

Abstract
Drug delivery to the central nervous system (CNS) is greatly limited by the blood-brain barrier (BBB). Physical and biochemical properties of the BBB have rendered treatment of CNS diseases, including those with a hypoxia/reoxygenation (H/R) component, extremely difficult. Targeting endogenous BBB transporters from the ATP-binding cassette (ABC) superfamily (i.e., P-glycoprotein (P-gp)) or from the solute carrier (SLC) family (i.e., organic anion transporting polypeptides (OATPs in humans; Oatps in rodents)) has been suggested as a strategy that can improve delivery of drugs to the brain. With respect to P-gp, direct pharmacological inhibition using small molecules or selective regulation by targeting intracellular signaling pathways has been explored. These approaches have been largely unsuccessful due to toxicity issues and unpredictable pharmacokinetics. Therefore, our laboratory has proposed that optimization of CNS drug delivery, particularly for treatment of diseases with an H/R component, can be achieved by targeting Oatp isoforms at the BBB. As the major drug transporting Oatp isoform, Oatp1a4 has demonstrated blood-to-brain transport of substrate drugs with neuroprotective properties. Furthermore, our laboratory has shown that targeting Oatp1a4 regulation (i.e., TGF-β signaling mediated via the ALK-1 and ALK-5 transmembrane receptors) represents an opportunity to control Oatp1a4 functional expression for the purpose of delivering therapeutics to the CNS. In this review, we will discuss limitations of targeting P-gp-mediated transport activity and the advantages of targeting Oatp-mediated transport. Through this discussion, we will also provide critical information on novel approaches to improve CNS drug delivery by targeting endogenous uptake transporters expressed at the BBB.
Keywords: blood-brain barrier, drug delivery, organic anion transporting polypeptides, P-glycoprotein, transporters
Introduction
Effective treatment of diseases with a hypoxia-reoxygenation (H/R) component (i.e., ischemic stroke, traumatic brain injury, obstructive sleep apnea, cardiac arrest), and CNS diseases in general, requires that drugs permeate the blood-brain barrier (BBB) and attain effective concentrations at discrete molecular targets in the brain. This therapeutic objective is highly dependent upon expression and activity of transporters in brain microvascular endothelial cells. Such endogenous BBB transporters are determinants of drug disposition in the brain and, by extension, CNS drug efficacy (1). Therefore, it is critical to thoroughly study localization, regulation, and functional expression of BBB transport proteins in order to develop novel approaches for CNS drug delivery. To date, considerable research has focused on studying transport mechanisms that limit BBB drug permeation by describing the role of ATP-binding cassette (ABC) transporters (i.e., P-glycoprotein (P-gp)) in restricting CNS drug uptake (2,3). It should be noted that several clinical trials targeting P-gp with small molecule first-generation (i.e., verapamil, cyclosporine A) and second-generation (i.e., valspodar) inhibitors have failed due to inhibitor toxicity and/or enhanced penetration of xenobiotics into non-CNS tissues (4,5). Current research suggests that the clinical problem of achieving effective CNS drug delivery can be overcome by targeting solute carrier (SLC) transporters that are endogenously expressed at the brain microvascular endothelium such as organic anion transporting polypeptides (OATP in humans; Oatp in rodents). At the human BBB, the primary drug transporting OATP isoform is OATP1A2, which has a rodent orthologue designated Oatp1a4 (6). Previous in vivo studies have reported that Oatp1a4 can facilitate blood-to-brain transport of opioid analgesic peptides (7) and atorvastatin, a 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase inhibitor that has neuroprotective properties in the setting of cerebral hypoxia and subsequent reoxygenation stress (8). These preclinical studies demonstrate the immense potential of targeting endogenous BBB uptake transporters to facilitate improved pharmacotherapy of CNS diseases.
Achieving the goal of developing Oatps as transporter targets for enhanced CNS drug delivery requires a thorough understanding of their molecular regulation. Therefore, characterizing intracellular signaling mechanisms that can modulate these transporters is paramount. One such pathway that has shown promise is transforming growth factor (TGF)-β signaling. At the BBB, TGF-β signaling is mediated by two distinct cell surface receptors designated activin receptor-like kinase (ALK)-1 and ALK-5 (9). Studies have previously shown that Oatp1a4 functional expression is regulated by pharmacological inhibition of TGF-β/ALK-5 signaling using the selective small molecule SB431542 (7) or activation of TGF-β/ALK-1 signaling using the established ALK-1 ligand bone morphogenetic protein (BMP)-9 (10). These results highlight the opportunity conferred by targeting TGF-β signaling to control functional expression of Oatp1a4 at the BBB and, ultimately, optimize CNS drug delivery.
In this review, we summarize current knowledge on challenges of targeting P-gp efflux activity and on therapeutic advantages of targeting Oatp influx transport to achieve optimal CNS drug delivery. In particular, we emphasize how identification and characterization of the TGF- β signaling pathway offers a unique opportunity to precisely control blood-to-brain drug transport mediated by Oatp isoforms at the BBB.
Blood-Brain Barrier
Perhaps the most significant obstacle to treatment of CNS diseases, including those with an H/R component, is the BBB. According to the World Health Organization, CNS disorders are the major medical challenge of the twenty-first century (11). The properties of the BBB are such that drug delivery is highly restricted due to reduced transcellular and paracellular transport of solutes. Illustrating this point is a review of greater than 6000 drugs in the Comprehensive Medicinal Chemistry database, which showed that only 6% of all currently marketed drugs are active in the CNS. Moreover, it is estimated that approximately 98% of small molecule therapeutics do not cross the BBB (12). Clearly, restriction of drug penetration to the brain by the BBB is a primary factor that reduces drug effectiveness for the treatment of CNS diseases.
The BBB is both a physical and biochemical barrier that separates the CNS from the systemic circulation (6) and, therefore, promotes neurological protection against potentially toxic xenobiotics and associated metabolites. Structurally, the BBB is comprised of a single layer of brain capillary endothelial cells that are linked by junctional protein complexes (13). Under physiological conditions, these junc-tional protein complexes render the BBB almost impermeable to passive influx of circulating substances with the exception of small, lipid-soluble molecules. The junctional complexes between endothelial cells are made up of (i) junction adhesion molecules (i.e., cadherin proteins) that span the plasma membrane and are linked to the cytoplasm by scaffolding proteins alpha, beta, and gamma catenin (14); and (ii) tight junction proteins (i.e., occludin and claudins), transmembrane proteins anchored to cytoskeletal filaments via accessory proteins known as zonula occluden (ZO) proteins (2). Tight junction protein complexes confer physical barrier properties by creating a high transendothelial resistance (TEER) of approximately 2000 Ωcm2, which is considerably higher than TEER values measured in other vascular tissues (i.e., 3–33 Ωcm2) (15). Additionally, the lack of fenestration, vesicular traffic, and pinocytosis in brain capillary endothelial cells contributes to reduced solute transport between brain interstitium and blood (13). The functional site of the BBB is the brain microvascular endo-thelium; however, enveloping these endothelial cells is the extracellular basement membrane, astrocytes, pericytes, neurons and microglia that comprise the neurovascular unit (NVU) (16). The NVU provides support to the BBB by releasing trophic factors that play a critical role in brain development and maintenance of the BBB phenotype (17,18).
Efflux transporters (i.e., P-gp, Multidrug Resistance Proteins (MRP in humans; Mrp in rodents), Breast Cancer Resistance Protein (BCRP in humans; Bcrp in rodents)) contribute to BBB biochemical properties. These transporters are highly expressed at the luminal membrane of endothelial cells (Fig. 1) and transport a diverse range of lipid-soluble compounds out of the brain capillary endothelium and CNS, thus limiting the ability of these substances to accumulate in brain parenchyma (14). Conversely, SLC transporters permit selective uptake of endogenous and exogenous substances, such as essential nutrients, across the BBB via carrier-mediated transport. An example of carrier-mediated transport is the glucose transporter (i.e., Glut-1) that is expressed both at the luminal and abluminal membranes of endothelial cells and facilitates transport as determined by the transmem-brane solute concentration gradient (19).
Fig. 1.
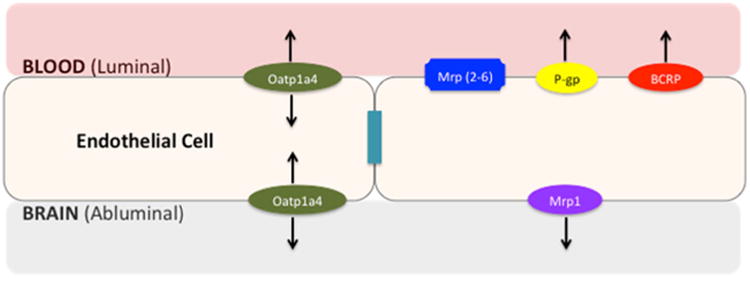
Proposed localization of ABC and SLC transporters at the blood-brain barrier. The blood-brain barrier consists of endothelial cells linked together by tight junction complexes, limiting paracellular diffusion of solutes. Efflux transporters (i.e., P-gp, MRPs/Mrps, BCRP/Bcrp) limit transcellular transport of xenobiotics while SLC uptake transporters (i.e., Oatp1a4) permit selective uptake of solutes into the brain
Overcoming the BBB is no small feat. Many strategies have been employed for the purpose of improving delivery of drugs to the brain. Physical BBB disruption using pharmacological agents and ultrasound has been attempted as a means for CNS drug delivery. While its application in treating CNS tumors has produced encouraging results, disruption of the BBB requires careful management because it permits entry of circulating toxic substances that are otherwise excluded from the CNS (20). Ultimately, to cross the BBB in pharmacologically effective concentrations, drugs must be lipid-soluble, have a molecular weight of less than 400 Da, and not be a substrate for active efflux protein pumps such as P-gp (21). However, chemical modifications made to improve a drug's ability to cross the BBB often results in loss of the desired CNS activity. Additionally, increasing lipophilicity of drugs can result in enhanced efflux transport via P-gp (22). It is now common practice to screen new drugs for P-gp mediated transport due to the well-established role of this transporter in limiting drug penetration into the brain (23).
P-Glycoprotein
The ATP-binding cassette (ABC) superfamily of proteins consists of membrane-bound, ATP-dependent transporters that extrude drugs and metabolites from cells (24). Members of this family include P-gp, MRPs/Mrps, and BCRP/Bcrp. ABC transporters play a significant role in modulating drug uptake, distribution and excretion, thus contributing to a considerable barrier to effective therapeutic delivery of drugs to various organ systems including the brain (2). In humans, 48 ABC genes have been identified and classified into seven subfamilies (25). ABC transporters have a wide range of substrates, from small inorganic and organic molecules, such as amino acids, sugars, and vitamins to large organic compounds such as peptides, lipid molecules, oligonucleo-tides, and polysaccharides (26). As a result, ABC transporters are responsible for conferring the multidrug resistance (MDR) phenotype, which first became evident during treatment of tumors with anticancer drugs (24,27). P-gp primarily transports cationic and neutral compounds while MRP/Mrp substrates are primarily anionic drugs and their glucuronidated, sulfated, and glutathione-conjugated metabolites. The substrate profile for BCRP/Bcrp has considerable overlap with P-gp and includes sulfoconjugated organic anions as well as hydrophobic and amphiphilic compounds (2). The pharmacology of MRP/Mrp isoforms and BCRP/ Bcrp and the role of these transporters at the BBB has been reviewed in detail elsewhere (24,28) and will not be discussed further in this review.
P-gp was first discovered by Dr. Victor Ling at the University of Toronto through the characterization of Chinese hamster ovary cells that were resistant to colchicine (29). Since this initial discovery, P-gp has been determined to be a 170-kDa transmembrane protein that possesses two homologous halves, each with an intracellular ATP-binding site (30). Efflux transport of P-gp substrates requires ATP-hydrolysis and follows a carrier-mediated primary active transport mechanism (31). P-gp can recognize and transport drugs with varying structures and sizes that primarily range from 250 to 1202 g/mol (32). Expression of P-gp has been demonstrated at the BBB in humans and in rodents where many studies have shown luminal membrane localization of P-gp on brain capillary endothelial cells (27). Furthermore, Western blot analysis using isolated rat brain endothelial cells showed strong enrichment of P-gp in brain capillary luminal membrane fractions when compared to brain capillaries and whole membranes, indicating that P-gp is enriched on the luminal membrane of the brain endothelium (30). In another study, magnetic microbeads cross-linked to anti-platelet endothelial cell adhesion molecule (PECAM)-1 antibody were used to isolate endothelial cells from rat brain and P-gp was strongly enriched in the endothelial cell fraction, which suggests a high level of P-gp protein expression in brain capillaries (33).
Changes in P-gp expression have been observed under several pathological conditions. For example, in post-mortem Alzheimer's disease (AD) brain, Aβ deposition was inversely correlated with P-gp expression in brain vasculature as assessed by immunohistochemistry. Additionally, in a mouse model of AD, the lack of P-gp expression exacerbates Aβ deposition and acute inhibition of P-gp increased Aβ levels in brain interstitial fluid within hours of treatment (34). Similarly, there is decreased P-gp activity in Parkinson's disease (PD) patients. Using positron emission tomography (PET), there was a significantly increased uptake of the P-gp substrate, [11C]-verapamil, in PD patients relative to control (35). Conversely, in patients with medically intractable epilepsy, P-gp is overexpressed in capillary endothelial cells and astrocytes. Many antiepileptic drugs (AEDs) are substrates for P-gp, thus contributing to the pharmacoresistance observed in these patients (3). Additionally, there is evidence that P-gp expression is increased in focal cerebral ischemia. Mice subjected to middle cerebral artery occlusion (MCAO), showed elevated P-gp staining, via immunohistochemistry, on endothelial cells in ipsilateral (i.e., ischemic) cerebral cortex tissue (36). Furthermore, Western blot studies in enriched brain microvessel fractions showed increased P-gp levels as early as 3 h and remained high at 24 h, post stroke which greatly reduced accumulation in the brain and efficacy of neuroprotectants (36). In another MCAO study using rats, Pgp expression at the BBB was elevated and resulted in a decreased concentration of P-gp substrates in the ischemic brain, within 4 h post stroke (37). Additionally, it is important to consider that drug treatment itself can directly modulate functional expression of P-gp at the BBB. Nuclear receptors, such as the pregnane-X-receptor (PXR) and constitutive androstane receptor (CAR), are ligand-activated transcription factors that are involved in P-gp regulation in several tissues, including the brain microvasculature (38–42). For example, CAR activation with the known activating ligand acetaminophen increased P-gp expression at the BBB and, subsequently, reduced CNS penetration of morphine (42). Taken together, these studies highlight the importance of understanding how disease and/or pharmacotherapy can modulate P-gp functional expression at the BBB, knowledge that is required to achieve optimal drug delivery to treat CNS disorders.
Modulation of P-gp Transport with Pharmacological Inhibitors
As noted above, previous studies have attempted to circumvent BBB efflux transport by pharmacological inhibition of P-gp using small molecules. However, this strategy is limited by system wide functional expression of this efflux transporter. P-gp is expressed in epithelial cells lining the colon, small intestine, pancreatic ductules, bile ductules, kidney proximal tubules and adrenal gland, as well as endothelial cells of the BBB (32). Since P-gp contributes to extrusion of toxic substances as a means of protecting vital organs, blocking basal P-gp functions will lead to unintended consequences, especially in treatment of chronic diseases. Studies in P-gp knockout mice resulted in an up to 100-fold increase in brain uptake of known P-gp substrate drugs (43). As such, it is recommended that P-gp inhibition should be relied upon only in short-term use to treat diseases such as brain tumors when the goal is to increase drug concentrations for a short period of time (16). Alternative strategies, such as targeting signaling pathways that regulate P-gp expression and/or activity, may be a better approach to improve drug delivery (4,44,45).
Several approaches have been employed to inhibit P-gp transport activity at the BBB. These include (i) blocking the substrate-binding site competitively, non-competitively or allosterically (ii) interfering with ATP hydrolysis (32), and (iii) reducing MDR1 gene mRNA levels (31). The overall concern with these approaches is toxicity and unpredictable pharmacokinetics. For example, first-generation P-gp modulators (i.e., verapamil, cyclosporine A, tamoxifen) have low binding affinities that require use of high doses that were proven to be toxic (31,32,46). In fact, such direct inhibition of P-gp has failed clinically due to multiple patient deaths (46). Second-generation P-gp inhibitors such as valspodar (PSC 833) were developed to possess a higher affinity for P-gp and lower systemic toxicity as compared to first-generation inhibitors; however, they also inhibit CYP3A4 metabolizing enzymes and other ABC transporters, effects that can dramatically increase plasma drug concentrations (32). For example, valspodar inhibits CYP3A4-mediated metabolism of paclitaxel, leading to potentially unsafe serum concentrations (46). As a result, significant considerations have to be made when using P-gp inhibitors because the risk of adverse events may outweigh the potential therapeutic benefits.
A logical approach to improve CNS delivery of drugs would be to target signaling pathways that regulate P-gp expression and/or transport activity. Selective regulation of P-gp at the BBB could promote improved permeability of CNS drugs into brain parenchyma, thus enabling a drug to reach its site of action at therapeutic concentrations (44,45). For example, Cannon and colleagues showed, in vivo, that targeting sphingolipid signaling with fingolimod, an clinically approved drug used in multiple sclerosis treatment, reduced basal P-gp activity and promoted improved CNS drug delivery (44). However, there are significant limitations to this approach because basal P-gp activity is crucial in keeping toxic substances out of the brain. An improved approach might be targeting endogenous SLC uptake transporters, (i.e., Oatps) expressed at the BBB. The potential of Oatps as a CNS drug delivery target has been validated from studies with established Oatp substrate drugs (i.e., statins) where neuroprotection in hypoxia and inflammatory disorders has been demonstrated (47).
Organic Anion Transporting Polypeptides (OATPs/Oatps)
Members of the SLC transporter family play a critical role in organic compound permeation across the BBB, with a broad range of substrate specificity as well as tissue localization. Thus far, 319 SLC genes have been identified that make up 43 families of SLC transporters (2). Of these, members of the SCLO (i.e., OATPs/Oatps) and SLC22 (OCT/Oct; OAT/ Oat) family are expressed at the BBB (48). Reviews of OAT and OCT transporters have been published elsewhere (12,49,50).
The SLCO family in humans and rodents consists of 14 members. Of these, Oatp1a4 (previously designated Oatp2 and a known drug transporting isoform) is expressed in the brain capillaries, liver, intestine, kidney, retina, and choroid plexus (2,50–52). Oatp1a4 was originally isolated from the rat brain with an apparent molecular weight of 92 kDa (48). Western blot analysis in brain capillary fractions has demonstrated expression of Oatp1a4 protein in rat brain (7,8) as well as retinal capillary endothelial cells that make up the blood-retinal barrier (53) while immunofluorescence staining of isolated microvessels from rat brain showed localization to the microvessel lumen (54). Double labeling with anti-Oatp and anti-GFAP immunofluorescence studies detected cellular localization of Oatp1a4 both at the luminal and abluminal membranes of the brain capillary endothelial cells (55). Immunofluorescence staining of human frontal cortex has demonstrated localization of OATP1A2, the human orthologue of Oatp1a4, in brain capillary endothelial cells (56). More recently, using paraffin-embedded sections from human cortex, cerebellum, and hippocampus, localization of OATP1A2 in neurons was discovered (57).
OATP/Oatp-mediated transport is predominantly facilitative or secondary-active. Transport of solutes via OATP/ Oatps is dictated by the electrochemical gradient across the plasma membrane or ion gradients generated by ATP-dependent pumps to transport substrates against the concentration gradient (58). Nevertheless, the energetics of OATP/ Oatp transport has not been fully characterized. For example, rat Oatp1 operates as an anion exchanger that ispartly driven by the glutathione (GSH) electrochemical gradient; however, in Oatp1a4 expressing oocytes, extracellular GSH had minimal effects on taurocholate and digoxin uptake, indicating transport is not directly coupled with GSH (59). Nevertheless, the consensus is that substrate drug in the cytoplasm, which is introduced into the cell via uptake transporters localized on the luminal membrane, reaches a critical threshold and triggers a change in direction of the transmembrane concentration gradient which results in solute efflux via transporters localized at the abluminal membrane (6).
Due to its expression in human brain endothelial cells, OATP1A2 is believed to play a major role in drug delivery and neuroactive peptides to the brain as well as removal of organic metabolites (51). Detailed characterization of Oatp1a4 has also been conducted in Xenopus laevis oocytes where substrates such as thyroxine, taurocholate, digoxin, DPDPE, and estradiol 17β-glucuronide were transported in a saturable manner (60). In another study, transport studies in human embryonic kidney cells expressing mouse Oatp1a4 identified that 3-hydroxy-3-methylglutaryl coenzyme A (HMG CoA) reductase inhibitors (i.e., pitavastatin, rosuvastatin, pravastatin) are Oatp1a4 substrates (61). Additionally, brain-to-blood transport of statins after microinjection into cerebral cortex was significantly reduced in Oatp1a4 (–/–) mice. Similarly, blood-to-brain transport of statins via in situ brain perfusion was significantly decreased in Oatp1a4 (–/–) mice as compared to the wild type controls (61). Oatp1a4-mediated transport was further demonstrated in our laboratory where in vivo experiments showed that CNS uptake of taurocholate and DPDPE were inhibited in the presence of Oatp1a4 inhibitors (i.e., digoxin, fexofenadine) (7). These studies demonstrate that Oatp1a4 can mediate transport of substrate drugs across the BBB, thus revealing Oatp1a4 as a promising target for CNS drug delivery.
TGF-β Signaling and Regulation of Oatp Transport Activity
Advancement of Oatps as transporter targets for optimization of CNS drug delivery requires identification and characterization of regulatory mechanisms that can be exploited to control blood-to-brain transport. Our laboratory has identified the TGF-β signaling pathway as a discrete molecular pathway involved in Oatp regulation at the BBB (7,8). Members of the TGF-β superfamily of signaling ligands, which include BMPs, activins, and TGF-βs, bind to two types of transmembrane serine-threonine kinase receptors known as type I and type II receptors (62,63). BMPs comprise the largest group of ligands within the TGF-β family (64) and play an essential role in cellular processes including differentiation, proliferation, angiogenesis, apoptosis, and bone formation (65,66). In mammals, seven BMP type I receptors, (i.e., ALK 1–7), and three BMP type II receptors (i.e., BMPR-II, ActR-II and ActR-IIB) have been identified (62). In the absence of ligands, type I and type II receptors exist as homodimers on the cell surface, where the latter is a constitutively active kinase (67). Upon activation, type II and type I receptors form a heteromeric complex, where the type II receptor kinase phosphorylates the intracellular GS (glycine-serine) rich region of type I receptor (63), initiating downstream signaling via mediators known as Smads. Smad proteins are divided into three distinct subfamilies. These include receptor-regulated Smads (R-Smads), common-partner Smads (Co-Smads), and inhibitory Smads (I-Smads) (68). The R-Smads are further divided into two groups (i.e., BMP-Smads, TGF-β-Smads). Smad1, Smad5, and Smad8 are phosphorylated by BMP-type receptors such as ALK-1, whereas Smad2 and Smad3 are phosphorylated by TGF-β type receptors such as ALK-5 (67). Structurally, R-Smads have highly conserved globular domains on the N-and C-terminal known as Mad homology (MH) 1 and MH2, respectively, and connected by a highly variable linker region (69) (Fig. 2).
Fig. 2.
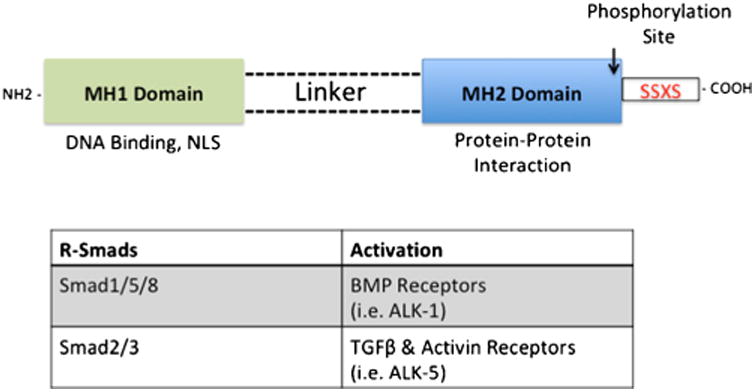
Structure of Smad Proteins, critical molecules involved in TGF-β Signaling. R-Smad consists of two highly conserved globular regions known as MH1 located on the N-terminus and MH2 located on the C-terminus, which are joined together by a variable linker region. Phosphorylation of R-Smads occurs on the SSXS motif located on the C-terminus. The MH1 region contains nuclear localization signal and plays a role in DNA binding, while the MH2 region is involved in protein-protein interaction, which includes dimerization with other Smad proteins and transcription factors
Activation of R-Smads by type I receptors occurs via phosphorylation of the Ser-Ser-X-Ser motif at the MH2 C-terminal (62). In addition to interacting with surface receptors, the MH2 region plays a role in oligomer formation with other Smads as well as DNA-binding proteins, while the MH1 domain is responsible for DNA binding activities and negative regulation of the MH2 domain (70,71). Furthermore, the MH1 region has a nuclear localization signal that directs the activated Smad complex into the nucleus (72), where the MH2 region interacts with transcription co-activators CREB binding protein (CBP) and p300. CBP and p300 have intrinsic acetyltransferase activity, which enables remodeling of the chromatin template (69). The common Smad (i.e., Smad4) further promotes transcription of target genes by stabilizing the interaction between R-Smads and p300/CBP (73).
In summary, TGF- β signaling via Smad proteins is initiated upon ligand binding at the type I/II cell surface receptors, which proceed to form a heterotetrameric complex consisting of two type I and two type II receptors. Subsequently, activated type I receptor intra-cellularly phosphorylates R-Smads at the MH2 region, allowing the formation of a heterotrimeric complex with Smad4 and translocates into the nucleus as guided by the nuclear localization sequence located in the MH1 region. Finally, the active Smad complex binds with DNA at the promoter site and interacts with other transcription factors to regulate gene expression (74,75).
The TGF-β pathway is one of the main components involved in vascular remodeling and angiogenesis mediated via ALK-1 and ALK-5 (76). ALK-1 is restricted to expression in endothelial cells and other highly vascularized tissues (77–79), while ALK5 is broadly expressed in other tissues, including endothelial cells (80). Both type I receptors have distinct and opposing functions where activation of ALK1, via Smad1/5 promotes activation phase of angiogenesis by inducing endothelial cell proliferation and migration, whereas ALK5 activation via Smad2/3 promotes the resolution phase by inhibiting endothelial cell proliferation and migration (80). Studies in human microvessel endothelial cells (HMVEC) showed ALK1 enhanced the formation of tube-like structures whereas ALK5 caused endothelial cell aggregation, suggesting ALK1 is involved in proliferation and migration while ALK5 mediates cell adhesion and extracellular matrix remodeling (81). In another study, pharmacological inhibition of ALK5 using SB43152 induced proliferation and sheet formation of mouse embryonic stem cells (82). Furthermore, microarray analysis performed in human umbilical vein endothelial cells (HUVEC) infected with constitutively active form of ALK1 and ALK5 revealed activation of different target genes. For example, there was significant upregulation of Id1 via ALK1 activation and upregulation of plasminogen activator inhibitor type 1 (PAI-1) (83). Id1 promotes EC proliferation and migration while PAI-1 is a negative regulator of EC migration and angiogenesis (80).
Expression of transporters at the BBB can be altered by stress of pathological conditions. For example, using a well established peripheral inflammatory pain model, rats injected with λ-carrageenan exhibited a higher expression of Oatp1a4 in brain microvessels during acute pain/inflammation, which correlated with increased brain uptake of Oatp1a4 substrates, taurocholate and DPDPE (7). In addition, functional expression of Oatp1a4 was also increased in a model of global cerebral hypoxia. Rats exposed to hypoxia/reoxygenation (H/R) stress showed a significant increase in Oatp1a4 in brain microvessels, which correlated with increased transport of its substrate, (i.e., atorvastatin) (8). These findings suggest that acute peripheral inflammatory pain and H/R stress modulate expression of transporters at the BBB, but more importantly, understanding the signaling and molecular mechanisms involved may be used to precisely regulate expression of transporters to enhance drug delivery. It is paramount to consider that pathological conditions (i.e., peripheral inflammatory pain, H/R stress) can alter tight junction integrity and increase paracellular permeability at the BBB (9,84,85). Such paracellular “leak” can greatly impact drug permeation at the BBB under pathological conditions; however, our laboratory has shown that functional expression of putative BBB membrane transporters can overcome these changes in paracellular permeability for drugs that are known transport substrates. For example, P-gp-mediated transport was observed to predominate over paracellular “leak” of morphine, an established P-gp substrate, under conditions of peripheral inflammatory pain (86). More recently, our group has shown that Oatp-mediated transport was the critical determinant of brain uptake of Oatp substrate drugs (i.e., taurocholate, atorvastatin, DPDPE) in in vivo models of both pain and global cerebral hypoxia (7,8). Taken together, these studies point towards the significant role for BBB transporters in determining CNS drug uptake under pathological conditions.
At the BBB, TGF-β signaling is mediated by ALK1 and ALK5 (9). To that end, our laboratory has shown Oatp1a4 functional expression in rat brain capillary endothelial cells is regulated via TGF-β/ALK5 signaling by in vivo treatment with the ALK5 inhibitor, SB431542 (7,8). Nevertheless, the role of ALK1 in regulation of BBB Oatp1a4 expression has not been elucidated. Awell-characterized agonist for ALK1 is bone morphogenetic protein (BMP)-9, which plays a role in several cellular processes including differentiation, proliferation, angiogenesis, and bone formation (65,66,87). BMP-9 is primarily produced by the liver and is present in the systemic circulation at a concentration of 5 ng/mL, which is above its EC50 (66) and has been shown to physiologically induce constitutive Smad1/5/8 phosphorylation in endothelial cells (88). In another study using human dermal microvascular endothelial cells, Smad1/5/8 activation was demonstrated in a dose-dependent manner with BMP-9. This BMP-induced response was inhibited by siRNA-mediated downregulation of ALK1 and furthermore was abolished with extracellular ALK1 domain (89). In our lab, we demonstrated in vivo treatment with a pharmacological dose of BMP-9, increased Oatp1a4 expression in rat brain microvessels (10). Specificity of the BMP-9 effect on Oatp1a4 expression was determined using LDN193189. LDN193198 is a dorsomorphin derivative that is an established ALK1 antagonist (90). Our experiments with LDN193189 showed that inhibition of the ALK1 receptor attenuates the BMP-9-induced increase in Oatp1a4 expression in rat brain microvessels (10). Taken together, our data implies that TGF-β/ALK1 signaling modulates Oatp1a4 protein expression at the BBB. These findings are critical because it implicates the ALK1 and ALK5 receptors as molecular targets that can be exploited to control drug transport mechanisms via Oatps at the BBB. Targeting Oatp1a4 at the BBB for drug delivery is thus a promising approach as it is a major drug transporter at the BBB (6) (Fig. 3).
Fig. 3.
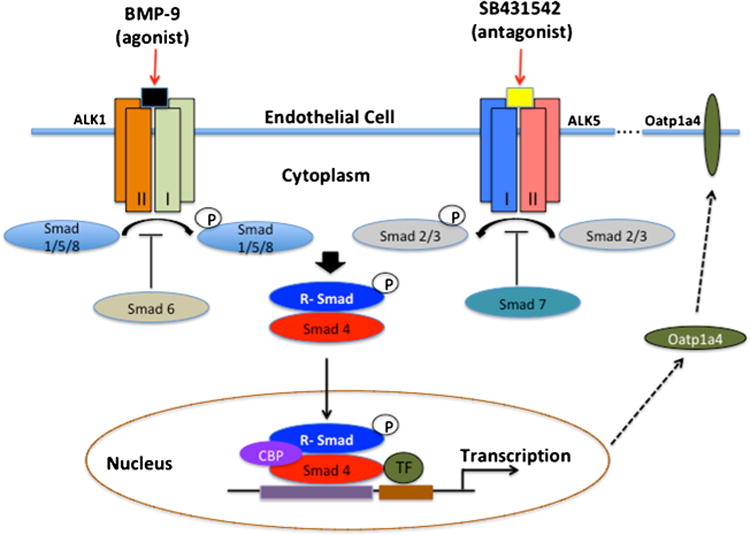
The TGF-β signaling pathway. At the BBB, TGF-β signaling is mediated by two distinct receptors designated ALK-1 and ALK-5. Activation of ALK-1 by binding of BMP-9 triggers phosphorylation of Smads 1, 5, and 8, while activation of ALK-5 via TGF-β, triggers phosphorylation of Smads 2 and 3. Once phosphorylated, these Smad signal-transducing proteins bind to the common Smad (i.e., Smad4) and form a complex that translocate into the nucleus and regulate transcription of target genes
Oatp1a4 is an attractive target for drug delivery due to its substrate profile, which includes HMG-CoA reduc-tase inhibitors (i.e., statins) (61). CNS delivery of statins offers an opportunity to improve treatment of diseases such as ischemic stroke. Clinical studies have shown that statins are associated with early neurological improvement following stroke that is related to the neuroprotective/ antioxidant effects of statins (91,92). Preclinical studies have demonstrated that atorvastatin reduces expression of oxidative stress markers and increases cerebral glutathione concentrations, suggesting reactive oxygen species scavenger activity in the brain (93,94). In an in vivo model of cerebral hypoxia, our lab has demonstrated neuroprotec-tive properties of statins following H/R stress via attenuation of poly (ADP-ribose) polymerase (PARP) cleavage, a biomarker of neuronal apoptosis, in rats dosed with atorvastatin (8).
Conclusion and Future Directions
Transporters are critical components of the BBB and represent discrete molecular components that can be targeted in order to improve CNS drug delivery. In this review, we have described the role of two endogenous BBB transporters (i.e., P-gp, Oatp1a4) in determining CNS uptake of drugs. Targeting P-gp with small molecules has been challenging because first and second P-gp inhibitors have exhibited toxicity issues as well as inhibition of CYP3A4 and other transporters, respectively (30,31,40). Future advances in targeting P-gp for CNS drug delivery are likely to involve targeting of regulatory pathways (95, 96) or transporter trafficking in brain microvascular endothelial cells (45). We propose an alternative approach to optimization of CNS drug delivery via targeting of endogenous SLC uptake transporters expressed at the BBB, such as Oatps. We have shown that Oatp1a4 is a discrete transporter target that can facilitate delivery of drugs with neuroprotective properties (i.e., statins) to the brain and identified members of the TGF-β superfamily as targets for functional regulation of Oatp1a4.
Overall, endogenous transporters at the BBB must be studied in detail in order to discern the optimal time course and the most effective routes of administration for centrally acting drugs. Illustrating this point is the fact that Oatp1a4 is expressed in multiple organs. For example, increased expression of Oatp1a4 in the liver may lead to increased metabolism of substrate drugs, such as statins, thereby limiting CNS penetration and efficacy. Additionally, crosstalk between signaling pathways must be considered when determining outcomes in drug transport across the BBB. For example, activation of ALK5 in brain endothelial cells (BECs) with TGF-β1 increases Abcb1 mRNA, corresponding to increased P-gp function. In the same study, activation of ALK1 with BMP-9 mimicked the TGF-β1 induced upregulation of P-gp function (18). This is especially important for promiscuous drugs that may be substrates for both Oatp1a4 and P-gp, since activation of ALK1 leads to functional upregulation of these transporters with opposing directions in solute transport.
Ultimately, information derived from BBB transport studies can be extended to improve discovery of new therapeutic agents to treat CNS diseases. For example, an examination of the chemical properties of statins that confer selectivity for Oatp-mediated transport can inform structure-based drug design of new chemical entities that are both effective neuroprotectants and good transport substrates for OATP1A2/Oatp1a4. Similar approaches can be employed to limit P-gp-mediated transport to further improve CNS penetration of novel therapeutic agents. Furthermore, a consideration of biological variables (i.e., age, sex) must be incorporated into future experimental design in order to develop an improved translational understanding of BBB transport mechanisms and, ultimately, more effective strategies for pharmacological treatment of CNS diseases.
Acknowledgments
This work was supported by grants from the National Institute of Health (R01-NS084941, R01-NS42652, and R01-DA11271). WA is supported by a pre-doctoral appointment to a National Institutes of Health Training Grant (T32-HL07244).
References
- 1.Qosa H, Mohamed LA, Alqahtani S, Abuasal BS, Hill RA, Kaddoumi A. Transporters as drug targets in neurological diseases. Clin Pharmacol Ther. 2016;100(5):441–53. doi: 10.1002/cpt.435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ronaldson PT, Davis TP. Targeting transporters: promoting blood-brain barrier repair in response to oxidative stress injury. Brain Res. 2015;1623:39–52. doi: 10.1016/j.brainres.2015.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Loscher W, Potschka H. Role of multidrug transporters in pharmacoresistance to antiepileptic drugs. J Pharmacol Exp Ther. 2002;30(1):7–14. doi: 10.1124/jpet.301.1.7. [DOI] [PubMed] [Google Scholar]
- 4.Potschka H. Modulating P-glycoprotein regulation: future perspective for pharmacoresistant epilepsies? Epilepsia. 2010;51(8):1333–47. doi: 10.1111/j.1528-1167.2010.02585.x. [DOI] [PubMed] [Google Scholar]
- 5.Palmeira A, Sousa E, Vasconcelos MH, Pinto MM. Three decades of P-gp inhibitors: skimming though several generations and scaffolds. Curr Med Chem. 2012;19(13):1946–2025. doi: 10.2174/092986712800167392. [DOI] [PubMed] [Google Scholar]
- 6.Ronaldson PT, Davis TP. Targeted drug delivery to treat pain and cerebral hypoxia. Pharmacol Rev. 2013;65:291–314. doi: 10.1124/pr.112.005991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ronaldson PT, Finch JD, DeMarco KM, Quigley CE, Davis TP. Inflammatory pain signals an increase in functional expression of organic anion transporting polypeptide 1a4 at the blood-brain barrier. J Pharm Exp Ther. 2011;336:827–39. doi: 10.1124/jpet.110.174151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Thompson BJ, Sanchez-Covarrubias L, Slosky LM, Zhang Y, Laracuente M, Ronaldson PT. Hypoxia/reoxygenation stress signals an increase in organic anion transporting polypeptide 1a4 (Oatp1a4) at the blood-brain barrier: relevance to CNS drug delivery. J Cereb Blood Flow Metab. 2014;34:699–707. doi: 10.1038/jcbfm.2014.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ronaldson PT, DeMarco KM, Sanchez-Covarrubias L, Solinsky CM, Davis TP. Transforming growth factor-β signaling alters substrate permeability and tight junction protein expression at the blood-brain barrier during inflammatory pain. J Cereb Blood Flow Metab. 2009;29(6):1084–98. doi: 10.1038/jcbfm.2009.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Abdullahi W, Brzica H, Ibbotson K, Davis TP, Ronaldson PT. Bone morphogenetic protein-9 increases functional expression of organic anion transporting polypeptide 1a4 at the blood-brain barrier via the activin receptor-like kinase (ALK)-1 receptor. J Cereb Blood Flow Metab. 2017 doi: 10.1177/0271678X17702916. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mahringer A, Ott M, Reimold I, Reichel V, Fricker G. The ABC of the blood-brain barrier—regulation of drug efflux pumps. Curr Pharm Des. 2011;17:2762–70. doi: 10.2174/138161211797440221. [DOI] [PubMed] [Google Scholar]
- 12.Pardridge WM. Drug transport across the blood-brain barrier. J Cereb Blood Flow Metab. 2012;32:1959–72. doi: 10.1038/jcbfm.2012.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee G, Dalla S, Hong M, Bendayan R. Drug transporters in the central nervous system: brain barriers and brain parenchyma considerations. Pharmacol Rev. 2001;53:569–96. [PubMed] [Google Scholar]
- 14.Abbott NJ, Patabendige AAK, Dolman DEM, Yusof SR, Begley DJ. Structure and function of the blood-brain barrier. Neurobiol Dis. 2010;37:13–25. doi: 10.1016/j.nbd.2009.07.030. [DOI] [PubMed] [Google Scholar]
- 15.Butt AM, Jones HC, Abbott NJ. Electrical resistance across the blood-brain barrier in anaesthetized rats: a developmental study. J Physiol. 1990;429:47–62. doi: 10.1113/jphysiol.1990.sp018243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen Y, Liu L. Modern methods for delivery of drugs across the blood-brain barrier. Adv Drug Deliv Rev. 2012;64:640–65. doi: 10.1016/j.addr.2011.11.010. [DOI] [PubMed] [Google Scholar]
- 17.Abbott NJ, Ronnback L, Hansson E. Astrocyte-endothelial interactions at the blood-brain barrier. Nat Rev Neurosci. 2006;7:41–53. doi: 10.1038/nrn1824. [DOI] [PubMed] [Google Scholar]
- 18.Baello S, Iqbal M, Bloise E, Javam M, Gibb W, Matthews SG. TGF-β1 regulation of multidrug resistance P-glycoprotein in the developing male blood-brain barrier. Endocrinology. 2014;155(2):475–84. doi: 10.1210/en.2013-1472. [DOI] [PubMed] [Google Scholar]
- 19.Sanchez-Covarrubias SLM, Thompson BJ, Davis TP, Ronaldson PT. Transporters at the CNS barrier sites: obstacles or opportunities for drug delivery? Curr Pharm Des. 2014;20(10):1422–49. doi: 10.2174/13816128113199990463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Banks WA. From blood-brain barrier to blood-brain interface: new opportunities for CNS drug delivery. Nat Rev Drug Discov. 2016;15:275–92. doi: 10.1038/nrd.2015.21. [DOI] [PubMed] [Google Scholar]
- 21.Pardridge WM. Molecular Trojan horses for the blood-brain barrier drug delivery. Curr Opin Pharmacol. 2006;6:494–500. doi: 10.1016/j.coph.2006.06.001. [DOI] [PubMed] [Google Scholar]
- 22.Gabathuler R. Approaches to transport therapeutic drugs across the blood-brain barrier to treat brain diseases. Neurobiol Dis. 2010;37:48–57. doi: 10.1016/j.nbd.2009.07.028. [DOI] [PubMed] [Google Scholar]
- 23.Borst P, Schinkel AH. P-glycoprotein ABCB1: a major player in drug handling by mammals. J Clin Invest. 2013;123(10):4131–3. doi: 10.1172/JCI70430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dallas S, Miller DS, Bendayan R. Multidrug resistance-associated proteins: expression and function in the central nervous system. Pharmacol Rev. 2006;58:140–61. doi: 10.1124/pr.58.2.3. [DOI] [PubMed] [Google Scholar]
- 25.Leslie EM, Deeley RG, Cole SPC. Multidrug resistance proteins: role of P-glycoprotein, MRP1, MRP2, and BCRP (ABCG2) in tissue defense. Toxicol Appl Pharmacol. 2005;204:216–37. doi: 10.1016/j.taap.2004.10.012. [DOI] [PubMed] [Google Scholar]
- 26.Wilkens S. Structure and mechanism of ABC transporters. F1000Prime Rep. 2015;7:14. doi: 10.12703/P7-I4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sun H, Dai H, Shaik N, Elmquist WF. Drug efflux transporters in the CNS. Adv Drug Deliv Rev. 2003;55:83–105. doi: 10.1016/s0169-409x(02)00172-2. [DOI] [PubMed] [Google Scholar]
- 28.Doyle LA, Ross DD. Multidrug resistance mediated by the breast cancer resistance protein BCRP (ABCG2) Oncogene. 2003;22:7340–58. doi: 10.1038/sj.onc.1206938. [DOI] [PubMed] [Google Scholar]
- 29.Ling V, Thompson LH. Reduced permeability in CHO cells as a mechanism of resistance to colchicine. J Cell Physiol. 1974;83(1):103–16. doi: 10.1002/jcp.1040830114. [DOI] [PubMed] [Google Scholar]
- 30.Beaulieu E, Demeule M, Ghitescu L, Beliveau R. P-glycoprotein is strongly expressed in the luminal membranes of the endothelium of blood vessels in the brain. Biochem J. 1997;326:539–44. doi: 10.1042/bj3260539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Srivalli KM, Lakshmi PK. Overview of P-glycoprotein inhibitors: a rational look. Braz J Pharm Sci. 2012;48(3):353–67. [Google Scholar]
- 32.Amin L. P-glycoprotein inhibition for the optimal drug delivery. Drug Target Insights. 2013;7:27–34. doi: 10.4137/DTI.S12519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Demeule M, Labelle M, Regina A, Berthelet F, Beliveau R. Isolation of endothelial cells from brain, lung, and kidney: expression of the multidrug resistance P-glycoprotein isoforms. Biochem Biophys Res Commun. 2001;281:827–34. doi: 10.1006/bbrc.2001.4312. [DOI] [PubMed] [Google Scholar]
- 34.Cirrito JR, Deane R, Fagan AM, et al. P-glycoprotein deficiency at the blood-brain barrier increases amyloid-β deposition in an Alzheimer disease mouse model. J Clin Invest. 2005;115:3285–90. doi: 10.1172/JCI25247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kortekaas R, Leenders KL, van Oostrom JCH, et al. Blood-brain barrier dysfunction in Parkinsonian midbrain in vivo. Ann Neurol. 2005;57:176–9. doi: 10.1002/ana.20369. [DOI] [PubMed] [Google Scholar]
- 36.Spudich A, Kilic E, Xing H, et al. Inhibition of multidrug resistance transporter-1 facilitates neuroprotective therapies after focal cerebral ischemia. Nat Neurosci. 2006;9(4):487–8. doi: 10.1038/nn1676. [DOI] [PubMed] [Google Scholar]
- 37.Cen J, Liu L, Li MS, et al. Alteration in P-glycoprotein at the blood-brain barrier in the early period of MCAO in rats. J Pharm Pharmacol. 2013;65:665–72. doi: 10.1111/jphp.12033. [DOI] [PubMed] [Google Scholar]
- 38.Bauer B, Hartz AM, Fricker G, Miller DS. Pregnane X receptor up-regulation of P-glycoprotein expression and transport function at the blood-brain barrier. Mol Pharmacol. 2004;66:413–9. doi: 10.1124/mol.66.3.. [DOI] [PubMed] [Google Scholar]
- 39.Bauer B, Yang X, Hartz AM, et al. In vivo activation of human pregnane X receptor tightens the blood-brain barrier to methadone through P-glycoprotein up-regulation. Mol Pharmacol. 2006;70:1212–9. doi: 10.1124/mol.106.023796. [DOI] [PubMed] [Google Scholar]
- 40.Wang X, Sykes DB, Miller DS. Constitutive androstane receptor-mediated up-regulation of ATP-driven xenobiotic efflux transporters at the blood-brain barrier. Mol Pharmacol. 2010;78:376–83. doi: 10.1124/mol.110.063685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chan GN, Hoque MT, Cummins CL, Bendayan R. Regulation of P-glycoprotein by orphan nuclear receptors in human brain microvessel endothelial cells. J Neurochem. 2011;118:163–75. doi: 10.1111/j.1471-4159.2011.07288.x. [DOI] [PubMed] [Google Scholar]
- 42.Slosky LM, Thompson BJ, Sanchez-Covarrubias L, et al. Acetaminophen modulates P-glycoprotein functional expression at the blood-brain barrier by a constitutive androstane receptor-dependent mechanism. Mol Pharmacol. 2013;84:774–86. doi: 10.1124/mol.113.086298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Loscher W, Potschka H. Blood-brain barrier active efflux transporters: ATP-binding cassette gene family. Neurotherapeutics. 2005;2:86–98. doi: 10.1602/neurorx.2.1.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cannon RE, Peart JC, Hawkins BT, Campos CR, Miller DS. Targeting blood-brain barrier sphingolipid signaling reduces basal P-glycoprotein activity and improves drug delivery to the brain. Proc Natl Acad Sci. 2012;109(39):15930–5. doi: 10.1073/pnas.1203534109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tome ME, Herndon JM, Schaefer CP, et al. P-glycoprotein traffics from the nucleus to the plasma membrane in rat brain endothelium during inflammatory pain. J Cereb Blood Flow Metab. 2016;36(11):1913–28. doi: 10.1177/0271678X16661728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Thomas H, Coley HM. Overcoming multidrug resistance in cancer: an update on the clinical strategy of inhibiting P-glycoprotein. Cancer Control. 2003;10(2):159–65. doi: 10.1177/107327480301000207. [DOI] [PubMed] [Google Scholar]
- 47.Liu H, Yu N, Lu S, et al. Solute carrier of the organic anion-transporting polypeptides 1A2- madin-darby canine kidney II: a promising in vitro system to understand the role of organic anion-transporting polypeptide 1A2 in blood-brain barrier drug penetration. Drug Metab Dispos. 2015;43:1008–18. doi: 10.1124/dmd.115.064170. [DOI] [PubMed] [Google Scholar]
- 48.Kusuhara H, Sugiyama Y. Active efflux across the blood-brain barrier: role of the solute carrier family. Neurotherapeutics. 2005;2:73–85. doi: 10.1602/neurorx.2.1.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Roth M, Obaidat A, Hagenbuch B. OATPs, OATs and OCTs: the organic anion and cation transporters of the SLCO and SLC22A gene superfamilies. Br J Pharmacol. 2012;165:1260–87. doi: 10.1111/j.1476-5381.2011.01724.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Stieger B, Gao B. Drug transporters in the central nervous system. Clin Pharmacokinet. 2015;54:225–42. doi: 10.1007/s40262-015-0241-y. [DOI] [PubMed] [Google Scholar]
- 51.Hagenbuch B, Meier PJ. The superfamily of organic anion transporting polypeptides. Biochim Biophys Acta. 2003;1609:1–18. doi: 10.1016/s0005-2736(02)00633-8. [DOI] [PubMed] [Google Scholar]
- 52.Wang P, Hata S, Xiao Y, Murray JW, Wolkoff AW. Topological assessment of oatp1a1: a 12-transmembrane domain integral membrane protein with three N-linked carbohydrate chains. Am J Physiol Gastrointest Liver Physiol. 2008;294:1052–9. doi: 10.1152/ajpgi.00584.2007. [DOI] [PubMed] [Google Scholar]
- 53.Akunuma S, Hirose S, Tachikawa M, Hosoya K. Localization of organic anion transporting polypeptide (Oatp) 1a4 and Oatp1c1 at the rat blood-retinal barrier. Fluids Barriers CNS. 2013;10(1):29–35. doi: 10.1186/2045-8118-10-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Roberts LM, Black DS, Raman C. Subcellular localization of transporters along the rat blood-brain barrier and blood-cerebral-spinal fluid barrier by in vivo biotinylation. Neuroscience. 2008;155:423–38. doi: 10.1016/j.neuroscience.2008.06.015. [DOI] [PubMed] [Google Scholar]
- 55.Gao B, Stieger B, Noe B, Fritschy JM, Meier PJ. Localization of the organic anion transporting polypeptide 2 (Oatp2) in capillary endothelium and choroid plexus epithelium of rat brain. J Histochem Cytochem. 1999;47(10):1255–63. doi: 10.1177/002215549904701005. [DOI] [PubMed] [Google Scholar]
- 56.Gao B, Hagenbuch B, Kullak-Ublick GA, Benke D, Aguzzi A, Meier PJ. Organic anion-transporting polypeptides mediate transport of opiod peptides across the blood-brain barrier. J Pharm Exp Ther. 2000;294:73–9. [PubMed] [Google Scholar]
- 57.Gao B, Vavricka SR, Meier PJ. Differential cellular expression of organic anion transporting peptides OATP1A2 and OATP2B1 in the human retina and brain: implications for carrier-mediated transport of neuropeptides and nerurosteroids in the CNS. Eur J Phys. 2015;467:1481–93. doi: 10.1007/s00424-014-1596-x. [DOI] [PubMed] [Google Scholar]
- 58.Lin L, Yee SW, Kim RB, Giacomini KM. SLC transporters as therapeutic targets: emerging opportunities. Nat Rev Drug Discov. 2015;14:543–60. doi: 10.1038/nrd4626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Li L, Meier PJ, Ballatori N. Oatp2 mediates bidirectional organic solute transport: a role for intracellular glutathione. Mol Pharmacol. 2000;58:335–40. doi: 10.1124/mol.58.2.335. [DOI] [PubMed] [Google Scholar]
- 60.Kakyo M, Sakagami H, Nishio T. Immunohistochemical distribution and functional characterization of an organic anion transporting polypeptide 2 (oatp2) FEBS let. 1999;445:343–6. doi: 10.1016/s0014-5793(99)00152-0. [DOI] [PubMed] [Google Scholar]
- 61.Ose A, Kusuhara H, Endo C, et al. Functional characterization of mouse organic anion transporting peptide 1a4 in the uptake and efflux of drugs across the blood-brain barrier. Drug Metab Dispos. 2010;38:168–76. doi: 10.1124/dmd.109.029454. [DOI] [PubMed] [Google Scholar]
- 62.Miyazono K, Kamiya Y, Morikawa M. Bone morphogenetic protein receptors and signal transduction. J Biochem. 2010;147(1):35–51. doi: 10.1093/jb/mvp148. [DOI] [PubMed] [Google Scholar]
- 63.Derynck R, Zhang YE. Smad-dependent and Smadindependent pathways in TGF-β family signaling. Nature. 2003;425(6958):577–84. doi: 10.1038/nature02006. [DOI] [PubMed] [Google Scholar]
- 64.Massague J, Chen YG. Controlling TGF-β signaling. Genes Dev. 2000;14:627–44. [PubMed] [Google Scholar]
- 65.Brown MA, Zhao Q, Baker KA, et al. Crystal structure of BMP-9 and functional interactions with pro-region and receptors. J Biol Chem. 2005;280(26):25111–8. doi: 10.1074/jbc.M503328200. [DOI] [PubMed] [Google Scholar]
- 66.Herrera B, Dooley S, Breitkopf-Heinlein K. Potential roles of bone morphogenetic (BMP)-9 in human liver diseases. Int J Mol Sci. 2014;15:5199–220. doi: 10.3390/ijms15045199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Itoh S, Itoh F, Goumans MJ, ten Dijke P. Signaling of transforming growth factor-β family members through Smad proteins. Eur J Biochem. 2000;267:6954–696. doi: 10.1046/j.1432-1327.2000.01828.x. [DOI] [PubMed] [Google Scholar]
- 68.Moustakas A, Souchelnytskyi S, Heldin CH. Smad regulation in TGF-β signal transduction. J Cell Sci. 2001;114:4359–69. doi: 10.1242/jcs.114.24.4359. [DOI] [PubMed] [Google Scholar]
- 69.Pouponnot C, Jayaraman L, Massague J. Physical and functional interaction of SMADs and p300/CBP. J Biol Chem. 1999;273:22865–8. doi: 10.1074/jbc.273.36.22865. [DOI] [PubMed] [Google Scholar]
- 70.Shi Y, Hata A, Lo RS, Massague J, Pavletich NP. A structural basis for mutational inactivation of the tumour suppressor Smad 4. Nature. 1997;388:87–93. doi: 10.1038/40431. [DOI] [PubMed] [Google Scholar]
- 71.Shi Y, Wang YF, Jayaraman L, Yang H, Massague J, Pavletich NP. Crystal structure of Smad MH1 domain bound to DNA: Insights on DNA binding in TGF-β signaling. Cell. 1998;94:585–94. doi: 10.1016/s0092-8674(00)81600-1. [DOI] [PubMed] [Google Scholar]
- 72.Xiao Z, Liu X, Henis YI, Lodish HF. A distinct nuclear localization signal in the N terminus of Smad 3 determines its ligand-induced nuclear translocation. Proc Natl Acad Sci. 2000;97(14):7853–8. doi: 10.1073/pnas.97.14.7853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lee PSW, Chang C, Liu D, Derynck R. Sumoylation of Smad4, the common Smad mediator of transforming growth factor-β family signaling. J Biol Chem. 2003;278(30):27853–63. doi: 10.1074/jbc.M301755200. [DOI] [PubMed] [Google Scholar]
- 74.Suzuki Y, Ohga N, Morishita Y, Hida K, Miyazono K, Watabe T. BMP-9 induces proliferation of multiple types of endothelial cells in vitro and in vivo. J Cell Sci. 2010;123:1684–92. doi: 10.1242/jcs.061556. [DOI] [PubMed] [Google Scholar]
- 75.Verrecchia F, Mauviel A. Transforming growth factor-β signaling through the Smad pathway: role in extracellular matrix gene expression and regulation. J Invest Dermatol. 2002;118(2):211–5. doi: 10.1046/j.1523-1747.2002.01641.x. [DOI] [PubMed] [Google Scholar]
- 76.Oh SP, Seki T, Goss KA, et al. Activin receptor-like kinase 1 modulates transforming growth factor-β1 signaling in the regulation of angiogenesis. Proc Natl Acad Sci. 2000;97(6):2626–31. doi: 10.1073/pnas.97.6.2626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Johnson DW, Berg JN, Baldwin MA, et al. Mutations in the activin receptor-like kinase 1 gene in hereditary haemorrhagic telangiectasia type 2. Nat Genet. 1996;13:189–95. doi: 10.1038/ng0696-189. [DOI] [PubMed] [Google Scholar]
- 78.Wei Z, Salmon RM, Upton PD, Morrell NW, Li W. Regulation of bone morphogenetic protein 9 (BMP9) by redox-dependent proteolysis. J Biol Chem. 2014;289(45):31150–9. doi: 10.1074/jbc.M114.579771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Li W, Salmon RM, Jiang H, Morrell NW. Regulation of ALK1 ligands, BMP9 and BMP10. Biochem Soc Trans. 2016;44:1135–41. doi: 10.1042/BST20160083. [DOI] [PubMed] [Google Scholar]
- 80.Lebrin F, Deckers M, Bertolino P, ten Dijke P. TGF-β receptor function in the endothelium. Cardiovasc Res. 2005;65:599–608. doi: 10.1016/j.cardiores.2004.10.036. [DOI] [PubMed] [Google Scholar]
- 81.Wu X, Ma J, Han JD, Wang N, Chen YG. Distinct regulation of gene expression in human endothelial cells by TGF-β and its receptors. Microvasc Res. 2006;71:12–9. doi: 10.1016/j.mvr.2005.11.004. [DOI] [PubMed] [Google Scholar]
- 82.Watabe T, Nishihara A, Mishima K, et al. TGF-β receptor kinase inhibitor enhances growth and integrity of embryonic stem cell-derived endothelial cells. J Cell Biol. 2003;163(6):1303–11. doi: 10.1083/jcb.200305147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ota T, Fujii M, Sugizaki T, et al. Targets of transcriptional regulation by two distinct type I receptors for transforming growth factor-β in human umbilical vein endothelial cells. J Cell Physio. 2002;193:299–318. doi: 10.1002/jcp.10170. [DOI] [PubMed] [Google Scholar]
- 84.Witt KA, Mark KS, Hom S, Davis TP. Effects of hypoxia-reoxygenation on rat blood-brain barrier permeability and tight junctional protein expression. Am J Physiol Heart Circ Physiol. 2003;285:H2820–31. doi: 10.1152/ajpheart.00589.2003. [DOI] [PubMed] [Google Scholar]
- 85.Witt KA, Mark KS, Sandoval KE, Davis TP. Reoxygenation stress on blood-brain barrier paracellular permeability and edema in the rat. Microvasc Res. 2008;75:91–6. doi: 10.1016/j.mvr.2007.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Seelbach MJ, Brooks TA, Egleton RD, Davis TP. Peripheral inflammatory hyperalgesia modulates morphine delivery to the brain: a role for P-glycoprotein. J Neurochem. 2007;102:1677–90. doi: 10.1111/j.1471-4159.2007.04644.x. [DOI] [PubMed] [Google Scholar]
- 87.Sieber C, Kopf J, Hiepen C, Knaus P. Recent advances in BMP receptor signaling. Cytokine Growth Factor Rev. 2009;20:343–55. doi: 10.1016/j.cytogfr.2009.10.007. [DOI] [PubMed] [Google Scholar]
- 88.Bidart M, Ricard N, Levet S, et al. BMP9 is produced by hepactocytes and circulates mainly in an active mature form complexed to its prodomain. Cell Mol Life Sci. 2012;69:313–24. doi: 10.1007/s00018-011-0751-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.David L, Mallet C, Mazerbourg S, Feige JJ, Bailly S. Identification of BMP9 as functional activators of the orphan activin receptor-like kinase 1 (ALK1) in endothelial cells. Blood. 2007;109(5):1953–61. doi: 10.1182/blood-2006-07-034124. [DOI] [PubMed] [Google Scholar]
- 90.Mayeur C, Kolodziej SA, Wang A, et al. Oral administration of a bone morphogenetic protein type I receptor inhibitor prevents the development of anemia of inflammation. Haematologica. 2015;100(2):68–71. doi: 10.3324/haematol.2014.111484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Montecucco F, Quercioli A, Mirabelli-Badenier M, Viviani GL, Mach F. Statins in the treatment of acute ischemic stroke. Curr Pharm Biotechnol. 2012;13:68–76. doi: 10.2174/138920112798868737. [DOI] [PubMed] [Google Scholar]
- 92.Sutherland BA, Minnerup J, Balami JS, Arba F, Buchan AM, Kleinschnitz C. Neuroprotection for ischaemic stroke: translation from the bench to the bedside. Int J Stroke. 2012;7:407–18. doi: 10.1111/j.1747-4949.2012.00770.x. [DOI] [PubMed] [Google Scholar]
- 93.Butterfield DA, Barone E, Di Domenico F. Atorvastatin treatment in a dog preclinical model of Alzheimer's disease leads to up-regulation of haem oxygenase-1 and is associated with reduced oxidative stress in brain. Int J Neuropsychopharmacol. 2012;15:981–7. doi: 10.1017/S1461145711001118. [DOI] [PubMed] [Google Scholar]
- 94.Barone E, Mancuso C, Di Domenico F, et al. Biliverdin reductase–A: a novel drug target for atorvastatin in a dog pre-clinical model of Alzheimer disease. J Neurochem. 2012;120:135–46. doi: 10.1111/j.1471-4159.2011.07538.x. [DOI] [PubMed] [Google Scholar]
- 95.Amin ML. P-glycoprotein inhibition for optimal drug delivery. Drug Target Insights. 2013;7:27–34. doi: 10.4137/DTI.S12519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Chan GN, Evans RA, Banks DB, et al. Selective induction of P-glycoprotein at the CNS barriers during a symptomatic stage of an ALS animal model. Neurosci Lett. 2016;639:103–13. doi: 10.1016/j.neulet.2016.12.049. [DOI] [PMC free article] [PubMed] [Google Scholar]