, Weiner MW, Mattsson N, Alzheimer’s Disease Neuroimaging, I. Cognitive and functional changes associated with Abeta pathology and the progression to mild cognitive impairment. Neurobiology of aging. 2016;48:172–81. doi: 10.1016/j.neurobiolaging.2016.08.017. [DOI] [PubMed] [Google Scholar]
- Knopman DS, Jack CR, Jr, Wiste HJ, Weigand SD, Vemuri P, Lowe V, Kantarci K, Gunter JL, Senjem ML, Ivnik RJ, Roberts RO, Boeve BF, Petersen RC. Short-term clinical outcomes for stages of NIA-AA preclinical Alzheimer disease. Neurology. 2012;78(20):1576–82. doi: 10.1212/WNL.0b013e3182563bbe. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laird NM, Ware JH. Random-effects models for longitudinal data. Biometrics. 1982;38(4):963–74. [PubMed] [Google Scholar]
- Mintun MA, Larossa GN, Sheline YI, Dence CS, Lee SY, Mach RH, Klunk WE, Mathis CA, DeKosky ST, Morris JC. [11C]PIB in a nondemented population: potential antecedent marker of Alzheimer disease. Neurology. 2006;67(3):446–52. doi: 10.1212/01.wnl.0000228230.26044.a4. [DOI] [PubMed] [Google Scholar]
- Morris JC. The Clinical Dementia Rating (CDR): current version and scoring rules. Neurology. 1993;43(11):2412–4. doi: 10.1212/wnl.43.11.2412-a. [DOI] [PubMed] [Google Scholar]
- Morris JC, Roe CM, Grant EA, Head D, Storandt M, Goate AM, Fagan AM, Holtzman DM, Mintun MA. Pittsburgh compound B imaging and prediction of progression from cognitive normality to symptomatic Alzheimer disease. Archives of neurology. 2009;66(12):1469–75. doi: 10.1001/archneurol.2009.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price JL, McKeel DW, Jr, Buckles VD, Roe CM, Xiong C, Grundman M, Hansen LA, Petersen RC, Parisi JE, Dickson DW, Smith CD, Davis DG, Schmitt FA, Markesbery WR, Kaye J, Kurlan R, Hulette C, Kurland BF, Higdon R, Kukull W, Morris JC. Neuropathology of nondemented aging: presumptive evidence for preclinical Alzheimer disease. Neurobiology of aging. 2009;30(7):1026–36. doi: 10.1016/j.neurobiolaging.2009.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price JL, Morris JC. Tangles and plaques in nondemented aging and “preclinical” Alzheimer’s disease. Annals of neurology. 1999;45(3):358–68. doi: 10.1002/1531-8249(199903)45:3<358::aid-ana12>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- Racine AM, Koscik RL, Berman SE, Nicholas CR, Clark LR, Okonkwo OC, Rowley HA, Asthana S, Bendlin BB, Blennow K, Zetterberg H, Gleason CE, Carlsson CM, Johnson SC. Biomarker clusters are differentially associated with longitudinal cognitive decline in late midlife. Brain: a journal of neurology. 2016;139(Pt 8):2261–74. doi: 10.1093/brain/aww142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rentz DM, Parra Rodriguez MA, Amariglio R, Stern Y, Sperling R, Ferris S. Promising developments in neuropsychological approaches for the detection of preclinical Alzheimer’s disease: a selective review. Alzheimer’s research & therapy. 2013;5(6):58. doi: 10.1186/alzrt222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satterthwaite FE. Synthesis of Variance. Psychometrika. 1941;16(5):309–16. [Google Scholar]
- Skoog I, Davidsson P, Aevarsson O, Vanderstichele H, Vanmechelen E, Blennow K. Cerebrospinal fluid beta-amyloid 42 is reduced before the onset of sporadic dementia: a population-based study in 85-year-olds. Dementia and geriatric cognitive disorders. 2003;15(3):169–76. doi: 10.1159/000068478. doi:68478. [DOI] [PubMed] [Google Scholar]
- Sperling RA, Aisen PS, Beckett LA, Bennett DA, Craft S, Fagan AM, Iwatsubo T, Jack CR, Jr, Kaye J, Montine TJ, Park DC, Reiman EM, Rowe CC, Siemers E, Stern Y, Yaffe K, Carrillo MC, Thies B, Morrison-Bogorad M, Wagster MV, Phelps CH. Toward defining the preclinical stages of Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s & dementia: the journal of the Alzheimer’s Association. 2011;7(3):280–92. doi: 10.1016/j.jalz.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperling RA, Rentz DM, Johnson KA, Karlawish J, Donohue M, Salmon DP, Aisen P. The A4 study: stopping AD before symptoms begin? Science translational medicine. 2014;6(228):228fs13. doi: 10.1126/scitranslmed.3007941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steenland K, Zhao L, Goldstein F, Cellar J, Lah J. Biomarkers for predicting cognitive decline in those with normal cognition. Journal of Alzheimer’s disease: JAD. 2014;40(3):587–94. doi: 10.3233/JAD-2014-131343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stomrud E, Hansson O, Blennow K, Minthon L, Londos E. Cerebrospinal fluid biomarkers predict decline in subjective cognitive function over 3 years in healthy elderly. Dementia and geriatric cognitive disorders. 2007;24(2):118–24. doi: 10.1159/000105017. [DOI] [PubMed] [Google Scholar]
- Storandt M, Grant EA, Miller JP, Morris JC. Longitudinal course and neuropathologic outcomes in original vs revised MCI and in pre-MCI. Neurology. 2006;67(3):467–73. doi: 10.1212/01.wnl.0000228231.26111.6e. [DOI] [PubMed] [Google Scholar]
- Twamley EW, Ropacki SA, Bondi MW. Neuropsychological and neuroimaging changes in preclinical Alzheimer’s disease. Journal of the International Neuropsychological Society: JINS. 2006;12(5):707–35. doi: 10.1017/S1355617706060863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vos SJ, Xiong C, Visser PJ, Jasielec MS, Hassenstab J, Grant EA, Cairns NJ, Morris JC, Holtzman DM, Fagan AM. Preclinical Alzheimer’s disease and its outcome: a longitudinal cohort study. The Lancet Neurology. 2013;12(10):957–65. doi: 10.1016/S1474-4422(13)70194-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wechsler D. Wechsler Adult Intelligence Scale-Third edition (WAIS-III) Harcourt Assessments; San Antonia, TX: 1997. [Google Scholar]
- Wechsler D, Stone C. Manual: Wechsler Memory Scale. Psychological Corporation; New York: 1973. [Google Scholar]
- Zwan MD, Rinne JO, Hasselbalch SG, Nordberg A, Lleo A, Herukka SK, Soininen H, Law I, Bahl JM, Carter SF, Fortea J, Blesa R, Teunissen CE, Bouwman FH, van Berckel BN, Visser PJ. Use of amyloid-PET to determine cutpoints for CSF markers: A multicenter study. Neurology. 2015 doi: 10.1212/WNL.0000000000002081. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.